

We report the discovery of a novel family of prohomeostatic and neuroprotective lipid mediators called elovanoids.
Abstract
We report the characterization of a novel class of lipid mediators termed elovanoids (ELVs) (ELV-N32 and ELV-N34), which are dihydroxylated derivatives of 32:6n3 and 34:6n3, respectively. The precursors of ELVs are made by elongation of a 22:6n3 fatty acid and catalyzed by ELOVL4 (elongation of very-long-chain fatty acids–4). The structure and stereochemistry of ELVs were established using synthetic compounds produced by stereocontrolled total synthesis. We report that ELV-mediated protection is induced in neuronal cultures undergoing either oxygen/glucose deprivation or N-methyl-d-aspartate receptor–mediated excitotoxicity, as well as in experimental ischemic stroke. The methyl ester or sodium salt of ELV-N32 and ELV-N34 resulted in reduced infarct volumes, promoted cell survival, and diminished neurovascular unit disruption when administered 1 hour following 2 hours of ischemia by middle cerebral artery occlusion. Together, our data reveal a novel prohomeostatic and neuroprotective lipid-signaling mechanism aiming to sustain neural cell integrity.
INTRODUCTION
The central nervous system (CNS) depicts unique enrichment in omega-3 (n-3) polyunsaturated fatty acids (PUFAs) as acyl chains of membrane phospholipids. Among the n-3 family, docosahexaenoic acid (DHA; 22:6n3) is the most abundant PUFA and serves as a precursor of enzymatically derived dihydroxylated derivatives, known as docosanoids, which include potent neuroprotective mediators made “on demand” when disruptions to homeostasis are impending (1, 2). Additionally, the elongase enzyme ELOVL4 (elongation of very-long-chain fatty acids–4) converts DHA or eicosapentaenoic acid (20:5n-3) into very-long-chain (≥C28) PUFAs (VLC-PUFAs,n-3) in the brain, retina, testes, and skin (3). In the skin, saturated VLC fatty acids predominantly occur.
VLC-PUFAs,n-3 are incorporated as acyl chains at the C1 position of phosphatidylcholines and in sphingolipids. These fatty acids are more abundant in the retina, where mutant ELOVL4 causes juvenile macular degeneration in autosomal dominant Stargardt’s disease 3 (STGD3) (3, 4). ELOVL4 is expressed in the CNS, including in hippocampal neurons of the subgranular layer of the dentate gyrus (DG), a locus for medial temporal lobe epilepsy. In the CNS, mutations in ELOVL4 lead to impaired neural development, neuronal dysfunction, hyperexcitability, and seizures (5). Although the occurrence of VLC-PUFAs,n-3 has been well documented for some time, their significance and possible conversion into bioactive mediators have not yet been uncovered.
We report here the characterization and bioactivities of a novel class of dihydroxylated lipid mediators that we termed elovanoids (ELVs), which are derived from VLC-PUFAs,n-3 with 32 or 34 carbons and likely even longer fatty acid chains, presumably through previously unknown pathways. By investigating brain phosphatidylcholine molecular species containing VLC-PUFAs,n-3, we also have uncovered and characterized a novel prohomeostatic and neuroprotective signaling mechanism that is set in motion upon imminent cellular disruption of homeostasis, such as uncompensated oxidative stress (UOS), oxygen/glucose deprivation (OGD) (as in stroke), N-methyl-d-aspartate (NMDA) receptor–mediated excitotoxicity (as in epilepsy and other neurological conditions), and experimental ischemic stroke, and at the onset of neurodegenerative diseases.
These findings are different from other endogenous prohomeostatic and neuroprotective mechanisms because they involve a phospholipid molecular species that is endowed with acyl chains with two different PUFA precursors of bioactive lipids. This unusual signaling encodes two PUFA-derived lipid mediators, the precursors of which are stored in specific phosphatidylcholine molecular species. Whereas the 22-carbon DHA, which is the first-described PUFA precursor of the stereoselective dihydroxylated lipid mediator neuroprotectin D1 (NPD1) (6–8), is located at the C2 position of the phospholipids, the VLC-PUFAs,n-3 are located at the C1 position of the phospholipids and are subject to alternatively or concomitantly regulated pathways. Therefore, the findings revealed here feature a different signal bifurcation prohomeostatic and neuroprotective mechanism that aims to sustain neural cell integrity. Because there are fatty acids longer than 34:6n-3 and products of other ELOVL enzymes, we anticipate that other ELVs might also be endogenously made to regulate cell function.
RESULTS
Structure and stereochemistry of ELV-N32 and ELV-N34 in mixed neuronal cultures
The complete structures and stereochemistry of the novel ELVs (ELV-N32 and ELV-N34) were established through a direct comparison with compounds prepared via stereocontrolled total organic synthesis by adapting our previously reported methodologies for the total synthesis of NPD1 (9). Further validation of these structural assignments was established by synthesizing deuterium-labeled derivatives for liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis.
ELV-N32 and ELV-N34 were prepared by stereocontrolled total chemical synthesis (Fig. 1A). The availability of these synthetic ELVs with fully defined structures and stereochemistry allowed us to determine the complete R/S configuration and the Z/E geometry of the double bonds in these mixed neuronal cell culture–derived ELVs. We also generated synthetic stereochemically pure deuterium-labeled ELVs, and by matching them with endogenously produced molecules by LC-MS/MS, we further confirmed their structure and stereochemistry.
Fig. 1. Discovery and structural characterization of ELVs (ELV-N32 and ELV-N34) in neuronal cell cultures.
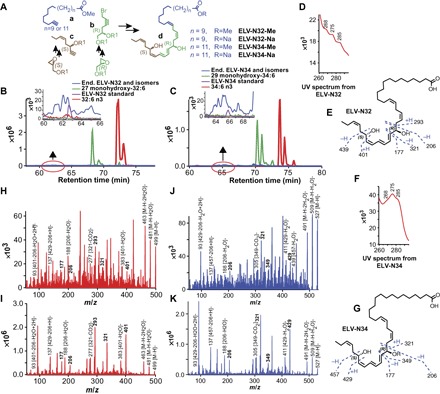
(A) The ELV structural framework was synthesized from three key intermediates a, b, and c, each of which was prepared from readily available starting materials. The stereochemistry of intermediates b and c was predefined by using enantiomerically pure epoxide starting materials. The final ELVs (d) were assembled via iterative couplings of intermediates a, b, and c, and they were isolated as the Me or Na. (B to K) Identification of ELVs in neuronal cell cultures. Cerebral-cortical mixed neuronal cells were incubated with 32:6n3 and 34:6n3 (10 μM each) under OGD conditions. In (B), 32:6n3 (red line), endogenous monohydroxy-32:6 (green line) and ELV-N32 (blue line) are shown with ELV-N32 standard (purple line) in the inset. MRM of ELV-N32 shows two large peaks eluted earlier than the peak when standard ELV-N32 is eluted, but they show the same fragmentation patterns, suggesting that they are isomers. In (C), the same features were shown in 34:6n3 and ELV-N34. (D) UV spectrum of endogenous ELV-N32 shows triene features, but these are not definite at this concentration. (E) Fragmentation pattern of ELV-N32. (F) UV spectrum of endogenous ELV-N34 showing triene features. (G) Fragmentation pattern of ELV-N34. (H) Full fragmentation spectra of endogenous ELV-N32 and (I) ELV-N32 standard show that all major peaks from the standard match to the endogenous peaks but are not perfectly matched; endogenous ELV-N32 has more fragments that do not show up in the standard, suggesting that it may contain isomers. (J) For ELV-N34 full fragmentation spectra, the endogenous ELV-N34 peaks match to the standard ELV-N34 (K), also suggesting the existence of ELV-N34 isomers.
