

Abstract
An important unresolved issue in microbial secondary metabolite production is the abundance of biosynthetic gene clusters that are not expressed under typical laboratory growth conditions. These so-called silent or cryptic gene clusters are sources of new natural products, but how they are silenced, and how they may be rationally activated are areas of ongoing investigation. We recently devised a chemogenetic high-throughput screening approach (“HiTES”) to discover small molecule elicitors of silent biosynthetic gene clusters. This method was successfully applied to a Gram-negative bacterium; it has yet to be implemented in the prolific antibiotic-producing streptomycetes. Herein we have developed a high-throughput transcriptional assay format in Streptomyces spp. by leveraging eGFP, inserted both at a neutral site and inside the biosynthetic cluster of interest, as a read-out for secondary metabolite synthesis. Using this approach, we successfully used HiTES to activate a silent gene cluster in Streptomyces albus J1074. Our results revealed the cytotoxins etoposide and ivermectin as potent inducers, allowing us to isolate and structurally characterize 14 novel small molecule products of the chosen cluster. One of these molecules is a novel antifungal, while several others inhibit a cysteine protease implicated in cancer. Studies addressing the mechanism of induction by the two elicitors led to the identification of a pathway-specific transcriptional repressor that silences the gene cluster under standard growth conditions. The successful application of HiTES will allow future interrogations of the biological regulation and chemical output of the countless silent gene clusters in Streptomyces spp.
Graphical Abstract
INTRODUCTION
Bacterial secondary metabolites have been an important source of pharmaceutical compounds and account for some of our most essential medicines.1–3 Among bacteria that are recognized as proven producers, actinomycetes, notably Streptomyces spp., are the most prolific.4–7 They have been responsible for ~50% of clinical antibiotics, as well as numerous anticancer agents. A more recent realization regarding their potential for secondary metabolite production has come from whole genome sequencing. These data have shown that the molecules discovered thus far merely represent the tip of the iceberg and that most secondary metabolite biosynthetic pathways are not active under typical laboratory growth conditions.4–7 Thus, methods that stimulate such pathways could profoundly impact natural products research and, thereby, drug discovery.
Secondary metabolites are generated by dedicated bio-synthetic gene clusters (BGCs), sets of usually contiguous genes, whose protein products assemble complex molecules from simple precursors. The BGCs that do not give rise to appreciable concentrations of a metabolite during standard laboratory growth have been described as “silent” or “cryptic”.8–12 While several approaches have been developed for finding the products of silent BGCs, they do not inform on when, why, and how a producing host activates a given cluster; that is, the existing methods are focused on identifying the product(s) of a BGC rather than uncovering the endogenous regulatory circuits that control cryptic metabolism. Microbial symbiotic interactions have in some cases been used to identify molecules from one symbiont that stimulate secondary metabolism in another. For free-living bacteria, however, exogenous signals or cues that elicit cryptic metabolites remain largely unknown.12
Given that bacteria employ secondary metabolites to communicate and compete with other microbes, it would stand to reason that they could serve as potential inducers of silent BGCs. To test this idea, we recently developed HiTES (high-throughput elicitor screens), a method that identifies signals necessary for eliciting cryptic metabolites.13–15 In this approach, a reporter gene is inserted inside the BGC of interest, and the resulting strain is screened against libraries of secondary metabolites in a high-throughput fashion to find candidate elicitors. Subsequently, both the small molecule products and the regulatory pathways that lead to induction can be investigated. In a proof-of-concept study, we demonstrated the feasibility of HiTES in the Gram-negative model bacterium Burkholderia thailandensis. Interestingly, all the elicitors that we identified from a library of structurally and functionally diverse molecules were well-known clinical antibiotics, suggesting that at subinhibitory concentrations, antibiotics serve as modulators of silent gene clusters and therefore can be used to find new secondary metabolites.13,16–19
Because Streptomyces spp. are an abundant source of silent BGCs and have a proven track record for generating pharmaceutically active compounds, application of HiTES to this genus would be especially beneficial. Herein we describe a high-throughput transcriptional reporter assay for adaptation of HiTES to Streptomyces spp. Using this methodology, we successfully activated a silent BGC in Streptomyces albus J1074 and identified ivermectin and etoposide, well-known anti-parasitic and antibiotic compounds, respectively, as chemogenetic modulators of secondary metabolism. Induction by these elicitors allowed us to isolate and structurally elucidate 14 novel secondary metabolites, some of which are bioactive as antifungal compounds or inhibitors of important enzymes. Moreover, we report that these elicitors impinge on multiple regulatory pathways, including a previously uncharacterized pathway-specific transcriptional regulator and possibly the SOS response, to elicit cryptic metabolite production. Our method is broadly applicable and will assist in unveiling the products and regulation of other silent BGCs in Streptomyces spp.
RESULTS AND DISCUSSION
Application of HiTES to Streptomycetes
In order to apply HiTES to Streptomyces spp., we first chose a model organism and a silent BGC. We selected S. albus J1074 (hereafter S. albus) as the target organism, because it is genetically tractable and serves as a commonly used heterologous host for expression of actinomycete genes and gene clusters.20–22 Along with Streptomyces coelicolor, S. albus serves as a model strain for this genus. Moreover, like other streptomycetes, S. albus harbors an excess of silent BGCs.22,23 Among these, we chose a large nonribosomal peptide synthetase (NRPS) gene cluster, to which a compound has not yet been associated (Figure 1A, Table S1). Very recent results by the Matsunaga group suggest it is homologous to a cluster in a marine streptomycete strain, and that it gives rise to the so-called surugamides, consistent with the MS-based results by Mohimani et al.24,25 We have named this cluster sur, based on previous results and those reported herein.
Figure 1.

