

Abstract
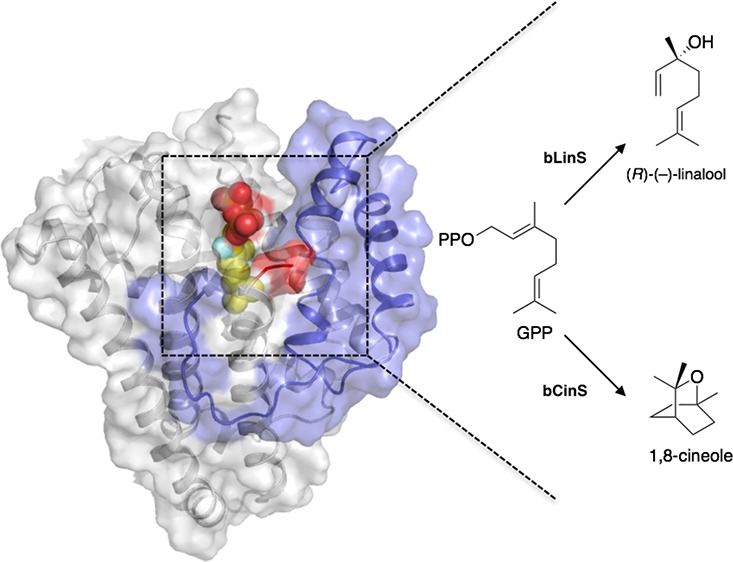
Terpenoids form the largest and stereochemically most diverse class of natural products, and there is considerable interest in producing these by biocatalysis with whole cells or purified enzymes, and by metabolic engineering. The monoterpenes are an important class of terpenes and are industrially important as flavors and fragrances. We report here structures for the recently discovered Streptomyces clavuligerus monoterpene synthases linalool synthase (bLinS) and 1,8-cineole synthase (bCinS), and we show that these are active biocatalysts for monoterpene production using biocatalysis and metabolic engineering platforms. In metabolically engineered monoterpene-producing E. coli strains, use of bLinS leads to 300-fold higher linalool production compared with the corresponding plant monoterpene synthase. With bCinS, 1,8-cineole is produced with 96% purity compared to 67% from plant species. Structures of bLinS and bCinS, and their complexes with fluorinated substrate analogues, show that these bacterial monoterpene synthases are similar to previously characterized sesquiterpene synthases. Molecular dynamics simulations suggest that these monoterpene synthases do not undergo large-scale conformational changes during the reaction cycle, making them attractive targets for structured-based protein engineering to expand the catalytic scope of these enzymes toward alternative monoterpene scaffolds. Comparison of the bLinS and bCinS structures indicates how their active sites steer reactive carbocation intermediates to the desired acyclic linalool (bLinS) or bicyclic 1,8-cineole (bCinS) products. The work reported here provides the analysis of structures for this important class of monoterpene synthase. This should now guide exploitation of the bacterial enzymes as gateway biocatalysts for the production of other monoterpenes and monoterpenoids.
Keywords: monoterpene synthase, monoterpenoids, sesquiterpene synthase, terpenes, protein crystallography, molecular dynamics simulations
Introduction
Terpenoids are the most abundant and largest class (>75000) of natural products. Most are commonly found in plants, and their biological roles range from interspecies communication to intracellular signaling and defense against predatory species.1 Their use is wide ranging as pharmaceuticals, herbicides, flavorings, fragrances, and biofuels.2 Despite the commercial interest in terpenoids, efforts to produce these in high yields have been hampered by lack of availability of sufficiently robust and high-activity terpene synthase enzymes, although efforts to synthesize terpenoids by synthetic biology routes have gathered pace in recent years.3−8
Terpenoids are synthesized from the isoprene building blocks dimethylallyl pyrophosphate (DMAPP) and isopentenyl pyrophosphate (IPP). Combination of DMAPP and IPP generates pyrophosphate substrates of varying carbon lengths, which are then utilized by terpene synthases to produce either monoterpenes (C10), sesquiterpenes (C15), diterpenes (C20), and others. Geranyl pyrophosphate (GPP), the substrate used by monoterpene synthases is formed by coupling one molecule of DMAPP with IPP, while farnesyl pyrophosphate (FPP), the substrate for sesquiterpenes, is synthesized by coupling three individual isoprene precursors.9
The class I terpene synthases share a common α-helical fold and use a cluster of three Mg2+ ions to assist with substrate ionization and release of the pyrophosphate moiety (PPi). This generates a reactive allylic carbocation and triggers a cyclization cascade that likely involves multiple carbocation intermediates.10 In many cases, substrate and Mg2+ binding lead to a closed active site conformation, which guides substrate orientation and protects the carbocation intermediates from premature quenching.11 The exact architecture and mobility of the active site is thought to control the cyclization cascade to the final carbocation intermediate with high fidelity. The latter is usually subject to deprotonation or addition of a water molecule, leading to formation of a single product. However, some natural terpene synthases and engineered variant forms have been shown to form multiple reaction products.12,13
To date, available crystallographic structures for the monoterpene cyclases/synthases (mTC/S) that accept GPP as the substrate has been derived only for plant enzymes. Structures have been reported for bornyl diphosphate synthase (Salvia officinalis),14 limonene synthase (Mentha spicata15 and Citrus sinensis),16,17 1,8-cineole synthase (Salvia fruticosa),18 and γ-terpinene synthase (Thymus vulgaris).19 Without exception, plant mTC/S contains two domains: a C-terminal α-helical catalytic domain that belongs to the class I terpenoid fold, and a N-terminal α-barrel domain with unclear function and that appears to be relictual. Though the overall sequence conservation is low, the structure of the α-helical fold is highly conserved. The active site has two conserved regions, the aspartate-rich (DDXX(X)(D,E)) motif and the NSE (NDXXSXX(R,K)(E,D)) triad, required for binding three catalytically essential Mg2+ ions. Structures of bornyl diphosphate synthase and limonene synthases have been solved in complex with substrate analogues. In each case, GPP-analogues bind with their pyrophosphate moieties coordinated by the Mg2+ ions and a network of residues that are proposed to assist with catalysis.
Recent reports have shown that terpene synthases are also widely distributed in bacteria, but the majority of these accept FPP as substrate and produce sesquiterpenes.20,21 Ohnishi and co-workers characterized two bacterial mTC/S from Streptomyces calvuligerus, namely, 1,8-cineole synthase22 and linalool/nerolidol synthase, which can accept either GPP or FPP as substrate, leading to linalool or nerolidol products, respectively.23 Heterologous expression of these enzymes in Streptomyces avermitilis resulted in 1,8-cineole synthase (bCinS) producing 1,8-cineole and linalool/nerolidol synthase (bLinS) producing only linalool, indicating that bLinS is likely to function only as a mTC/S in this host.20 The sequences of both bCinS and bLinS reveal they comprise ∼330 amino acids in a single catalytic domain and lack the additional N-terminal α-barrel domain characteristic of plant enzymes. Surprisingly, no closely related homologues of both enzymes have been found in other bacteria.24 The bacterial mTC/S 2-methylisoborneol synthase is present in many bacteria. It accepts 2-methyl-GPP as substrate to produce 2-methylisoborneol. Unlike the bacterial mTC/S reported here, 2-methylisoborneol synthase has a considerably longer amino acid sequence (∼400–500), and crystal structures have revealed a N-terminal proline-rich domain that is disordered along with a class I terpenoid fold C-terminal domain.25
Linalool is mainly used as a fragrance material in 60–80% of perfumed hygiene products. It is widely used in cosmetic products like perfumes, lotions, soaps, and shampoos and also in noncosmetic products like detergents and cleaning agents. Furthermore, during the manufacturing process of Vitamin E, linalool is a vital intermediate. As an important ingredient in a wide range of commercial products, the worldwide use of linalool exceeds 1000 metric tonnes per annum.26 Both R and S isomers of linalool are found in nature with R-(−)-linalool being the most widely distributed in plant and flower extracts. To our knowledge, for industrial use as a fragrance, the isomeric mixture is used. 1,8-Cineole (also called eucalyptol) is used as a flavoring in food products, in cosmetics, and also has medicinal properties.27
This study integrates synthetic biology with biocatalysis and analysis of enzyme structures and mechanisms. Here we describe high-resolution crystal structures of bLinS and bCinS from Streptomyces calvuligerus and complexes with fluorinated substrate analogues. These structures define the active site architectures required to steer reactive carbocation intermediates to the desired product outcomes. Expression of bLinS and bCinS in E. coli monoterpene-producing strains leads to improved production of linalool and 1,8-cineole compared with plant monoterpene synthases, and the structures help to both rationalize product outcomes and guide future exploitation of these enzymes in monoterpene/monoterpenoid production.
