

Abstract
The aim of the study is to characterize the clinical ocular phenotype with congenital fibrosis of the extraocular muscles type 1 (CFEOM1) and to confirm whether the kinesin family member 21A (KIF21A) mutation was the pathogenic gene in this Chinese family.
Three affected individuals and 2 asymptomatic kinsfolk from a Chinese family underwent comprehensive ophthalmic examinations, orbital computerized tomography (CT), and postoperative histological examinations were performed in the proband. All the recruited members were screened for 3 exons (8, 20, and 21) of KIF21A mutations using the polymerase chain reaction (PCR) amplification and direct sequencing of corresponding PCR products.
All patients shared the clinical characteristics including bilateral ophthalmoplegia, blepharoptosis, hypertropic, and exotropic position with inability to raise either eye above the midline and a chin-up head position. Direct DNA sequence analysis from the affected members revealed a missense mutation in KIF21A (c.2860C>T, p.R954W). The unaffected members did not harbor the p.R954W mutation. The candidate mutation was not present in multiple web-accessible and in-house exome databases.
The p.Arg954Trp mutation of KIF21A was the causative mutation in this Chinese pedigree with CFEOM1.
Keywords: clinical characteristic, congenital fibrosis of the extraocular muscles, direct sequence, KIF21A, mutation
1. Introduction
Congenital fibrosis of the extraocular muscles (CFEOM) is a genetic ocular motility disorder, which is characterized clinically by nonprogressive restrictive ophthalmoplegia with or without ptosis.[1] The reported prevalence of CFEOM in England is 1 in 230,000.[2] Based on the clinical features and genetic components, CFEOM can be currently classified into 5 major types: CFEOM1 (OMIM 135700), CFEOM2 (OMIM 602078), CFEOM3 (OMIM 600638; OMIM 608283; OMIM 609384), CFEOM4 (OMIM 609428), and CFEOM5 (OMIM 616219).
CFEOM1, the classic form of CFEOM, is characterized by bilateral blepharoptosis and nonprogressive ophthalmoplegia with the eyes fixed in an infraducted position about 20° to 30° below the horizontal midline. CFEOM1 is an autosomal dominant trait caused by mutations in the KIF21A gene (OMIM 608283) on chromosome 12q12.[3] CFEOM2 is caused by the mutations in the ARIX gene (OMIM 602753) on chromosome 11q13.4. The typical phenotypes of CFEOM2 includes bilateral ptosis and ophthalmoplegia with eyes fixed in an exotropic position. The clinical manifestations of patients in families with CFEOM3 are similar to those with CFEOM1, whereas it can be diagnosed even if no patient exhibits the classic clinical signs. The subtypes of CFEOM3 could be classified into CFEOM3A, CFEOM3B, and CFEOM3C. Mutations in the TUBB3 gene (OMIM 602661) on the chromosome 16q24 and KIF21A gene on chromosome 12q12 are responsible for CFEOM3A and CFEOM3B, respectively.[4] The causative gene for CFEOM3C maps to chromosome 13q. CFEOM4, also known as the Tukel syndrome, its causative gene was mapped to chromosome 21q22.[5] Patients with the Tukel syndrome have nonprogressive restrictive ophthalmoplegia with blepharoptosis of the right eye and postaxial oligodactyly of the hands. CFEOM5 is caused by mutation in the COL25A1 gene (OMIM 610004) on chromosome 4q25. Patients with CFEOM5 presented with variable abnormal ocular motility without other systemic defects. Collectively, molecular diagnosis may help to improve the diagnostic accuracy of CFEOM.
2. Methods
2.1. Clinical subject
This study conformed to the tenets of the Declaration of Helsinki and was approved by the Ethics Committee of the Eye Hospital, Wenzhou Medical University. Written informed consent was obtained from all participating individuals or their guardians. Patients in this study originating from the remote mountainous area of Mianyang, Sichuan Province, China. Medical history was obtained and the pedigree chart is shown in Fig. 1. The proband underwent orbital CT and comprehensive ophthalmic examinations including slit-lamp examination, best-corrected visual acuity, intraocular pressure, fundus examination, extraocular motilities, and synoptophore testing. The proband underwent 2 strabismus surgeries, entropion surgery, and frontalis suspension surgery. The proband also had postoperative histological examinations of the bilateral superior recti with hematoxylin-eosin staining. The ophthalmologist performed detailed ocular examinations on the other 5 individuals.
Figure 1.