Both ELVs and their precursors were detected in cells under OGD stress (Fig. 1, B to K, and fig. S2). We used mass/charge ratio (m/z) multiple reaction monitoring (MRM) transitions of 499→93 and 499→401 for ELV-N32 detection (Fig. 1B) and m/z MRM transitions of 527→93 and 527→429 for ELV-N34 detection (Fig. 1C). For their corresponding monohydroxy precursors, we used m/z 483→385 for 27-hydroxyl-C32:6n3 (Fig. 1B) and m/z 511→413 for 29-hydroxyl-C34:6n3 (Fig. 1C). For further identification, we performed full fragmentation on ELVs and found good matches to the synthetically produced standards. Both ELVs had ultraviolet (UV) maxima at 275 nm, which are consistent with a conjugated triene structure (Fig. 1, D and F).
Following matching of synthetic ELVs with biogenic ELVs derived from mixed neuronal cells in culture, the complete structures and stereochemistry of ELV-N32 and ELV-N34 were established. The structures of ELV-N32 (ELV derived from a 32-carbon n-3 PUFA) and ELV-N34 (ELV derived from a 34-carbon n-3 PUFA) were determined to be (14Z,17Z,20R,21E,23E,25Z,27S,29Z)-20,27-dihydroxydo-triaconta-14,17,21,23,25,29-hexaenoic acid (Fig. 1E) and (16Z,19Z,22R,23E,25E,27Z,29S,31Z)-22,29-dihydroxytetra-triaconta-16,19,23,25,27,31-hexaenoic acid (Fig. 1G), respectively.
Neuroprotection by ELVs in UOS, OGD, or NMDA receptor–induced excitotoxicity
We found that, at a concentration of 200 nM, the sodium salt ELV-N34 Na or the methyl ester ELV-N34 Me evoked neuroprotection to cerebral-cortical mixed neuronal cells in culture exposed to UOS for 12 hours, which was induced by the addition of tumor necrosis factor–α (TNFα) (10 ng/ml) and H2O2 (50, 100, or 200 μM). There was a dose-dependent increase in apoptotic nuclei that was counteracted by ELV-N34 Na or ELV-N34 Me (Fig. 2I).
Fig. 2. ELV-N32 and ELV-N34 elicit protection of cerebral-cortical mixed neuronal cell or hippocampal mixed neuronal cultures exposed to OGD or UOS.
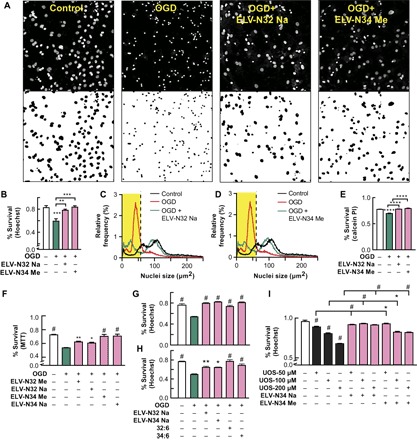
(A) Representative images of cerebral-cortical mixed neuronal cultures (DIV 12) challenged with 90-min OGD. The cells were fixed and stained with Hoechst 33258 after 12-hour treatment with ELV-N32 Na or ELV-N34 Me at a concentration of 500 nM, showing pyknosis as a result of OGD and neuroprotection elicited by ELV-N32 Na or ELV-N34 Me. (B) Summary of data from (A) [**P < 0.001 and ***P < 0.0001, one-way analysis of variance (ANOVA), followed by Holm-Sidak’s multiple comparisons test; n = 9]. (C and D) An unbiased image analysis method was applied to count Hoechst-positive nuclei, and the percentage of relative frequency distribution of pyknotic versus nonpyknotic nuclei is shown in the presence of OGD + ELV-N32 Na (C) or OGD + ELV-N34 Me (D), respectively. When the cells were subjected to OGD stress, they underwent pyknosis, as shown by the leftward shift of the nuclear peak. Again, upon treatment with either ELV-N32 Na or ELV-N34 Me, there was a positive rightward shift toward the control nuclear population peak, indicating that cellular survival was elicited by these novel lipid mediators. The nuclear size cutoff for defining pyknotic versus nonpyknotic nuclei is represented by black dashed lines and highlighted by a yellow rectangle. (E) Neuroprotection elicited by ELV-N32 Na or ELV-N34 Me, as assessed by calcein-positive cell counting after exposure of cerebral-cortical mixed neuronal cultures (DIV 12) challenged with 90-min OGD (****P < 0.0001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 3). PI, propidium iodide. (F) Cell survival as assessed by MTT assay after exposure of cerebral-cortical mixed neuronal cell cultures (DIV 12) challenged with 90-min OGD, followed by treatment with ELV-N32 Me, ELV-N32 Na, ELV-N34 Me, or ELV-N34 Na at a concentration of 1 μM. (#P < 0.0001, *P < 0.05, and **P < 0.001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 9). (G and H) Neuroprotection elicited by ELV-N32 Na, ELV-N34 Na, 32:6, or 34:6, as assessed by an unbiased image analysis followed by Hoechst-positive nuclei counting after hippocampal mixed neuronal culture (DIV 12), is subjected to OGD stress in the presence or absence of ELV-N32 Na or ELV-N34 Me at a concentration of 500 nM; 32:6 or 34:6 when added at a concentration of 500 nM also showed neuroprotection (#P < 0.0001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 3) (G) or cerebral-cortical mixed neuronal cell culture (DIV 28) (#P < 0.0001, *P < 0.05, and **P < 0.001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 3) (H), respectively, were subjected to 90-min OGD. (I) Neuroprotection elicited by ELV-N34 Na or ELV-N34 Me at a concentration of 200 nM, as assessed by an unbiased image analysis, followed by Hoechst-positive nuclei counting after cerebral-cortical mixed neuronal cell culture (DIV 12), were subjected to 12-hour UOS induced by the addition of TNFα (10 ng/ml) and H2O2 (50, 100, or 200 μM) (#P < 0.0001 and *P < 0.001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 9).
To determine the neuroprotective bioactivity of ELV-N32 or ELV-N34 against OGD-induced neuronal cell death, we exposed cerebral-cortical mixed neuronal cells in culture or hippocampal mixed neurons in culture to OGD for 90 min (10–13). After 2 hours of reoxygenation, ELV-N32 or ELV-N34 were added at a concentration of either 200 nM, 500 nM, or 1 μM, and cell viability was then assessed by either Hoechst-positive nuclei counting, calcein-positive cell counting, or MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assays. Under all different conditions and concentrations, it was found that ELV-N32 Na, ELV-N32 Me, ELV-N34 Na, or ELV-N34 Me elicited neuroprotection, as compared to cells exposed to OGD alone (Fig. 2, A to H, and figs. S1, E to H, S5, A and B, S6, E to G, and S7, C and D). Moreover, our results also indicate that the precursor 34:6 could elicit neuroprotection at a concentration as low as 250 nM when added after OGD exposure (fig. S1H). This implies conversion into ELV-N34 because the experimental conditions were similar to those shown in Fig. 1.