The sur biosynthetic gene cluster and associated reporter constructs. (A) The sur cluster; surA–surD encode nonribosomal peptide synthetases, surE encodes an α,β-hydrolase, and surR encodes a GntR-type transcriptional regulator. The black arrow represents the promoter. (B) Genetic constructs utilized in this study. Triple eGFP driven by Psur inserted at a neutral site (attB::Psur-eGFPx3) or in place of surE (surE::eGFPx3). (C) Reporter assays measuring signal amplification with one, two, or three copies of eGFP driven by PermE.
Selection of a Reporter System in S. albus J1074
Bioinformatic analysis of the sur cluster identified only one promoter sequence upstream of surE (Psur, Figure 1A). While additional promoters maybe present, we selected Psur for further study. To apply HiTES using this promoter, we considered two reporter systems, XylE and eGFP, both of which have previously been employed in S. coelicolor.26,27 The frequently used lacZ reporter gene is not applicable to streptomycetes as many members encode endogenous β-galactosidase activities.28 We created two constructs for each reporter gene, one in which xylE or eGFP was driven by Psur (Psur-eGFP and Psur-xylE) and a second as a positive control, where each reporter gene was driven by the constitutively active erythromycin resistance gene promoter, PermE (PermE-eGFP and PermE-xylE, Tables S2 and S3). Both constructs were inserted into an attB neutral site in the S. albus chromosome and examined using the catechol assay (XylE) or fluorescence (eGFP). Rapid and reliable results proved difficult in our hands with attB::PermE-xylE. By contrast, attB::PermE-eGFP gave reproducible, albeit weak, signal.
The advantage of reporter genes such as xylE and lacZ, in comparison to eGFP, is that the signal is amplified by enzymatic activity. On the other hand, a benefit of eGFP is that addition of substrate or cell lysis is not required prior to readout. To achieve signal amplification with eGFP, we created two additional genetic constructs, one containing two and another containing three copies of eGFP (PermE-eGFPx2 and PermE-eGFPx3). AttP-mediated neutral site insertion, followed by fluorescence assays resulted in significantly increased signal and a good Z′ score of 0.51 with attB::PermE-eGFPx3 (Figure 1B,C), which is sufficient for reliable results in a high-throughput assay format.13 The high-throughput screens described below were thus conducted with reporter constructs containing three copies of eGFP.
Activation of sur via HiTES
Two different S. albus reporter strains were created for activating the sur cluster: attB::Psur-eGFPx3, where the reporter is inserted at an attB neutral site, as described above, as well as surE::eGFPx3, in which the reporter genes replace surE and are therefore directly downstream of Psur (Figures 1B, S1). The latter construct was generated to avoid possible polar effects that can arise from regulatory elements that may be adjacent to a given attB site.29 With these two constructs in hand, HiTES was carried out using a ~500-member natural products library (see SI Methods). Each S. albus construct was grown in a 96-well format, supplemented with the compound library, and the effect of each compound on the silent BGC was determined using eGFP-derived fluorescence. Interference from naturally fluorescent compounds in the library was eliminated by subtracting the fluorescence measurements at 60 h from those collected at t = 0, immediately after addition of the library compounds.
Results for both genetic constructs are shown (Figure 2A). Each bar represents the level of expression of sur in the presence of a single candidate elicitor. The positive (attB::PermE-eGFPx3) and negative (attB::Psur-eGFPx3 and surE::eGFPx3 in the absence of compounds) controls are marked. Hits were considered those that gave at least a 2.5-fold induction of eGFP-derived fluorescence emission. The results from the two reporter strains were largely congruous though some differences can be observed (Figure 2A). For example, some hits were only observed in attB::Psur-eGFPx3 but not in surE::eGFPx3, possibly because of the aforementioned polar effects associated with attB sites. In both cases, up to 5-fold induction of sur expression could be identified. Comparison of the two assays and identification of molecules that induced the sur cluster in both reporter strains provided the following best hits: the natural product-derived anticancer agent etoposide (1, Figure 2B), the famous antiparasitic agent and Streptomyces avermitilis-derived ivermectin (2), the immunosuppressant cyclosporine A (3), the nootropic dihydroergocristine (4), plant-derived piceattanol (5), and bromolaudanosine (6). These results indicate that sur expression is subject to the presence and nature of exogenous small molecules in the media. They also suggest that activation of sur using HiTES is efficient as a number of elicitors could be identified from a relatively small library.
Figure 2.

High-throughput elicitor screen to induce the sur cluster. (A) Relative expression of a neutral site reporter, attB::Psur-eGFPx3 (top), or targeted sur reporter, surE::eGFPx3 (bottom), as a function of exogenously supplied small molecules. Each bar represents the response to a specific molecule from a library of 502 natural products, acquired as a single replicate. Plus signs denote the positive control, attB::PermE-eGFPx3. Minus signs denote the negative controls, attB::Psur-eGFPx3 (top) or surE::eGFPx3 (bottom) in the absence of any metabolites. The expression has been normalized to the positive control. Compounds that gave the top eight hits are marked. (B) Structures of the top six elicitors, observed in both HiTES assays, shown in panel A.