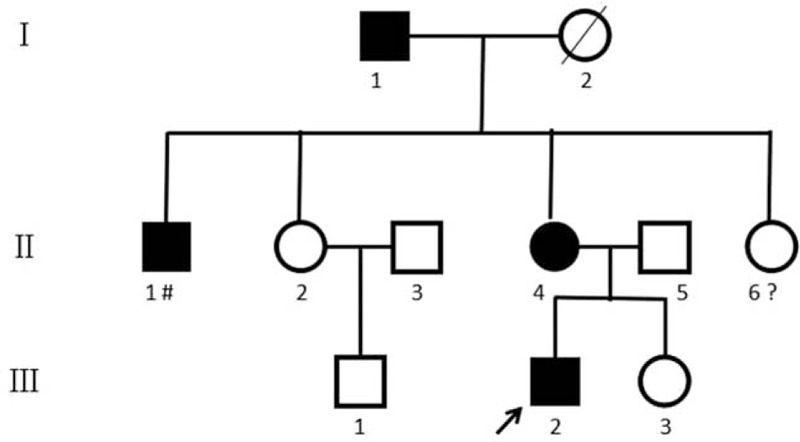
Pedigree of a Chinese family affected with CFEOM1. Normal individuals are shown as empty symbols. Affected males and females are indicated by filled-in squares and circles, respectively. The proband of the pedigree is indicated by an arrow. Deceased individuals are indicated by a slash (/). The normal individual who lost contact with her family at 4 years old is indicated by a question mark (?). The affected male who was unable to be examined due to his occupation as a migrant worker is indicated by octothorpe (#). CFEOM1 = congenital fibrosis of the extraocular muscles type 1.
2.2. DNA isolation
The proband and 5 members of the pedigree donated a 5 mL of whole venous blood sample for genomic DNA isolation. Ethylenediaminetetraacetic acid (EDTA) was used to treat the tubes used for blood collection. Genomic DNA was extracted by standard techniques. A NanoDrop ND-2000 spectrophotometer and 0.6% agarose gel electrophoresis was used to concentrate and purify the DNA. Genomic DNA samples and PCR amplification templates were stored at –20°C.
2.3. Mutation analysis
ABI9700-PCR Amplifier (ABI, America) was used to perform standard PCR. Each reaction tube contained 200 ng of genomic DNA, 0.2 μM of each primer, 0.2 mM dNTP, 10×buffer 5 μL, and 0.2U TapDNA Polymerase (Dingguo Changsheng Biotechnology Limited Company, Beijing, China). Because the frequently reported mutations are in Exon 8, 20, and 21, which are the mutation-rich exons, we consider screening strategy allowing analysis of these 3 exons with first priority. The fragments harboring corresponding exons were amplified by PCR. The initial denaturation PCR reaction was carried out for 30 cycles at 94°C for 5 minutes, denatured at 94°C for 40 seconds, annealed at 60°C for 30 seconds, elongated at 72°C for 40 seconds, and followed by 10 minutes of extension at 72°C. Table 1 lists the specific conditions for each PCR primer. The PCR product was then amplified in 1.5% agarose gel electrophoresis for 30 minutes. Afterwards, the PCR products were purified with Amicon purification columns and then used as templates for direct sequencing by the ABI 3100 Genetic Analyzer (Perkin-Elmer, America).
Table 1.
KIF21A PCR primers.

3. Results
3.1. Clinical findings
Three affected individuals presented with similar CFEOM1 phenotypes of nonprogressive bilateral ophthalmoplegia, bilateral blepharoptosis, hypertropic and exotropic position with inability to raise either eye above the midline, and compensatory chin-up head position.
Patient III:2, the 17-year-old proband, was born with bilateral blepharoptosis after an uneventful full-term pregnancy. Presenting visual acuities were 3/20 in the right eye and 6/20 in the left eye. His intraocular pressures were normal in both eyes. The width of the palpebral fissures measured 1 mm in the right eye and 2 mm in left eye. Severe bilateral blepharoptosis and compensatory chin-up head position was observed. The primary position of each eye was fixed in an infraducted position about 20° to 30° of vertical deviation and 40° to 50° of horizontal deviation. He was not capable of raising either eye above the horizontal midline. Forced duction testing during strabismus surgery demonstrated both eyes could only abduct with resistance in all other directions. Bilateral inferior entropion and blepharoptosis were also observed (Fig. 2). Orbital CT showed a reduced size of the superior rectus and slight abnormality of the optic nerve course. After vertical strabismus diorthosis and horizontal strabismus diorthosis, the fixating eye could hold a primary position with no deviation, with a 15°to 20° horizontal deviation of the other eye. The width of the postoperative palpebral fissure was much wider than that of the preoperative palpebral fissure, and the upper eyelid edge now reached the upper edge of the pupil (Fig. 3). Postoperative histological examination of the bilateral superior recti revealed atrophy and fibrosis. Vascular proliferation, hemorrhage, and hyperemia were observed in the affected areas. An increase in myocyte nuclei was also found (Fig. 4).