Furthermore, we found that NMDA exposure at a concentration of 25, 50, or 100 μM for 12 hours induced neuronal death in cerebral-cortical mixed neuronal cells and hippocampal mixed neurons in culture (Fig. 3, A to H, and figs. S1, A to D, S5, C and D, S6, B to D, S7, A and B), which was compensated for by adding either ELV-N32 (Na or Me) or ELV-N34 (Na or Me) at a concentration of either 200 or 500 nM, when added simultaneously along with NMDA. We also found that there was a dose-dependent increase in apoptotic nuclei when cells were exposed to NMDA at a concentration of either 25, 50, or 100 μM, which was compensated for in the presence of ELV-N32 Na or ELV-N34 Me. For one experiment (Fig. 3F), we tested whether NMDA excitotoxicity can be attenuated by the addition of the noncompetitive NMDA receptor antagonist MK801 maleate (dizocilpine; 10 μM). Of all the treatment combinations tested in this experiment, we found that ELV-N32 Na (by itself) provided the maximum neuroprotection to the cerebral-cortical mixed neurons subjected to 100 μM NMDA–induced excitotoxicity. However, we also found that the NMDA excitotoxicity can be overcome by the addition of the noncompetitive NMDA receptor antagonist MK801 maleate (dizocilpine; 10 μM). The addition of MK801 and ELV-N32 Me together elicited a more potent neuroprotection than that elicited by ELV-N32 Me alone. In addition, the precursor 34:6 at a concentration of 500 nM attenuates NMDA receptor–mediated excitotoxicity (Fig. 3H). Similar to the case above, this also implies conversion into ELV-N34 because the experimental conditions were similar to those shown in Fig. 1.
Fig. 3. ELV-N32 and ELV-N34 induce protection of cerebral-cortical mixed neuronal cell cultures exposed to NMDA excitotoxicity.
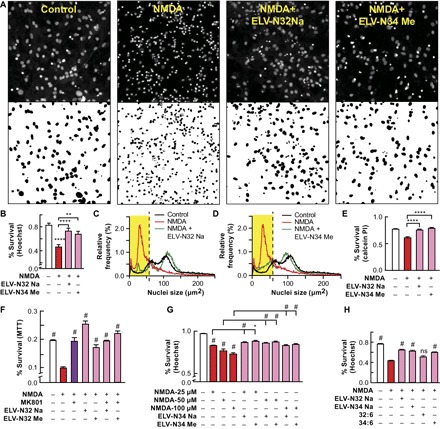
(A) Representative images of cerebral-cortical mixed neuronal cell cultures (DIV 12) subjected to 12-hour NMDA excitotoxicity. The cells were fixed and stained with Hoechst 33258 after 12-hour treatment with ELV-N32 Na or ELV-N34 Me at a concentration of 500 nM along with NMDA at a concentration of 100 μM, showing pyknosis as a result of NMDA excitotoxicity and neuroprotection elicited by ELV-N32 Na or ELV-N34 Me. (B) Summary of data from (A) (****P < 0.0001 and **P < 0.05, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 9). (C and D) An unbiased image analysis method was applied to count Hoechst-positive nuclei, and the percentage of relative frequency distribution of pyknotic versus nonpyknotic nuclei is shown in the presence of NMDA + ELV-N32 Na (C) or NMDA + ELV-N34 Me (D), respectively. When the cells were subjected to NMDA excitotoxicity, they underwent pyknosis, as shown by the leftward shift of the nuclear peak. Again, upon treatment with either ELV-N32 Na or ELV-N34 Me, there was a positive rightward shift toward the control nuclear population peak, indicating cellular survival elicited by these ELVs. The nuclear size cutoff for defining pyknotic versus nonpyknotic nuclei is represented by black dashed lines and highlighted by a yellow rectangle. (E) Neuroprotection elicited by ELV-N32 Na or ELV-N34 Me, as assessed by calcein-positive cell counting after exposure of cerebral-cortical mixed neuronal cell cultures (DIV 12) challenged with 12-hour NMDA at a concentration of 100 μM (****P < 0.0001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 3). (F) Cell survival as assessed by MTT assay after exposing cerebral-cortical mixed neurons in culture (DIV 12) to NMDA (100 μM) excitotoxicity in the presence of the noncompetitive NMDA receptor antagonist MK801 maleate (dizocilpine) (10 μM), ELV-N32 Na (200 nM), or ELV-N32 Me (200 nM) (#P < 0.001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 6). (G) Neuroprotective effects of ELV-N32 Na or ELV-N32 Me at a concentration of 200 nM after exposure of cerebral-cortical mixed neurons (DIV 12) to NMDA excitotoxicity (25, 50, or 100 μM). Cell survival was assessed by unbiased image analysis and counting of Hoechst-positive nuclei. (#P < 0.0001 and *P < 0.05, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 9). (H) Cerebral-cortical mixed neurons in culture (DIV 28) exposed to NMDA (50 μM) in the presence or absence of ELV-N32 Na or ELV-N34 Na or 32:6 or 34:6 (500 nM), as assessed by Hoechst staining and cell counting. (#P < 0.0001, one-way ANOVA, followed by Holm-Sidak’s multiple comparisons test; n = 3). ns, not significant.
ELV-induced sustained neurological improvement and protection after ischemic stroke
Focal ischemic stroke leads to impaired sensorimotor and cognitive functions, with 70 to 80% of patients displaying hemiparesis immediately after stroke (14). We administered the ELVs by stereotaxically implanting infusion cannulas into the right lateral ventricle 1 hour after 2 hours of middle cerebral artery occlusion (MCAo). Functional deficits in rodents following MCAo resemble sensorimotor deficits, and because the ultimate goal of any stroke therapy is the restoration of neurological/behavioral functions, two tests of the sensorimotor battery were used to detect neurological deficits following experimental ischemic stroke (15). All ELV-treated animals greatly improved neurologic scores in a sustained fashion up to the 7-day survival period compared to the cerebrospinal fluid (CSF) group (Fig. 4A). CSF-treated rats continued to exhibit severe impairments through this period. T2-weighted imaging (T2WI) revealed large lesions, and T2 hyperintensities were observed in the ischemic core and penumbra of CSF-treated rats, consistent with edema formation (Fig. 4, B and C). In contrast, ischemic core and penumbra volumes (computed from T2WI) were significantly reduced by all ELV treatments (Fig. 4B). Total lesion volumes were significantly reduced by ELV-N32 Na, ELV-N32 Me, ELV-N34 Na, and ELV-N34 Me compared to the CSF-treated group (by 60, 56, 99, and 91%, respectively) (Fig. 4B). Three-dimensional (3D) lesion volumes were computed from T2WI on day 7 after MCAo (Fig. 4D). Lesion volume was markedly reduced with ELV treatment and was mostly localized only in the subcortical areas of the brain (Fig. 4D).
Fig. 4. ELV-N32 and ELV-N34 improve neurological/behavioral score, protect the penumbra, and reduce MRI lesion volumes after ischemic stroke.
