

Significance
Superantigen toxins were defined over 25 years ago for their ability to activate T cells in a T-cell receptor β-chain variable domain-dependent manner. This “Vβ-specific” T-cell activation is the hallmark feature of the superantigen, and although these toxins can mediate dangerous human disease such as toxic shock syndrome, mechanisms that explain why bacteria produce superantigens have remained enigmatic. Herein, we provide evidence that Streptococcus pyogenes utilizes superantigens to target functional, Vβ-specific T cells to promote a state of colonization providing a mechanism that helps explain why bacteria produce toxins that specifically activate T cells of the adaptive immune system. This work also implicates the superantigen exotoxins as potential vaccine candidates against this globally important, human-specific pathogen.
Keywords: superantigen, Streptococcus pyogenes, T cells, infection, nasopharynx
Abstract
The globally prominent pathogen Streptococcus pyogenes secretes potent immunomodulatory proteins known as superantigens (SAgs), which engage lateral surfaces of major histocompatibility class II molecules and T-cell receptor (TCR) β-chain variable domains (Vβs). These interactions result in the activation of numerous Vβ-specific T cells, which is the defining activity of a SAg. Although streptococcal SAgs are known virulence factors in scarlet fever and toxic shock syndrome, mechanisms by how SAgs contribute to the life cycle of S. pyogenes remain poorly understood. Herein, we demonstrate that passive immunization against the Vβ8-targeting SAg streptococcal pyrogenic exotoxin A (SpeA), or active immunization with either wild-type or a nonfunctional SpeA mutant, protects mice from nasopharyngeal infection; however, only passive immunization, or vaccination with inactive SpeA, resulted in high-titer SpeA-specific antibodies in vivo. Mice vaccinated with wild-type SpeA rendered Vβ8+ T cells poorly responsive, which prevented infection. This phenotype was reproduced with staphylococcal enterotoxin B, a heterologous SAg that also targets Vβ8+ T cells, and rendered mice resistant to infection. Furthermore, antibody-mediated depletion of T cells prevented nasopharyngeal infection by S. pyogenes, but not by Streptococcus pneumoniae, a bacterium that does not produce SAgs. Remarkably, these observations suggest that S. pyogenes uses SAgs to manipulate Vβ-specific T cells to establish nasopharyngeal infection.
The globally prominent bacterial pathogen Streptococcus pyogenes (also commonly referred to as the group A Streptococcus) exists primarily as a colonizer within the human upper respiratory tract and skin, but is also capable of causing some of the most aggressive and invasive infections known. Indeed, up to 12% of some adolescent populations may be colonized asymptomatically by S. pyogenes (1); yet, this pathogen remains responsible for over 700 million superficial infections, and at least 500,000 deaths, primarily due to invasive infections and acquired autoimmune manifestations in resource-poor settings (2). Despite this enormous impact on human populations, there are currently no vaccines available against this pathogen (3).
S. pyogenes encodes an impressive repertoire of virulence factors that primarily function to disrupt multiple facets of the host innate immune response (4). However, one family of toxins secreted by this organism, known as superantigens (SAgs) (5), function to specifically target and activate both CD4+ and CD8+ T cells of the adaptive immune system (6). SAgs function by bridging lateral surfaces of the MHC class II (MHC-II) molecule on antigen-presenting cells with the T-cell receptor (TCR) on T cells, in a TCR variable β-chain (Vβ)-dependent manner. Indeed, Vβ-specific T-cell activation is the defining feature of the SAg (7) and these unconventional interactions explain how SAgs can activate such a large percentage of the total T-cell population (8). In rare cases, systemic T-cell activation by SAgs can lead to the streptococcal toxic shock syndrome (9), which in the context of invasive streptococcal disease is extremely dangerous, with a mortality rate of over 30% (10).
The role of SAgs in severe human infections has been well established (5, 11, 12), and specific MHC-II haplotypes are known risk factors for the development of invasive streptococcal disease (13), an outcome that has been directly linked to SAgs (14, 15). However, how these exotoxins contribute to superficial disease and colonization is less clear. Using experimental murine models established to mimic acute nasopharyngeal infection (16), the expression of HLAs and that of a specific SAg [i.e., streptococcal pyrogenic exotoxin A (SpeA)], were absolutely required for productive infection (17). As the upper respiratory tract is a major niche for S. pyogenes (18), this provided one explanation as to why this pathogen produces SAgs. Immunization with an MHC-II binding site mutant of SpeA also provided initial evidence that anti-SAg antibodies could mediate protection from nasopharyngeal infection (17).
Herein, we provide evidence that passive immunization, or vaccination with a further-attenuated SpeA toxoid, affords antibody-mediated protection in a murine model of S. pyogenes nasopharyngeal infection. Furthermore, our vaccination experiments also uncovered an antibody-independent protection phenotype whereby vaccination with fully functional SAg induced Vβ-specific T-cell unresponsiveness. Remarkably, T cells were required for efficient S. pyogenes infection. Productive infection resulted in a T-cell–dependent proinflammatory cytokine microenvironment, which may be beneficial to S. pyogenes, although T-cell depletion did not impact the upper respiratory tract bacterial burden of a non-SAg secreting organism, Streptococcus pneumoniae. This work supports the use of toxoid SAgs as potential vaccine candidates against S. pyogenes nasopharyngeal infection and indicates that SAgs specifically target and manipulate Vβ-specific T-cell subsets to promote the initiation of infection.
Results
Passive Immunization with SAg-Neutralizing Antibodies Protects Mice from S. pyogenes Nasopharyngeal Infection.
The human upper respiratory tract represents the major ecological niche for many strains of S. pyogenes (18), and intranasal inoculation of mice has been used to model this environment (16, 19). Previously, we demonstrated that mouse expression of HLA class II molecules (referred to as B6HLA mice), and S. pyogenes MGAS8232 expression of SpeA, were critical host and bacterial factors, respectively, that enhanced nasopharyngeal infection by up to four orders of magnitude (17). It was also demonstrated that vaccination of these mice with a SpeA MHC-II binding mutant (SpeAY100A) was protective during nasopharyngeal challenge with S. pyogenes MGAS8232, a phenotype that was linked to anti-SpeA antibodies (17).
To confirm the protective nature of the anti-SAg humoral response, we passively immunized B6HLA mice with antiserum prepared in rabbits that had been vaccinated with SpeA (Fig. 1A). As a control, we passively immunized B6HLA mice with anti-SpeC rabbit serum since deletion of speC from S. pyogenes MGAS8232 had no measurable impact on nasopharyngeal infection (17). Following treatment with anti-SpeA serum, quantitating bacterial colony-forming units (cfus) from the complete nasal turbinates (cNTs) demonstrated a dramatic reduction in bacterial burden compared with the control anti-SpeC serum group (Fig. 1B). Furthermore, Western blot analysis demonstrated that the anti-SAg sera were specific for their intended toxin (Fig. 1C), and SAg-specific antibodies were recovered from the serum of treated mice as determined by ELISA (Fig. 1 D and E). These data indicate that humoral immunity against specific SAgs can be protective during experimental S. pyogenes nasopharyngeal infection.
Fig. 1.

Passive immunization with anti-SpeA serum reduces the burden of S. pyogenes in the nasopharynx. (A) Passive immunization schedule. (B) Nasal challenge of B6HLA mice with ∼108 cfus of S. pyogenes MGAS8232 after passive immunization with rabbit anti-SpeA (red) or anti-SpeC (green) serum. Data points represent cfus from the complete nasal turbinates (cNTs) of individual mice at 48 h. Horizontal bars represent the geometric mean. The horizontal dotted line indicates the theoretical limit of detection. (C) Recombinant SAg (SDS/PAGE; Top) and Western blot experiments (Bottom two panels) to demonstrate specificity of rabbit polyclonal immune serum to specific SAg proteins. (D and E) Serum IgG antibody titers determined using ELISA from B6HLA mice passively immunized with indicated treatment (anti-SpeA, red; anti-SpeC, green). Bars represent the mean ± SEM. Significance was determined by unpaired Student’s t test (*P < 0.05; **P < 0.01).