Validation of HiTES Results
To validate the effects of these putative elicitors on the sur cluster, we performed RT-qPCR. Upon exposure of wt S. albus to each elicitor or DMSO (control), total RNA was isolated and converted to cDNA, and the surA and surC transcripts were quantified by qPCR (Figure S2). The results show a 3-fold induction of surA and surC by ivermectin, etoposide, and dihydroergocristine, consistent with the HiTES results (Figure 3A). More modest effects were observed with the other three elicitors. We thus focused our efforts on the top two elicitors, etoposide (1, Figure 2B) and ivermectin (2). To further verify the effect of 1 and 2, a dose–response assay was carried out. With both elicitors, a concentration-dependent induction of sur was observed, yielding optimal upregulation at ~23 μM (etoposide) and ~30 μM (ivermectin) (Figure 3B). These concentrations induced a ~10-fold (etoposide) and 4.5-fold (ivermectin) upregulation of surC. Etoposide had antibacterial effects on S. albus and exhibited a half-maximal inhibitory concentration (IC50) of 10 μg/mL. The diminished response in induction of sur observed at high concentrations correlated with antibiosis. Thus, as reported with trimethoprim and other antibiotics, etoposide exhibits hormetic effects, stimulating metabolism at subinhibitory concentrations, while killing S. albus at higher concentrations.13–19 Ivermectin did not inhibit S. albus growth at the concentrations employed.
Figure 3.
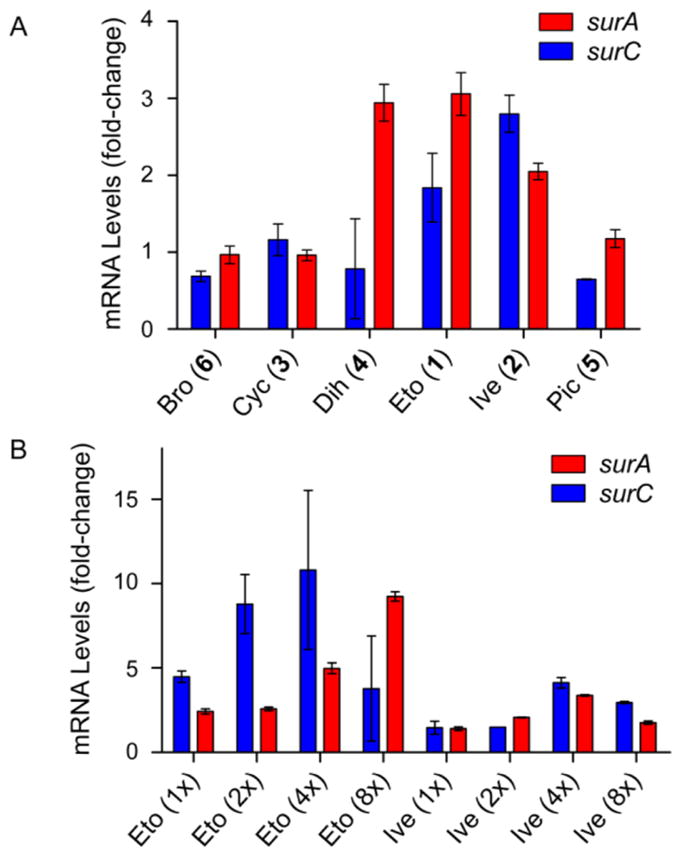
Effect of selected elicitors identified by HiTES on the expression of the sur BGC. (A) RT-qPCR analysis of the effects of the top six elicitors on sur expression. Shown is the observed fold-change in surA and surC mRNA levels compared to the DMSO negative control. (B) Concentration-dependence of the induction of sur by the top two elicitors (1 and 2), determined by RT-qPCR. Shown is the observed fold-change in mRNA levels with respect to the DMSO negative control. Note that 1x corresponds to a final concentration of 5.7 μM and 7.6 μM for 1 and 2, respectively.
Finally, we examined the effect of 1 and 2 on the S. albus secondary metabolome. S. albus was treated with each elicitor and cultured for 4 days, and cell-free supernatants were analyzed by HPLC-Qtof-MS. The results show a remarkable induction of a large array of secondary metabolites by etoposide and ivermectin (Figure 4A). Despite their disparate structures, 1 and 2 stimulated production of very similar metabolomes, with many of the same peaks observed in both cultures. These results corroborate the screening results and indicate that etoposide and ivermectin are inducers of cryptic metabolite production in S. albus. Moreover, they highlight the ability of HiTES in identifying structurally divergent molecules that are functionally similar in inducing a given silent BGC.
Figure 4.

Effect of 1 and 2 on the secondary metabolome of S. albus. (A) HPLC-MS base-peak chromatogram analysis of the secondary metabolome of S. albus in response to 1 (blue trace) and 2 (red trace), compared to the DMSO negative control (black trace). (B) Targeted extracted-ion chromatograms for selected products of the sur cluster. The color-coding in each panel is the same as that in panel A. Albucyclone A and acyl-surugamide A are induced, while production of surugamide I is vastly enhanced.
Discovery of Secondary Metabolites Biosynthesized by sur
We next set out to identify the cryptic metabolites of the sur cluster. To facilitate the identification of these products, two insertional gene inactivation mutants were generated, surA::apr (ΔsurA) and surB::apr (ΔsurB), where the surA or surB gene was replaced by the apramycin resistance marker (apr) (Figure S1). Comparison of the supernatants of wt S. albus with the two mutants, in the presence of ivermectin, allowed identification of compounds generated by sur (Figure S3). High-resolution (HR) MS analysis indicated that these compounds fall into four categories, suggesting that sur has a diverse output. Within each category, we isolated the most abundant member(s) and solved their structures by HR-MS, HR tandem-MS, and 1D and 2D NMR. These compound families are described in turn below.
