

Abstract
A review of efforts that have provided total syntheses of vancomycin and related glycopeptide antibiotics, their agylcons, and key analogues is provided. It is a tribute to developments in organic chemistry and the field of organic synthesis that not only can molecules of this complexity be prepared today by total synthesis, but that such efforts can be extended to the preparation of previously inaccessible key analogues that contain deep-seated structural changes. With the increasing prevalence of acquired bacterial resistance to existing classes of antibiotics and with the emergence of vancomycin resistant pathogens (VRSA and VRE), the studies pave the way for the examination of synthetic analogues rationally designed to not only overcome vancomycin resistance, but to provide the foundation for the development of even more powerful and durable antibiotics.
Keywords: Vancomycin, Orienticin C, Teicoplanin, and Ristocetin A, Enzymatic Glycosylation, Glycopeptide Antibiotics, Vancomycin Analogues, Complestatin, Chloropeptin
Graphical abstract
1. Introduction
An important development in the field of glycopeptide antibiotics occurred in the late 1990s when three groups independently achieved the total synthesis of vancomycin. Given the sheer structural complexity of the natural product, this series of synthetic accomplishments was remarkable and at the frontiers of the field of organic synthesis at that time. With reports of the rapid increase in resistant bacterial strains by health officials, this effort was driven not only by the challenge of developing an effective route to the complex natural product, but also to pave the way for biological interrogation of previously inaccessible synthetic analogues. Herein, we review only work completing total syntheses of members of the vancomycin related glycopeptide antibiotics, their aglycons, and synthetic analogues. Work on their semisynthetic modifications1,2 and methodological studies are not reviewed as they have been covered elsewhere.
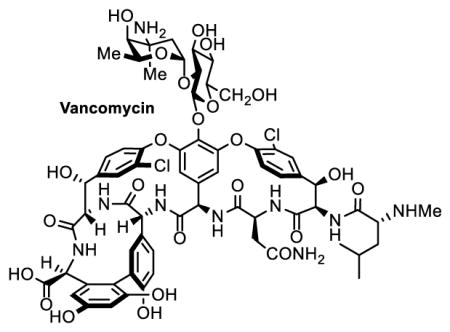
The glycopeptide antibiotics are currently among the leading members of the clinically important natural products discovered through the isolation of bacterial metabolites. They possess a broad spectrum of antibacterial activity against Gram-positive pathogens with manageable side-effects. Since their clinical introduction, the glycopeptide antibiotics vancomycin (1) and teicoplanin (6) have become the drugs of ‘last resort’ when resistant bacterial infections are encountered (Figure 1). With the emergence of methicillin-resistant Staphylococcus aureus (MRSA), vancomycin (1) has been widely used in the clinic as the ‘go to’ treatment.3,4 Originally restricted to hospitals, today more than 60% of both ICU (intensive care unit) and community acquired S. aureus infections are MRSA,5,6 and are responsible for upwards of 12,000 deaths in the United States in 2011 alone.7 Moreover, infectious diseases (e.g. influenza and pneumonia), complicated by additional bacterial infections often requiring treatment with vancomycin, are ranked among the leading causes of death in the U.S.. The glycopeptide antibiotics are also recommended for use with patients allergic to β-lactam antibiotics, undergoing cancer chemotherapy or ongoing dialysis therapy.8 Consequently, the importance and clinical use of vancomycin continues to steadily increase since its introduction 60 years ago.9 As vancomycin resistant bacteria have been observed in the clinic in both enterococci (VRE, 1987)10 and S. aureus (VRSA, 2002)11–17 and as the prevalence of antibiotic resistant pathogens has increased, discovery of the next generation durable antibiotics capable of addressing such bacterial infections has become an increasely urgent problem.18
Figure 1.