Active Vaccination with Wild-Type or Toxoid SAg Reduces S. pyogenes Nasopharyngeal Infection.
Our previous experiments demonstrated that SpeAY100A could elicit protection when used as a vaccine; however, this SpeA mutant still maintained residual superantigenic activity in vitro at high concentrations (i.e., 1 µg mL−1) (17). We therefore desired to generate a fully inactive SpeA toxoid. Previous research implicated two leucines (Leu41 and Leu42) as critical residues for the interaction of SpeA with the MHC-II α-chain, and mutants containing substitutions at these positions have been used in vaccination studies (20, 21). Consistent with this, a model of SpeA in complex with HLA-DQ8 predicted Tyr100 would hydrogen bond with the conserved MHC-II α-chain Lys39 (Fig. 2A), while the SpeA side chains of Leu41 and Leu42 were predicted to extend into a pocket formed by the MHC-II α1-domain (Fig. S1). Based on this analysis, we generated a triple mutant containing alanine substitutions at all three positions (SpeAL41A/L42A/Y100A), henceforth known as SpeATRI (Fig. 2B). SpeATRI was attenuated at all concentrations tested for activating B6HLA mouse splenocytes compared with wild-type SpeA (Fig. 2C). Next, we used SpeATRI in our vaccination regimen (Fig. 2D) in parallel with wild-type SpeA, or a vehicle (sham) control. Interestingly, mice vaccinated with wild-type SpeA and SpeATRI were both protected from nasopharyngeal infection compared with sham-vaccinated mice (Fig. 2E); however, only SpeATRI-vaccinated mice generated significant anti-SpeA IgG antibody titers (Fig. 2F). Low levels of anti-SpeA IgM were only detected in the SpeATRI-vaccinated mice, while anti-SpeA IgA were not detectable from any group (Fig. S2). The SpeATRI-vaccinated mice supported our previous conclusion that anti-SAg antibody could be protective, yet the lack of anti-SpeA antibodies in the wild-type SpeA-vaccinated mice was puzzling. Knowing that SAgs target T cells based on expression of specific Vβ T-cell receptors, we hypothesized that protection in the wild-type SpeA-vaccinated mice may be independent of humoral immunity but related to the T-cell response to the vaccination. To test this idea, we used wild-type staphylococcal enterotoxin B (SEB), a SAg that targets mouse Vβ8+ TCRs (22), similar to SpeA (23). As an additional control, we used wild-type SpeC, a SAg that does not activate mouse T cells (24). Recombinant SEB and SpeC were purified (Fig. 2B), and it was demonstrated that SEB could stimulate B6HLA splenocytes similar to wild-type SpeA, while SpeC was unable to do so (Fig. 2C). Following vaccination, mice that received SEB had significantly reduced S. pyogenes bacterial numbers, whereas SpeC-treated mice were comparable to sham-treated mice (Fig. 2E). As expected, SEB or SpeC vaccination did not elicit detectable anti-SpeA antibodies (Fig. 2F), further indicating that SEB-induced protection was not mediated by humoral immunity.
Fig. 2.

Vaccination with specific SAg proteins induces antibody-mediated, and antibody-independent protection from nasopharyngeal infection by S. pyogenes. (A) Ribbon diagram model of SpeA (blue) in complex with the TCR (α-chain, light blue; β-chain, yellow) and MHC-II (α-chain, red; β-chain, green). Inset image shows amino acid residues mutated in SpeATRI (blue) and the conserved lysine 39 on MHC-II (red). (B) Recombinant SAgs visualized on a 15% SDS/PAGE. (C) SAg activation of B6HLA mouse splenocytes (2 × 105 cells per well) using SpeA (red), SpeATRI (pink), SEB (blue), and SpeC (green) at the indicated concentrations using murine IL-2 as a readout. Bars represent the mean ± SEM. (D) SAg vaccination protocol. (E) Nasal challenge of B6HLA mice with ∼108 cfus of S. pyogenes MGAS8232 postvaccination with indicated treatments (control, black; SpeA, red; SpeATRI, pink; SEB, blue; and SpeC, green). Data points represent cfus from the complete nasal turbinates (cNTs) of individual mice at 48 h. Horizontal bars represent the geometric mean. The horizontal dotted line indicates theoretical limit of detection. (F) Serum IgG antibody titers determined using ELISA from B6HLA mice vaccinated with indicated treatment (control, black; SpeA, red; SpeATRI, pink; SEB, blue; and SpeC, green). Bars represent the mean ± SEM. Significance was determined by one-way ANOVA with Dunnett’s multiple comparison post hoc test (**P < 0.05; ***P < 0.01).
Fig. S1.

Molecular model of SpeA in complex with the mouse Vβ8 TCR and HLA-DQ8. Shown on the Left is the ribbon diagram as depicted in Fig. 2A and generated as described in SI Materials and Methods. Shown on the Right is a stereo (cross-eyed) view of the HLA-DQ8 surface with SpeA amino acid side chains shown for Leu41, Leu42, and Tyr100. Both side chains of Leu41 and Leu42 are predicted to fit within a pocket formed by the α-1 domain of HLA-DQ8.
Fig. S2.

Anti-SpeA IgM and IgA titers in B6HLA mice vaccinated with specific SAg proteins. Serum IgM and IgA antibody titers were determined using ELISA from B6HLA mice vaccinated with the indicated protein. Bars represent the mean ± SEM (n = 4–6 per group). Significance was determined by one-way ANOVA with Dunnett’s multiple comparison post hoc test (***P < 0.01).
Wild-Type SpeA- and SEB-Vaccinated Mice Have Poorly Responsive Vβ8+ T Cells.
Since the protective phenotype from wild-type SpeA- and wild-type SEB-vaccinated mice was not likely due to neutralizing antibodies, we examined if this protective phenotype stemmed from an impact on the specific T-cell subset that is targeted by both SpeA and SEB (i.e., Vβ8+ T cells). To assess this, B6HLA mice were vaccinated with either a vehicle control (sham), wild-type SpeA, or wild-type SEB, killed on day 43 (without infection), and splenocytes were harvested (Fig. 3A). Using flow cytometry, we assessed CD3+ lymphocytes for expression of Vβ8+ TCRs, and used CD3+Vβ3+ lymphocytes as an internal control (Fig. 3B). There was no difference in percentages of CD3+Vβ3+ lymphocytes between groups; however, there was a clear reduction of CD3+Vβ8+ lymphocytes in SpeA- and SEB-vaccinated mice compared with the sham control (Fig. 3C). This result is likely due to Vβ-specific T-cell death and/or TCR down-regulation, which are known to occur following SAg exposure in mice (25, 26). Next, splenocytes were stimulated with increasing concentrations of either Vβ8-targeting SAgs (SpeA or SEB) or the Vβ11-targeting SAg streptococcal mitogenic exotoxin Z (SmeZ) (27) as an internal control. Compared with control-vaccinated mice, splenocytes from SpeA- or SEB-vaccinated mice were poorly responsive to Vβ8-targeting SAgs, requiring 100- to 1,000-fold higher concentration of SAg to reach comparable activity with the sham-vaccinated splenocytes (Fig. 3 D and E). However, SmeZ could activate splenocytes similarly for all three groups, where SEB-vaccinated mice were actually more responsive than sham-vaccinated mice (Fig. 3F). These data demonstrate that detectable CD3+Vβ8+ T cells were reduced in both wild-type SpeA and wild-type SEB-vaccinated mice. Furthermore, these splenocytes were highly impaired for activation by Vβ8-targeting SAgs (i.e., SpeA and SEB), but not to a Vβ11-targeting SAg (i.e., SmeZ), and this phenotype correlated with protection from nasopharyngeal infection by S. pyogenes (Fig. 2E).
Fig. 3.