Experimental Section
Expression and Purification of bCinS and bLinS
The full-length genes coding for 1,8-Cineole synthase (bCinS; WP_003952918) and Linalool synthase (bLinS; WP_0003957954) from Streptomyces clavuligerus ATCC 27064 were codon optimized and synthesized from GeneArt (Life Technologies). The genes were amplified using PCR and subcloned into pETM11 vector digested with NcoI and XhoI using Infusion cloning (Clontech). The final construct coded for either 1,8-Cineole synthase (bCinS) or Linalool synthase (bLinS) with a 6X-His tag followed by a TEV protease cleavage site at the N-terminus. The expression and purification method explained below was identical for both the proteins. The plasmid was transformed into E. coli ArcticExpress (DE3) cells (Agilent), and a few colonies were inoculated into 100 mL of 2X-YT media containing 40 μg/mL of kanamycin and 20 μg/mL of gentamycin and grown for 3–4 h at 37 °C. The culture was diluted into 3 L of fresh 2X-YT media containing 40 μg/mL of kanamycin and allowed to grow at 37 °C until the OD at 600 nm reached 0.6–0.8. At this stage, the temperature was reduced to 16°C and 0.1 mM Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added and incubated for 14–18 h. The cells were harvested by centrifugation at 6000g for 10 min, and the pellet was resuspended in buffer A (25 mM Tris pH 8.0, 150 mM NaCl, 1 mM DTT, 4 mM MgCl2, and 5% (v/v) glycerol). The cells were lysed by sonication, and the debris was removed by centrifugation at 30 000g for 30 min. The supernatant was filtered through a 0.2 μm filter and loaded onto a 5 mL HisTrap column (GE Healthcare) pre-equilibrated with buffer A. The column was washed with buffer A containing 10 mM imidazole (pH 8.0) and increasing up to 40 mM imidazole by step gradients with 3 column volume for each concentration. Increasing the concentration of imidazole to 200–500 mM eluted the protein. The purified protein was desalted using a Centripure P100 column (emp Biotech GmbH) equilibrated with buffer A. To remove the His tag, TEV protease was added (1:1000 (w/w)) to the protein and incubated at 4 °C with gentle mixing for 24 h. The TEV protease was removed by passing the protein mixture through a 5 mL HisTrap column, and the flow through was collected. The His-tag removed protein was concentrated and loaded onto a Hiload Superdex (26/60) S75 column (GE Healthcare) pre-equilibrated with buffer A. Pure fractions from the gel filtration column were concentrated to 13–15 mg/mL and stored at −80 °C as aliquots. Samples for EPR experiments were prepared as explained above except buffer A was lacking MgCl2.
Biotransformations
Biotransformation reactions (0.25 mL) were prepared using buffer A and set up in glass vials containing 2 mM GPP and 20 μM of bCinS or bLinS. The vials were incubated at 25 °C with gentle shaking for 16 h. The vials were cooled to 4 °C, and 0.25 mL of ethyl acetate containing 0.01% (v/v) sec-butyl benzene as internal standard was added. The samples were vortexed for 2 min and then spun at 18 000g for 5 min. Supernatant fractions containing the ethyl acetate layer were removed and dried over anhydrous magnesium sulfate. Samples were analyzed by GC-MS.
Monoterpenoid Production in E. coli
Both bLinS and bCinS genes, including RBS, were amplified from their respective pETM-11 expression vectors using primers pET_IF_Fw (5′-CAT CCC CAC TAC TGA GAA TC-3′) and pET_IF_Rv (5′- GGT GGT GGT GCT CGA GTT A-3′) and cloned using InFusion (Takara) into plasmid pGPPSmTC/S15 (Table S1), which was PCR linearized using the primer pair Vector_IF_Fw (5′-TAA CTC GAG CAC CAC CAC CAC C-3′) and Vector_IF_Rv (5′-TCA GTA GTG GGG ATG TCG TAA TCG-3′) resulting in plasmids pGPPSmTC/S38 and pGPPSmTC/S39, respectively (Table S1). Correct insertion was confirmed by automated sequencing (Eurofins).
For monoterpenoid production, the pGPPSmTC/S plasmids were cotransformed with pMVA into E. coli DH5α and grown as described before.3 Briefly, expression strains were inoculated in terrific broth (TB) supplemented with 0.4% glucose in glass screw capped vials, and induced for 72 h at 30 °C with 50 μM IPTG and 25 nM anhydro-tetracycline. A 20% n-nonane layer was added to capture the volatile terpenoids products. After induction, the nonane overlay was collected, dried over anhydrous MgSO4, and mixed at a 1:1 ratio with ethyl acetate containing 0.1% (v/v) sec-butyl benzene as internal standard.
GC-MS Analysis
Samples were injected onto an Agilent Technologies 7890B GC equipped with an Agilent Technologies 5977A MSD. The products were separated on a DB-WAX column (30 m × 0.32 mm i.d., 0.25 μm film thickness, Agilent Technologies). The injector temperature was set at 240 °C with a split ratio of 20:1 (1 μL injection). The carrier gas was helium with a flow rate of 1 mL/min and a pressure of 5.1 psi. The following oven program was used: 50 °C (1 min hold), ramp to 68 °C at 5 °C/min (2 min hold), and ramp to 230 °C at 25 °C/min (2 min hold). The ion source temperature of the mass spectrometer (MS) was set to 230 °C, and spectra were recorded from m/z 50 to m/z 250. Compound identification was carried out using authentic standards and comparison to reference spectra in the NIST library of MS spectra and fragmentation patterns as described previously.3
GC Analysis
To determine the chirality of linalool and nerolidol produced by bLinS, samples were analyzed by gas chromatography on an Agilent Technologies 7890A GC system equipped with an FID detector, a 7693 autosampler, and a CP-Chirasil-DEX-CB column (25 m × 0.25 mm i.d., 0.25 μm film thickness). The biotransformation samples and isomers of linalool and nerolidol standards were analyzed using GC. In this method, the injector temperature was at 180 °C, and 1 μL of sample was injected split-less. The carrier gas was helium with a flow rate of 1 mL/min and a pressure of 11.3 psi. For nerolidol-containing samples, the program began at a temperature of 70 °C and then increased to 150 °C at 8 °C/min (2 min hold). This was followed by an increase in temperature to 190 °C at 10 °C/min (3 min hold). For linalool-containing samples, the program began at a temperature of 70 °C which was then increased to 90 °C at 8 °C/min. This was followed by an increase in temperature to 150 °C at a rate of 2 °C/min and then to 190 °C at 40 °C/min (1 min hold). The FID detector was maintained at a temperature of 200 °C with a flow of hydrogen at 30 mL/min.
Chemical Synthesis of Fluorinated Substrate Analogues
Unless otherwise stated, all reactions were carried out in oven-dried glassware. Reactions were monitored by thin-layer chromatography (TLC) on silica gel 60 F254 plates, visualized with phosphomolybdic acid stain (10 g of phosphomolybdic acid in 100 mL of ethanol). Column chromatography was performed on Merck silica Gel 60 (particle size 40–63 μm). 1H NMR, 13C NMR, 31P, and 19F spectra were obtained using a combination of 400 and 500 MHz spectrometers and are reported as chemical shift on the parts per million scale. Multiplicity abbreviated (br = broad, s = singlet, d = doublet, dd = double doublet, t = triplet, m = multiplet, etc.) and coupling constants were obtained in Hertz. Assignments were aided by COSY and HSQC. All mass spectrometry results are reported as the mass to charge ratio and are reported with % abundance against the base peak (100%).
Synthesis of 2-Fluorogeraniol and 2-Fluoronerol
Sodium hydride (538 mg, 60% dispersion, 13.5 mmol) was washed with petroleum ether and suspended in THF (40 mL). The suspension was cooled to 0 °C, and a solution of the ethyl (diethoxyphosphoryl) fluoroacetate (2.48 mL, 12.2 mmol) in THF (13.4 mL) was added dropwise over 10 min. The reaction was stirred for 30 min before adding 6-methyl-5-hepten-2-one (1.5 mL, 10.2 mmol) dropwise over 30 min. The reaction was stirred overnight at room temperature. The reaction was cooled back to 0 °C and quenched by pouring on to ice water. The product was extracted with diethyl ether (3 × 30 mL), dried over MgSO4 and then reduced to dryness. The crude product was then dissolved in THF (64 mL), cooled to 0 °C, and LiAlH4 (541 mg, 14.3 mmol) was added. The reaction was stirred at room temperature for 3 h then quenched with the addition of saturated aqueous NH4Cl. The solution was extracted with diethyl ether (3 × 30 mL) and the subsequent combined organic phases were washed with brine (30 mL). The product was purified by column chromatography (hexane/diethyl ether, 95/5, v/v) to give 2-fluorogeraniol (783 mg, 41%) and 2-fluoronerol (856 mg, 45%) with a total yield of 86%25,28 (Scheme S1). 2-fluorogeraniol: 1H NMR (400 MHz, CDCl3) δ 5.17–5.05 (m, 1H, H7), 4.24 (dd, J = 22.3, 3.2 Hz, 2H, H1), 2.10 (m, 4H, H5, H6), 1.68 (s, 3H, H4), 1.67 (d, J = 2.9 Hz, 3H, H9/10), 1.61 (s, 3H, H9/10). 13C NMR (101 MHz, CDCl3) δ 132.2, 123.6, 116.1 (d, J = 16.2 Hz), 58.2 (d, J = 29.3 Hz), 29.8 (d, J = 7.07 Hz), 25.9 (d, J = 4.0 Hz), 25.7, 17.6, 15.4 (d, J = 6.01 Hz). 19F NMR (471 MHz, CDCl3) δ −121.3 (t, J = 22.4 Hz) (Figure S1a–e). 2-fluoronerol:1H NMR (400 MHz, CDCl3) δ 5.16–5.02 (m, 1H, H7), 4.19 (dd, J = 22.9, 5.3 Hz, 2H, H1), 2.16–2.00 (m, 4H, H5, H6), 1.70 (s, 3H, H4), 1.60 (s, 3H, H9/10), 1.57 (s, 3H, H9/10). 13C NMR (101 MHz, CDCl3) δ 133.3, 123.5, 115.9 (d, J = 14.1 Hz), 58.0 (d, J = 29.2 Hz), 31.9 (d, J = 5.1 Hz), 26.6 (d, J = 5.1 Hz), 25.8, 17.9, 13.7 (d, J = 9.1 Hz). 19F NMR (471 MHz, CDCl3) δ – 119.4 (t, J = 22.6 Hz) (Figure S2a–e).