Structures of Vancomycin and Representative Related Glycopeptide Antibiotics.
Since the establishment of the structures of glycopeptide antibiotics, extensive synthetic efforts have been made through both semisynthetic and total synthesis means. These studies have laid the foundation for ongoing structure–function studies of the antibiotics, aiding in the definition of their mechanism(s) of action. They have also elucidated unanticipated new roles for added non-naturally occurring functionality that have led to the discovery of improved or rationally designed glycopeptide antibiotics.
This review summarizes the total synthesis of the vancomycin related glycopeptide antibiotics, along with occasional discussions of their use in key studies to define specific mechanisms of action responsible for their continued effective clinical use for decades. Lastly, a summary of recent total synthesis efforts aimed at the redesign of vancomycin through rational structural modifications to address vancomycin resistant bacterial infections is provided.
2. Glycopeptide Antibiotic Background
2.1 Structure: Isolation and Characterization
Vancomycin was isolated in the early 1950s at Eli Lilly and disclosed in 1956, and other glycopeptide antibiotics within this family followed.19 Even though vancomycin was approved for clinical use in 1958, its structure was established only 25 years later. Early chemical degradation studies,20–22 followed by the subsequent seminal NMR studies of Williams,23 and a pioneering X-ray crystal structure by Sheldrick of the degradation product CDP-124 provided an initial structure. This was followed by iterative corrections to the assigned structure that resulted from an unrecognized atropisomer isomerization25 and an asparagine to isoaspartate rearrangement26,27 under conditions of deglycosylation and provide a rich history to the full structural assignment of vancomycin by Harris that appeared in 1982.26 This was followed by full structural assignments for ristocetin, teicoplanin and a series glycopeptide antibiotics. The comprehensive reviews of Perkins,28 Williams,29,30 Nicolaou,31 Courvalin,32,33 Walsh34,35 and Kahne36 and others37–39 provide tabular accounts of the known glycopeptide antibiotics, a summary of the structure elucidation studies, and discussions of their biosynthesis, mechanism of action, and mechanisms of resistance.
The complex structures, the strained cyclic peptide subunits that contain biaryl and diaryl ether linkages interwoven into the intricate bi-, tri- and tetracyclic heptapeptide frameworks of their underlying rigid structures, the varied glycosylation patterns, and the unusual centers of axial or planar chirality (atropisomers) of the glycopeptide antibiotics present formidable synthetic challenges that were addressed in the efforts summarized herein.
2.2 Mechanism of Action
The seminal studies that defined the mechanism of action of the glycopeptide antibiotics include: (1) the initial Strominger demonstration that they inhibit bacterial cell wall biosynthesis,40 (2) the insightful and comprehensive studies of Perkins that established antibiotic binding to the D-Ala-D-Ala terminus of precursor peptidoglycans,28,41,42 (3) the pioneering NMR studies of Williams that defined the structures of antibiotic bound complexes with model D-Ala-D-Ala ligands,43 (4) the later confirmation of the structures and their complexes by X-ray crystallographic studies conducted by Sheldrick44 and (5) the beautiful studies of Walsh–Courvalin45 that unraveled the molecular mechanism of vancomycin resistance. Each of these developments constitute rich chapters in our understanding of the glycopeptide antibiotics today.
The glycopeptides antibiotics are unusual in that they do not target a specific protein or nucleic acid within bacteria like most other antibiotic classes (e.g. β-lactams), but instead they bind the peptidoglycan precursors necessary for construction of the cell wall. The binding to D-Ala-D-Ala sequesters a substrate for the enzyme-catalyzed bacterial cell wall cross-linking reaction (transpeptidase) and impacts the transglycosylase catalyzed incorporation of lipid intermediate II into the polysaccharide cell wall backbone (Figure 2). It is difficult for bacteria to make single genetic alterations that result in resistant conferring changes within these precursors, thus allowing the glycopeptide antibiotics to remain effective for nearly 60 years.9
Figure 2.