SpeA- and SEB-vaccinated mice have poorly functional Vβ8+ T cells. (A) Vaccination protocol. (B and C) Flow cytometric analysis of splenocytes at day 43 postsuperantigen vaccination (n = 4 for each group). (B) Representative flow plots for each treatment group stained for CD3 (APC) and either Vβ3 or Vβ8 (FITC). Staining of Vβ3 and Vβ8 are from the same mouse. Each sample was first gated on lymphocyte population based on forward scatter and side scatter before gating on CD3+Vβ+ population. (C) Percentage of CD3+Vβ3+ or CD3+Vβ8+ T-cell subset for each treatment group (control, black; SpeA, red; and SEB, blue). Data are shown as mean ± SEM. Significance was determined by two-way ANOVA with Dunnett’s multiple comparison post hoc test (*P < 0.05; ***P < 0.001). (D–F) B6HLA mouse splenocyte IL-2 activation assay postvaccination with control (black circle), SpeA (red circle), or SEB (blue triangle) (n = 3 for each group). Treated mouse splenocytes were stimulated with increasing concentrations of SAgs targeting specific T-cell variable β-chain (Vβ) subsets (D) SpeA, Vβ8; (E) SEB, Vβ8; and (F) SmeZ, Vβ11. Stimulation occurred for 18 h and culture supernatants were analyzed for IL-2 using ELISA as a readout for T-cell activation. Data are shown as the mean ± SEM. Significance was determined by two-way ANOVA with Tukey’s post hoc test on the highest (106 pg mL−1) concentration tested (**P < 0.01; ***P < 0.001).
T Cells Are Required for Efficient Nasopharyngeal Infection by S. pyogenes MGAS8232.
Since our wild-type SAg vaccination studies suggested a role for SAg-responsive T cells during S. pyogenes infection, we sought to deplete T cells from the murine infection model and determine the impact on nasopharyngeal infection. We used a previously described T-cell depletion protocol (28) to deplete CD4+ or CD8+ T cells, or both T-cell subsets concurrently, followed by nasopharyngeal infection with S. pyogenes MGAS8232 (Fig. 4A). T-cell depletion was confirmed by flow cytometric analysis of the lymphocyte population from cervical lymph nodes compared with the isotype control-treated mice (Fig. 4 B and C). Removal of either CD8+ T cells alone, or the removal of both CD4+ and CD8+ T cells, significantly reduced the nasopharyngeal burden of S. pyogenes MGAS8232 in B6HLA mice (Fig. 4D). We also evaluated Streptococcus pneumoniae, which is another human pathogen of the upper respiratory tract that is not known to produce SAgs. First, we tested nasopharyngeal infection in both conventional B6 mice and B6HLA mice, and S. pneumoniae infected both mice backgrounds at similar levels (Fig. 4E). This further suggests S. pneumoniae does not produce a human-specific SAg, whereas S. pyogenes cannot efficiently infect B6 mice lacking human MHC-II (17). Next, we tested nasopharyngeal infection with S. pneumoniae in isotype-treated, and CD4/CD8 T-cell–depleted mice. Removal of both T-cell subsets did not significantly alter recovered cfus, although in contrast to S. pyogenes, there was a trend for increased cfus recovered in the T-cell–depleted mice (Fig. 4E). These data indicate that in the absence of T cells, S. pyogenes MGAS8232 is highly impaired for the ability to infect the nasopharynx of B6HLA mice whereas S. pneumoniae is unaffected.
Fig. 4.

T-cell–dependent nasopharyngeal infection is specific to S. pyogenes. (A) T-cell depletion protocol. (B and C) Flow cytometric analysis of cervical lymph node populations at day 0 post T-cell depletion (n = 3 per group). (B) Representative flow plots for each treatment group stained for CD4 (APC-eFluor 780) and CD8 (PE). Each sample had the lymphocyte population first gated upon using forward scatter (FSC) and side scatter (SSC). (C) Percentage of CD4+ and CD8+ cells to total lymphocyte population in both treatment groups. Data are shown as mean ± SEM. Significance was determined by Student’s t test (***P < 0.001). (D) Nasal challenge with ∼108 cfus of S. pyogenes MGAS8232 of B6HLA mice with indicated treatments [isotype control (LTF-2), black; CD4 depleted (GK1.5), gray; CD8 depleted (YTS169.4), purple; T-cell depleted (GK1.5 + YTS169.4), pink]. (E) Nasal challenge with 107 cfus of S. pneumoniae P1121 of B6 (triangles) or B6HLA mice (squares) with either no treatment (open symbols), isotype control (LTF-2) (black symbols) or T-cell depleted (GK1.5 + YTS169.4) pink]. Data points represent cfus from the complete nasal turbinates (cNTs) of individual mice 48 h postinfection. Horizontal bars represent the geometric mean. The horizontal dotted line indicates limit of detection. Significance was determined by one-way ANOVA with Dunnett’s multiple comparisons post hoc test (*P < 0.05; **P < 0.01).
SAg-Responsive T Cells Are Required for Nasopharyngeal Inflammation by S. pyogenes, but Not S. pneumoniae.
We previously demonstrated that nasopharyngeal infection by S. pyogenes induces a SAg-driven inflammatory environment at 24 h within the cNT that appears to promote infection (17). To further assess differences between the T-cell–depleted mice, we conducted a cytokine/chemokine array from cNT homogenates. As predicted, in uninfected mice there was no apparent inflammatory signature (Fig. 5A and Fig. S3), whereas infection in the presence of T cells (isotype control) generated a proinflammatory environment that correlated with high bacterial load (Fig. 5B and Fig. S3). However, depletion of CD4+ or CD8+ T cells reduced the inflammatory signature, while remarkably, depletion of both CD4+ and CD8+ T cells largely resembled uninfected control mice (Fig. 5B and Fig. S3). Interestingly, infection with S. pneumoniae induced a comparatively moderate inflammatory environment, which was exaggerated in T-cell–depleted mice (Fig. 5C and Fig. S3). To confirm these findings, we also conducted the cytokine/chemokine array from mice vaccinated with wild-type SpeA or wild-type SEB. Similar to the T-cell depletion experiments, sham-vaccinated mice induced a strong inflammatory signature, whereas both SAg-vaccinated groups resembled the uninfected control group (Fig. 5D). Remarkably, these collective results indicate that S. pyogenes MGAS8232 requires T cells to efficiently infect nasopharyngeal tissue, and additionally, the presence of SAg-responsive T cells results in a proinflammatory environment, whereas S. pneumoniae could persist in the nasopharynx regardless of T cells.
Fig. 5.

Heat map of multiplex cytokine array from S. pyogenes- and S. pneunomiae-infected mice. B6HLA mice were either uninfected (A), underwent T-cell–depleting antibody treatment (B and C), or vaccinated (D) and infected with either ∼108 cfus of S. pyogenes MGAS8232 (B and D) or ∼107 cfus of S. pneumoniae P1121 (C) for 48 h. Mice were killed and supernatant from cNT homogenates was procured for cytokine and chemokine analysis. Data shown represent the mean cNT cytokine response that displayed significant differences between any groups. Values for each row were normalized to have the highest cytokine response as 100% (n ≥ 3 mice per group). Corresponding quantitative data and statistical analyses are shown in Fig. S3.
Fig. S3.

Cytokine response from B6HLA mice in the complete nasal turbinates during streptococcal infection after specified treatment. Mice were uninfected (open bars) or infected intranasally with either 108 cfus of S. pyogenes MGAS8232 (solid bars) or 107 cfus of S. pneumoniae P1121 (striped bars). Mice were treated as indicated with T-cell–depleting antibodies [α-CD4 (GK1.5), α-CD8 (YTS169.4) or combination], an isotype control (LTF-2), or wild-type SAg vaccination (SpeA or SEB) or control (sham). Mice were killed 48 h later and complete nasal turbinate (cNT) homogenates were analyzed for multiple cytokines and chemokines [(A), Th1-type cytokines; (B), Th2-type cytokines (C), Treg-type cytokines (D), Th17-type cytokines (E), chemokines (F) growth factors]. Data represent the mean ± SEM of cNT cytokine/chemokine concentration (pg mL−1) (n ≥ 3 mice per group). Significance was determined by one-way ANOVA with Dunnett’s multiple comparison post hoc test (S. pyogenes) or Student’s t test (S. pneumoniae) (*P < 0.05; **P < 0.01; ***P < 0.001).