Synthesis of the first group of metabolites required surA but not surB (Figure 5). It consisted of compounds with m/z 912, 989, 884, 870, 856, and 842 (9–14, Figure 6A), all of which lacked characteristic UV–visible absorption features (Figure S4, Table S4). Relevant NMR correlations used to solve the structure of the most abundant variant are shown (13, Scheme 1). Within this family, two compounds were identical to the octapeptides surugamide A (9) and D (10), which were previously reported by Takada et al. from a marine streptomycete.30 The other four analogs, however, were new. We call these surugamide G, H, I, and J (11–14); their production was vastly increased in the presence of elicitor 1 or 2 (Figure 4B and Figure 5). Complete structural assignment by NMR revealed that they are comprised of the same 8mer cyclic scaffold as surugamide A with variations in the amino acid sequence as shown in Figure 6A (see Figures S5–S8 and Tables S5–S7). The stereochemistry was assigned based on an analysis of the biosynthetic gene cluster using bioinformatic methods (see SI). It is identical to that of surugamide A, which was determined experimentally.30 These additional metabolites expand the diversity of the surugamide natural products.
Figure 5.
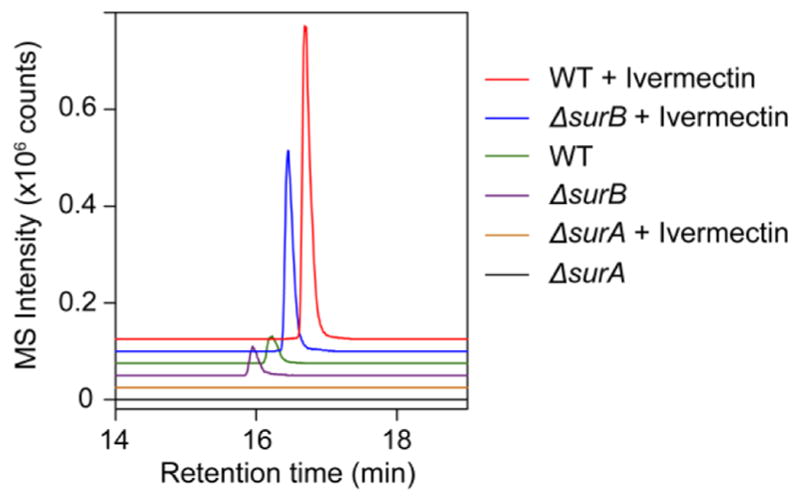
Production of surugamide I requires surA but not surB. Shown are extracted ion chromatograms for surugamide I in wt, ΔsurA, and ΔsurB cultures grown in the presence or absence of ivermectin.
Figure 6.

Small molecule products of the sur cluster induced by etoposide and ivermectin. (A) Structures of the surugamides. Variants A and D were previously elucidated;30 derivatives G, H, I, and J are new analogs identified in this study. (B, C) Structures of the cryptic and novel acyl-surugamide A (B) and albucyclones (C). The source of structural variability in the albucyclones is shown. (D) Structures of albuquinone A, mansouramycin C, and a mansouramycin analog. Compounds 22 and 23 are elicited by etoposide or ivermectin, and are likely produced by a separate gene locus in S. albus. (E) Structures of surugamide F and variants. Analog F was solved previously;24 variants F2 and F3 were found in this study and their proposed structures are supported by HR-MS and tandem HR-MS. Note that the stereochemistries are based on bioinformatic analyses (Figure S21) and on results from Streptomyces sp. JAMM992.24,30
Scheme 1.

Relevant NMR Correlations Used to Solve the Structures of Metabolites Elicited by 1 and 2
A second group of metabolites, whose production was only observed in the presence of ivermectin or etoposide and was abolished in the ΔsurA mutant, consisted of m/z 982, 968, 954, 940, and 926 (Figure S3, Table S4). All variants are new metabolites as determined by HR-MS analysis and comparison to a database of known natural products. We call these acyl-surugamides A–E. The structure of variant A was determined by NMR (15, Scheme 1, Figures S9–S10). 1H and COSY data showed that it consists of two spin systems, an octapeptide scaffold as well as a butyryl group. Analysis of TOCSY, HMBC, and NOESY data, which showed correlations between the butyryl 1H’s and the lysine side-chain protons, clearly pointed to acylation of the lysine side-chain amine of surugamide and thus completed the structural assignment (15, Figure 6B, Table S8). This subgroup of metabolites is a novel, side-chain-modified variant of surugamides and indicates that additional tailoring of the octapeptide scaffold further amplifies the output of the sur cluster in the presence of elicitors (Figure 4B).
We also found a family of six metabolites that were significantly larger than those discussed above (m/z 1126, 2 × 1112, 2 × 1084, and 1070) and only produced in the presence of elicitors 1 and 2 (Figure 4B, Table S4). Much like surugamides and acyl-surugamides, their production was dependent on SurA but not SurB (Figure S3). These compounds consisted of two spin systems, one of which was the surugamide scaffold. The lysine residue in the surugamide portion was again found to be acylated, in this case with an unusual second spin system with a small 1H/13C ratio, as shown by HMBC, NOESY, and HR-MS data (Figures S11–S17, Tables S5, S9–11). Further, the UV–vis spectrum of this set of compounds exhibited features typical of quinones, consisting of a broad absorption band with a λmax of 470 nm (Figure S4). Analysis of a full set of 1D and 2D NMR data ultimately showed that the second spin system consists of a novel isoquinoline quinone moiety (16, Scheme 1). We call these metabolites albucyclones A–F (16–21, Figures 6C, 4B). Within this family, we were able to assign structures to all six variants. They all contain the unusual isoquinoline quinone acyl group (Figure S12) but vary in the sequence of the octapeptide. Albucyclones are a novel combination of two natural products that likely arises from cross-talk between two different BGCs. The induction of albucyclones further emphasizes that under stimulatory conditions, the output of the sur cluster can be amplified to generate new hybrid molecules.