Synthesis of 2-Fluorogeranyl Pyrophosphate (FGPP) and 2-Fluoroneryl Pyrophosphate (FNPP)
Acetonitrile (60 mL) was added to 2-fluorogeraniol or 2-fluoronerol (400 mg, 2.3 mmol). To this, trichloroacetonitrile (2 mL) was added followed by H3PO4(Et3N)2 salt (2.1 g). The reaction was stirred overnight. It was then poured on to diethyl ether (50 mL) and washed with concentrated aqueous ammonia (3 × 100 mL). The ammonia washes were combined and washed once with diethyl ether (50 mL). The aqueous phase was reduced to dryness. The crude product was loaded on to a silica gel column, and the starting material was recovered using petroleum ether/diethyl ether (9/1, v/v). The eluent system was then switched to propanol/concentrated aqueous ammonia/water (7/2/1, v/v/v) to isolate mono and pyrophosphate derivatives (Scheme S1). When 2-fluorogeraniol was used, 2-fluorogeranyl monophosphate (93 mg, 0.37 mmol, 16%) and 2-fluorogeranyl pyrophosphate (74 mg, 0.2 mmol, 10%) were obtained. When 2-fluoronerol was used, 2-fluoroneryl monophosphate (117 mg, 0.47 mmol, 20%) and 2-fluoro neryl pyrophosphate (200 mg, 0.54 mmol, 27%) were obtained. 2-fluorogeraniol pyrophosphate: 1H NMR (500 MHz, D2O) δ 5.29–5.15 (m, 1H, H7), 4.59 (dd, J = 23.6, 6.2 Hz, 2H, H1), 2.22–2.09 (m, 4H, H5, H6), 1.72 (d, J = 2.8 Hz, 3H, H4), 1.69 (s, 3H, H9/10), 1.62 (s, 3H, H9/10). 13C NMR (126 MHz, D2O) δ 133.9 (s), 123.7 (s), 119.5 (d, J = 15.0 Hz), 60.9 (dd, J = 31.6, 5.0 Hz), 29.1 (d, J = 6.4 Hz), 25.1 (s), 24.8 (s), 16.9 (s), 14.6 (d, J = 4.9 Hz). 31P NMR (162 MHz, D2O) δ – 8.37 (d, J = 21.5 Hz), – 10.95 (d, J = 21.6 Hz). 19F NMR (471 MHz, D2O) δ −120.44 (t, J = 23.6 Hz). HRMS ESI C10H18FO7P2 [M–H]− calculated: 331.0512, found: 331.0517 (Figure S3a–g). Data was found to be in accordance with the literature.16,292-fluoronerol pyrophosphate: 1H NMR (400 MHz, D2O) δ 5.14–5.06 (m, 1H, H7), 4.50 (dd, J = 23.6, 6.1 Hz, 2H, H1), 2.07 (br s, 4H, H5, H6), 1.62 (s, 6H, H4, H9/10), 1.54 (s, 3H, H9/10). 13C NMR (126 MHz, D2O) δ 134.3 (s), 123.3 (s), 119.1 (d, J = 13.2 Hz), 60.4 (dd, J = 31.0, 4.3 Hz), 30.8 (d, J = 4.6 Hz), 25.7 (d, J = 2.7 Hz), 24.8 (s), 16.9 (s), 12.8 (d, J = 8.6 Hz) 31P NMR (162 MHz, D2O) δ – 7.64 (d, J = 21.4 Hz), – 10.93 (d, J = 20.9 Hz) 19F NMR (471 MHz, D2O) δ – 119.13 (t, J = 23.1 Hz). HRMS ESI C10H18FO7P2 [M–H]− calculated: 331.0512, found: 331.0517 (Figure S4a–h). Data was found to be in accordance with the literature.16,29
Crystallization of bCinS and bLinS
Crystallization trials containing 200 nl of protein and 200 nl of precipitant solution were set up in 3-well swissci plates using a mosquito robot (TTP Labtech). Five commercial screens, namely, Morpheus I and II, JCSG+, PACT premier and SG1 (Molecular Dimensions Ltd.) were used in initial trails. For both enzymes, three distinct samples were screened: the apo-enzyme, the enzyme in the presence of 2 mM FGPP, and the enzyme in the presence of 2 mM FNPP. The bCinS-FNPP crystallized in Morpheus II A4 condition (90 mM of LiNaK (0.3 M lithium sulfate, 0.3 M sodium sulfate, 0.3 M potassium sulfate), 0.1 M of buffer system 4 (1 M MOPSO, 1 M Bis-Tris) pH 6.5 and 50% precipitant mix 8 (10% PEG 20000, 50% trimethylpropane, 2% NDSB 195)). The bLinS-FGPP crystallized in Morpheus D7 condition (0.12 M Alcohols (0.2 M 1,6-hexanediol, 0.2 M 1-nutanol, 0.2 M 1,2-propanediol, 0.2 M 2-propanol, 0.2 M 1,4-butanediol, 0.2 M 1,3-propanediol), 0.1 M Buffer System 2 (1.0 M sodium HEPES, MOPS (acid)) pH 7.5 and 50% v/v precipitant Mix 3 (40% v/v glycerol, 20% w/v PEG 4000)). The apo-LinS crystallized in SG1 E2 condition (25% w/v PEG3350). Although apo-bCinS crystallized, optimization of growth conditions failed to produce single crystals of sufficient size for further study. In an attempt to obtain the bCinS-FGPP structure, bCinS-FNPP crystals were soaked overnight in the presence of 2 mM FGPP prior to cryo-cooling. The apo-bLinS crystals were cryo-protected by soaking in mother liquor supplemented with 20% glycerol. For all FGPP and FNPP complexes, the ligands were included in the cryo-solution.
Structure Solution
All data were collected at Diamond Light Source (DLS). Diffraction images were integrated and scaled by xia230 automated data processing pipeline, using XDS31 and XSCALE. Crystals of bCinS contained two molecules in the asymmetrical unit and belonged to P1 space group. Crystals of bLinS belonged to the tetragonal system (spacegroup I4) and also contained two molecules in asymmetrical unit. The bLinS structures (apo-bLinS and bLinS-FGPP) were solved by molecular replacement using the Pentalenene synthase structure (PDB: 1PS1(32)) as the search model in Phaser.33 The bCinS-FNPP structure was solved by model replacement using the apo-bLinS structure as the search model. The apo-bLinS, bLinS-FGPP, bCinS-FNPP and bCinS-FNPP/FGPP models were built using Autobuild in Phenix.34 The structures were completed using iterative rounds of manual model building in coot35 and refinement in phenix.refine.36 The structures were analyzed using PDB_REDO37 and validated using molprobity tools.38 The refinement statistics are provided in Table 1. The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 5NX4, 5NX5, 5NX6 and 5NX7.
Table 1. X-ray Data Collection and Refinement Statistics.
| bLinS (apo) | bLinS-FGPP | bCinS-FNPP | bCinS-FNPP/FGPP | |
|---|---|---|---|---|
| data collection | ||||
| space group | I4 | I4 | P1 | P1 |
| unit cell dimensions | a = b = 140.15 Å, c = 87.18 | a = b = 139.37 Å, c = 86.06 | a = 60.81 Å, b = 60.83 Å, | a = 60.75 Å, b = 60.83 Å, |
| Å; α = β = γ = 90° | Å; α = β = γ = 90° | c = 64.10 Å; α = 90.04°, | c = 64.26 Å; α = 92.67°, | |
| β = 92.89°, γ = 101.98° | β = 89.98°, γ = 101.77° | |||
| X-ray source | DLS I04-1 | DLS I04 | DLS I04 | DLS I04 |
| wavelength (Å) | 0.92819 | 0.99 | 0.99 | 0.9795 |
| resolution range (Å) | 50.90–2.38 (2.42–2.38) | 36.62–1.82 (1.85–1.82) | 32.27–1.63 (1.66–1.63) | 64.19–1.51 (1.53–1.51) |
| multiplicity | 4.5 (3.9) | 6.8 (6.9) | 1.8 (1.8) | 2 (2) |
| I/σ I | 14.6 (1.7) | 18.2 (1.4) | 6.7 (1.2) | 10 (2.1) |
| completeness (%) | 99.7 (99.7) | 100 (100) | 96 (94.6) | 95.9 (93.4) |
| Rmerge | 0.082 (0.766) | 0.057 (1.371) | 0.09 (0.639) | 0.035 (0.315) |
| Rmeas | 0.093 (0.889) | 0.062 (1.483) | 0.127 (0.904) | 0.047 (0.428) |
| Rpim | 0.043 (0.44) | 0.024 (0.563) | 0.09 (0.639) | 0.032 (0.287) |
| CC1/2 | 0.998 (0.576) | 0.999 (0.51) | 0.987 (0.479) | 0.999 (0.824) |
| total observations | 152818 (6641) | 502331 (25222) | 193529 (9539) | 270436 (13510) |
| total unique | 33849 (1697) | 73739 (3654) | 107492 (5258) | 136704 (6737) |
| refinement | ||||
|---|---|---|---|---|
| R-work | 0.1850 | 0.1667 | 0.1662 | 0.1492 |
| R-free | 0.2240 | 0.1950 | 0.20 | 0.1830 |
| RMS (bonds) | 0.002 | 0.009 | 0.013 | 0.008 |
| RMS (angles) | 0.45 | 0.94 | 1.15 | 0.96 |
| average B-factor (Å2) | 52.5 | 42.8 | 24.8 | 27.8 |
| Ramachandran plot | ||||
|---|---|---|---|---|
| favored | 98.94 | 99.29 | 98.98 | 98.45 |
| allowed | 1.06 | 0.71 | 1.02 | 1.35 |
| outliers | 0 | 0 | 0 | 0.2 |
EPR Spectroscopy
Electron paramagnetic resonance (EPR) measurements were carried out using a Bruker ELEXSYS-500 X-band EPR spectrometer operating in both cw and pulsed modes, equipped with an Oxford variable-temperature unit and ESR900 cryostat with Super High-Q resonator. All EPR samples were prepared in the quartz capillary tubes (outer diameter; 4.0 mm, inner diameter 3.0 mm) and frozen in liquid N2. The X-band EPR tubes were then transferred into the EPR probe head, which was precooled to 20 K. The low-temperature EPR spectra were measured at 20 K as a frozen solution. A microwave power of 36 dB (50 mW) and modulation of 5 G appear to be optimal for recording the EPR spectrum of the bLinS and bCinS protein samples prepared using various ratios of protein to Mn2+ concentration in the presence of 10-fold excess of FGPP. The concentrations of the proteins (bLinS and bCinS) and FGPP in all the samples were 0.400 mM and 1.5 mM, respectively, whereas the ratio to the Mn2+ concentration was systematically varied from 1 to 6. The low-temperature EPR spectra were acquired using the following conditions: sweep time of 84 s, microwave power of 50 mW, time constant of 41 ms, and modulation amplitude of 5 G. All the spectra have been normalized to account for the different numbers of scans accumulated for each sample. The data analysis was performed using EasySpin toolbox for the Matlab program package.