Discussion
T lymphocytes are central components of the adaptive immune system, and through the extreme diversity of TCRs, these cells can recognize a virtually unlimited assortment of microbial peptides when presented by MHC molecules. Despite the variability of TCRs through variable (V), diversity (D), and joining (J) segment [V(D)J] recombination, and the polygenic and polymorphic nature of MHC-II molecules, the SAg exotoxins have managed to evolve to recognize both of these highly diverse adaptive immune receptors, forcing the activation of numerous Vβ-specific T cells, and thus altering the course of the immune response. However, mechanisms by which SAg-mediated manipulation of the adaptive immune system contributes to the benefit of S. pyogenes, and other SAg-producing microbes, is not well understood. Herein, we present evidence that S. pyogenes requires functional, Vβ-specific T-cell populations to promote an environment that dramatically enhances the early stages of nasopharyngeal infection by this globally important pathogen.
Not surprisingly, T lymphocytes are beneficial to the host in numerous infection models including Mycobacterium tuberculosis (29), Haemophilus influenzae (30), Salmonella enterica serovar Typhimurium (31), and Listeria monocytogenes (28). Although active immunity to S. pneumoniae nasopharyngeal infection has been shown to be dependent upon CD4+ T cells (32), our control T-cell depletion experiments did not overtly influence S. pneumoniae cfus by 48 h (Fig. 4E). This was expected as the mice were naïve to S. pneumoniae, and this bacterium is not known to produce SAgs. However, in the absence of functional Vβ-specific T cells (Fig. 3 D–F), or in the absence of both CD4+ and CD8+ T cells (Fig. 4 B and C), cfus of S. pyogenes MGAS8232 were dramatically reduced by approximately three orders of magnitude (Figs. 2E and 4D). Additionally, removal of CD8+ T cells alone impaired nasopharyngeal infection. As SAgs activate both CD4+ and CD8+ T cells in a Vβ-specific manner, we suspect that although both cells likely contribute to the phenotype, CD8+ T cells may be more numerically dominant within this environment (Fig. 4C). Alternatively, CD8+ T cells may be functionally more important for this phenotype. To assess how general this T-cell–dependent phenotype is for different S. pyogenes strains, we evaluated two additional strains that encode speA, including S. pyogenes 5448 and MGAS315. The M1 serotype S. pyogenes 5448, surprisingly, did not efficiently infect the B6HLA mice (Fig. S4A), although we could not detect SpeA expression from this background (Fig. S4B), likely due to degradation from high levels of the SpeB cysteine protease produced by this strain (33). S. pyogenes MGAS315, however, which does produce SpeA (Fig. S4B), infected higher than MGAS8232, although depletion of T cells from the B6HLA mice trended to reduce infection by only ∼1 log (Fig. S4A). The B6HLA mouse infection model, accordingly, does have limitations where the majority of the streptococcal SAgs are not functionally active (Fig. S5), and similarly to SpeC (24), we believe this is due to the inability of most streptococcal SAgs to target mouse Vβs. Thus, although all S. pyogenes isolates may not require SAg-responsive T cells in this mouse model, we do predict that SAgs other than SpeA would likely contribute to human nasopharyngeal infection, and it remains to be determined if and which SAgs when targeted would afford the most protection in diverse human populations.
Fig. S4.

Infection phenotypes of B6HLA mice and T-cell–depleted B6HLA mice with S. pyogenes 5448 and MGAS315. (A) Nasal challenge with ∼108 cfus of S. pyogenes 5448 or MGAS315 of B6HLA mice with indicated treatments. Data points represent cfus from the complete nasal turbinates (cNTs) of individual mice 48 h postinfection. Horizontal bars represent the geometric mean. The horizontal dotted line indicates limit of detection. Differences between treatments (isotype versus α-CD4/CD8) were not significantly different (P > 0.05) by Students t test. (B) 12% SDS/PAGE (Top), and anti-SpeA Western blot analysis (Bottom) of recombinant SpeA protein, and concentrated supernatant samples from the indicated strains. Molecular weight markers are labeled (Left) in kilodaltons and SpeA and the pro-SpeB zymogen and mature SpeB (produced by strain 5448) are indicated with arrows.
Fig. S5.

Dose-dependent IL-2 responses from B6 and B6HLA mouse splenocytes in response to recombinant streptococcal SAgs. (A) Fifteen percent SDS/PAGE of recombinant streptococcal SAgs. Molecular weight markers are labeled (Left) in kilodaltons. (B and C) SAg activation of B6 and B6HLA mouse splenocytes (2 × 105 cells per well) using the recombinant SAgs shown in A. Mouse IL-2 was measured after 18 h at the indicated SAg concentrations and bars represent the mean ± SEM.
SAgs have long been recognized for the ability to suppress antibody production (34, 35), which occurs in part through T-cell- and Fas–FasL-dependent apoptosis of B cells (36, 37). Although the lack of anti-SpeA antibodies in the wild-type SpeA-vaccinated mice was therefore not unexpected (Fig. 2F), we were initially surprised by the low cfus in wild-type SpeA-vaccinated mice (Fig. 2E). However, as we have detected activation of SpeA-targeted Vβ8+ T cells in vivo during nasopharyngeal infection by S. pyogenes (38), and since SAg exposure is known to induce Vβ-specific T-cell unresponsiveness (25, 39), we reasoned that S. pyogenes may require Vβ-specific T cells to promote nasopharyngeal infection. This prediction was supported by two different experimental approaches, including the wild-type SEB vaccination experiments (Fig. 2E), and the T-cell depletion experiments (Fig. 4D). These findings are also entirely consistent with our previous work where host expression of human MHC-II (HLA-DQ8), and expression of SpeA (17), were similarly critical for efficient infection by S. pyogenes MGAS8232.
Cytokine and chemokine analysis demonstrated that in the absence of T-cell function, when the S. pyogenes bacterial load was high (Fig. 4D), the nasopharyngeal environment was rich in proinflammatory cytokines and chemokines (Fig. 5 B and D and Fig. S3). Remarkably, in wild-type SpeA- or SEB-vaccinated mice (Fig. 5D), or CD4/CD8-depleted mice (Fig. 5B), the cytokine/chemokine profile phenocopied the uninfected control mice (Fig. 5A). T-cell depletion did not impact significantly on nasopharyngeal S. pneumoniae cfus, although an increased trend was noted in the T-cell–depleted mice (Fig. 4E) that was accompanied by an enhanced proinflammatory cytokine signature (Fig. 5C). Thus, the inflammatory signature was entirely consistent with the relative cfus obtained from either pathogen. If a pathogen can avoid mucociliary clearance mechanisms, one of the first steps for nasopharyngeal colonization is attachment to the underlying epithelial surfaces (40). However, binding to epithelial surfaces would be expected to engage multiple pattern recognition receptors, resulting in cytokine production (41). Thus, it appears that in the absence of SAg-driven T-cell activation, S. pyogenes cannot initiate even the earliest steps of nasopharyngeal colonization. It is tempting to speculate that this inflammatory response, per se, could provide a suitable environment that allows S. pyogenes to survive and proliferate, at least in an acute setting.