The presence of the unusual moiety in the albucyclones indicated to us that the free acid derivative of the isoquinoline quinone must be generated separately. To assess this hypothesis, we examined the supernatants of ivermectin-treated S. albus cultures for the predicted isoquinoline quinone side-chain. Indeed, we identified a family of low molecular weight compounds that were only induced by ivermectin and displayed the expected quinone UV–vis spectrum (Figure S4). We were able to elucidate the structures of two compounds with m/z of 219 and 203 (22, Scheme 1, Figure 6D, Tables S4, S12). The former is a novel compound; we have given it the trivial name albuquinone A. It is similar to the side chain in the albucyclones. The second compound (23, m/z 203, 3-methyl-7-methylamino-5,8-isoquinolinedione) was previously identified.31 Both 22 and 23 are similar to mansouramycin C (24), a metabolite previously isolated from a marine streptomycete (Figure S18).32 Compound 23 has broad and potent anticancer activity with IC50 values ranging from 0.2–50 μM against 36 diverse cancer cell lines, with a mean IC50 of 3.5 μM. Together, these results suggest that ivermectin induces other silent BGCs that generate bioactive metabolites; one these BGCs, in cross-talk with the sur cluster, gives rise to the albucyclones.
The last group of cryptic metabolites produced by the sur cluster consisted of m/z 1056, 1042, and 1070 (Table S4). This was the only set of compounds that was dependent on surB but not surA (Figure S3). We were not able to isolate sufficient material for NMR analysis and instead investigated their structures by HR-MS/MS (Table S5). This analysis showed they were linear decapeptides and revealed nearly all b and y ion fragments (25–27, Figures 6E, S19–S20, Table S5). One compound (m/z 1056) was identical to the previously reported surugamide F,24 as determined by HR-MS and HR-MS/MS (Table S5). The second and third are new analogs, which we call surugamide F2 and F3. As with the F variant, HR-MS/MS data are consistent with the presence of an unusual β-amino acid (Tables S4, S5); surugamide F2 contains a Leu-to-Val substitution at position 2 relative to surugamide F, while surugamide F3 has a Val-to-Leu/Ile substitution at position 7 (Figure 6E).
In sum, we were able to solve the structures of 14 novel metabolites, which fall into five categories. Our results indicate that the sur pathway is not only capable of generating a divergent set of molecules from relaxed specificities in the adenylation domains of the NRPSs (see below), which lead to point mutations in the peptides, but that it can also engage in cross-talk with other gene clusters or genes to generate hybrid metabolites. This leads to further diversification and amplification in the output of the sur BGC, to which at least 20 compounds can be attributed. Lastly, the application of HiTES, aside from unveiling the small molecule products of sur, also led to characterization of the new albuquinone A and the mansouramycin-variant, indicating that ivermectin and etoposide activate other BGCs in S. albus.
A Biosynthetic Model for Surugamides and Analogs
The results above show that the output of sur in S. albus is similar to that previously reported from the marine strain Streptomyces sp. JAMM992.24 In both cases, the BGC produces a decapeptide as well as a cyclic octapeptide. A biosynthetic model for how sur produces these varied peptides is proposed (Figure S21). Backed by our mutagenesis data and bioinformatic analyses (Figures 5, S3), the synthesis of the octapeptides requires SurA and SurD (Figure 1A, red nrps genes), while the linear surugamides F, F2, and F3 are generated by SurB and SurC (Figure 1A, blue nrps genes), analogous to previous results by Ninomiya et al.24 The synthesis of two distinct compounds from the same gene cluster is highly unusual and, in part, explains the diverse output of the sur cluster in S. albus.33,34
Bioactivity of Cryptic Metabolites Induced by Ivermectin or Etoposide
As alluded to above, streptomycete secondary metabolites are often endowed with exquisite, sometimes useful, bioactivities. A series of bioassays were carried out to examine the activities of the cryptic metabolites above (Table 1). Surugamides have been shown to harbor inhibitory activity against cathepsin B, a cysteine protease and anticancer target.30 Surugamides G–J were investigated in a cathepsin B assay and showed good inhibitory activity, in line with those for surugamide A.30 Notably, surugamide I was a strong cathepsin B inhibitor with an IC50 of 9.0 μM. We also examined acyl-surugamide A, albucyclones, and albuquinone A in antibacterial and antifungal assays. While albucyclones and albuquinone did not reveal significant antibiotic activity against the strains tested, acyl-surugamide A exhibited good antifungal activity with an IC50 of 3.5 μM against Saccharomyces cerevisiae. These results underline the utility of HiTES in uncovering cryptic, bioactive metabolites.
Table 1.
Half-Maximal Inhibitory Concentrations (IC50, μM) for Products of the sur Cluster in Selected Antibacterial, Antifungal, and Cathepsin B Inhibition Assays
| assays | 11 | 12 | 13 | 14 | 15 | 16 | 21 | 22 |
|---|---|---|---|---|---|---|---|---|
| Cathepsin B Inhibition | >100 | 12.4 | 9.0 | >100 | 43.3 | >100 | >100 | a |
| Staphylococcus aureus Newman | >100 | a | a | >100 | 27.4 | a | >100 | >100 |
| Bacillus subtilis 168 | >100 | a | a | >100 | 21.7 | a | >100 | >100 |
| Enterococcus faecalis OG1RF | >100 | a | a | >100 | >100 | a | >100 | >100 |
| Saccharomyces cerevisiae | >100 | a | a | >100 | 3.5 | a | >100 | >100 |
| Saccharomyces pombe | >100 | a | a | >100 | 32.5 | a | >100 | >100 |
| E. coli K12 | >100 | a | a | >100 | >100 | a | >100 | >100 |
| Pseudomonas aeruginosa PAO1 | >100 | a | a | >100 | >100 | a | >100 | >100 |
Not determined.