Simulations of Apo-bCinS and bLinS
Molecular dynamics (MD) simulations of apo-bCinS and bLinS were carried out in AMBER14 using the CHARMM27 force field.39,40 The protonation states of titratable residues were estimated using the PDB 2PQR server with proPKA, and the enzymes were solvated using a box of minimum 12 Å around the protein with counterions added. Two sets of isothermal–isobaric ensemble (NPT) MD simulations were performed at 298 K for each enzyme, using different starting velocities, following the system setup. Langevin dynamics was used for temperature control (collision frequency of 5 ps–1 for equilibration and 2 ps–1 for production), and pressure was controlled by coupling to an external bath (AMBER14 default settings) for NPT conditions. The system setup consisted of: (i) energy minimization of the solvent; (ii) 50 ps of (NPT) solvent equilibration; (iii) energy minimization of the entire system with positional restraints of 5 kcal mol–1 Å–2 applied to all Cα atoms; (iv) canonical ensemble (NVT) thermalisation to 298 K over 20 ps with positional restraints of 5 kcal mol–1 Å–2 on Cα atoms; (v) 40 ps of NPT equilibration with decreasing restraints on the Cα atoms; (vi) 1 ns unconstrained NPT equilibration; (vii) 100 ns production simulation. Average linkage hierarchical clustering (after alignment of structures based on Cα positions) was used to identify representative structures to illustrate protein conformational sampling during the simulations.
Simulations of the Ternary Complexes of bCinS with Three Mg2+ Ions and GPP or NPP
The protonation states of titratable residues were estimated using PropKA3.1,41,42 and the enzyme was solvated using a box of TIP3P43 water molecules (with a minimum buffer or 13 Å around the protein) using the solvate plugin of the VMD package.44 Counterions were added to neutralize the system using autoionize plugin of VMD.44 The CHARMM27 forcefield39 was used to describe the protein with parameters for GPP and NPP that were adapted from those used for FPP in the work of van der Kamp et al.45 The position of the GPP or NPP substrate was based on the position of the fluorinated analogue resolved in the crystal structure. Due to the minimal differences in the structure of the inhibitor and substrate (F vs H), the position in the crystal structure was considered a suitable starting point for the simulations. It has been suggested that many terpene cyclase/synthase structures contain substrates bound in unreactive conformations;46,47 however, structures containing the larger and more flexible FPP, the building block for sesquiterpenes are more prevalent than monoterpenes. The parameter set developed by Allner et al.48 was used to describe the three Mg2+ ions. The setup of the model consisted of the following: (i) minimization of the positions of the hydrogen atoms (all heavy atoms fixed); (ii) minimization of the solvent (with all protein heavy atoms fixed); (iii) energy minimization of the entire system with positional restraints of 5 kcal mol–1 Å–2 applied to all Cα atoms; (iv) canonical ensemble (NVT) thermalisation to 300 K over 20 ps with positional restraints of 5 kcal mol–1 Å–2 on Cα atoms; (v) thermal equilibration at 300 K for 100 ps with positional restraints of 5 kcal mol–1 Å–2 on Cα atoms; (vi) 140 ps of NPT equilibration with decreasing restraints on the Cα atoms; (vi) 100 ns production simulation. Two sets of isothermal–isobaric ensemble (NPT) MD simulations were performed at 300 K for each enzyme, repeating steps (iv)–(vi) to obtain two models with different initial conditions. MD simulations were carried out on GPUs using the PMEMD code49 of AMBER16.50 Langevin dynamics was used for temperature control (collision frequency of 5 ps–1 for equilibration and 2 ps–1 for production), and pressure was controlled by coupling to an external bath (AMBER16 default settings) for NPT conditions. Average linkage hierarchical clustering (after alignment of structures based on positions of active site residues) was carried out using the CPPTRAJ utility of AMBERTOOLS 1650 to identify representative structures of the ternary complex over the course of the simulations.
Simulations of the Ternary Complexes of bLinS with Three Mg2+ Ions and GPP or FPP
The models of bLinS were built from the coordinates of chain B of the protein, with positions of the Mg2+ ions determined on the basis of alignment with the structures of sesquiterpene synthases aristocholene synthase (ATAS, PDB 4KUX51) and Epi-isozizaene synthase (PDB 3KB9(52)). GPP was built into the model on the basis of the position of the phosphate ion observed in the bLinS chain B structure and using the geometry of FGPP observed in the bCinS-FGPP structure. The FPP model was generated on the basis of the position of farnesyl thiolodiphosphate FSPP in ATAS.51 Some positional restraints were then applied to the Mg2+ ions and coordinating protein residues in the NSD and DDXXD motifs in order to form the correct binding pattern. The Mg2+ to oxygen atom distance (for Asn218, Ser222, Asp226 and Asp79) was restrained to a value of 2.3 Å with a force constant k = 20 kcal mol–1 Å–2. The same procedure as used for the bCinS models was then followed to perform the MD simulations of bLinS with GPP and FPP.
Results and Discussion
Linalool and 1,8-Cineole Production in E. coli
Biotransformation reactions showed that purified bLinS and bCinS produced linalool and 1,8-cineole, respectively, when supplied with GPP. No byproducts were observed when analyzed by GC-MS (Figure 1). To investigate the suitability of both enzymes for monoterpenoid production in engineered E. coli strains, bLinS and bCinS were inserted in an E. coli “plug-and-play” monoterpenoid production platform, which consists of two gene modules.3 The first module (pMVA) contains a hybrid mevalonate (MVA) pathway under regulation of IPTG-inducible promoters,53 and the second (plasmid series pGPPSmTC/S, Table S1) comprises a refactored, N-terminally truncated geranyl diphosphate synthase (GPPS) gene from Abies grandis (AgtrGPPS2) followed by an mTC/S gene (in this case bLinS or bCinS, respectively) under control of a tetracycline-inducible promoter. Strains containing both the pMVA and pGPPS-bLinS or pGPPS-bCinS plasmids, respectively, were grown in a two-phase shake flask system using glucose as the feedstock and n-nonane as an organic phase to facilitate product capture. Products accumulated in the organic phase were identified and quantified by GC-MS analysis.
Figure 1.

GC-MS analysis of bCinS and bLinS. (A) bCinS product profile when inserted in an engineered E. coli strain capable of overproducing GPP. (B) bCinS conversion of GPP (2 mM) in vitro. (C) bCinS conversion of NPP (2 mM) in vitro. D) 1,8-cineole standard (0.1 mg/mL). E) bLinS product profile when inserted in an engineered E. coli strain capable of overproducing GPP. F) bLinS conversion of GPP (2 mM) in vitro. G) R-(−)-linalool standard (0.1 mg/mL). H) cis- and trans-nerolidol standards (0.1 mg/mL). IS = internal standard (sec-butyl benzene).
Product profiles and titers obtained with bLinS and bCinS were compared with previously obtained profiles using mTC/S enzymes obtained from plants (Figure 1), i.e. LinS from Artemisia annua (RLinS_Aa) and CinS from Salvia fruticosa (CinS_Sf), Arabidopsis thaliana (CinS_At), and Citrus unshiu (CinS_Cu).3 Both bacterial enzymes outperformed the plant enzymes: bLinS produced about 300-fold more linalool than RLinS_Aa (363.3 ± 57.9 versus 1.3 mg Lorg–1). With bCinS, 1,8-cineole was produced in considerably purer form compared to that produced using the plant enzymes. Strains containing bCinS produced 116.8 ± 36.4 mg Lorg–1 (96% pure); this compares to 118.2 mg Lorg–1 (67% pure) for CinS_Sf, 46.6 mg Lorg–1 (42% pure) for CinS_At, and 18.2 (63% pure) for CinS_Cu for the strains containing the corresponding plant CinS enzymes.