Bacterial Cell Wall Precursors of Gram-positive Bacteria, and Vancomycin Binding to D-Ala-D-Ala.
In fact, the only clinically significant resistance to the glycopeptide antibiotics, which first emerged in enterococci in 1987 (VanA and VanB VRE),10 was not independently evolved by pathogenic bacteria. Rather, it was co-opted from nonpathogenic vancomycin producing organisms that use this intricate mechanism of resistance to protect themselves while producing the glycopeptide antibiotic.46 In short, it entails detection of a glycopeptide challenge and an orchestrated response that results in late stage remodeling of the peptidoglycan precursor N-terminus from D-Ala-D-Ala to D-Ala-D-Lac.45,47 This change, which represents a single atom exchange in the precursors, reduces glycopeptide antibiotic binding 1000-fold, rendering them ineffective.48
Thus, extension of the efforts on the total synthesis of the glycopeptide antibiotics to the preparation of analogues that contain compensatory single atom exchanges have been disclosed, which now exhibit dual D-Ala-D-Ala/D-Ala-D-Lac binding and activity against both vancomycin sensitive and resistant organisms. These efforts, along with peripheral modifications to the glycopeptide antibiotics that introduce additional mechanisms of action, are summarized in Section 7. These studies have provided extraordinarily potent analogues that display especially durable antimicrobial activity.
3. Total Synthesis of Orienticin C Aglycon
3.1 Evans Synthesis
In route to the development of a total synthesis of vancomycin itself, Evans reported the total synthesis of the orienticin C aglycon.49,50 This natural product aglycon is nearly identical to that found in vancomycin, lacking only the C and E ring aryl chlorides on the aglycon.51 This difference removes the challenge of controlling two of the three centers of axial or planar (atropisomer) chirality found in vancomycin, but addresses the key bonds formed in assembling the core structure. Moreover, the late stage intermediates prepared in these efforts conceivably could be used to also prepare the vancomycin aglycon.
Key highlights of the route include two biomimetic oxidative cyclization reactions of an acyclic tetrapeptide containing the ABCD rings,52–58 a late stage thermal equilibration about the AB biaryl center of axial chirality to access the natural atropisomer, along with a final stage SNAr diaryl ether macrocyclization reaction for DE ring closure (Figure 3).59,60 They additionally utilized the powerful Evans chiral oxazolidone-based imide enolate functionalization reactions and aldol methodology to obtain the unnatural amino acid subunits with high enantio- and diastereoselectivity.61–64 For details on the preparation of the individual unnatural amino acid subunits (e.g. Rings A–E, 7–12), the reader is referred to their original reports.49,50
Figure 3.

Evans Retrosynthetic Analysis of Orienticin C Aglycon.
Beginning with the acyclic tetrapeptide 13, thallium(III) nitrate-promoted formation of the CD macrocycle with formation of the diaryl ether was accomplished through a modified Yamamura oxidative cyclization55–58 of the CD ring system with subsequent in situ reduction of an intermediate p-quinol (Scheme 1). The free phenol of the E ring was protected as an acid-stable mesylate, which increased the oxidation potential of ring D rendering it unreactive toward the conditions of a subsequent VOF3-mediated oxidative biaryl coupling.53,54 Protecting group exchange at the N-terminus, from N-Boc to trifluoroacetamide, afforded 14 in 46% overall yield from 13. A second key macrocyclization with formation of the AB ring system was accomplished by oxidative biaryl coupling of 14 mediated by VOF3 to yield the unnatural atropisomer53,54 15 of the bicyclic tetrapeptide that by design also cleaved the B ring benzyl ether. Notably, the unnatural AB atropisomer in 15, as well as in the isolated AB ring system, adopts a conformation that bears a central trans amide. Conversion of this released phenol to a triflate and its reductive cleavage65 was conducted to remove the extraneous B ring phenol originally needed for biaryl coupling and that served to control the cyclization atropodiastereoselectivity. This latter reaction also unexpectedly debrominated the D ring, necessitating re-halogenation in a subsequent step. Next, global demethylation enlisting AlBr3–NaI (vs AlCl3, ClCH2CH2Cl) yielded 16 in 88% yield. The natural atropisomer 17 was obtained by thermal atropisomerization of 16 in MeOH (55 °C) over two days, at which time no starting unnatural atropisomer 16 was observed by 1H NMR analysis. The stability of the precursor methyl ether towards atropisomerization as well as the thermodynamic preference for the (P)-atropisomer of the AB ring system and its preferential adoption of the central cis amide were defined both with 16/17 as well as with the isolated AB ring system.49,50,53,54 Exhaustive benzylation of the three phenols, reductive removal of the trifluoroacetamide with NaBH4, mesylate cleavage with methyl magnesium chloride, and Boc protection of the N-terminus amine, followed by regioselective iodination of the D ring with NIS afforded 18 in 57% overall yield from 17. Lastly, the ring D free phenol was protected as its allyl ether (85%), and the missing D ring phenol required for down-stream macrocyclization was installed via lithium–halogen exchange, trapping the resulting anion with triethyl borate followed by oxidation (19, 53%).
Scheme 1.