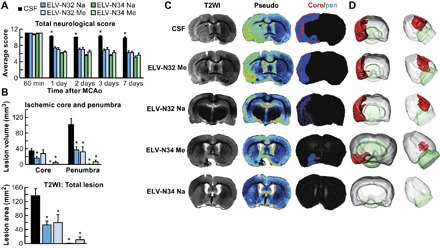
(A) Total neurological score (normal score, 0; maximal score, 12) during MCAo (60 min) and at various times after treatment. At 60 min of MCAo, all animals had a score of 11 (of a possible 12). ELV-treated rats had significantly improved neurological scores on days 1, 3, and 7 compared to the vehicle (CSF)–treated group. (B) Ischemic core, penumbra, and total lesion volumes, computed from T2WI on day 7, were significantly reduced by ELV treatment compared to the vehicle group. (C) Representative T2WI, pseudo images, core/penumbra, and (D) 3D infarct volumes computed from T2WI on day 7. Core and penumbra were extracted from the entire brain. Core (red) and penumbral (blue) tissues were automatically identified in vehicle- and ELV-treated animals using the computational MRI method Hierarchical Region Splitting for penumbra identification. T2 hyperintensities were observed in the ischemic core and penumbra of vehicle-treated rats, consistent with edema formation. In contrast, ELV-treated animals had smaller lesion sizes. 3D reconstructions are from the same animal in each group on day 7. Values shown are means ± SD (n = 5 to 6 per group) (*P < 0.05, versus CSF group; repeated-measures ANOVA, followed by Bonferroni tests).
ELV-attenuated cellular damage, blood vessel integrity, and NVU disruption
Neurons, astrocytes, and blood vessels implicated in cerebral infarction were examined using immunohistochemistry on day 7. CSF-treated rats exhibited large lesions involving cortical and subcortical regions, characterized by loss of neuronal, glial, and vascular elements (Fig. 5, A and B). In contrast, ELV-treated rats showed less infarction with an increased number of neuron-specific neuronal nuclear antigen (NeuN)– and glial fibrillary acidic protein (GFAP)–positive cells and SMI-71–positive vessels in the cortex compared to the CSF-treated group. SMI-71, an endothelial barrier antigen, is a marker for blood-brain barrier (BBB) integrity. Cellular counts for NeuN, SMI-71, and GFAP (regions delineated in the diagram in Fig. 5C) demonstrated that all ELV treatments increased NeuN-positive neurons and GFAP-positive reactive astrocytes and protected blood vessel integrity (Fig. 5C). As a result of almost all ELV treatments (except for ELV-N32 Na), blood vessel density (SMI-71) was increased within the penumbral tissues, with parallel formation of denser GFAP-rich scar tissue. Thus, enhancement of blood vessel density likely facilitates neurogenesis and synaptogenesis, which, in turn, contribute to improved repair and, ultimately, improved functional recovery.
Fig. 5. ELV-N32 and ELV-N34 attenuate experimental ischemic stroke–induced neuronal and astrocyte cellular damage.
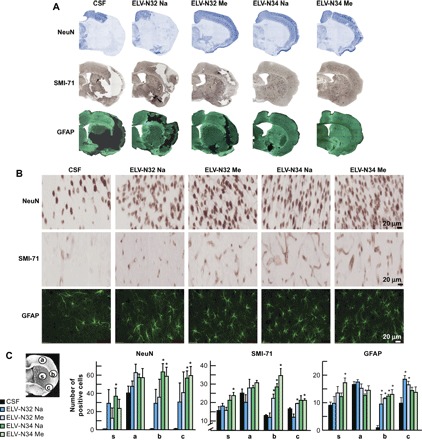
(A and B) Representative NeuN (blue), SMI-71 (brown), and GFAP (dark green) stained brain sections from all groups. Vehicle (CSF)–treated rats showed extensive neuronal loss, reduction of GFAP-reactive astrocytes, and SMI-71–positive vessels. In contrast, treatment with ELVs increased NeuN-, SMI-71-, and GFAP-positive cells. (C) Coronal brain diagram (bregma, +1.2 mm) showing locations of regions for NeuN-, SMI-71-, and GFAP-positive cell counts in the cortex (a, b, and c) and striatum (s). Numbers of NeuN-positive neurons, SMI-71–positive vessels, and GFAP-positive astrocytes, increased by ELV treatment in the ischemic core (s) and different penumbral areas (a, b, and c), are shown. Values shown are means ± SD (*P < 0.05, significantly different from vehicle; repeated-measures ANOVA, followed by Bonferroni tests; n = 5 to 6 per group).
Ischemic disruption of the neurovascular unit (NVU) was measured initially by infiltration of endogenous immunoglobulin G (IgG) into the brain parenchyma (Fig. 6, A and B). IgG staining intensity was observed in the ipsilateral hemisphere after MCAo (Fig. 6A). Staining intensity at 7 days was similar among the CSF-, ELV-N32 Na–, and ELV-N32 Me–treated groups. In contrast, treatment with ELV-N34 Na and ELV-N34 Me showed significantly less IgG staining in the cortex; staining was mostly localized in the core of infarction (subcortex). In addition, IgG immunoreactivity from the whole hemisphere (total) was reduced (all animals survived uneventfully) (Fig. 6B). Brains from CSF-treated rats exhibited a pannecrotic lesion involving both cortical and subcortical regions of the right hemisphere (Fig. 6C). By contrast, infarct size in the rats treated with ELV compounds showed less extensive damage, mostly in the subcortical area. ELV-mediated protection was extensive in the frontal-parietal cortex (tissue was salvaged by 57 to 96%) and subcortex (73 to 75%) compared to the CSF-treated group (Fig. 6D). Total infarct volume, corrected for brain swelling, was markedly reduced in all ELV-treated groups by 55 to 91% (Fig. 6D).
Fig. 6. ELV-N32 and ELV-N34 diminish NVU disruption and reduce brain infarction after ischemic stroke.
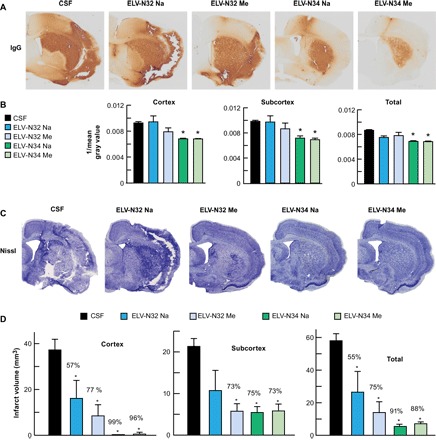
(A) NVU breakdown was assessed by immunodetection of IgG within the parenchyma. IgG staining (brown) indicates NVU breakdown. Vehicle (CSF)–treated rats displayed increased IgG immunoreactivity in the cortex and subcortex. Treatment with ELV-N34 Na or ELV-N34 Me showed less IgG staining in the cortex and was mostly localized in the core of infarction (subcortex). (B) Bar graph shows that ELV-N34 Na and ELV-N34 Me significantly reduced IgG immunoreactivity in the cortex, subcortex, and whole hemisphere (total). Values shown are means ± SD (*P < 0.05, versus the vehicle group; repeated-measures ANOVA, followed by Bonferroni tests; n = 5 to 6 per group). (C) Nissl-stained brain sections from rats treated with vehicle and ELVs. Vehicle-treated rats show large cortical and subcortical infarction. In contrast, rats treated with ELVs show less extensive damage, mostly in the subcortical area. (D) Cortical, subcortical, and total corrected infarct volumes. All ELV treatments markedly reduced cortical, subcortical, and total infarct volumes compared to the vehicle-treated group. Values shown are means ± SD (*P < 0.05, significantly different from vehicle; repeated-measures ANOVA, followed by Bonferroni tests; n = 5 to 6 per group).