Mechanism of Induction of sur by Ivermectin or Etoposide
Having identified inducers of the cryptic sur cluster and characterized its various products, we next explored the mechanism of induction by ivermectin and etoposide using a forward genetic approach. The sur cluster appears to express a pathway-specific transcriptional regulator of the GntR family,35–38 which we have named SurR (Figure 1A). We imagined that one mechanism of induction by the elicitors could involve modulation of the expression of surR. To test this hypothesis, we examined the level of expression of this transcriptional regulator by RT-qPCR upon exposure of wt S. albus to DMSO (control), ivermectin, or etoposide. We found that both elicitors induced a 2–2.5-fold down-regulation of surR expression (Figure 7A).
Figure 7.

Involvement of surR in silencing of the sur gene cluster. (A) RT-qPCR analysis of the effect of etoposide and ivermectin on the expression of surR. Shown is the observed fold-change in surR mRNA levels compared to the DMSO negative control. (B) HPLC-MS base-peak chromatogram analysis of the secondary metabolome of wt S. albus (gray trace), ΔsurR (black trace), ΔsurR + 1 (blue trace), and ΔsurR + 2 (red trace). (C) Targeted extracted-ion chromatograms for the products of the sur cluster in ΔsurR (black trace), ΔsurR + 1 (blue trace), and ΔsurR + 2 (red trace). Production of all three products is enhanced upon addition of 1 or 2 to the ΔsurR mutant.
If SurR is a transcriptional repressor and thus silences the sur gene cluster, the down-regulation by the elicitors could explain their stimulatory activities. We tested this idea by deleting surR using insertional mutagenesis (surR::apr, referred to as ΔsurR). Subsequently, we compared the secondary metabolome of wt S. albus with that of ΔsurR. Consistent with our idea, the mutant displayed a striking overproduction of surugamides, acyl-surugamides, albucyclones, and albuquinone A (Figure 7B, black trace). In fact, the secondary metabolome of ΔsurR was very similar to that induced by ivermectin or etoposide in wt S. albus. These results are entirely consistent with a role for surR as a repressor or silencer of the sur BGC. They further imply that activation of sur by ivermectin and etoposide functions, in part, through down-regulation of surR.
To examine whether surR expression is the only regulatory pathway, by which 1 and 2 induce sur, we treated ΔsurR with each elicitor and assessed the resulting secondary metabolomes by HPLC-Qtof-MS. We observed a remarkable induction of a variety of metabolites, including many described above (Figure 7B,C). The combination of elicitors and ΔsurR appeared to have a synergistic effect on secondary metabolite biosynthesis, exhibiting increased production levels compared to ΔsurR alone or ivermectin/etoposide treatment of the wt strain. These results indicate that the two elicitors are pleiotropic and, aside from modulating the transcription of surR, also impinge on other regulatory pathways that result in induction of sur. Treatment of the ΔsurR strain with ivermectin or etoposide results in a >35-fold increase in the production of surugamide I. We estimate at least a >40-fold and >50-fold upregulation of the cryptic acyl-surugamide A and albucyclone A, respectively, based on the lower limits of detection of our HPLC-Qtof-MS.
It has previously been shown that etoposide inhibits bacterial, and more specifically, actinobacterial DNA gyrase, which leads to induction of the SOS response.39–42 Specifically, low concentrations of DNA gyrase inhibitors induce both the recombinase recA as well as the regulator lexA. Stress has been linked to activation of secondary metabolism though a detailed molecular mechanism is in most cases not known. We examined whether recA and lexA in the SOS response pathway were induced by etoposide, by determining their expression levels via RT-qPCR. We observed a ~2-fold induction of both genes in response to 1 (Figure S22); no changes were observed with ivermectin. Similar levels of induction were previously reported in other bacteria.42 While additional studies are necessary, these data suggest that the SOS response may be partially involved in activating secondary metabolism in the presence of elicitor 1.
Our studies addressing the mode of induction by 1 and 2 have identified a pathway-specific regulator and suggest that multiple pathways, perhaps including SOS response in case of 1, can lead to induction of sur. The treatment of ΔsurR with 1 or 2 will facilitate efficient production and further characterization of the products of sur in the future. These results also set the stage for investigating the detailed regulatory circuits that the elicitors affect to induce sur.
CONCLUSIONS
The secondary metabolomes of bacteria still comprise countless mysteries, with a significant fraction remaining hidden or silenced via largely unknown mechanisms. By performing the first application of HiTES in a streptomycete, we provide insights into the products and regulation of the silent sur BGC in S. albus. Induction of the sur cluster allowed us to characterize 14 novel secondary metabolites, including several that arise from cross-talk between sur and another BGC. One of these products, acyl-surugamide A, displays antifungal activity. With our previous use of HiTES in B. thailandensis, we demonstrate that this approach is broadly applicable to silent BGCs in both Gram-negative and Gram-positive bacteria.
Several largely complementary approaches have been developed for inducing silent gene clusters in bacteria.9–12,43–49 The advantages of our approach are that any given cryptic gene cluster can be activated in a targeted fashion within genetically tractable strains. By varying the elicitors or their concentrations or both, the level of activation can be tuned, and activation levels of up to ~150-fold have been achieved, thus greatly enhancing secondary metabolite synthesis from a given silent BGC.13 Combination of HiTES with traditional gene deletions, that is chemical genetics with classical genetics, further augments the diversity and quantity of cryptic metabolites, as we show with the treatment of ΔsurR with 1 or 2. Moreover, the elicitors can be pleiotropic and give rise to numerous other secondary metabolites, thus facilitating small molecule discovery in a global and pathway-specific manner simultaneously.13,14 Lastly, with small molecule probes at hand to modulate the expression of a cluster, the regulatory pathways can be examined.