Evans Synthesis of the ABCD Ring System of Orienticin C Aglycon.
After Boc deprotection, EDCI-promoted peptide coupling of 21 with the tripeptide 2252 assembled the heptapeptide framework that contains the E ring in a reaction where competitive epimerization has consistently challenged most other efforts (Scheme 2). The final DE macrocycle was closed in a SNAr reaction upon treatment with CsF in DMSO at room temperature, which provided the product both in high yield (90%) and with a kinetic diastereoselection of 7:1 in favor of the natural atropisomer 23. Although this planar chirality was removed in subsequent steps for orienticin C, this result demonstrated that, unlike the isolated ring system,60 the DE ring system could be closed atropodiastereoselectively when attached to the intact ABCD ring system. This had important implications for subsequent work on the total synthesis of vancomycin. Reduction of the nitro group with Zn and acetic acid followed by formation and in situ reductive removal of the diazonium salt yielded 24.66 Notably, initial extensive efforts to implement a third biomimetic inspired oxidative cyclization reaction for closure of the DE ring system with formation of the diaryl ether, enlisting modifications of the Yamamura protocols, provided more modest results (ca. 20% yield). This led Evans to adopt the SNAr macrocyclization reaction introduced and developed by Boger60 and Beugelmans.67
Scheme 2.

Final Stages of the Evans Total Synthesis of Orienticin C Aglycon.
Next, the N-methylamide, which served admirably to prevent C-terminus epimerization throughout the total synthesis, was selectively nitrosated with dinitrogen tetraoxide (N2O4).68,69 This nitrosated amide could be cleaved under mild conditions with LiOOH, unmasking the C-terminus carboxylic acid. The final three steps entailed palladium-catalyzed deallylation, hydrogenolysis removal of the aryl chlorides as well as the benzyl and Ddm groups, and Boc deprotection of the N-terminus amine to afford the orienticin C aglycon (27).
4. Total Synthesis of Vancomycin
4.1 Total Synthesis of Vancomycin Aglycon
Vancomycin is a rigid tricyclic heptapeptide with three macrocyclic ring systems embedded in the framework, one possessing axial chirality and two containing elements of planar chirality. The central phenol of the aglycon is attached to a disaccharide, consisting of glucose and vancosamine. Given the complexity of vancomycin, the development of synthetic strategies to construct its skeleton attracted many groups, three of which reported total syntheses in 1998–1999: David A. Evans, K. C. Nicolaou, and Dale L. Boger. This transpired roughly 45 years after its isolation, 40 years after its introduction into the clinic, and nearly 25 years after its structure determination. Within this section, the three separate reports are discussed and key differences in their strategies are outlined.
4.1.1 Evans Synthesis
The Evans total synthesis of the vancomycin aglycon70,71 was based largely on their previous work with orienticin C. The same key disconnections were envisioned apart from the disconnection of the CD ring system (Figure 4). Here, the C–O bond of the C ring was broken rather than to the D ring C–O bond and its synthetic formation now relied on a SNAr diaryl ether bond formation60 rather than a biomimetic inspired oxidative cyclization. Major differences are also found within the synthesis of the ABCD system where the AB macrocycle was formed first at the tripeptide stage, followed by the coupling addition of the D ring and construction of the CD macrocycle. This contrasts the efforts on orienticin C,49,50 where the ABCD acyclic tetrapeptide was prepared before sequentially closing the macrocycles in the reverse order, forming the CD ring macrocycle prior to formation of the AB ring system. Thus, the overarching strategy adopted for the total synthesis of the vancomycin aglycon entailed a macrocyclization order in which the AB, CD and DE ring systems were sequentially introduced and relied on empirically defined substrate control of the kinetic atropodiastereoselectivity of the three key macrocyclization reactions. As with orienticin C, the C-terminus carboxylic acid was masked as a N-methylamide to prevent epimerization at this base-sensitive center.
Figure 4.

Evans Retrosynthetic Analysis of Vancomycin Aglycon.
After Boc deprotection of the ring A precursor 29, attachment of the C ring was achieved by EDCI-promoted amide coupling and afforded 31 in 72% yield (Scheme 3). The B ring (30) was attached to the AC fragment with EDCI, after a base-mediated ring opening of the oxazolidinone and Boc deprotection of the N-terminus amine of 31. After a protecting group exchange at the N-terminus amine (trifluoroacetamide for N-Boc), the 12-membered AB macrocycle 34 was formed, using VOF3 to mediate an oxidative biaryl coupling of 33 that selectively formed the unnatural atropisomer (>95:5). The N-terminus amine of 34 was deprotected with NaHCO3, and the D ring 35 was subsequently attached with HATU–HOAt in 65% yield. Silyl ether cleavage of the TBS protected D ring phenol was achieved with HF–pyridine to provide 36 in 85% yield. It is noteworthy that the triphenolic D ring was orthogonally protected as the O-allyl, O-TBS, and O-Ms derivatives avoiding problematic debromination experienced in the total synthesis of orienticin C. The CD ring system and its 16-membered diaryl ether 37 were formed in a base promoted, room temperature SNAr reaction60 between the D ring phenol with the C ring o-nitrofluoroarene in which the atropodiastereoselectivity favored the natural atropisomer disposition of the chloro substituent in a 5:1 ratio. Remarkably, the cyclization could be achieved even without added base at a reasonable rate simply upon dissolution in polar, aprotic solvents (e.g. NMP). Both this more reactive chloro substituted C ring precursor 28 as well as the corresponding dechloro substrate were examined. The ring closure of the initially examined unsubstituted (dechloro) precursor provided a 10:1 atropodiastereoselectivity favoring the unnatural disposition of the nitro substituent. That led to the examination of the chloro substituted substrate 36 where the nitro group stereochemical disposition still dominated and the adoption of a strategy in which the activating nitro group was reductively removed rather than additionally serving as a precursor for the chloro substituent. Conversion of the B ring phenol to a triflate 37, nitro group reduction, and aniline diazotization/reduction66 followed by a Pd-catalyzed hydrogenolysis of the aryl triflate65 and allyl ether cleavage yielded 38 in 77% over three steps.
Scheme 3.