DISCUSSION
We have identified previously unknown lipid mediators derived from VLC-PUFAs,n-3 that protect neural cells. We demonstrated that ELV-N32 (Na or Me forms) or ELV-N34 (Na or Me forms), when applied to cerebral-cortical mixed neuronal cells or hippocampal mixed neuronal cells in culture, can overcome the damaging effects of UOS, NMDA-induced neuronal excitotoxicity, or OGD. Most of the strokes are ischemic in nature (16), and deprivation of oxygen and glucose leads to a cascade of events involving mitochondrial damage, which ultimately leads to neuronal death. Therefore, the in vitro OGD model provides an opportunity for teasing out the cellular events and putative underlying neuroprotective signaling pathways in which ELVs participate. We showed that both ELV-N32 and ELV-N34 elicit neuroprotection and overcome neuronal cytotoxicity. We also showed that the 34-carbon n-3 VLC-PUFA (C34:6n3) precursor of ELVs, when applied at a dose of 250 nM after 2 hours of reoxygenation phase following 90 min of OGD insult, could provide neuroprotection to cerebral-cortical neurons. In conclusion, the endogenously generated ELVs (ELV-N32 or ELV-N34) ameliorated neuronal injury induced by several stressors, such as NMDA receptor activation, UOS, or OGD in cerebral-cortical mixed neuronal and hippocampal mixed neuronal cultures. These novel bioactive lipids belong to a new class of lipid mediators, termed ELVs, which are derived from phospholipid molecular species having two different PUFAs at positions C1 and C2.
We have shown here that all ELV treatments, delivered at 1 hour after 2 hours of experimental ischemic stroke, improved neurological recovery throughout the 7-day survival period. We also used magnetic resonance imaging (MRI), a highly sensitive tool for the detection of changes in water content and diffusion, both of which characterize acute ischemic stroke (17). The rapid induction of brain edema following focal ischemia is the leading cause of morbidity and death after stroke (18). Maximum protection was detected in the cortex (the penumbral area) and also in the subcortical area. Histopathology revealed smaller infarcts in cortical and subcortical areas with less pancellular damage, denser eosinophilic areas, and shrunken neurons along the infarct margin, all of which were detected in ELV-treated rats.
Cerebral ischemia initiates a complex cascade of cellular, molecular, and metabolic events that lead to irreversible brain damage (19–21). Dead neurons and injured tissue are scavenged by activated resident microglia and/or macrophages that invade the injured tissue from the bloodstream. Surviving astrocytes and activated microglia in the penumbra may facilitate restoration of neuronal integrity by producing growth factors, cytokines, and extracellular matrix molecules involved in repair mechanisms (22). Our results demonstrate that ELV treatment increased the number of NeuN-positive neurons and GFAP-positive reactive astrocytes and the SMI-71–positive blood vessel density in the cortex. Blood vessel integrity facilitates neurogenesis and synaptogenesis, which, in turn, contribute to improved functional recovery.
Neurogenesis continues after birth and can be reactivated as a response to injury (23). The generation of new neurons from progenitors occurs in the subgranular zone of the DG, the subventricular zone (SVZ) of some cortical areas, the substantia nigra, and the periinfarcted areas (24). Neural stem cells (NSCs) persist in the forebrain SVZ within a niche containing endothelial cells that might stimulate NSC expansion and neurogenesis. Experimental stroke increases neurogenesis and angiogenesis, but how endothelial cells influence stroke-induced neurogenesis is unknown. The leading process of the migrating neural progenitor cells (NPCs) is closely associated with blood vessels, suggesting that this interaction provides directional guidance to the NPCs (23). The origin of newly formed vessels and the significance of neovascularization and neurogenesis are important unanswered issues in the understanding of stroke pathophysiology and their contribution to recovery.
After cerebral ischemia, the integrity of the NVU is compromised, allowing uncontrolled entry of molecules into the brain parenchyma that worsens damage caused by ischemia (25). In patients, a loss of NVU integrity is associated with a worse stroke outcome (26). Here, we measured ischemic disruption of the NVU by infiltration of endogenous IgG into the brain parenchyma. Treatment with ELV-N34 Na and ELV-N34 Me attenuated NVU disruption induced by focal cerebral ischemia.
We showed here that the newly identified ELVs protected neurons undergoing OGD or NMDA receptor–mediated excitotoxicity. Moreover, ELVs attenuated infarct volumes, rescued the ischemic core and penumbra, diminished NVU damage, and promoted cell survival accompanied with neurological/behavioral recovery. It is reasonable to propose that novel ELV therapies have the potential to treat focal ischemic stroke and other conditions that engage inflammatory/homeostatic disruptions.
MATERIALS AND METHODS
Primary cultures of neurons
All animals were handled in compliance with National Institutes of Health (NIH) guidelines, and the experimental protocols were approved by the Institutional Animal Care and Use Committee for Animal Research of Louisiana State University Health New Orleans. Primary cultures of cortical and hippocampal neurons were harvested from 18-day-old embryos (E18) taken from timed-pregnant, 2-month-old Sprague-Dawley (SD) rats (Charles River Laboratories). Briefly, timed-pregnant SD rats were euthanized, and embryos were collected under sterile conditions. Embryonic brains were dissected out on ice by forceps and placed in a petri dish containing ice-cold Hanks’ balanced salt solution (HBSS) (Gibco) (without Ca2+) containing 10 mM Hepes (pH 7.2) (Gibco), 0.2 mM glutamine, and penicillin (100 U/ml) and streptomycin (100 μg/ml) (Pen Strep) (Gibco). Meninges were removed under a dissecting microscope, and cortical or hippocampal tissues were chopped into small pieces using micro-spring scissors. These tissues were transferred to 15-ml tubes containing oxygenated trypsin-EDTA (0.025% in HBSS) and deoxyribonuclease I. The tubes were incubated in 37°C chamber for 15 min and were agitated every 5 min. After stopping the trypsinization with 5 ml of 10% fetal bovine serum, these tissues were triturated 15 times using a fire-polished Pasteur pipette. Then, cell clumps were left to settle for 2 min to allow debris to settle down, and the supernatant was transferred to a 15-ml Eppendorf tube. The supernatant was filtered through a 70-μm pore size filter (Corning cell strainer) and centrifuged in a tube for 5 min at 1000 rpm. The cells were then resuspended in Neurobasal medium (Gibco) containing 2% B27 (Gibco) and 2% N-2 (Gibco) supplements, along with 0.5 mM glutamine and Pen Strep (50 U/ml) (Gibco). Cells were counted using a Neubauer hemocytometer. Cells (1 × 106) were seeded on a poly-d-lysine–coated 12-well cell culture dish (Corning) and cultured in an incubator (37°C, 5% CO2). The culture medium was first replaced after 24 hours, and half of the medium was then replaced with fresh medium every 3 days for the first week and every other day thereafter. As a result, approximately 90% purity of neurons was obtained, as determined by class III β-tubulin, GFAP, and Hoechst 33258 staining (fig. S4B). Cells were maintained in culture for about 2 weeks until they were exposed to UOS, OGD, or NMDA excitotoxicity. The representative images showing the morphology of cerebral-cortical mixed neuronal cell cultures and hippocampal mixed neuronal cultures [12 days in vitro (DIV 12)] are shown in fig. S4A, whereas representative images showing the morphology of cerebral-cortical mixed neuronal cell cultures (DIV 28) are shown in fig. S4 (C and D).