This work supports the development and testing of toxoid SAgs as vaccine candidates. The majority of previous streptococcal SAg vaccine research has focused on the generation of anti-SAg antibodies for protection against sepsis and toxic shock syndrome (20, 21, 42). This concept has had clinical implications, whereby administration of i.v. immunoglobulins, which contains SAg-neutralizing antibodies (43), have been demonstrated to reduce patient mortality in some settings (44–46). The passive immunization experiments show conclusively that anti-SAg antibodies can be protective against experimental S. pyogenes nasopharyngeal infection (Fig. 1). However, the current most promising S. pyogenes vaccines target the M protein, a surface-anchored virulence determinant and multiple variations are currently in early clinical trials (3). However, an impediment for these vaccines is the hypervariability of the M protein with over 200 streptococcal emm types and differential distributions worldwide (47), making a universally protective vaccine based solely on this molecule challenging. S. pyogenes SAgs are usually encoded on mobile, or putatively mobile, bacteriophage elements and thus different strains of S. pyogenes often encode different combinations of SAgs (48). Although streptococcal SAgs, in most cases, are immunologically distinct (17), this repertoire to date appears to be limited to 14 SAgs (5). Consequently, we believe that SAgs should receive renewed consideration for inclusion within a multicomponent vaccine.
Many important upper respiratory tract pathogens exist predominantly within a state of asymptomatic colonization (40), and thus a number of bacterial “virulence” factors have likely evolved under selective pressures outside circumstances of overt disease, and may more accurately function as “colonization” factors. Our data provide a mechanism whereby SAgs target and activate Vβ-specific T cells to remodel the nasopharyngeal environment to promote the earliest stages of colonization. Indeed, in the absence of a functional SAg, an appropriate MHC-II receptor, or functional Vβ-specific T cells, S. pyogenes fails to colonize and multiply. The specific immunological changes induced by SAgs that are beneficial to S. pyogenes infection remain to be characterized, although we favor a T-cell–driven inflammatory environment necessary for colonization that may allow for the exposure of host cells’ bindings sites, impairment of innate immune responses, and/or enhanced acquisition of nutrients in the nutrient-poor nasopharyngeal environment. Overall, this work further supports SAgs as prophylactic vaccines to target the carriage state of this important and human-specific pathogen, as well as furthers our understanding of these toxins outside of the context of severe and invasive disease.
Materials and Methods
Bacteria.
S. pyogenes strains MGAS8232, 5448, and MGAS315, and S. pneumoniae strain P1121, were used for the nasal infection experiments. Further experimental details are provided in SI Materials and Methods.
Mice.
C57BL/6 mice expressing human major histocompatibility complex II molecules (HLA-DQ8, HLA-DR4/DQ8) have been previously described (14, 49, 50). HLA-DQ8 and HLADR4/DQ8 mice were infected equally well with S. pyogenes MGAS8232 compared with C57BL/6 (Fig. S6) and henceforth, both were used in experiments and labeled B6HLA. Further experimental details on mouse experiments are provided in SI Materials and Methods.
Fig. S6.
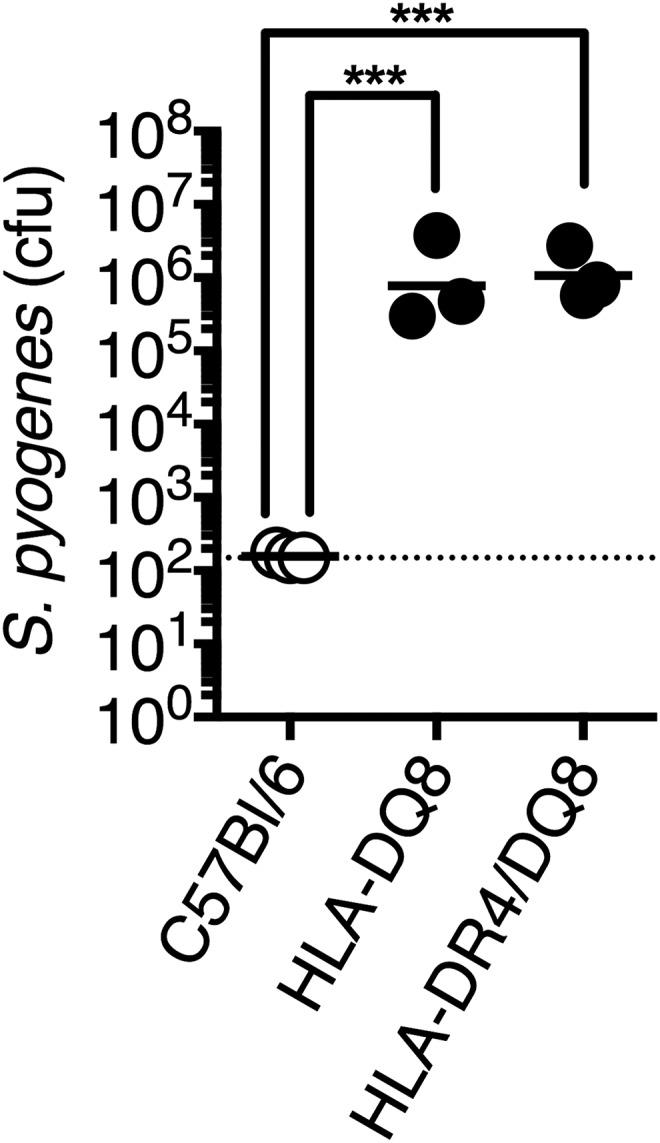
B6 mice expressing HLA-DQ8 and HLA-DR4/DQ8 have similar S. pyogenes infection phenotypes. Nasopharyngeal infection of conventional B6 mice, B6 mice expressing HLA-DQ8 only, or B6 mice expressing both HLA-DR4 and DQ8 alleles were infected with ∼108 cfus of S. pyogenes MGAS8232. Data points represent cfus from the complete nasal turbinates (cNTs) of individual mice 48 h postinfection. Horizontal bars represent the geometric mean. The horizontal dotted line indicates the limit of detection. Significance was determined by one-way ANOVA with Tukey’s post hoc test (***P < 0.001).
Recombinant SAg and Antibody Production.
Details on protein expression and purification, and antibody production, are provided in SI Materials and Methods.
Molecular Modeling.
Details of the molecular modeling are provided in SI Materials and Methods.
Flow Cytometry.
Details of flow cytometry analysis are provided in SI Materials and Methods.
Mouse Cytokine/Chemokine Array.
Details of the cytokine/chemokine array experiments are provided in SI Materials and Methods.
Statistical Analysis.
All statistical analysis was completed using Prism software (GraphPad). Significance was calculated using, where indicated, the Student’s t test and one-way or two-way ANOVA with Dunnett’s or Tukey’s multiple comparisons post hoc test. A P value less than 0.05 was determined to be statistically significant.
SI Materials and Methods
Bacteria.
Escherichia coli strains XL1-blue and BL21 (DE3) were used for cloning and protein expression, respectively. E. coli strains were grown in Luria-Bertani (LB) broth or on 1.5% (wt/vol) LB agar at 37 °C with appropriate antibiotics. S. pyogenes MGAS8232 is an M18 serotype, rheumatic fever isolate that has been previously sequenced and annotated (51). The genome of MGAS8232 encodes for six distinct SAgs (SpeA, SpeC, SpeG, SpeL, SpeM, and SmeZ) (51) and, although only production of SpeA, SpeC, and SpeL were detectable by Western blot analysis, SpeA was the only SAg important for the nasopharyngeal infection phenotype (17). S. pyogenes 5448 is an M1 serotype that was isolated from an invasive infection (52). This strain encodes four distinct SAgs (SpeA, SpeG, SpeJ, and SmeZ) and is a well-studied representative of the “M1T1” clone. S. pyogenes MGAS315 in an M3 serotype isolated from a patient with streptococcal toxic shock syndrome (53) and this strain encodes four distinct SAgs (SpeA, SpeG, SpeK, and SSA) as well as a truncated SmeZ gene (54). S. pneumoniae P1121 is a serotype 23F derived from nasopharyngeal human carriage of strain P833, which was obtained from a child with otitis media (55). S. pyogenes was routinely grown statically at 37 °C in Todd Hewitt media supplemented with 1% (wt/vol) yeast extract and S. pneumoniae was grown in tryptic soy broth statically at 37 °C in 5% CO2.