Figure 2.
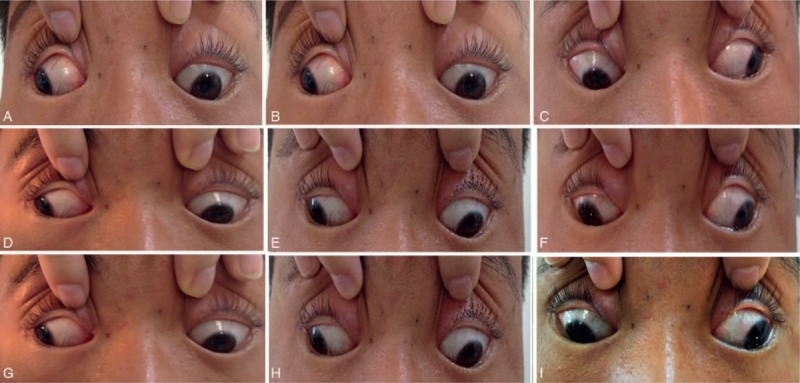
Photographs of the proband. Photographs were taken in the primary position (E) and the 8 cardinal gaze positions (A, B, C, D, F, G, H, and I). The photos show typical ophthalmoplegia, hypertropia, and exotropia in the primary position with the inability to raise either eye above the horizontal midline. This patient also had ptosis in both eyes (not shown).
Figure 3.

Postoperation photographs of the proband's vertical strabismus diorthosis and horizontal strabismus diorthosis. After surgery, the primary position of the fixating eye shows no deviation with a 15° to 20° horizontal deviation of the other eye (A). The upper eyelid margin reached the upper edge of the pupil with reduced chin-up head position (B).
Figure 4.
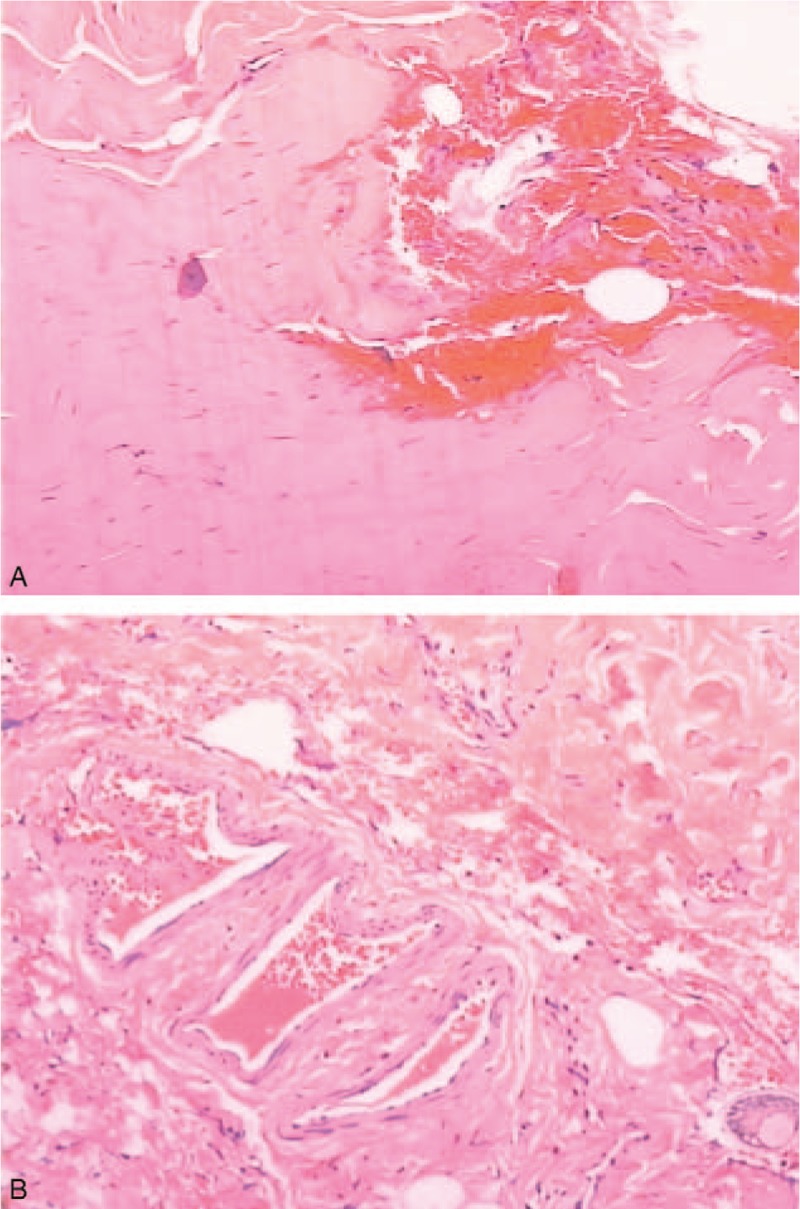
Photographs of postoperative histological examination. Bilateral superior recti showed atrophy and fibrosis. Vascular proliferation, hemorrhage, and hyperemia were observed in local areas (A). An increase in myocyte nuclei was also found (B).