OGD, NMDA excitotoxicity, or UOS exposure
An in vitro OGD model was established (10–13). Primary cortical or hippocampal neurons were cultured from SD rat embryos. On DIV 12, cells were washed with phosphate-buffered saline (PBS) and incubated with glucose-free Neurobasal medium (Gibco) for 30 min. After that, the cells were placed in a modular incubator chamber (Billups-Rothenberg Inc.) and incubated in an anaerobic chamber (95% N2 and 5% CO2) for OGD for 90 min at 37°C. After 90 min of OGD exposure, cells were returned to the original medium [Neurobasal medium (Gibco) containing 2% B27 (Gibco) and 2% N-2 (Gibco) supplements, along with 0.5 mM glutamine and Pen Strep (50 U/ml) (Gibco)] and placed in a normoxic chamber (37°C, 5% CO2) for 2 hours. Then, the medium was changed with a medium containing either ELV-N32 or ELV-N34 (500 nM) and maintained in a normoxic chamber (37°C, 5% CO2) for 12 hours, after which the cells were sampled and assayed for cell viability using different methods, as previously described (27–31). For the normoxic (control) conditions, neurons were washed with PBS but maintained in a regular medium [Neurobasal medium (Gibco) containing 2% B27 (Gibco) and 2% N-2 (Gibco) supplements, along with 0.5 mM glutamine and Pen Strep (50 U/ml) (Gibco)] during the course of 120 min when the other cells were subjected to OGD stress. Following this, the control cells were subjected to two subsequent regular medium changes to match the timings of the cells that were OGD-stressed. Cerebral-cortical mixed neuronal cells or hippocampal mixed neuronal cells in culture were exposed to either NMDA or UOS for 12 hours by the addition of either NMDA (at a concentration of 25, 50, or 100 μM) or TNFα (10 ng/ml) and H2O2 (50, 100, or 200 μM). Cell viability and neuroprotection in the presence of ELV-N32, ELV-N34, 32:6, or 34:6 were assayed after 12 hours.
Hoechst staining and unbiased image analysis
Cells were washed with 1X Dulbecco’s PBS containing no calcium or magnesium (Gibco) and fixed for 10 min using ice-cold 4% paraformaldehyde (PFA), followed by a 15-min incubation with 100% methanol. Then, the cells were washed with 1X PBS (pH 7.4) (Gibco) and incubated in PBS containing 20 μM Hoechst 33258 (Molecular Probes) for 20 min. After that, the cells were washed three times with 1X PBS and stored in 1X PBS at 4°C until they were imaged for microscopy. One 4 × 4 tile mosaic was acquired from the center of each well using a Zeiss 510 Meta laser confocal microscope and the LSM 510 Meta software. The size of the images was 3600 μm × 3600 μm with a resolution of 1.1378 pixels/μm and a pixel size of 0.879 μm × 0.879 μm. Each of the images had anywhere from 8000 to 15,000 cells, based on different experimental conditions and from one preparation of culture to the other. We imaged at least three separate wells per condition per experiment and then analyzed them for cell survival. Images were imported into the ImageJ image analysis software (NIH, Bethesda, MD) and batch-processed using custom macros. An Otsu auto threshold was applied to each image of Hoechst-stained nuclei, and the area of each detected object was recorded, as previously described (27, 28). To estimate the percentage of nonpyknotic nuclei, we chose a size cutoff value above which objects were assumed to be nonpyknotic. The size cutoff value for pyknosis was located at the mode of the corresponding NMDA/OGD-only treatment group, and all objects below that value were assumed to be pyknotic, except for those with areas of <10 μm2, which were assumed to represent cellular debris and were excluded from the analysis. The results were exported into Microsoft Excel and analyzed. We noticed that there were small but unavoidable changes in the distribution of object sizes that would occur from preparation to preparation and slightly shift the appropriate cutoff value. Hence, the cutoff is not the same for all the experiments. The nuclear size cutoff for defining pyknotic versus nonpyknotic nuclei is now represented by black dashed lines and yellow boxes in Figs. 2 (C and D) and 3 (C and D) and fig. S5 (A to D). For different experiments, the cutoff value varied within a range from 48 to 62 μm2, with most of the pyknotic nuclei measuring about 30 to 50 μm2, whereas most of the nonpyknotic nuclei ranging from 80 to 140 μm2.
Calcein AM–propidium iodide live/dead assay and MTT assay
A 10-ml solution was prepared by combining both the components of the live/dead cytotoxicity kit (Invitrogen) using 20 μl of component A (calcein AM) and 20 μl of component B (propidium iodide). Calcein acetoxymethyl ester (calcein AM) is a cell-permeant dye that can be used to determine cell viability in most eukaryotic cells. In live cells, the nonfluorescent calcein AM is converted to green fluorescent calcein after acetoxymethyl ester hydrolysis by intracellular esterases. On each well of a 12-well cell culture plate, 50 μl of this solution was added, and the cells were incubated in a normoxic chamber (37°C, 5% CO2) for 1 to 2 hours. Then, the cells were imaged using an Olympus FluoView laser confocal microscope. Images were imported into the NIH ImageJ image analysis software, and green and red channels were separated. Then, using the cell counter, the images were counted to determine the number of live cells (green) and dead nuclei (red). The results were exported into Microsoft Excel and analyzed. The representative images of calcein-stained neurons are shown in fig. S3. The MTT assay is based on the cleavage of the yellow tetrazolium salt MTT to purple formazan crystals by metabolically active cells. The assay was performed to measure the viability of primary cortical neurons in each treatment group. Briefly, MTT (5 mg/ml and 100 μl per well) (Sigma-Aldrich) was added to the cells in 12-well plates and incubated in a normoxic chamber (37°C, 5% CO2) for 2 hours. Then, the generated blue formazan reduction product, due to the action of succinate dehydrogenase in living cells on the dye, was dissolved in 1 ml of isopropyl alcohol and transferred to triplicate wells in a 96-well plate, and its absorbance was read at 490 nm using a Molecular Probes Spectramax microplate reader. The results were expressed as the percentage of cell survival.
MCAo and cannula implantation into the right lateral ventricle
All studies were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center. Male SD rats (Charles River Laboratories), weighing 280 to 340 g, were fasted overnight but allowed free access to water. Atropine sulfate [0.5 mg/kg intraperitoneally (ip)] was injected 10 min before anesthesia. Anesthesia was induced with 3% isoflurane in a mixture of 70% nitrous oxide and 30% oxygen. All rats were orally intubated and mechanically ventilated. During ventilation, the animals were paralyzed with pancuronium bromide (0.6 mg/kg ip). The catheters were implanted into the right femoral artery and vein for blood sampling and infusion of drug. Serial analyses of arterial blood gases, plasma glucose, arterial blood pressure, and heartbeat rate were conducted before and during surgical procedure. Rectal (CMA 150 Temperature Controller and CMA Microdialysis AB) and cranial (temporal muscle; Omega Engineering) temperatures were closely monitored before, during, and after MCAo. Rectal temperature and body weight were monitored daily until sacrifice.
Rats underwent 2 hours of right MCAo by an intraluminal filament, as previously described (15). Briefly, the right common carotid artery bifurcation was exposed through a midline neck incision, and the occipital artery branches of the external carotid artery were isolated, ligated, and dissected. After careful isolation of the internal carotid artery (ICA), a 3-0 monofilament coated with poly-l-lysine was advanced through the ICA to the middle cerebral artery until mild resistance was felt. The neck incision was closed with a silk suture, and the animals were then allowed to recover. After 2 hours of MCAo, rats were reanesthetized with the same anesthetic combination. Temperature probes were reinserted, and intraluminal sutures were carefully removed. The animals were then allowed to survive for 7 days with free access to food and water.
Thirty minutes after suture removal, a brain infusion cannula was implanted into the right lateral ventricle for treatment administration in each rat. Briefly, rats were anesthetized with 3% isoflurane and were secured to a stereotaxic apparatus with skull leveled between bregma and lambda. A sterile stainless steel cannula (5 mm long) was implanted into the lateral ventricle using the stereotaxic coordinates (0.2 mm caudal to bregma, 2 mm lateral to midline, and 5 mm below the dura). Cannulas were removed after the treatment was completed.