As well as GPP formation catalyzed by the heterologous GPPS, the engineered E. coli strains also produce the sesquiterpene precursor farnesyl diphosphate (FPP) from native host encoded enzymes.54 Strains containing bLinS were able to convert FPP to nerolidol (159.1 ± 7.3 mg Lorg–1), indicating that bLinS acts as both a monoterpene and sesquiterpene synthase. We demonstrated that bLinS makes R-(−)-linalool and trans-nerolidol with GPP and FPP, respectively (Figure S5a–f). In contrast, no sesquiterpene products were detected with E. coli strains containing bCinS indicating it is restricted to the production of monoterpene products. With each of the strains, geraniol and farnesol (and their derivatives) were detected in organic overlays of cultures alongside the expected terpenoids. An unidentified endogenous E. coli pathway has previously been shown to convert both GPP and FPP into geraniol and farnesol respectively,3 which are subsequently converted into oxidative byproducts by endogenous dehydrogenation and isomerization reactions.55 In particular, E. coli PhoA phosphatase was implicated in converting GPP to geraniol56 and two integral membrane phosphatases (PgpB and YbjG) were shown to convert FPP to farnesol.57
The reported product profiles and yields suggest that bacterial monoterpene synthases are better suited compared to the corresponding plant enzymes for monoterpenoid production using engineered E. coli strains. Armed with this information we set out to determine the structures of bLinS and bCinS, in both ligand-free and complexed with fluorinated substrate analogues, with the objective of informing on mechanism, and guiding future engineering/exploitation in biocatalysis and metabolic engineering programmes.
Structure of the bCinS FNPP Complex
Crystals of bCinS were obtained when cocrystallized with 2-fluoro neryl pyrophosphate (FNPP), a fluorinated GPP isomer. Unfortunately, bCinS crystallized poorly when not bound to a substrate analogue. This suggests a conformational change occurs between an open (apo)-form and a closed (substrate-inhibitor bound) complex similar to that seen with other terpene cyclases.45,58 Previous studies have indicated that some terpene cyclases/synthases can also accept neryl pyrophosphate (NPP) as substrate.16 In the case of bCinS, incubation with NPP also leads to 1,8 cineole (Figure 1). As observed with other terpene synthases, fluorination of the substrate blocks the key ionization step, blocking diphosphate release and formation of the geranyl/neryl cation.28 The bCinS-FNPP structure was determined to 1.63 Å and reveals the enzyme is a dimer of a typical class I terpenoid α-helical domain, with the active sites oriented in an antiparallel fashion (Figure 2a). Analysis of the bCinS dimer revealed a total buried surface area of 4114 Å2, indicating the oligomeric state is biologically relevant (using PISA59). Both monomers are similar in structure (rmsd of 0.25 Å over 315 Cα atoms), with residues that constitute one of the loops close to the active site disordered. The bound FNPP is clearly defined in the electron density of both active sites, with no significant differences in conformation between both monomers (Figure 2b). The pyrophosphate moiety of FNPP makes extensive interactions with residues in the active site, in addition to coordination by two Mg2+ ions and interactions with several water molecules. While one Mg2+ is bound by the conserved NSE motif (Mg2+ B), the other is bound by the aspartate rich motif (Mg2+ A). No clear density could be observed that corresponds to the location of the third metal ion (Mg2+ C).
Figure 2.

Structure of bCinS in complex with FNPP. (A) Cartoon representation of the bCinS dimer with the solvent accessible surface shown color coded per monomer. (B) Stereoview of the FNPP-Mg2+ ion binding site. Key polar interactions are shown by dotted lines. The electron density indicates multiple positions of the diphosphate moiety as well as several Mg2+ binding residues. (C) Stereoview of the FNPP hydrophobic binding pocket. A single water molecule is present, located close to the C6 atom.
EPR Reveals Binding of 3 Mn2+ Ions to bCinS
To ascertain whether bCinS binds to two or three Mg2+ ions, we employed EPR spectroscopy by titrating bCinS purified in the absence of MgCl2 with Mn2+. The Mn2+ ion serves as a valuable probe of the Mg2+ ion binding sites.60−62 This substitution allowed application of cw-EPR spectroscopy to investigate the number of potential metal binding sites in bCinS. Comparison of the EPR spectra of the aqueous MnCl2 and bCinS with and without the inhibitor FGPP indicates that the spectrum of the 1:1 bCinS-FGPP:Mn2+ sample contains a highly resolved multiplet structure (Figure 3a; red spectrum). This multiplet structure is the55Mn hyperfine coupling which is due to the interaction of electron spin (S = 5/2) of the Mn2+ ion with the nuclear spin (I = 5/2) of 55 Mn nucleus. It is a characteristic signature of binding of FGPP to Mn2+ ion, which is centered at g ∼ 2.0. This multiplet feature increases in intensity only until the ratio of Mn2+ ion concentration relative to bCinS-FGPP reaches 3. However, where the relative concentration of Mn2+ ion is greater than 3, the EPR spectra show overall increase in intensity due to the contribution from free/unbound Mn2+ ion. The EPR spectrum of the 1:6 bCinS-FGPP:Mn2+ sample (Figure 3a; magenta spectrum) can be simulated (Figure 3a; cyan spectrum) by 1:1 addition of the EPR spectra of 1:3 bCinS-FGPP:Mn2+ sample with (Figure 3a; blue spectrum) the 1:3 bCinS:Mn2+ (Figure 3a; black spectrum). This indicates that there are 3 potential metal binding sites available in bCinS. Detailed analysis and assignment of the various transitions in the EPR spectra (Figures S6a,b and S7a,b) are provided in the Supporting Information.
Figure 3.

EPR confirms binding of 3 Mn2+ in solution. cw-EPR spectra of ‘Mn2+’ substituted bCinS (A) and bLinS (B) protein samples with varying equivalents of Mn2+ concentration with or without FGPP measured as a frozen solution along with standard MnCl2. The plot shows the multiplet EPR signal arise from the Mn2+ ion around the g = 2 region (from 250 to 400 mT).
Structure of the bCinS-FGPP Complex and bCinS Mechanism
Soaking of the bCinS-FNPP crystals with FGPP led to the partial exchange of the inhibitor in both monomers (structure determined to 1.51 Å resolution). Besides the obvious reorientation of the carbon skeleton, the presence of FGPP does not lead to active site reconfiguration. However, the soaking protocol used has led to clear electron density of a partially occupied third Mg2+ ion (Mg2+ C; Figure 4). This in turn is accompanied by a modest change in conformation of the E155 region (Figure 4a), bringing the E155 side chain into close contact with water molecules ligating Mg2+ C. Given the partial occupancy of the inhibitors and of the E155/Mg2+ C, it is unclear whether there is a direct link between the nature of the ligand bound in the active site and the binding of Mg2+ C. However, as both GPP and NPP act as substrates for bCinS, presumably both requiring binding of three Mg2+ ions, it seems plausible the soaking procedure used is responsible for the observed changes in the E155 region and the associated Mg2+ C binding.
Figure 4.

Structure of bCinS in complex with FGPP. (A) Cartoon representation of an overlay of the bCinS-FNPP complex structure (in gray) with the bCinS-FGPP/FNPP structure obtained by soaking bCinS-FNPP with GFPP (in blue). A loop region C-terminal to D85 as well as the region surrounding E155 adopt distinct conformations in response to binding of Mg2+ C, these regions are colored in red. (B) Stereoview of the FGPP/FNPP-Mg2+ ion binding site. Key polar interactions are shown by dotted lines.