Patient I:1 and Patient II:4 had the same phenotypes as the proband (Fig. 5). Patient I:1, the proband's grandfather, suffered from severe bilateral blepharoptosis, with the palpebral fissure widths measuring about 1 mm in both eyes. Bilateral restrictive ophthalmoplegia, hypertropic and exotropic position, inability to raise either eye above the horizontal midline, and marked chin-up head position were observed. Patient II:4, the proband's mother, presented similar features to Patient I:1 and the proband.
Figure 5.
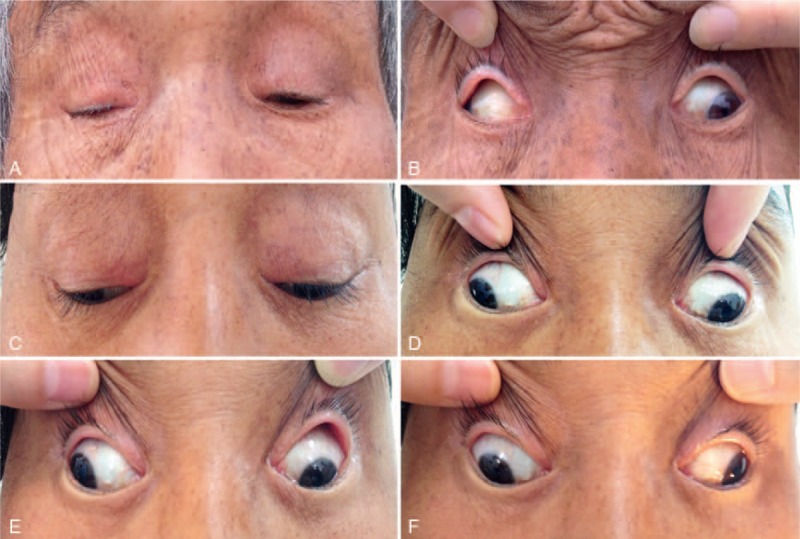
Photographs of Patient I:1 and Patient II:4. Photos of Patient I:1 were taken in primary gaze (A and B). Photos of Patient II:4 were taken in primary gaze (C and D), in right gaze (E), and left gaze (F).
3.2. Molecular genetic analysis
Direct DNA sequence analysis of 3 exons of KIF21A gene showed the same heterozygous base substitution c.2860C>T (GenBank accession No. AY368079) in all 3 patients, which resulted in substitution of the Arginine residue at codon 954 by a tryptophan residue (p.Arg954Trp). The mutation was not present in the other 2 unaffected family individuals. The direct sequence results can be seen in Fig. 6.
Figure 6.

Gene sequence analysis. The gene sequences from the proband, Patient I:1, and Patient II:4 (A, B, and C, respectively). Analysis showed c.2860C>T mutation. Normal gene sequences were obtained from Individual II:5 and Individual III:3 (D and E, respectively).
The effects of the mutation on protein function were assessed by 4 online softwares, including SIFT (http://sift.jcvi.org/), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), Pmut (http://mmb2.pcb.ub.es:8080/PMut/), and Panther (http://www.pantherdb.org/tools/csnpScoreForm.jsp). The prediction results are Deleterious, Probably damaging, Disease and Probably damaging. We searched these mutations in multiple databases, including the dbSNPs (v130), HapMap, 1000 Genome, and 702 in-house exome data. The candidate mutation was not present in these databases. All these data showed p.Arg954Trp is a causative mutation in this family.