Treatment
ELVs as Na or Me were dissolved in artificial CSF and administered into the right lateral ventricle 1 hour after 2 hours of MCAo. The following ELVs were used: ELV-N32 Na, ELV-N32 Me, ELV-N34 Na, and ELV-N34 Me (5 μg/50 μl), or CSF (50 μl). All treatments were administered by a researcher blinded to the treatment groups.
Neurological/behavioral tests
Behavioral tests were performed by an observer blinded to the treatment groups at 60 min (during MCAo) and then on days 1, 2, 3, and 7 after MCAo. The battery consisted of two tests that had been used previously to evaluate various aspects of neurologic function: (i) the postural reflex test to examine upper body posture while the animal is suspended by the tail and (ii) the forelimb placing test to examine sensorimotor integration in forelimb placing responses to visual, tactile, and proprioceptive stimuli. Neurological function was graded on a scale of 0 to 12 (normal score, 0; maximal score, 12), as previously described (15). Rats that did not demonstrate high-grade contralateral deficit (score, 10 to 11) at 60 min during MCAo were excluded from further study.
MRI acquisition and analysis of total lesion, core, and penumbra volumes
High-resolution ex vivo MRI was performed on 4% PFA–fixed brains on day 7 using an 11.7-T Bruker Advance 8.9-cm horizontal bore instrument equipped with an 89-mm receiver coil (Bruker Biospin). T2WI, diffusion-weighted images (DWI), 3D volumes, and apparent diffusion coefficient (ADC) maps were collected, as we previously described (32). T2 and ADC maps were computed from T2WI and DWI, respectively. We used Hierarchical Region Splitting to automatically identify core and penumbra volumes (total lesion = core + penumbra) from T2 relaxation and water mobility (ADC), as we have published previously (32). Our penumbral tissue determination by Hierarchical Region Splitting was confirmed by the use of perfusion-weighted imaging (PWI)/DWI subtractions at each brain level, as we have performed previously (32). The penumbra was defined as the difference between the PWI and abnormal ADC (diffusion-perfusion mismatch) (2 SD elevation or reduction compared to normal tissues).
Histopathology and immunohistochemistry
Animals were allowed to survive for 7 days after MCAo. Then, rats were reanesthetized with 3% isoflurane, 70% nitrous oxide, and a balance of oxygen and transcardially perfused with 0.9% saline, followed by 4% PFA. Brains were then removed and embedded in a gelatin matrix using MultiBrain Technology (NeuroScience Associates), as previously described (33). To quantitate infarct volume, we digitized histological sections (MCID core imaging software, InterFocus Imaging Ltd.) at nine standardized coronal levels (bregma levels: +5.2, +2.7, +1.2, −0.3, −1.3, −1.8, −3.8, −5.0, and −7.3 mm) using a charge-coupled device camera (QICAM Fast 1394, QImaging) (33). Brain sections were imaged on a motorized microscope BX61VS (Olympus) at 10× objective. An investigator blinded to the experimental groups then outlined the zone of the cortical and subcortical infarcts as well as the left and right hemispheres of each section. Infarct volume was calculated as the integrated product of the cross-sectional area and intersectional distance and corrected for brain swelling. Brain edema was measured by the differences of ipsilateral and contralateral hemispheres (15). Immunohistochemical procedures were performed on the adjacent sections to identify specific vascular and neuronal elements in the ischemic core and penumbra. The following antibodies were used: rat BBB (SMI-71, BioLegend) as a vascular marker, GFAP (Agilent Technologies) to label reactive astrocytes, and NeuN (Chemicon/Millipore) and biotinylated anti-rat IgG antibody (BioLegend) to detect NVU breakdown. The number of positive cells and immunopositive vessels was counted in the cortex and striatum at the level of the central lesion (bregma level, −0.3 mm). Data were expressed as numbers of positive cells and vessels per high-power microscopic field (magnification, ×40). The images of sections were obtained using confocal laser microscope (LSM 510, Carl Zeiss MicroImaging) following the specific experimental protocols. The images were acquired with a dimension of 212.3 μm × 212.3 μm using Zen software (Carl Zeiss MicroImaging). Image analysis was conducted using ImageJ software. Analyses were conducted by an investigator blinded to the experimental conditions. IgG staining intensity was calculated and averaged at the same levels as assessed for ischemic damage, as previously described (25, 34). To calculate the intensity of IgG staining, we converted the images to gray scale and recorded and compared the mean gray values. ImageJ software assigns black pixels for the numerical value of “0” and white pixels for the numerical value of “1.” Gradations of gray were assigned the numerical values in between, increasing with pixel lightness and decreasing with pixel darkness. As such, IgG staining intensity values were expressed as the reciprocal of mean gray for graphical clarity. All sections were imaged at the same time with the same settings and with no adjustment to brightness or contrast. IgG staining intensity was measured in the entire contralateral and ipsilateral hemispheres, as well as the cortex and striatum.
Statistical analysis
For cell cultures
All results were expressed as means ± SEM. Data from all experiments were evaluated using one-way ANOVA, followed by Holm-Sidak’s multiple comparisons post hoc test. Statistical analyses were performed using GraphPad Prism software (version 7.02). A value of P < 0.05 was considered statistically significant.
For ischemic stroke
Values are presented as means ± SD. Repeated-measures ANOVA, followed by Bonferroni procedures to correct for multiple comparisons, were used for intergroup comparisons of neurobehavioral scores over time and infarct areas across coronal levels. Two-tailed Student’s t tests were used for two-group comparisons. Differences at P < 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We would like to thank NeuroScience Associates Inc. for performing immunohistochemical staining. Funding: This work was supported by the National Institute of Neurological Disorders and Stroke (grant NS046741 to N.G.B.) and the National Institute of General Medical Sciences (grant GM103340 to N.G.B.). This work was also funded by the Arthur C. Cope Scholar Fund administered by the American Chemical Society (to N.A.P.). Author contributions: N.G.B. and N.A.P. conceived the project and wrote the manuscript. N.G.B., S.B., B.J., and L.B. designed the experiments. S.B., B.J., J.H., M.-A.K., A.O., H.M., S.J.M., R.Y., and L.K. performed the experiments and collected and analyzed the data. All authors discussed the results and edited the manuscript. Competing interests: N.G.B. and N.A.P. are inventors on a patent application related to this work filed by the Board of Supervisors for Louisiana State University and Agricultural and Mechanical College and University of Southern California (Patent Cooperation Treaty application no. PCT/US16/17112, 9 February 2016; U.S. patent application no. 15/549,676, 9 August 2017). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1700735/DC1
fig. S1. ELV-N32 and ELV-N34 elicit protection of cerebral-cortical mixed neuronal cell cultures exposed to OGD or NMDA.
fig. S2. ELV-N32 and ELV-N34 in neuronal cell cultures.
fig. S3. Representative images of calcein-stained cerebral-cortical mixed neuronal cells exposed to OGD or NMDA.
fig. S4. Representative bright-field and fluorescent images of cerebral-cortical mixed neuronal and hippocampal mixed neuronal cultures.
fig. S5. Absolute frequency histograms of cerebral-cortical mixed neuronal or hippocampal mixed neuronal cultures exposed to OGD or NMDA.
fig. S6. Percent relative frequency histograms.
fig. S7. Clustered histograms of percent relative frequencies.