Evans Synthesis of the Vancomycin ABCD Ring System.
Before equilibrating the AB ring system to the natural atropisomer, the D ring phenol was converted to a pivalate ester and the N-terminus amine was deprotected and converted to the trifluoroacetamide. Lastly, global demethylation of the A and B ring methyl ethers with AlBr3–EtSH yielded 41. Upon warming the bicyclic tetrapeptide 41 in MeOH (55 °C), clean atropisomerization to the natural P-configuration was achieved, providing 42 in 54% yield (in 96 h) from 40.53,54
In preparation for the final peptide coupling, the three phenols were converted to their benzyl ethers, the pivalate group was removed and the resulting phenol re-protected as an allyl ether, and both the mesylate and trifluoroacetamide protecting groups were removed, yielding 43 in 65% over 5 steps.
The final peptide coupling brought together the ABCD tetrapeptide 43 and the acyclic tripeptide 44 and was conducted with EDCI, remarkably without detectable epimerization (Scheme 4). The final macrocyclization of the 16-membered DE ring system was accomplished with a second room temperature SNAr reaction for diaryl ether formation, using CsF (DMSO) and providing selective formation of the natural P atropisomer 45 (5:1 ratio) in high yield (95%).59,60,67 After subsequent reduction of the nitro group to the aniline, the diastereomers could be separated by column chromatography. The appropriately functionalized E ring 46 was formed through a Sandmeyer substitution reaction upon CuCl and CuCl2 treatment of the aniline-derived diazonium tetrafluoroborate salt.72–74 Next, the masked C-terminus N-methylamide was nitrosated with dinitrogen tetraoxide (N2O4) and subsequently hydrolyzed with LiOOH in 68% yield.68 Despite the potential nitrosation at other amide sites, the documented steric effects of such a competitive reaction with amides were defined,68,69 aiding in the designed and implemented selectivity first explored in the total synthesis of the orienticin aglycon.49,50 Allyl ether cleavage followed by hydrogenolysis cleavage of the benzyl ethers, using transfer hydrogenation conditions (Pd/C and 1,4-cyclohexadiene) to avoid dechlorination, afforded 49.75 The final step in the conversion to vancomycin aglycon (50) was achieved by acid-catalyzed N-terminus Boc and asparagine residue Ddm removal (83%).
Scheme 4.

Final Stages of the Evans Total Synthesis of Vancomycin Aglycon.
The total synthesis of eremomycin aglycon was also disclosed in these efforts although no details were reported.70,71 In these efforts, the nitro group of the predominant unnatural atropisomer derived from cyclization of the unsubstituted (dechloro) substrate related to 36 was removed and carried through an analogous synthesis to provide the eremomycin aglycon.
4.1.2 Nicolaou Synthesis
Concurrent with the Evans report, Nicolaou published three back-to-back papers76–78 describing work culminating in a total synthesis of vancomycin aglycon (Figure 5 for retrosynthetic analysis). All three total syntheses are based on formation of the linking diaryl ethers in the key CD and DE ring system macrocyclization reactions, and Nicolaou developed a new reaction for this bond construction. Through a triazene appended to the D ring with o,o’-dibromo substitution, copper bromide successfully promoted a metal-activated SNAr reaction for both CD and DE diaryl ether formation.79 An additional difference from the Evans approach was the use of the Sharpless asymmetric dihydroxylation80 reaction to access the individual subunits with the required absolute stereochemistry in the A, D and E rings, along with use of a Sharpless asymmetric aminohydroxylation81 reaction for asymmetric synthesis of the the C ring subunit. Details on the design and development of the methodology involved in the approach can be found in a full account of this work.82–84
Scheme 5.