Interestingly, two widely used drugs, ivermectin, derived from the natural product avermectin, and a well-known anticancer drug, etoposide, were discovered as inducers of sur. Investigations into the mode of induction by these elicitors led to identification of a pathway-specific repressor, SurR, which silences the sur cluster, thus explaining why little or no products are observed under standard growth conditions. While ivermectin and etoposide clearly modulate the expression of surR, the details of how these elicitors exert a striking stimulatory effect on the sur cluster remains to be established. Unlike ivermectin, etoposide mildly induces recA and lexA and inhibits S. albus growth, affording another example in which growth-inhibitory molecules elicit cryptic metabolism.16,18 This provides further support for the notion that old antibiotics may be used to find new (cryptic) ones. But in general, our understanding of the many mechanisms by which subinhibitory concentrations of antibiotics modulate microbes is very much in its infancy.50,51 Further applications of HiTES can shed light on many issues in this area. Indeed, with the successful use of HiTES in S. albus, the chemical output and biological regulation underlying many silent BGCs in Streptomyces spp. can now be investigated.
EXPERIMENTAL SECTION
A detailed description of all the methods employed in this work, including creation of genetic constructs of S. albus, high-throughput elicitor screens, validation by RT-qPCR and HPLC-MS, and isolation and structural elucidation of secondary metabolites, as well as bioactivity assays, is given in the Supporting Information.
Supplementary Material
Acknowledgments
We thank Istvan Pelczer for assistance with acquisition of NMR spectra, as well as the Searle Scholars Program, the Princeton University IP Accelerator Fund, and the National Institutes of Health (Grant DP2-AI-124786 to M.R.S.) for generous support of this work.
Footnotes
Notes
The authors declare no competing financial interest.
Detailed descriptions of materials and methods employed, including genetic manipulation of S. albus, high-throughput elicitor screens, RT-qPCR, isolation of secondary metabolites and structural elucidation, bioactivity assays, and experiments with ΔsurR; NMR, UV–vis, and HR-MS data for all secondary metabolites; HPLC-MS profiles of wt S. albus vs ΔsurA and ΔsurB. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b02716.
(PDF)
References
- 1.Newman DJ, Cragg GM. J Nat Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 2.Cragg GM, Newman DJ. Biochim Biophys Acta, Gen Subj. 2013;1830:3670–3695. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cragg GM, Grothaus PG, Newman DJ. Chem Rev. 2009;109:3012–3043. doi: 10.1021/cr900019j. [DOI] [PubMed] [Google Scholar]
- 4.Berdy J. J Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- 5.Bentley SD, et al. Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- 6.Oliynyk M, Samborskyy M, Lester JB, Mironenko T, Scott N, Dickens S, Haydock SF, Leadlay PF. Nat Biotechnol. 2007;25:447–453. doi: 10.1038/nbt1297. [DOI] [PubMed] [Google Scholar]
- 7.Ikeda H, Ishikawa J, Hanamoto A, Shinose M, Kikuchi H, Shiba T, Sakaki Y, Hattori M, Omura S. Nat Biotechnol. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- 8.Nett M, Ikeda H, Moore BS. Nat Prod Rep. 2009;26:1362–1384. doi: 10.1039/b817069j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rutledge PJ, Challis GL. Nat Rev Microbiol. 2015;13:509–523. doi: 10.1038/nrmicro3496. [DOI] [PubMed] [Google Scholar]
- 10.Zerikly M, Challis GL. ChemBioChem. 2009;10:625–633. doi: 10.1002/cbic.200800389. [DOI] [PubMed] [Google Scholar]
- 11.Ochi K, Hosaka T. Appl Microbiol Biotechnol. 2013;97:87–98. doi: 10.1007/s00253-012-4551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu H, Sandiford SK, van Wezel GP. J Ind Microbiol Biotechnol. 2014;41:371–386. doi: 10.1007/s10295-013-1309-z. [DOI] [PubMed] [Google Scholar]
- 13.Seyedsayamdost MR. Proc Natl Acad Sci U S A. 2014;111:7266–7271. doi: 10.1073/pnas.1400019111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Okada BK, Wu Y, Mao D, Bushin LB, Seyedsayamdost MR. ACS Chem Biol. 2016;11:2124–2130. doi: 10.1021/acschembio.6b00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craney A, Ozimok C, Pimentel-Elardo SM, Capretta A, Nodwell JR. Chem Biol. 2012;19:1020–1027. doi: 10.1016/j.chembiol.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 16.Okada BK, Seyedsayamdost MR. FEMS Microbiol Rev. 2017;41:19–33. doi: 10.1093/femsre/fuw035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yim G, Wang HH, Davies J. Philos Trans R Soc, B. 2007;362:1195–1200. doi: 10.1098/rstb.2007.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yoon V, Nodwell JR. J Ind Microbiol Biotechnol. 2014;41:415–424. doi: 10.1007/s10295-013-1387-y. [DOI] [PubMed] [Google Scholar]