REFERENCES AND NOTES
- 1.Serhan C. N., Dalli J., Colas R. A., Winkler J. W., Chiang N., Protectins and maresins: New pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim. Biophys. Acta 1851, 397–413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bazan N. G., Molina M. F., Gordon W. C., Docosahexaenoic acid signalolipidomics in nutrition: Significance in aging, neuroinflammation, macular degeneration, Alzheimer’s, and other neurodegenerative diseases. Annu. Rev. Nutr. 31, 321–351 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cameron D. J., Tong Z., Yang Z., Kaminoh J., Kamiyah S., Chen H., Zeng J., Chen Y., Luo L., Zhang K., Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int. J. Biol. Sci. 3, 111–119 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Agbaga M.-P., Different mutations in ELOVL4 affect very long chain fatty acid biosynthesis to cause variable neurological disorders in humans. Adv. Exp. Med. Biol. 854, 129–135 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Aldahmesh M. A., Mohamed J. Y., Alkuraya H. S., Verma I. C., Puri R. D., Alaiya A. A., Rizzo W. B., Alkuraya F. S., Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am. J. Hum. Genet. 89, 745–750 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mukherjee P. K., Marcheselli V. L., Serhan C. N., Bazan N. G., Neuroprotectin D1: A docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 101, 8491–8496 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marcheselli V. L., Hong S., Lukiw W. J., Tian X. H., Gronert K., Musto A., Hardy M., Gimenez J. M., Chiang N., Serhan C. N., Bazan N. G., Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 278, 43807–43817 (2003). [DOI] [PubMed] [Google Scholar]
- 8.Serhan C. N., Gotlinger K., Hong S., Lu Y., Siegelman J., Baer T., Yang R., Colgan S. P., Petasis N. A., Anti-inflammatory actions of neuroprotectin D1/protectin D1 and its natural stereoisomers: Assignments of dihydroxy-containing docosatrienes. J. Immunol. 176, 1848–1859 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Petasis N. A., Yang R., Winkler J. W., Zhu M., Uddin J., Bazan N. G., Serhan C. N., Stereocontrolled total synthesis of neuroprotectin D1/protectin D1 and its aspirin-triggered stereoisomer. Tetrahedron Lett. 53, 1695–1698 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tasca C. I., Dal-Cim T., Cimarosti H., In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol. Biol. 1254, 197–210 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Chen H., Lin W., Zhang Y., Lin L., Chen J., Zeng Y., Zheng M., Zhuang Z., Du H., Chen R., Liu N., IL-10 promotes neurite outgrowth and synapse formation in cultured cortical neurons after the oxygen-glucose deprivation via JAK1/STAT3 pathway. Sci. Rep. 6, 30459 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lo E. H., Dalkara T., Moskowitz M. A., Mechanisms, challenges and opportunities in stroke. Nat. Rev. Neurosci. 4, 399–415 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Eltzschig H. K., Eckle T., Ischemia and reperfusion—From mechanism to translation. Nat. Med. 17, 1391–1401 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thrift A. G., Thayabaranathan T., Howard G., Howard V. J., Rothwell P. M., Feigin V. L., Norrving B., Donnan G. A., Cadilhac D. A., Global stroke statistics. Int. J. Stroke 12, 13–32 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Belayev L., Alonso O. F., Busto R., Zhao W., Ginsberg M. D., Middle cerebral artery occlusion in the rat by intraluminal suture. Neurological and pathological evaluation of an improved model. Stroke 27, 1616–1622 (1996). [DOI] [PubMed] [Google Scholar]
- 16.Shi S.-h., Qi Z.-f., Luo Y.-m., Ji X.-m., Liu K. J., Normobaric oxygen treatment in acute ischemic stroke: A clinical perspective. Med. Gas Res. 6, 147–153 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bazan N. G., Eady T. N., Khoutorova L., Atkins K. D., Hong S., Lu Y., Zhang C., Jun B., Obenaus A., Fredman G., Zhu M., Winkler J. W., Petasis N. A., Serhan C. N., Belayev L., Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp. Neurol. 236, 122–130 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Posada-Duque R. A., Barreto G. E., Cardona-Gomez G. P., Protection after stroke: Cellular effectors of neurovascular unit integrity. Front. Cell. Neurosci. 8, 231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iadecola C., Anrather J., The immunology of stroke: From mechanisms to translation. Nat. Med. 17, 796–808 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moskowitz M. A., Lo E. H., Iadecola C., The science of stroke: Mechanisms in search of treatments. Neuron 67, 181–198 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratan R. R., Beyond neuroprotection to brain repair: Exploring the next frontier in clinical neuroscience to expand the therapeutic window for stroke. Transl. Stroke Res. 1, 71–73 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panickar K. S., Norenberg M. D., Astrocytes in cerebral ischemic injury: Morphological and general considerations. Glia 50, 287–298 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Font M. A., Arboix A., Krupinski J., Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr. Cardiol. Rev. 6, 238–244 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arvidsson A., Collin T., Kirik D., Kokaia Z., Lindvall O., Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 8, 963–970 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Haley M. J., Lawrence C. B., The blood–brain barrier after stroke: Structural studies and the role of transcytotic vesicles. J. Cereb. Blood Flow Metab. 37, 456–470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brouns R., Wauters A., De Surgeloose D., Mariën P., De Deyn P. P., Biochemical markers for blood-brain barrier dysfunction in acute ischemic stroke correlate with evolution and outcome. Eur. Neurol. 65, 23–31 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Stark D. T., Bazan N. G., Synaptic and extrasynaptic NMDA receptors differentially modulate neuronal cyclooxygenase-2 function, lipid peroxidation, and neuroprotection. J. Neurosci. 31, 13710–13721 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeshita K., Ogawa H. I., Maeda T., Structural chromosome aberrations cause swelling of the nucleus. Genes Environ. 38, 10.1186/s41021-016-0047-7(2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hardingham G. E., Bading H., Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 11, 682–696 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hardingham G. E., Fukunaga Y., Bading H., Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5, 405–414 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Iadecola C., Niwa K., Nogawa S., Zhao X., Nagayama M., Araki E., Morham S., Ross M. E., Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 98, 1294–1299 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obenaus A., Dilmac N., Tone B., Tian H. R., Hartman R., Digicaylioglu M., Snyder E. Y., Ashwal S., Long-term magnetic resonance imaging of stem cells in neonatal ischemic injury. Ann. Neurol. 69, 282–291 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson S. N., Gibson T. R., Thompson B. M., Deng Y., Hall E. D., Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp. Neurol. 201, 253–265 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Readnower R. D., Chavko M., Adeeb S., Conroy M. D., Pauly J. R., McCarron R. M., Sullivan P. G., Increase in blood–brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 88, 3530–3539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/9/e1700735/DC1
fig. S1. ELV-N32 and ELV-N34 elicit protection of cerebral-cortical mixed neuronal cell cultures exposed to OGD or NMDA.
fig. S2. ELV-N32 and ELV-N34 in neuronal cell cultures.
fig. S3. Representative images of calcein-stained cerebral-cortical mixed neuronal cells exposed to OGD or NMDA.
fig. S4. Representative bright-field and fluorescent images of cerebral-cortical mixed neuronal and hippocampal mixed neuronal cultures.
fig. S5. Absolute frequency histograms of cerebral-cortical mixed neuronal or hippocampal mixed neuronal cultures exposed to OGD or NMDA.
fig. S6. Percent relative frequency histograms.
fig. S7. Clustered histograms of percent relative frequencies.