Nicolaou Synthesis of the Vancomycin ABCD Ring System.
The chiral benzooxaborolol 54 was coupled with the B ring aryl iodide 55 under Suzuki cross-coupling reaction conditions to obtain 58 with modest atropodiastereoselectivity (2:1), favoring the desired atropisomer (Scheme 5). A Mitsunobu reaction was used for the introduction of an azido group with stereochemical inversion to provide (S)-59 in high yield. Attachment of the C ring 51 mediated by EDCI was achieved in 85% yield after hydrolysis of the precursor methyl ester. After N-Boc deprotection to afford 61 in 90% yield, the central amino acid 52 was coupled to the tripeptide 61 with EDCI to obtain the tetrapeptide 62 in good yield. Copper bromide activated formation of the 16-membered diaryl ether through triazene chelation and promoted the SNAr reaction (MeCN, reflux, 20 min) to provide 63 as a separable 1:1 mixture of atropisomers in a combined yield of 60%.79 With the monocyclic tetrapeptide in hand, the TBS ether was cleaved with Bu4NF (80%), the azido group was reduced with triethylphosphine (71%), and the ethyl ester was hydrolyzed with LiOH (68%). The resulting free amine and carboxylic acid were coupled in a macrolactamization reaction mediated by FDPP (71%), affording the bicyclic ABCD ring system.85,86 Lastly, the ABCD vancomycin ring system 64 was prepared for subsequent peptide attachment by TBS protection of the β-hydroxy group and Boc deprotection of the N-terminus amine, providing the bicyclic tetrapeptide 66.
After obtaining 53 in high enantio- and diastereopurity in a 4-step synthetic sequence, 53 was coupled with N-Boc-N-methylleucine (57, EDCI–HOBt) and was followed by ethyl ester hydrolysis with lithium hydroxide to afford 67 (Scheme 6). Next, attachment of the Ddm-protected asparagine methyl ester (EDCI–HOAt) gave the tripeptide in 82% yield. Subsequent TBS protection of the free alcohol and cleavage of the benzyl ether yielded 68 in 80% (2 steps). The desired E ring tripeptide was obtained after phenol o-chlorination with sulfuryl chloride and hydrolysis of the methyl ester, affording 69 for peptide coupling with the ABCD ring system.
Scheme 6.

Nicolaou Synthesis of the E Ring Tripeptide.
The heptapeptide backbone was assembled by coupling 66 and 69 (EDCI–HOAt). Final ring closure mediated by copper bromide provided the 16-membered DE ring system 70 in 74% (MeCN, reflux, 2 h) as a 1:3 mixture of atropisomers, providing predominantly the undesired isomer (Scheme 7). Following protocols disclosed in and central to the efforts of Boger,72–74,87 the major undesired isomer was re-equilibrated to a 1:1 mixture of DE ring system atropisomers in 1,2-dichlorobenzene under thermal conditions that do not impact the AB or CD atropisomer stereochemistry. Repeating this re-equilibration of the unnatural atropisomer led to a >90% recycled and recovered yield. The next challenge was the conversion of the triazene group to the corresponding phenol. Although the initial report by Nicolaou successfully achieved this transformation,78 the conditions presented in Scheme 7 represent an improved route disclosed shortly thereafter.84 Conversion of the triazene to the aryl iodide (NaI, I2, and TMSCl) was followed by formation of the corresponding aryl Grignard reagent, its trap with B(OMe)3 and subsequent oxidation to provide 72 in 32% yield from 71.
Scheme 7.