- 19.Romero D, Traxler MF, López D, Kolter R. Chem Rev. 2011;111:5492–5505. doi: 10.1021/cr2000509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Makitrynskyy R, Rebets Y, Ostash B, Zaburannyi N, Rabyk M, Walker S, Fedorenko V. J Ind Microbiol Biotechnol. 2010;37:559–566. doi: 10.1007/s10295-010-0701-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baltz RH. J Ind Microbiol Biotechnol. 2010;37:759–772. doi: 10.1007/s10295-010-0730-9. [DOI] [PubMed] [Google Scholar]
- 22.Zaburannyi N, Rabyk M, Ostash B, Fedorenko V, Luzhetskyy A. BMC Genomics. 2014;15:97. doi: 10.1186/1471-2164-15-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olano C, García I, González A, Rodriguez M, Rozas D, Rubio J, Sánchez-Hidalgo M, Brana AF, Méndez C, Salas JA. Microb Biotechnol. 2014;7:242–256. doi: 10.1111/1751-7915.12116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ninomiya A, Katsuyama Y, Kuranaga T, Miyazaki M, Nogi Y, Okada S, Wakimoto T, Ohnishi Y, Matsunaga S, Takada K. ChemBioChem. 2016;17:1709–1712. doi: 10.1002/cbic.201600350. [DOI] [PubMed] [Google Scholar]
- 25.Mohimani H, Gurevich A, Mikheenko A, Garg N, Nothias LF, Ninomiya A, Takada K, Dorrestein PC, Pevzner PA. Nat Chem Biol. 2016;13:30–37. doi: 10.1038/nchembio.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingram C, Brawner M, Youngman P, Westpheling J. J Bacteriol. 1989;171:6617–6621. doi: 10.1128/jb.171.12.6617-6624.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun J, Kelemen GH, Fernández-Abalos JM, Bibb MJ. Microbiology. 1999;145:2221–2227. doi: 10.1099/00221287-145-9-2221. [DOI] [PubMed] [Google Scholar]
- 28.Sánchez J, Arias ME, Hardisson C. Experientia. 1981;37:469. doi: 10.1007/BF01986139. [DOI] [PubMed] [Google Scholar]
- 29.Combes P, Till R, Bee S, Smith MC. J Bacteriol. 2002;184:5746–5752. doi: 10.1128/JB.184.20.5746-5752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takada K, Ninomiya A, Naruse M, Sun Y, Miyazaki M, Nogi Y, Okada S, Matsunaga S. J Org Chem. 2013;78:6746–6750. doi: 10.1021/jo400708u. [DOI] [PubMed] [Google Scholar]
- 31.Dowell RI, Hadley EM. J Med Chem. 1992;35:800–804. doi: 10.1021/jm00083a001. [DOI] [PubMed] [Google Scholar]
- 32.Hawas UW, Shaaban M, Shaaban KA, Speitling M, Maier A, Kelter G, Fiebig HH, Meiners M, Helmke E, Laatsch H. J Nat Prod. 2009;72:2120–2124. doi: 10.1021/np900160g. [DOI] [PubMed] [Google Scholar]
- 33.Rea MC, Sit CS, Clayton E, O’Connor PM, Whittal RM, Zheng J, Vederas JC, Ross RP, Hill C. Proc Natl Acad Sci U S A. 2010;107:9352–9357. doi: 10.1073/pnas.0913554107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pulsawat N, Kitani S, Nihira T. Gene. 2007;393:31–42. doi: 10.1016/j.gene.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 35.Haydon DJ, Guest JR. FEMS Microbiol Lett. 1991;63:291–295. doi: 10.1016/0378-1097(91)90101-f. [DOI] [PubMed] [Google Scholar]
- 36.Suvorova IA, Korostelev YD, Gelfand MS. PLoS One. 2015;10:e0132618. doi: 10.1371/journal.pone.0132618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rigali S, Schlicht M, Hoskisson P, Nothaft H, Merzbacher M, Joris B, Titgemeyer F. Nucleic Acids Res. 2004;32:3418–3426. doi: 10.1093/nar/gkh673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van Wezel GP, McDowall KJ. Nat Prod Rep. 2011;28:1311–1333. doi: 10.1039/c1np00003a. [DOI] [PubMed] [Google Scholar]
- 39.Chatterji M, Unniraman S, Mahadevan S, Nagaraja V. J Antimicrob Chemother. 2001;48:479–485. doi: 10.1093/jac/48.4.479. [DOI] [PubMed] [Google Scholar]
- 40.Couturier M, Bahassi EM, Van Melderen L. Trends Microbiol. 1998;6:269–275. doi: 10.1016/s0966-842x(98)01311-0. [DOI] [PubMed] [Google Scholar]
- 41.Elsea SH, Westergaard M, Burden DA, Lomenick JP, Osheroff N. Biochemistry. 1997;36:2919–2924. doi: 10.1021/bi962488f. [DOI] [PubMed] [Google Scholar]
- 42.Sioud M, Boudabous A, Cekaite L. Int J Mol Med. 2009;23:33–39. [PubMed] [Google Scholar]
- 43.Clardy J, Fischbach MA, Walsh CT. Nat Biotechnol. 2006;24:1541–1550. doi: 10.1038/nbt1266. [DOI] [PubMed] [Google Scholar]
- 44.Schmidt EW. Nat Chem Biol. 2008;4:466–473. doi: 10.1038/nchembio.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pettit RK. Appl Microbiol Biotechnol. 2009;83:19–25. doi: 10.1007/s00253-009-1916-9. [DOI] [PubMed] [Google Scholar]
- 46.Netzker T, Fischer J, Weber J, Mattern DJ, König CC, Valiante V, Schroeckh V, Brakhage AA. Front Microbiol. 2015;6:299. doi: 10.3389/fmicb.2015.00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ramadhar TR, Beemelmanns C, Currie CR, Clardy J. J Antibiot. 2014;67:53–58. doi: 10.1038/ja.2013.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang R, Seyedsayamdost MR. Nat Rev Chem. 2017;1:0021. [Google Scholar]
- 49.Seipke RF, Kaltenpoth M, Hutchings MI. FEMS Microbiol Rev. 2012;36:862–876. doi: 10.1111/j.1574-6976.2011.00313.x. [DOI] [PubMed] [Google Scholar]
- 50.Davies J. J Ind Microbiol Biotechnol. 2006;33:496–499. doi: 10.1007/s10295-006-0112-5. [DOI] [PubMed] [Google Scholar]
- 51.Davies J. Curr Opin Chem Biol. 2011;15:5–10. doi: 10.1016/j.cbpa.2010.11.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.