On the basis of the bCinS-FNPP and bCinsS-FGPP/FNPP structures, a mechanism for the bacterial 1,8-cineole synthesis can be proposed, by analogy to observations made with plant monoterpene synthases18 (Figure 5). Unlike FGPP, the carbon chain conformation of FNPP (and by extension the NPP substrate) is compatible with cyclization of the initial carbocation (in this case linalyl) derived from substrate ionization to form the (R)-terpinyl cation. Indeed, the FNPP C1 and C6 atoms are placed at a distance of ∼3.6 Å. In contrast, steric constraints require the FGPP carbon skeleton to undergo an isomerization step following substrate ionization and geranyl carbocation formation prior to cyclization. For other monoterpene cyclase enzymes, this has been proposed to occur via transient formation of linalyldiphosphate and concomitant change from the transoid to cisoid configuration.14,15 A second substrate ionization step then generates the linalyl carbocation species, which can proceed to the cyclization step. The fact that both GPP and NPP result in the same product suggests the exact configuration of the respective linalyl carbocation species (GPP versus NPP derived) and resulting terpinyl carbocation are similar, resembling the carbon chain configuration of the FNPP inhibitor. However, recent solution studies using labeled GPP have suggested bCinS proceeds via the (S)-terpinyl cation, in contrast to the (R)-terpinyl configuration proposed on the basis of the bCinS-FNPP crystal structure.63 Following formation of the terpinyl carbocation, conversion via the final cyclization step to the 1,8-cineol product is proposed to occur via a syn addition.63 With the exception of a single water molecule, coordinated by Trp58 and Asn305, the hydrophobic binding pocket is devoid of solvent. This water molecule is placed at a distance of ∼3.6 Å to the C6 of FNPP (Figure 2c), and thus appears the most likely candidate for nucleophilic attack on the terpinyl cation. MD simulations show that this water molecule remains at an average distance of 3.84 (±0.45 run1, (±0.53 run2) Å from C7 of GPP throughout the 100 ns simulation (Figure 6). The water molecule interacts with Asn305, but no longer interacts with Trp58. Figure 6A–C show the different positions of the hydrocarbon tail of GPP and NPP in the representative structure from the dominant cluster for the 100 ns simulations. The hydrocarbon tail of NPP occupies the position adopted by the side chain of Phe77 in the simulations of bCinS with GPP. There are more water molecules near to C7 of NPP and the shortest distance is not with a single water molecule throughout the entire simulation, as was observed for GPP. However, simulations with NPP show that the average position of the water molecule is more distant than in the bCinS/GPP system with an average C7–WAT O distance of 4.26 ± 0.59 Å run1 and 4.42 ± 0.59 Å in run2 (Figure 6d,e). Formation of the neutral α-terpineol through deprotonation is avoided by the lack of any suitable acid–base group in close proximity of this water molecule. Production of the bicyclic 1,8-cineole from the protonated α-terpineol species is proposed to occur via intramolecular proton transfer to C2, followed by C2–O bond formation leading to formation of the second cycle. Considering the relative position of the water molecule and the C2 atom in the FNPP structure, this scenario will require some conformational changes to occur. This is distinct from the proposed mechanism for the plant 1,8 cineole synthase, for which a syn addition of water is proposed, requiring no significant conformational changes prior the ensuing heterocyclization step.64
Figure 5.

Mechanistic proposal for bCinS. A schematic outline of a putative mechanism for the conversion of GPP and NPP to the 1,8 cineole product by bCinS. Taking into account the observed position and orientation of the bCinS ligands and adjacent water molecules, we propose that the (R)-terpinyl carbocation intermediate is formed, followed by the anti-addition of water, requiring a rotation step prior to hetercyclization.
Figure 6.

MD of bCinS with three Mg2+ ions and GPP or NPP. (A) Active site of bCinS in the dominant cluster from MD simulations with of bCinS/GPP (purple) and bCinS/NPP (cyan). Water molecules within 5 Å of GPP are shown in stick form and the closest water molecule to C7, the site of attack by the water molecule, is colored in purple for bCinS/GPP and cyan for bCinS/NPP. (B) and (C) show the same view of bCinS/GPP and bCinS/NPP alone, respectively. (D) The distance between C7 and O of WAT364 over the course of two 100 ns MD simulations of bCinS/GPP. (E) The distance between C7 and O of the closest water molecule to C7 of NPP over the course of two 100 ns MD simulations.
Structures of Apo-bLinS and bLinS-FGPP Complex
The bLinS could be crystallized in both the apo form (2.4 Å) as well as in complex with the substrate analogue FGPP (1.82 Å, Table 1). The bLinS structure reveals a dimer in the asymmetric unit, but the monomer interface is distinct to that observed for the bCinS enzyme (Figure 7a). The individual bLinS monomers overlay with rmsd of 0.83 Å for 293 Cα atoms, with a small shift in position of the N-terminal region encompassing the first two alpha helices (residues 1–62) located furthest away from the dimer interface. Co-crystallization with FGPP leads to crystals with similar packing. Unexpectedly, clear electron density corresponding to FGPP is only present in monomer A (Figure 7b). In contrast, electron density occupying the active site of monomer B is weak, and only a single phosphate ion could be modeled that might be associated with a disordered binding of the FGPP diphosphate moiety (Figure 7c).
Figure 7.

Structure of bLinS in apo and FGPP complex form. (A) Cartoon representation of the apo bLinS dimer, with one monomer colored in green and the second monomer in blue. The latter is overlaid with the first monomer (in gray) revealing small changes in conformation occur distant from the dimer interface. (B) Cartoon representation of monomer A bound to FGPP. The N-terminal region is shown in blue and the (here disordered) C-terminal region in red. (C) Cartoon representation of the monomer B bound to phosphate. Orientation and color coding are as in panel B. The ordering of the C-terminus and the partial closing of the N-terminal regions occludes the phosphate from solvent, in contrast to the solvent exposed nature of the diphosphate group in monomer A. (D) Stereoview of the active site of bLinS monomer A in complex with FGPP. Key polar interactions are shown by black dotted lines. (E) Stereoview of the active site of bLinS monomer B (in blue) in overlay with the active site of bCinS in complex with FGPP/FNPP (in gray).
The FGPP is bound to the bLinS active site of chain A in an extended conformation compared to the FGPP/FNPP configuration observed in the bCinS structures (Figure 7d). Only one Mg2+ ion coordinating the pyrophosphate moiety could be unambiguously modeled. This Mg2+ ion sits on the concave side of the PPi moiety and hydrogen bonds with Asp80 of the aspartate-rich motif in helix D. The direct interactions between the diphosphate moiety of FGPP and bLinS are limited to a polar interaction with Lys225. The hydrophobic moiety of FGPP is located in a predominantly hydrophobic pocket at the core of the bLinS structure, with a polar interaction observed between Asn218 of the Mg2+ B binding NSE motif and the FGPP fluorine atom. The lack of Mg2+ binding to the NSE motif, and the unusual position of the diphosphate moiety, suggests the FGPP is bound in a noncatalytic mode. We again used EPR to establish bLinS binds to three Mn2+ ions (and by extension three Mg2+) in solution, similar to other terpene synthases (Figure 3b). While the electron density in the active site of bLinS monomer B corresponds to a disordered species, the position of the single phosphate that is visible is more akin to what can be expected for the catalytic binding mode when superimposing bLinS on the bCinS ligand complex structures (Figure 7e). The phosphate in the bLinS monomer B establishes a network of polar contacts with the C-terminal region (R308, Y309) that is disordered in the apo-bLinS structure. It is furthermore positioned adjacent to the NSE motif, although a Mg2+ B ion could not be unambiguously located in this area. The ordering of the C-terminal region is incompatible with crystal packing for bLinS monomer A, possibly contributing to the noncatalytic conformation observed for the bound FGPP in the corresponding active site. A comparison with the apo-bLinS structure reveals the overall conformation for both monomers is similar, with the notable exception of the C-terminal region. However, class I terpenoid synthase structures have been found to alternate between an “open” state (i.e., apo) and a “closed” (i.e., ligand) bound state58.65 This calls into question whether the apo-bLinS structure is reflective of the open state, or whether the bLinS-FGPP complex corresponds to the closed state. The fact neither of the monomers in the bLinS-FGPP complex binds to the required three metal ions strongly suggests both apo bLinS and the bLinS-FGPP structures are in the open state, possibly stabilized by crystal packing contacts.
MD simulations suggest that, unlike other terpene synthases, neither bLinS nor bCinS undergo major conformational changes to between “open” and “closed” states, (Figures S8a–c and S9a–c). Although bLinS-FGPP is likely to correspond to the open state, the carbon chain of the bound FGPP (in monomer A) occupies a similar region as observed in the bCinS-FGPP/FNPP complexes (Figure 7e). This likely indicates the position of the active site hydrophobic pocket in bLinS, and might even reflect the corresponding conformation of carbon chain of the bound FGPP in the closed state. As linalool is an acyclic monoterpene product, the bLinS catalytic mechanism does not require a cyclization process. Instead, the geranyl cation attacks a nearby water molecule leading to linalool following deprotonation (Figure 8a). In the bLinS-FNPP structure, several water molecules are located within a distance of ∼4.5 Å from the FGPP (Figure 7d), and representing likely candidates for this process in case the FGPP carbon chain conformation is reflective of the catalytically relevant species. In contrast to the closed nature of the bCinS structure, the bLinS is relatively open, and we cannot rule out further closure might occur upon substrate binding in solution. Keeping this caveat in mind, the most likely candidate for the water attack is the molecule that is coordinated by Asp79 and Arg172 and is at a distance of 3.6 Å from C3 of FGPP. The position of the water molecule with respect to FGPP suggests production of R-(−)-linalool, which matches with the biochemical characterization. MD simulations show that the closest water molecule to C3 of GPP remains at an average distance of 3.29 (±0.21) Å in run1 and 3.93 (±0.52) Å in run2 (Figure 8c). MD simulations of bLinS in complex with FPP (Figure S9), the precursor to sesquiterpenes, shows that the active site is sufficiently large to accommodate a sesquiterpene, explaining the fact bLinS also accepts FPP as a substrate.23
Figure 8.

Mechanistic proposal for bLinS and MD of bLinS with GPP and three Mg2+ ions. (A) Schematic outline representing the conversion of GPP to the linalool product by bLinS. (B) Active site of bLinS in the dominant cluster from MD simulations with GPP and three Mg2+ ions. GPP is shown with cyan carbon atoms, bLinS is shown in purple in cartoon form and the Mg2+ ions are shown as green spheres. Water molecules within 5 Å of GPP are shown in stick form, with the water molecule closest to C3 of GPP shown in red. (C) The distance between C3 and O of WAT430 over the course of two 100 ns MD simulations.