Mice.
All animal experiments were conducted in accordance with the Canadian Council on Animal Care Guide to the Care and Use of Experimental Animals. Animal protocols (2009–038 and 2011–088) were approved by the Animal Use Subcommittee at Western University. These mice were bred in a barrier facility at Western University and routinely genotyped for appropriate transgenes. In circumstances where mice were intranasally infected with S. pyogenes as previously described (17, 38), or S. pneumoniae, they were first administered 2 mg mL−1 neomycin sulfate in their drinking water ad libitum 48 h before infection to inhibit the endogenous nasal microbiota. For murine intranasal infections with S. pyogenes, the bacteria were grown to early exponential phase (OD600 0.2–0.3), cells were washed and resuspended in Hank’s buffered saline solution (HBSS; ∼1 × 108 cfus per 15 µL) and 7.5 µL was inoculated into each nostril of a Flurane (isoflurane, USP; Baxter)-anesthetized mouse. For S. pneumoniae infections, bacteria was prepared as above, with indicated differences in resuspension (∼1 × 107 cfus per 10 µL) and inoculation (5 µL per nostril). Mice were killed 48 h postinfection and the complete nasal turbinates (cNTs) (including nasal-associated lymphoid tissue, nasal turbinates, and maxillary sinuses), were removed, homogenized (in HBSS), serially diluted, and plated on trypticase soy agar with 5% sheep’s blood plates (Becton Dickinson) for bacterial enumeration. Counts less than 25 cfus per 100 µL were considered below the theoretical limit of detection. Mice used in passive immunization and T-cell depletion experiment were 12–14 wk old, whereas mice used in the vaccination study were 6–8 wk old.
Adoptive Serum Transfer and Antibody Titration.
B6HLA transgenic mice were administered 500 µL of indicated rabbit anti-SAg serum via i.p. injection 24 h and 2 h before infection with S. pyogenes. Along with cNT acquisition for bacterial enumeration at the 48-h endpoint, serum was collected for antibody titer analysis. Antibody titers were determined by direct ELISA against 1 µg per well of wild-type SAg and calculated as the reciprocal of the lowest serum dilution with readings fourfold above control serum background.
SAg Vaccination Protocol.
B6HLA mice were injected s.c. on their back with 1 µg per 200 µL (100 µL 150 mM NaCl and 100 µL Imject alum adjuvant; Thermo Fisher Scientific) of recombinant SAg every 2 wk for a total of three injections. Two weeks following the last injection, mice were bled via saphenous vein and serum was collected for antibody titer analysis using ELISA. Twenty-four hours postbleed, mice were either infected with S. pyogenes as indicated above or killed for procurement of splenocytes for ex vivo activation assays or flow cytometry analysis.
Recombinant Superantigen and Antibody Production.
Genes encoding wild-type SpeA, SpeC, SmeZ, and SEB, lacking predicted signal peptides, were previously cloned and expressed as described (17). For the generation of SpeAL41/41A/Y100A (SpeATRI), PCR amplification was completed using speAY100A as template and primers that flanked both sides of the amino acids to be substituted (L41A/L42A) and contained the coding sequence of the desired substitutions: (5′–3′) forward TCTGTTGATCAAGCAGCATCTCACGATTTA; reverse TAAATCGTGAGATGCTGCTTGATCAACAGA (bold indicates altered nucleotides). Recombinant proteins were analyzed for purity on 12% or 15% separating SDS/PAGE and Western blots were visualized on a LI-COR Odyssey (LI-COR Biosciences) using IRDye800-conjugated donkey anti-rabbit IgG as the secondary antibody (Rockland). Lyophilized SAgs were used to generate polyclonal rabbit antibodies from a commercial source (ProSci).
Ex Vivo Splenocyte Stimulation with SAgs.
Splenocytes were treated with ACK lysing buffer (Gibco Life Technologies) and suspended (2 × 105 cells per well) in RPMI medium 1640 supplemented with 10% heat-inactivated FBS (Sigma-Aldrich), 0.1 mM MEM nonessential amino acids, 2 mM l-glutamine, 1 mM sodium pyruvate, 100 units mL−1 penicillin, 100 µg mL−1 streptomycin, and 5 mM Hepes buffer [complete RPMI (cRPMI); all from Gibco Life Technologies]. SAg were added at the indicated concentrations to mouse splenocytes (2 × 105 cells per well) and mouse IL-2 was determined after 18 h (at 37 °C, 5% CO2) by ELISA, according to the manufacturer’s instructions (eBioscience).
In Vivo T-Cell Depletion.
T cells were depleted as previously described (28). Briefly, B6HLA mice were injected with 300 µg per 500 µL of T-cell depleting antibodies in PBS [anti-CD4 (clone GK1.5); anti-CD8 (clone YTS169.4); or both, at 150 µg each] or isotype control (clone LTF-2) (all from BioXCell) on days −7, −6, and −1. On day zero, mice were either infected (S. pyogenes or S. pneumoniae) as indicated above or killed for cervical lymph node procurement, cell isolation, and flow cytometry analysis.
Flow Cytometry.
Isolated cells (from cervical lymph nodes or spleen) were passed through a 40-µm nylon cell strainer to remove tissue debris and resuspended in ACK lysing buffer (Gibco Life Technologies) to remove red blood cells if required (spleens). Cells were washed in PBS and resuspended in cRPMI. Live cells were counted using Trypan blue exclusion and aliquoted to a final concentration of 500,000 cells per tube. Cells were washed with PBS supplemented with 2% FBS and 0.01% sodium azide three times. Fc block (hybridoma clone 2.4G2) was added before cell staining. Splenocytes from the SAg exposure assay had the following panel administered: anti–CD3-APC (clone 145–2C11), anti-TCR variable β-chain 8.1 and 8.2-FITC (clone KJ16), and anti-TCR variable β-chain 3-FITC (clone KJ25). Cervical lymph nodes from T-cell–depleted mice were stained with anti–CD4-APC eFluor780 (clone GK1.5) and anti–CD8a-PE (clone 53–6.7). Stained cells were washed three more times as indicated above and evaluated on a BD LSR II Flow Cytometer (BD Biosciences). Data were analyzed using FlowJo v.10.0 (TreeStar).
Mouse Cytokine/Chemokine Array.
Cytokine and chemokine concentrations were determined from B6HLA cNT homogenates from mice treated with LTF-2 (isotype control), GK1.5 (anti-mouse CD4+), YTS169.4 (anti-mouse CD8+), or a combination of GK1.5 and YTS169.4 and infected with S. pyogenes MGAS8232 or S. pneumoniae P1121. Infected mice from the wild-type SpeA and wild-type SEB vaccination groups, and sham group, were also assessed. Uninfected B6HLA cNT homogenates were tested as a background control. Multiplex arrays were performed by Eve Technologies and heat maps were generated using matrix2png algorithm (56), and the data are shown as average cytokine response from three to four mice per group normalized to the highest concentration as 100%.
Molecular Modeling.
To generate the SAg model in complex with TCR and MHC-II, SpeA (PDB: 1FNU) (57) was superpositioned onto the known SEB:TCR:low-affinity MHC-II (PDB: 4C56) (58) with HLA-DQ8 (PDB: 1JK8) (59) and the murine TCR variable β-chain 8.2 (PDB: 1SBB) (22) replaced endogenous MHC-II and TCR, respectively. The TCR α-chain from 4C56 was used. The image was visualized using the PyMOL Molecular Graphics System, version 1.3 Schrödinger (www.pymol.org/).
Acknowledgments
We thank Dr. Dawn Bowdish (McMaster University) for providing the S. pneumoniae P1121 strain and Dr. Allison McGeer (University of Toronto) for providing the S. pyogenes 5448 strain. This work was supported by Canadian Institutes of Health Research Operating Grant MOP-142137 (to J.K.M.) and an internal award from Western University (Medical and Health Sciences Research Board 36819). J.J.Z. was supported by an Ontario graduate scholarship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Commentary on page 10000.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1700858114/-/DCSupplemental.