Final Stages of the Nicolaou Total Synthesis of Vancomycin Aglycon.
The final stages of the total synthesis involved reductive cleavage of the C-terminus benzyl ether (94%), methylation of the D ring free phenol (94%), and stepwise oxidation of the primary alcohol to the corresponding carboxylic acid.88,89 Carboxylic acid esterification with diazomethane (84%), and TBS ether deprotection followed by global deprotection (62%) with AlBr3–EtSH provided vancomycin agylcon (50).
4.1.3 Boger Synthesis
Shortly after the first two reports of the total synthesis of the vancomycin aglycon, Boger disclosed a complementary convergent strategy.87,90 In efforts leading up to the total synthesis, Boger established that the CD and DE ring systems were most effectively closed through formation of the diaryl ether linkage,91 and explored,92–94 implemented,60,72–74 and improved95 methodology to be used for ring closure of the CD and DE ring systems. An effective macrolactamization for closure of the 12-membered biaryl AB ring system was also disclosed by Boger, defining subtle features key to its unusual success, and an indirect strategy for controlling the three stereochemical elements of atropisomerism.72–74,95–98 Thus, two SNAr macrocyclizations of the 16-membered diaryl ethers, enlisting phenol nucleophilic substitution reactions of an o-nitrofluoroaromatic, were used for sequential CD and DE ring formations.60,72–74 A key macrolactamization reaction was employed for cyclization of the AB ring system, and the defined order of CD, AB and DE ring closures permitted selective thermal atropisomerism of the newly formed ring systems or their intermediate precursors (Figure 6). In addition to any diastereoselection that was achieved in the ring closures, this order permitted the recycling of any undesired atropisomer for each ring system and provided predictable control of the stereochemistry, dependably funneling all synthetic material into the one of eight atropodiastereomers found in the natural product. Key to recognition of this order was the establishment of the disparate thermodynamic parameters of atropisomerism: DE ring system Ea = 18.7 kcal/mol, 1:1 natural-P:M < AB biaryl precursor90 Ea = 25.1 kcal/mol, 3:1 natural-P:M < CD ring system Ea = 30.4 kcal/mol, 1:1.1 natural-P:M (Figure 6).72,95–97 Prior to these studies, the assumption was that the barrier to isomerization of either the CD or DE ring system atropisomers was too large to permit their observation.25
Figure 6.

Atropisomer Equilibration and Experimental Ea of the Individual Vancomycin Ring Systems and Defined Order of Ring System Introductions.
Similarly, the preference for a cis amide bond in the natural atropisomer of the isolated 12-membered AB ring system was defined. Prior to this work, the assumption was that the surrounding CD ring system in vancomycin induced adoption of this cis amide. Given this redefined intrinsic preference, it is now thought that it is the rigid AB ring system that dominates the conformational properties of the surrounding CD ring system. In contrast, the isolated or imbedded unnatural AB atropisomer adopts a conformation that bears a central trans amide.
Unlike the Evans synthesis in which the AB, CD, and DE ring systems were sequentially assembled, but like that of Nicolaou, the order of ring closures entailed CD, AB, and DE macrocyclizations. Two aromatic nucleophilic substitution reactions were used for formation of the CD and DE ring system diaryl ethers,60,72–74 and a distinguishing macrolactamization reaction was used to close the AB ring system. Unlike the Nicolaou approach, the stage at which the axial chirality of AB ring system was introduced permitted adjustment of atropisomer stereochemistry within both the CD and AB ring systems.
The absolute stereochemistry within each unnatural amino acid subunit was installed with use of the Sharpless asymmetric aminohydroxylation reaction (phenylglycine rings A, B and D99), or a diastereoselective Schöllkopf aldol-like addition reaction (β-hydroxylphenylalanine rings C and E) (Figure 7). Improvements in the synthesis of the E ring β-hydroxylphenylalanine, enlisting an asymmetric aldol reaction of a glycine imine bearing an α-hydroxypinanone chiral auxiliary, was also later disclosed.100 For details on the synthesis of each subunit, the reader is directed to the original reports.90
Figure 7.