4. Discussion
CFEOM, one of the congenital cranial dysinnervation disorders, is an autosomal dominant strabismus disorder associated with the absence of the superior division of the oculomotor nerve and corresponding midbrain motor neurons. These results in severe atrophy of the 2 muscles normally innervated by this nerve: the levator palpebrae superior and superior rectus muscles. These muscles elevate the eyelid and the eyeball. The primary clinical characteristics of CFEOM1 can be seen if these muscles are dysfunctional.[6]
Database searching has revealed 45 kinesin superfamily proteins (KIFs) in both mice and humans.[7] For neurons, intracellular transport is crucial for neuronal morphogenesis and functioning. Neurons use dynein and kinesin microtubule-dependent motor proteins to transport essential cellular components along dendritic and axonal microtubules. Dyneins and minus end-directed KIFs transport retrograde cargo to the cell body, whereas plus end-directed KIFs transport anterograde cargo to the synapse.[8] KIFs are characterized by a motor domain, consisting of a microtubule-binding domain and an ATP-binding motif. KIF21A, encoded by the KIF21A gene, belongs to the family of plus end-directed KIFs. Predicated KIF21A protein contains 4 domains: an N-terminal motor domain, coiled-coil regions of the stalk, alternatively spliced regions, and 7 WD40 repeats in the C-terminus. The KIF21A gene spans 150 kb of genomic DNA and consists of 38 exons. Most KIF21A gene mutations cluster in a coiled-coil region. Arg954 of KFI21A is located in a coiled-coil region, which is essential for the association and stability of the kinesin dimeric structure. Although Cheng et al suggests that gain-of-function mutations underlie CFEOM1 and that homozygous and heterozygous mutant dimers can disrupt KIF21A autoinhibition, the molecular mechanism of CFEOM1 remains elusive.[9] P.Arg954Trp mutation may lead to an inability to transfer necessary cargo to ocular motor axons, neuromuscular junctions, or extraocular muscles.
Mutational analysis of KIF21A in CFEOM1 revealed a total of 13 different missense mutations (c.84C>G, c.1056C>G, c.1067T>C, c.2830G>C, c.2839A>G, c.2840T>G, c.2840T>C, c.2841G>A, c.2860C>T, c.2861G>A, c.2861G>T, c.3022G>C, c.3029T>C).[4,9–15] Of these mutations, 1 (c.2860 C>T) had been associated with both CFEOM1 and CFEOM3, 1 (c.2841 G>A) had been found in a patient with CFEOM3, and the others have been found in patients with CFEOM1. Mutation c.84C>G was located in exon 2, mutation c.1056C>G and c.1067T>C located in exon 8, mutations c.2830G>C, c.2839A>G, c.2840T>C, c.2840T>G, and c.2841G>A located in exon 20, and mutations c.2860C>T, c.2861G>A, c.2861G>T, c.3022G>C, and c.3029T>C located in exon 21. All these identified mutations and clinical types of CFEOM indicate it is clinically and genetically heterogeneous disease.[16] As far as know, the 2860C>T mutation was the most commonly identified mutation of CFEOM 1 in every race.[17] Yang[18] and Zhang et al[19] also reported the 2860C>T mutation in Chinese, respectively.
In conclusion, we describe a recurrent (c.2860C>T) missense KIF21A mutation identified in a Chinese family with CFEOM1 phenotypes. Our study adds to the knowledge about the genotype and phenotype of KIF21A and supports the genetic heterogeneity of CFEOM1. This report will be helpful to guide genetic study and clinical diagnosis of CFEOM1. However, the specific molecular consequence of the mutation in KIF21A is not clear. We did not study the expression and functional proteomics and establish an animal model, which ascribed to many limitations. Further studies are needed to provide insights into the detailed molecular pathogenesis of p.Arg954Trp mutation.
Acknowledgments
The authors are thankful for the participation of the family from Sichuan Province, China.
Footnotes
Abbreviations: CFEOM1 = congenital fibrosis of the extraocular muscles type 1, CT = computerized tomography, EDTA = ethylenediaminetetraacetic acid, KIF21A = Kinesin family member 21A, PCR = polymerase chain reaction.
Funding: This work was supported by grants from the Bureau of Science and Technology of WenZhou (grant no. Y20150075, ZD. Z.), the Natural Science Foundation of China (81201181, F.G.; 81473295 & 81670882, ZM. S.), Science Technology project of Zhejiang Province (2017C37176, F.G.).