Bacterial mTC/S Are Structurally Similar to Sesquiterpene Synthases
The bLinS and bCinS are single domain (α) enzymes, whereas the plant mTC(S) typically contain two domains (α and β). This makes them structurally more similar to the sesquiterpene synthases (Figure 9a), which are also usually composed of only a single class I terpenoid fold domain.10 It is notable that genome mining for bacterial terpene synthase-like genes followed by heterologous expression revealed the majority of these enzymes made sesquiterpenes as products.20 So far, bLinS and bCinS are the only characterized bacterial mTC(S) that accept GPP as substrate and thus lead to monoterpene formation. The bCinS-FNPP complex is specifically compared to the structures of plant limonene synthases15,16 and bornyl diphosphate synthase14 for which complexes with the substrate analogues are available. When comparing the corresponding C-terminal catalytic domains with the bCinS complexes, it is clear that the orientation of GPP/NPP analogue in bCinS is such that the beta phosphate occupies the location comparable to the alpha phosphate binding site in the plant enzymes and vice versa, and resembles the orientation observed in sesquiterpene synthase complex. For the functionally analogous plant 1,8-cineole synthase (Sf-CinS1), only the apoenzyme structure is available. Furthermore, superimposition of the bCinS and Sf-CinS1 reveals distinct active site architectures. In Sf-CinS1, Asn338, which coordinates a water molecule, was found to be crucial for the synthesis of 1,8-cineole.18 Mutation of Asn338 to Ile resulted in the formation of sabinene as the major product but no α-terpineol and 1,8-cineole, establishing the role of Asn338 in water capture. In bCinS, as mentioned before, residues Trp58 and Asn305 coordinate the water molecule proposed to be involved in the water attack. Though Asn305 in bCinS resides in a different helix and region of the active site compared to Asn338 in Sf-CinS1, Asn305 might play a similar role to that proposed for the plant enzyme (Figure S10). Analysis using DALI66 and PDBefold67 servers showed many sesquiterpene synthases including pentalenene synthase (PDB 1 ps1),32 germacradienol synthase (PDB 5i1u),68 hedycaryol synthase (PDB 4mc3),69 geosmin synthase (PDB 5dz2),70 epi-isozizaene synthase (PDB 4ltv),65 selinadiene synthase (40kz),58 and aristolochene synthase (PDB 4kwd)51 are very similar to bLinS and bCinS structures (Table S2).
Figure 9.

bLinS and bCinS are related to bacterial sesquiterpene synthases. (A) Stereoview of a cartoon representation of a structural overlay of bLinS (monomer B; in green), bCinS FGPP/FNPP (in blue) and aristolochene synthase (ATAS) (PDB code 4KUX) in complex with a C15 substrate analogue (in magenta). (B) Active site overlay of bCinS and ATAS, color coding as in panel A. (C) Active site overlay of bLinS (monomer B) and ATAS, color coding as in panel A.
Two sesquiterpene synthase structures have been reported in complex with substrate analogues: Aspergillus terreus aristocholene synthase (ATAS) with farnesyl thiolodiphosphate (FSPP; PDB 4KUX) and selinadiene synthase (SdS) with dihydrofarnesyl diphosphate (DHFPP; PDB 4OKZ). A comparison of these structures with bLinS and bCinS might allow pinpointing of those active site differences that play a role in determining substrate specificity (C10 versus C15). Since the Mg2+ and pyrophosphate binding regions are highly conserved, most variations in the active site architecture are restricted to hydrophobic cavity surrounding the substrate carbon chain. In bCinS, two phenylalanines (Phe 77 and Phe 179) constrict the substrate-binding site when compared to the ATAS-FSPP and SdS-DHFPP structures, and they would clash with a putative FPP substrate (Figure 9b). Phe179 resides in the kink region of the helix G1/2 of bCinS, and is replaced by Gly174 in ATAS and Ala183 in SdS. The bCinS Phe77 resides in helix D and is homologous to Leu80 in ATAS/Leu78 in SdS, with the latter both adopting a conformation that is pointing away from the active site. This suggests bCinS evolved from a sesquiterpene synthase by restricting active site volume.
Interestingly, bLinS contains nonaromatic residues at positions equivalent to bCinS Phe77 and Phe179 (Thr75 and Cys177 in bLinS), and thus resembles ATAS and SdS (Figure 9c). This provides a rationale for the fact bLinS can accept both GPP and FPP as substrates but bCinS can only convert GPP.22,23
Conclusions
We have shown that expression of Streptomyces clavuligerus linalool synthase and 1,8-cineole synthase in an E. coli geranyl diphosphate producing strain leads to higher levels of production (linalool) or more enriched product profiles (1,8-cineole) than previously reported. Crystal structures of both S. clavuligerus monoterpene synthases reveal the bacterial monoterpene synthases are more similar to previously characterized sesquiterpene synthases. A comparison with the sesquiterpene synthases allowed identification of key residues that can be exploited for rational design and switching of activity between the two classes. These results provide a basis for application of the bacterial monoterpene synthases to generate diverse monoterpene scaffolds and employ synthetic biology approaches for large-scale monoterpenoid production.
Acknowledgments
We thank Colin Levy, Manchester Protein Structure Facility (MPSF) for help with X-ray data collection, Matthew J. Cliff for NMR analysis of synthetic compounds and Derren J. Heyes for project management. We thank Diamond Light Source for access to beamlines I04 and I04-1 (proposal number mx12788). This work was funded by the U.K. Biotechnology and Biological Sciences Research Council (BBSRC; BB/M000354/1, BB/M017702/1 and BB/L027593/1:ERA-IB2 TERPENOSOME). D.L. is a Royal Society Wolfson Merit Award holder. M.W.v.d.K. is a BBSRC David Phillips Fellow. N.S.S. was a Royal Society Wolfson Merit Award holder and is an Engineering and Physical Sciences Research Council (EPSRC; EP/J020192/1) Established Career Fellow.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b01924.
Extended EPR results and discussion, Scheme S1, Figures S1–S10, Tables S1–S2 (PDF)
The authors declare the following competing financial interest(s): Patent pending for the use of bLinS and bCinS in production of monoterpenes using biocatalysis and synthetic biology methods.
Supplementary Material
References
- Tholl D. Adv. Biochem. Eng./Biotechnol. 2015, 148, 63–106. 10.1007/10_2014_295. [DOI] [PubMed] [Google Scholar]
- George K. W.; Alonso-Gutierrez J.; Keasling J. D.; Lee T. S. Adv. Biochem. Eng./Biotechnol. 2015, 148, 355–89. 10.1007/10_2014_288. [DOI] [PubMed] [Google Scholar]
- Leferink N. G. H.; Jervis A. J.; Zebec Z.; Toogood H. S.; Hay S.; Takano E.; Scrutton N. S. Chemistryselect 2016, 1, 1893–1896. 10.1002/slct.201600563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralta-Yahya P. P.; Zhang F.; del Cardayre S. B.; Keasling J. D. Nature 2012, 488, 320–8. 10.1038/nature11478. [DOI] [PubMed] [Google Scholar]
- Jongedijk E.; Cankar K.; Buchhaupt M.; Schrader J.; Bouwmeester H.; Beekwilder J. Appl. Microbiol. Biotechnol. 2016, 100, 2927–38. 10.1007/s00253-016-7337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zebec Z.; Wilkes J.; Jervis A. J.; Scrutton N. S.; Takano E.; Breitling R. Curr. Opin. Chem. Biol. 2016, 34, 37–43. 10.1016/j.cbpa.2016.06.002. [DOI] [PubMed] [Google Scholar]
- Wang X.; Ort D. R.; Yuan J. S. Plant Biotechnol J. 2015, 13, 137–46. 10.1111/pbi.12343. [DOI] [PubMed] [Google Scholar]
- Formighieri C.; Melis A. Photosynth. Res. 2016, 130, 123–135. 10.1007/s11120-016-0233-2. [DOI] [PubMed] [Google Scholar]