References
- 1.Shaikh N, Leonard E, Martin JM. Prevalence of streptococcal pharyngitis and streptococcal carriage in children: A meta-analysis. Pediatrics. 2010;126:e557–e564. doi: 10.1542/peds.2009-2648. [DOI] [PubMed] [Google Scholar]
- 2.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 3.Dale JB, et al. Current approaches to group A streptococcal vaccine development. In: Ferretti JJ, Stevens DL, Fischetti VA, editors. Streptococcus pyogenes : Basic Biology to Clinical Manifestations. University of Oklahoma Health Sciences Center; Oklahoma City: 2016. pp. 937–983. [Google Scholar]
- 4.Walker MJ, et al. Disease manifestations and pathogenic mechanisms of Group A Streptococcus. Clin Microbiol Rev. 2014;27:264–301. doi: 10.1128/CMR.00101-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Commons RJ, et al. Streptococcal superantigens: Categorization and clinical associations. Trends Mol Med. 2014;20:48–62. doi: 10.1016/j.molmed.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Herrmann T, Baschieri S, Lees RK, MacDonald HR. In vivo responses of CD4+ and CD8+ cells to bacterial superantigens. Eur J Immunol. 1992;22:1935–1938. doi: 10.1002/eji.1830220739. [DOI] [PubMed] [Google Scholar]
- 7.Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science. 1990;248:705–711. doi: 10.1126/science.2185544. [DOI] [PubMed] [Google Scholar]
- 8.Sundberg EJ, Deng L, Mariuzza RA. TCR recognition of peptide/MHC class II complexes and superantigens. Semin Immunol. 2007;19:262–271. doi: 10.1016/j.smim.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cone LA, Woodard DR, Schlievert PM, Tomory GS. Clinical and bacteriologic observations of a toxic shock-like syndrome due to Streptococcus pyogenes. N Engl J Med. 1987;317:146–149. doi: 10.1056/NEJM198707163170305. [DOI] [PubMed] [Google Scholar]
- 10.Stevens DL. Streptococcal toxic shock syndrome associated with necrotizing fasciitis. Annu Rev Med. 2000;51:271–288. doi: 10.1146/annurev.med.51.1.271. [DOI] [PubMed] [Google Scholar]
- 11.McCormick JK, Yarwood JM, Schlievert PM. Toxic shock syndrome and bacterial superantigens: An update. Annu Rev Microbiol. 2001;55:77–104. doi: 10.1146/annurev.micro.55.1.77. [DOI] [PubMed] [Google Scholar]
- 12.Norrby-Teglund A, et al. Host variation in cytokine responses to superantigens determine the severity of invasive group A streptococcal infection. Eur J Immunol. 2000;30:3247–3255. doi: 10.1002/1521-4141(200011)30:11<3247::AID-IMMU3247>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 13.Kotb M, et al. An immunogenetic and molecular basis for differences in outcomes of invasive group A streptococcal infections. Nat Med. 2002;8:1398–1404. doi: 10.1038/nm1202-800. [DOI] [PubMed] [Google Scholar]
- 14.Nooh MM, El-Gengehi N, Kansal R, David CS, Kotb M. HLA transgenic mice provide evidence for a direct and dominant role of HLA class II variation in modulating the severity of streptococcal sepsis. J Immunol. 2007;178:3076–3083. doi: 10.4049/jimmunol.178.5.3076. [DOI] [PubMed] [Google Scholar]
- 15.Llewelyn M, et al. HLA class II polymorphisms determine responses to bacterial superantigens. J Immunol. 2004;172:1719–1726. doi: 10.4049/jimmunol.172.3.1719. [DOI] [PubMed] [Google Scholar]
- 16.Park HS, Francis KP, Yu J, Cleary PP. Membranous cells in nasal-associated lymphoid tissue: A portal of entry for the respiratory mucosal pathogen group A Streptococcus. J Immunol. 2003;171:2532–2537. doi: 10.4049/jimmunol.171.5.2532. [DOI] [PubMed] [Google Scholar]
- 17.Kasper KJ, et al. Bacterial superantigens promote acute nasopharyngeal infection by Streptococcus pyogenes in a human MHC class II-dependent manner. PLoS Pathog. 2014;10:e1004155. doi: 10.1371/journal.ppat.1004155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bessen DE. Tissue tropisms in group A Streptococcus: What virulence factors distinguish pharyngitis from impetigo strains? Curr Opin Infect Dis. 2016;29:295–303. doi: 10.1097/QCO.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alam FM, Turner CE, Smith K, Wiles S, Sriskandan S. Inactivation of the CovR/S virulence regulator impairs infection in an improved murine model of Streptococcus pyogenes naso-pharyngeal infection. PLoS One. 2013;8:e61655. doi: 10.1371/journal.pone.0061655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ulrich RG. Vaccine based on a ubiquitous cysteinyl protease and streptococcal pyrogenic exotoxin A protects against Streptococcus pyogenes sepsis and toxic shock. J Immune Based Ther Vaccines. 2008;6:8. doi: 10.1186/1476-8518-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roggiani M, et al. Toxoids of streptococcal pyrogenic exotoxin A are protective in rabbit models of streptococcal toxic shock syndrome. Infect Immun. 2000;68:5011–5017. doi: 10.1128/iai.68.9.5011-5017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, et al. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity. 1998;9:807–816. doi: 10.1016/s1074-7613(00)80646-9. [DOI] [PubMed] [Google Scholar]
- 23.Sundberg EJ, et al. Structures of two streptococcal superantigens bound to TCR beta chains reveal diversity in the architecture of T cell signaling complexes. Structure. 2002;10:687–699. doi: 10.1016/s0969-2126(02)00759-1. [DOI] [PubMed] [Google Scholar]
- 24.Li PL, Tiedemann RE, Moffat SL, Fraser JD. The superantigen streptococcal pyrogenic exotoxin C (SPE-C) exhibits a novel mode of action. J Exp Med. 1997;186:375–383. doi: 10.1084/jem.186.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacDonald HR, Baschieri S, Lees RK. Clonal expansion precedes anergy and death of V β 8+ peripheral T cells responding to staphylococcal enterotoxin B in vivo. Eur J Immunol. 1991;21:1963–1966. doi: 10.1002/eji.1830210827. [DOI] [PubMed] [Google Scholar]
- 26.Niedergang F, et al. The Staphylococcus aureus enterotoxin B superantigen induces specific T cell receptor down-regulation by increasing its internalization. J Biol Chem. 1995;270:12839–12845. doi: 10.1074/jbc.270.21.12839. [DOI] [PubMed] [Google Scholar]
- 27.Rajagopalan G, et al. Evaluating the role of HLA-DQ polymorphisms on immune response to bacterial superantigens using transgenic mice. Tissue Antigens. 2008;71:135–145. doi: 10.1111/j.1399-0039.2007.00986.x. [DOI] [PubMed] [Google Scholar]
- 28.Sirard JC, et al. Intracytoplasmic delivery of listeriolysin O by a vaccinal strain of Bacillus anthracis induces CD8-mediated protection against Listeria monocytogenes. J Immunol. 1997;159:4435–4443. [PubMed] [Google Scholar]
- 29.Kupz A, Zedler U, Stäber M, Kaufmann SHE. A mouse model of latent tuberculosis infection to study intervention strategies to prevent reactivation. PLoS One. 2016;11:e0158849. doi: 10.1371/journal.pone.0158849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foxwell AR, Kyd JM, Karupiah G, Cripps AW. CD8+ T cells have an essential role in pulmonary clearance of nontypeable Haemophilus influenzae following mucosal immunization. Infect Immun. 2001;69:2636–2642. doi: 10.1128/IAI.69.4.2636-2642.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Z, et al. Small intestinal intraepithelial lymphocytes expressing CD8 and T cell receptor γδ are involved in bacterial clearance during Salmonella enterica serovar typhimurium infection. Infect Immun. 2012;80:565–574. doi: 10.1128/IAI.05078-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malley R, et al. CD4+ T cells mediate antibody-independent acquired immunity to pneumococcal colonization. Proc Natl Acad Sci USA. 2005;102:4848–4853. doi: 10.1073/pnas.0501254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aziz RK, et al. Invasive M1T1 group A Streptococcus undergoes a phase-shift in vivo to prevent proteolytic degradation of multiple virulence factors by SpeB. Mol Microbiol. 2004;51:123–134. doi: 10.1046/j.1365-2958.2003.03797.x. [DOI] [PubMed] [Google Scholar]