The authors have no conflicts of interest to disclose.
References
- [1].Brown H. JH A. Congenital structural muscle anomalies. Strabismus Ophthalmic Symposium. St Louis: CV Mosby Company; 1950. 205–36. [Google Scholar]
- [2].Reck AC, Manners R, Hatchwell E. Phenotypic heterogeneity may occur in congenital fibrosis of the extraocular muscles. Br J Ophthalmol 1998;82:676–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yamada K, Chan WM, Andrews C, et al. Identification of KIF21A mutations as a rare cause of congenital fibrosis of the extraofcular muscles type 3 (CFEOM3). Invest Ophthalmol Vis Sci 2004;45:2218–23. [DOI] [PubMed] [Google Scholar]
- [4].Tukel T, Uzumcu A, Gezer A, et al. A new syndrome, congenital extraocular muscle fibrosis with ulnar hand anomalies, maps to chromosome 21qter. J Med Genet 2005;42:408–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Engle EC, Goumnerov B, McKeown CA, et al. Oculomotor nerve and muscle abnormalities in congenital fibrosis of the extraocular muscles. Ann Neurol 1997;41:314–25. [DOI] [PubMed] [Google Scholar]
- [6].Marszalek JR, Weiner JA, Farlow SJ, et al. Novel dendritic kinesin sorting identified by different process targeting of two related kinesins: KIF21A and KIF21B. J Cell Biol 1999;145:469–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yamada K, Andrews C, Chan WM, et al. Heterozygous mutations of the kinesin KIF21A in congenital fibrosis of the extraocular muscles type1 (CFEOM1). Nat Genet 2003;35:318–21. [DOI] [PubMed] [Google Scholar]
- [8].Yamada K, Hunter DG, Andrews C, et al. Novel KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Marcus Gunn Jaw Winking phenomenon. Arch Ophthalmol 2005;123:1254–9. [DOI] [PubMed] [Google Scholar]
- [9].Cheng L, Desai J, Miranda CJ, et al. Human CFEOM1 mutations attenuate KIF21A autoinhibition and cause oculomotor axon stalling. Neuron 2015;82:334–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chan WM, Andrews C, Dragan L, et al. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet 2007;8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Uyama E, Yamada K, Kawano H, et al. A Japanese family with FEOM1-linked congenital fibrosis of the extraocular muscles type 1 associated with spinal canal stenosis and refinement of the FEOM1 critical region. Neuromuscul Disord 2003;13:472–8. [DOI] [PubMed] [Google Scholar]
- [12].Wang P, Li S, Xiao X, et al. KIF21A novel deletion and recurrent mutation in patients with congenital fibrosis of the extraocular muscles. Int J Mol Med 2011;28:973–5. [DOI] [PubMed] [Google Scholar]
- [13].Ali Z, Xing C, Anwar D, et al. A novel de novo KIF21A mutation in a patient with congenital fibrosis of the extraocular muscles and Möbius syndrome. Mol Vis 2014;20:368–75. [PMC free article] [PubMed] [Google Scholar]
- [14].Miki H, Setou M, Kaneshiro K, et al. All kinesin superfamily protein, KIF, genes in mouse and human. Proc Natl Acad Sci USA 2001;98:7004–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shimizu S, Okinaga A, Maruo T. Recurrent mutation of the KIF21A gene in Japanese patients with congenital fibrosis of the extraocular muscles. Jpn J Ophthalmol 2006;49:443–7. [DOI] [PubMed] [Google Scholar]
- [16].Lu S, Zhao C, Zhao K, et al. Novel and recurrent KIF21A mutations in congenital fibrosis of the extraocular muscles type 1 and 3. Arch Ophthalmol 2008;126:388–94. [DOI] [PubMed] [Google Scholar]
- [17].Chan WM, Andrews C, Dragan L, et al. Three novel mutations in KIF21A highlight the importance of the third coiled-coil stalk domain in the etiology of CFEOM1. BMC Genet 2007;8:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang X, Yamada K, Katz B, et al. KIF21A mutations in two Chinese families with congenital fibrosis of the extraocular muscles (CFEOM). Mol Vis 2010;16:2062–70. [PMC free article] [PubMed] [Google Scholar]
- [19].Zhang XQ, Peng JH, Tang ZH, et al. Mutation p.Arg954Trp of KIF21A causes congenital fibrosis of the extraocular muscles in a Chinese family. Yi Chuan Xue Bao 2006;33:685–91. [DOI] [PubMed] [Google Scholar]