- Oldfield E.; Lin F. Y. Angew. Chem., Int. Ed. 2012, 51, 1124–37. 10.1002/anie.201103110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Honzatko R. B.; Peters R. J. Nat. Prod. Rep. 2012, 29, 1153–75. 10.1039/c2np20059g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaron J. A.; Christianson D. W. Pure Appl. Chem. 2010, 82, 1585–1597. 10.1351/PAC-CON-09-09-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srividya N.; Davis E. M.; Croteau R. B.; Lange B. M. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3332–7. 10.1073/pnas.1501203112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. C.; Croteau R. Arch. Biochem. Biophys. 2005, 439, 222–33. 10.1016/j.abb.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Whittington D. A.; Wise M. L.; Urbansky M.; Coates R. M.; Croteau R. B.; Christianson D. W. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 15375–80. 10.1073/pnas.232591099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D. C.; Youn B.; Zhao Y.; Santhamma B.; Coates R. M.; Croteau R. B.; Kang C. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 5360–5. 10.1073/pnas.0700915104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R. P.; Morehouse B. R.; Matos J. O.; Malik K.; Lin H.; Krauss I. J.; Oprian D. D. Biochemistry 2017, 56, 1716–1725. 10.1021/acs.biochem.7b00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morehouse B. R.; Kumar R. P.; Matos J. O.; Olsen S. N.; Entova S.; Oprian D. D. Biochemistry 2017, 56, 1706–1715. 10.1021/acs.biochem.7b00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampranis S. C.; Ioannidis D.; Purvis A.; Mahrez W.; Ninga E.; Katerelos N. A.; Anssour S.; Dunwell J. M.; Degenhardt J.; Makris A. M.; Goodenough P. W.; Johnson C. B. Plant Cell 2007, 19, 1994–2005. 10.1105/tpc.106.047779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph K.; Parthier C.; Egerer-Sieber C.; Geiger D.; Muller Y. A.; Kreis W.; Muller-Uri F. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2016, 72, 16–23. 10.1107/S2053230X15023043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.; Kuzuyama T.; Komatsu M.; Shin-Ya K.; Omura S.; Cane D. E.; Ikeda H. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 857–62. 10.1073/pnas.1422108112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.; Arima S.; Nagamitsu T.; Johmoto K.; Uekusa H.; Eguchi T.; Shin-ya K.; Cane D. E.; Ikeda H. J. Antibiot. 2015, 68, 385–94. 10.1038/ja.2014.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano C.; Kim H. K.; Ohnishi Y. ChemBioChem 2011, 12, 1988–91. 10.1002/cbic.201100330. [DOI] [PubMed] [Google Scholar]
- Nakano C.; Kim H. K.; Ohnishi Y. ChemBioChem 2011, 12, 2403–7. 10.1002/cbic.201100501. [DOI] [PubMed] [Google Scholar]
- Dickschat J. S. Nat. Prod. Rep. 2016, 33, 87–110. 10.1039/C5NP00102A. [DOI] [PubMed] [Google Scholar]
- Koksal M.; Chou W. K.; Cane D. E.; Christianson D. W. Biochemistry 2012, 51, 3011–20. 10.1021/bi201827a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapczynski A.; Letizia C. S.; Api A. M. Food Chem. Toxicol. 2008, 46, S190–2. 10.1016/j.fct.2008.06.087. [DOI] [PubMed] [Google Scholar]
- Seol G. H.; Kim K. Y. Adv. Exp. Med. Biol. 2016, 929, 389–398. 10.1007/978-3-319-41342-6_18. [DOI] [PubMed] [Google Scholar]
- Karp F.; Zhao Y.; Santhamma B.; Assink B.; Coates R. M.; Croteau R. B. Arch. Biochem. Biophys. 2007, 468, 140–6. 10.1016/j.abb.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit V. M.; Laskovics F. M.; Noall W. I.; Poulter C. D. J. Org. Chem. 1981, 46, 1967–1969. 10.1021/jo00322a060. [DOI] [Google Scholar]
- Winter G.; Lobley C. M.; Prince S. M. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1260–73. 10.1107/S0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–32. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesburg C. A.; Zhai G.; Cane D. E.; Christianson D. W. Science 1997, 277, 1820–1824. 10.1126/science.277.5333.1820. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–21. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 486–501. 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P. V.; Grosse-Kunstleve R. W.; Echols N.; Headd J. J.; Moriarty N. W.; Mustyakimov M.; Terwilliger T. C.; Urzhumtsev A.; Zwart P. H.; Adams P. D. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 352–67. 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R. P.; Long F.; Murshudov G. N.; Perrakis A. IUCrJ 2014, 1, 213–20. 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R. L.; Evanseck J. D.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha S.; Joseph-McCarthy D.; Kuchnir L.; Kuczera K.; Lau F. T. K.; Mattos C.; Michnick S.; Ngo T.; Nguyen D. T.; Prodhom B.; Reiher W. E.; Roux B.; Schlenkrich M.; Smith J. C.; Stote R.; Straub J.; Watanabe M.; Wiorkiewicz-Kuczera J.; Yin D.; Karplus M. J. Phys. Chem. B 1998, 102, 3586–3616. 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- Foloppe N.; MacKerell A. D. J. Comput. Chem. 2000, 21, 86–104. . [DOI] [Google Scholar]
- Olsson M. H. M.; Sondergaard C. R.; Rostkowski M.; Jensen J. H. J. Chem. Theory Comput. 2011, 7, 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Sondergaard C. R.; Olsson M. H. M.; Rostkowski M.; Jensen J. H. J. Chem. Theory Comput. 2011, 7, 2284–2295. 10.1021/ct200133y. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- van der Kamp M. W.; Sirirak J.; Zurek J.; Allemann R. K.; Mulholland A. J. Biochemistry 2013, 52, 8094–105. 10.1021/bi400898k. [DOI] [PubMed] [Google Scholar]
- Major D. T.; Freud Y.; Weitman M. Curr. Opin. Chem. Biol. 2014, 21, 25–33. 10.1016/j.cbpa.2014.03.010. [DOI] [PubMed] [Google Scholar]
- O’Brien T. E.; Bertolani S. J.; Tantillo D. J.; Siegel J. B. Chem. Sci. 2016, 7, 4009–4015. 10.1039/C6SC00635C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allner O.; Nilsson L.; Villa A. J. Chem. Theory Comput. 2012, 8, 1493–1502. 10.1021/ct3000734. [DOI] [PubMed] [Google Scholar]
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; Le Grand S.; Walker R. C. J. Chem. Theory Comput. 2012, 8, 1542–1555. 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Cerutti D. S.; Cheatham T. E. III; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; Izadi S.; Janowski P.; Kaus J.; Kovalenko A.; Lee T. S.; LeGrand S.; Li P.; Lin C.; Luchko T.; Luo R.; Madej B.; Mermelstein D.; Merz K. M.; Monard G.; Nguyen H.; Nguyen H. T.; Omelyan I.; Onufriev A.; Roe D. R.; Roitberg A.; Sagui C.; Simmerling C. L.; Botello-Smith W. M.; Swails J.; Walker R. C.; Wang J.; Wolf R. M.; Wu X.; Xiao L.; Kollman P. A.. Amber; University of California: San Francisco, 2016.
- Chen M.; Al-lami N.; Janvier M.; D’Antonio E. L.; Faraldos J. A.; Cane D. E.; Allemann R. K.; Christianson D. W. Biochemistry 2013, 52, 5441–53. 10.1021/bi400691v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aaron J. A.; Lin X.; Cane D. E.; Christianson D. W. Biochemistry 2010, 49, 1787–1797. 10.1021/bi902088z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso-Gutierrez J.; Chan R.; Batth T. S.; Adams P. D.; Keasling J. D.; Petzold C. J.; Lee T. S. Metab. Eng. 2013, 19, 33–41. 10.1016/j.ymben.2013.05.004. [DOI] [PubMed] [Google Scholar]
- Fujisaki S.; Nishino T.; Katsuki H. J. Biochem. 1986, 99, 1327–37. 10.1093/oxfordjournals.jbchem.a135600. [DOI] [PubMed] [Google Scholar]
- Zhou J.; Wang C.; Yoon S. H.; Jang H. J.; Choi E. S.; Kim S. W. J. Biotechnol. 2014, 169, 42–50. 10.1016/j.jbiotec.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Liu W.; Zhang R.; Tian N.; Xu X.; Cao Y.; Xian M.; Liu H. Bioengineered 2015, 6, 288–93. 10.1080/21655979.2015.1062188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. L.; Park J. E.; Choi E. S.; Kim S. W. Biotechnol. J. 2016, 11, 1291–1297. 10.1002/biot.201600250. [DOI] [PubMed] [Google Scholar]
- Baer P.; Rabe P.; Fischer K.; Citron C. A.; Klapschinski T. A.; Groll M.; Dickschat J. S. Angew. Chem., Int. Ed. 2014, 53, 7652–6. 10.1002/anie.201403648. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. J. Mol. Biol. 2007, 372, 774–97. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Morrissey S. R.; Horton T. E.; DeRose V. J. J. Am. Chem. Soc. 2000, 122, 3473–3481. 10.1021/ja992989z. [DOI] [Google Scholar]
- DeRose V. J. Curr. Opin. Struct. Biol. 2003, 13, 317–24. 10.1016/S0959-440X(03)00077-0. [DOI] [PubMed] [Google Scholar]
- Stich T. A.; Lahiri S.; Yeagle G.; Dicus M.; Brynda M.; Gunn A.; Aznar C.; Derose V. J.; Britt R. D. Appl. Magn. Reson. 2007, 31, 321–341. 10.1007/BF03166263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinkel J.; Rabe P.; Zur Horst L.; Dickschat J. S. Beilstein J. Org. Chem. 2016, 12, 2317–2324. 10.3762/bjoc.12.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise M. L.; Urbansky M.; Helms G. L.; Coates R. M.; Croteau R. J. Am. Chem. Soc. 2002, 124, 8546–7. 10.1021/ja0265714. [DOI] [PubMed] [Google Scholar]
- Li R.; Chou W. K.; Himmelberger J. A.; Litwin K. M.; Harris G. G.; Cane D. E.; Christianson D. W. Biochemistry 2014, 53, 1155–68. 10.1021/bi401643u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L.; Rosenstrom P. Nucleic Acids Res. 2010, 38, W545–W549. 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2256–68. 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Grundy D. J.; Chen M.; Gonzalez V.; Leoni S.; Miller D. J.; Christianson D. W.; Allemann R. K. Biochemistry 2016, 55, 2112–21. 10.1021/acs.biochem.6b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer P.; Rabe P.; Citron C. A.; de Oliveira Mann C. C.; Kaufmann N.; Groll M.; Dickschat J. S. ChemBioChem 2014, 15, 213–6. 10.1002/cbic.201300708. [DOI] [PubMed] [Google Scholar]
- Harris G. G.; Lombardi P. M.; Pemberton T. A.; Matsui T.; Weiss T. M.; Cole K. E.; Koksal M.; Murphy F. V. t.; Vedula L. S.; Chou W. K.; Cane D. E.; Christianson D. W. Biochemistry 2015, 54, 7142–55. 10.1021/acs.biochem.5b01143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.