- 34.Poindexter NJ, Schlievert PM. Suppression of immunoglobulin-secreting cells from human peripheral blood by toxic-shock-syndrome toxin-1. J Infect Dis. 1986;153:772–779. doi: 10.1093/infdis/153.4.772. [DOI] [PubMed] [Google Scholar]
- 35.Lussow AR, MacDonald HR. Differential effects of superantigen-induced “anergy” on priming and effector stages of a T cell-dependent antibody response. Eur J Immunol. 1994;24:445–449. doi: 10.1002/eji.1830240227. [DOI] [PubMed] [Google Scholar]
- 36.Hofer MF, et al. Differential effects of staphylococcal toxic shock syndrome toxin-1 on B cell apoptosis. Proc Natl Acad Sci USA. 1996;93:5425–5430. doi: 10.1073/pnas.93.11.5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stohl W, Elliott JE, Lynch DH, Kiener PA. CD95 (Fas)-based, superantigen-dependent, CD4+ T cell-mediated down-regulation of human in vitro immunoglobulin responses. J Immunol. 1998;160:5231–5238. [PubMed] [Google Scholar]
- 38.Zeppa JJ, et al. Nasopharyngeal infection of mice with Streptococcus pyogenes and in vivo detection of superantigen activity. Methods Mol Biol. 2016;1396:95–107. doi: 10.1007/978-1-4939-3344-0_8. [DOI] [PubMed] [Google Scholar]
- 39.Rellahan BL, Jones LA, Kruisbeek AM, Fry AM, Matis LA. In vivo induction of anergy in peripheral V beta 8+ T cells by staphylococcal enterotoxin B. J Exp Med. 1990;172:1091–1100. doi: 10.1084/jem.172.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siegel SJ, Weiser JN. Mechanisms of bacterial colonization of the respiratory tract. Annu Rev Microbiol. 2015;69:425–444. doi: 10.1146/annurev-micro-091014-104209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsatsaronis JA, Walker MJ, Sanderson-Smith ML. Host responses to group A Streptococcus: Cell death and inflammation. PLoS Pathog. 2014;10:e1004266. doi: 10.1371/journal.ppat.1004266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCormick JK, et al. Development of streptococcal pyrogenic exotoxin C vaccine toxoids that are protective in the rabbit model of toxic shock syndrome. J Immunol. 2000;165:2306–2312. doi: 10.4049/jimmunol.165.4.2306. [DOI] [PubMed] [Google Scholar]
- 43.Norrby-Teglund A, et al. Relative neutralizing activity in polyspecific IgM, IgA, and IgG preparations against group A streptococcal superantigens. Clin Infect Dis. 2000;31:1175–1182. doi: 10.1086/317423. [DOI] [PubMed] [Google Scholar]
- 44.Kaul R, et al. The Canadian Streptococcal Study Group Intravenous immunoglobulin therapy for streptococcal toxic shock syndrome: A comparative observational study. Clin Infect Dis. 1999;28:800–807. doi: 10.1086/515199. [DOI] [PubMed] [Google Scholar]
- 45.Norrby-Teglund A, et al. Successful management of severe group A streptococcal soft tissue infections using an aggressive medical regimen including intravenous polyspecific immunoglobulin together with a conservative surgical approach. Scand J Infect Dis. 2005;37:166–172. doi: 10.1080/00365540410020866. [DOI] [PubMed] [Google Scholar]
- 46.Linnér A, Darenberg J, Sjölin J, Henriques-Normark B, Norrby-Teglund A. Clinical efficacy of polyspecific intravenous immunoglobulin therapy in patients with streptococcal toxic shock syndrome: A comparative observational study. Clin Infect Dis. 2014;59:851–857. doi: 10.1093/cid/ciu449. [DOI] [PubMed] [Google Scholar]
- 47.Steer AC, Law I, Matatolu L, Beall BW, Carapetis JR. Global emm type distribution of group A streptococci: Systematic review and implications for vaccine development. Lancet Infect Dis. 2009;9:611–616. doi: 10.1016/S1473-3099(09)70178-1. [DOI] [PubMed] [Google Scholar]
- 48.Banks DJ, Beres SB, Musser JM. The fundamental contribution of phages to GAS evolution, genome diversification and strain emergence. Trends Microbiol. 2002;10:515–521. doi: 10.1016/s0966-842x(02)02461-7. [DOI] [PubMed] [Google Scholar]
- 49.Nabozny GH, et al. HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: A novel model for human polyarthritis. J Exp Med. 1996;183:27–37. doi: 10.1084/jem.183.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cheng S, et al. Expression and function of HLA-DQ8 (DQA1*0301/DQB1*0302) genes in transgenic mice. Eur J Immunogenet. 1996;23:15–20. doi: 10.1111/j.1744-313x.1996.tb00260.x. [DOI] [PubMed] [Google Scholar]
- 51.Smoot JC, et al. Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks. Proc Natl Acad Sci USA. 2002;99:4668–4673. doi: 10.1073/pnas.062526099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kansal RG, McGeer A, Low DE, Norrby-Teglund A, Kotb M. Inverse relation between disease severity and expression of the streptococcal cysteine protease, SpeB, among clonal M1T1 isolates recovered from invasive group A streptococcal infection cases. Infect Immun. 2000;68:6362–6369. doi: 10.1128/iai.68.11.6362-6369.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Musser JM, et al. Streptococcus pyogenes causing toxic-shock-like syndrome and other invasive diseases: Clonal diversity and pyrogenic exotoxin expression. Proc Natl Acad Sci USA. 1991;88:2668–2672. doi: 10.1073/pnas.88.7.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Beres SB, et al. Genome sequence of a serotype M3 strain of group A Streptococcus: Phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc Natl Acad Sci USA. 2002;99:10078–10083. doi: 10.1073/pnas.152298499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McCool TL, Cate TR, Moy G, Weiser JN. The immune response to pneumococcal proteins during experimental human carriage. J Exp Med. 2002;195:359–365. doi: 10.1084/jem.20011576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pavlidis P, Noble WS. Matrix2png: A utility for visualizing matrix data. Bioinformatics. 2003;19:295–296. doi: 10.1093/bioinformatics/19.2.295. [DOI] [PubMed] [Google Scholar]
- 57.Earhart CA, Vath GM, Roggiani M, Schlievert PM, Ohlendorf DH. Structure of streptococcal pyrogenic exotoxin A reveals a novel metal cluster. Protein Sci. 2000;9:1847–1851. doi: 10.1110/ps.9.9.1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rödström KEJ, Elbing K, Lindkvist-Petersson K. Structure of the superantigen staphylococcal enterotoxin B in complex with TCR and peptide-MHC demonstrates absence of TCR-peptide contacts. J Immunol. 2014;193:1998–2004. doi: 10.4049/jimmunol.1401268. [DOI] [PubMed] [Google Scholar]
- 59.Lee KH, Wucherpfennig KW, Wiley DC. Structure of a human insulin peptide-HLA-DQ8 complex and susceptibility to type 1 diabetes. Nat Immunol. 2001;2:501–507. doi: 10.1038/88694. [DOI] [PubMed] [Google Scholar]