Boger Retrosynthetic Analysis of Vancomycin Aglycon.
Peptide coupling of 76 with 80 (EDCI–HOBt) afforded the dipeptide 83 in 81% yield (Scheme 8). Boc deprotection of 83 with TBSOTf proceeded in 99% yield and coupling with 77 afforded 85. Using improved reaction conditions discussed in Section 7.1, treatment of 85 with K2CO3–CaCO3 closed the 16-membered macrocycle (85%) in a key SNAr reaction that proceed with little atropodiastereoselectivity (1:1.1, natural-P:M). In this reaction, the added CaCO3 served to scavenge the liberated fluoride as insoluble CaF2, preventing inadvertent silyl ether deprotection and competitive retro aldol cleavage of intact CD ring system. The isolated unnatural M atropisomer was converted to the natural P atropisomer upon heating at 140 °C in a solution of o-dichlorobenzene.95 Although this equilibration provides in a 1:1 mixture of M and P atropisomers, each atropisomer can be isolated and (M)-86 recycled through this procedure, funneling all material into the total synthesis. Conversion of the aryl nitro group to the corresponding chloride, achieved by nitro group reduction, aniline diazotization, and Sandmeyer substitution, afforded 88 in 89% yield.95 The barrier for atropisomer interconversion of the newly installed chloride is higher than that of the aryl nitro intermediate 87, further enhancing its stability toward a later atropisomer equilibration. A Suzuki reaction introduced the A ring (88%) and enlisted a powerful ligand-catalyst combination (Pd2(dba)3 and (o-tolyl)3P) to promote an otherwise challenging coupling of a hindered, electron-rich aryl bromide with an especially hindered aryl boronic acid.101 The kinetic atropodiastereoselectivity was modest (1:1.3 P:M), but thermal equilibration converted the material to a mixture preferentially favoring natural (P)-89 (3:1, P:M). Separation of the biaryl atropisomers and thermal re-equilibration and recycling of unnatural (M)-90 (Ea = 25.1 kcal/mol) under conditions that do not impact the preinstalled CD ring system atropisomers (Ea = 30.4 kcal/mol) funneled all material into the synthesis of 50. Removal of the TBS group (87%) facilitated subsequent hydrolysis of the methyl ester to the carboxylic acid (99%), and Cbz cleavage (99%) afforded 93. Lastly, to complete the ABCD ring system, a key macrolactamization was used to form the second macrocycle 94 (EDCI) in 62% yield. The C-terminus carboxylic acid was carried through this sequence as a MEM-protected hydroxymethyl group. This not only avoided inadvertent epimerization throughout the synthesis, but it also improved the macrolactamization reaction, which proceeded at both a faster rate and in higher yields than the corresponding methyl ester.
Scheme 8.

Boger Synthesis of the Vancomycin ABCD Ring System.
After Boc deprotection of the N-terminus amine, tripeptide 96 was coupled to residue 4 with EDCI–HOAt in 61% from 94 (Scheme 9). The final 16-membered macrocycle was formed in a room temperature nucleophilic aromatic substitution reaction promoted by CsF (DMSO), closing the ring with a kinetic atropodiastereoselectivity (8:1) favoring the natural P-isomer 97. Conversion of the nitro group to the corresponding aryl chloride afforded 98 in 60% yield from 97. Bis-TBS protection of the secondary alcohols under neutral conditions was conducted with use of N-TBS-N-methyl-trifluoroacetamide to avoid retro-aldol cleavage of the CD and DE ring systems. Cleavage of the MEM ether with B-bromocatecholborane (BCB), followed by stepwise Dess–Martin and Pinnick oxidation of the released primary alcohol and esterification of the carboxylic acid provided the methyl ester 101. Finally, stepwise nitrile hydration (H2O2, K2CO3, 85%), TBS ether cleavage under conditions that avoid competitive retro-aldol reaction of the released secondary alcohols (Bu4NF–HOAt, 81%), and global deprotection of the remaining four methyl ethers, the methyl ester, and N-terminus Boc group with AlBr3–EtSH (50%) in a single step afforded the vancomycin aglycon (50).
Scheme 9.

Final Stages of the Boger Total Synthesis of Vancomycin Aglycon.
4.2 Carbohydrate Introduction
Carbohydrate introduction on the vancomycin aglycon has been accomplished by two complementary approaches: (1) chemical glycosylation102–107; and (2) in vitro enzymatic glycosylation.108–115 An advantage of the chemical strategy is the broad substrate scope that allows use of a variety of modified carbohydrate coupling partners compared to the enzymatic glycosylation. The enzymatic reaction has the advantage that it is not necessary to protect nucleophilic functionalities in either the aglycon or the sugar, directly provides vancomycin in two steps from the fully deprotected aglycon, and avoids the chemoselectivity issues of the non-enzymatic means. The stable expression of the recombinate enzymes and the commericial or synthetic availability of the UDP-based cosubstrates today make the enzymatic glycosylation as scalable as the chemical glycosylation. Over the past few decades, base and acid promoted chemical glycosylation methods for selective glycosyl bond formation have been developed. However, base-mediated SN2 substitution of an anomeric halide is not suitable for use with vancomycin and related derivative due to competitive aglycon epimerization. The use of an acid-promoted glycosylation eliminates such issues.
4.2.1 Chemical Glycosylation
4.2.1.1 Kahne Synthesis
Kahne developed an approach in which activated glycosyl donors are sequentially coupled with the protected aglycon 104 via the acid promoted glycosylation strategy.102–104 A C2 ester functional group was used to assist in the selective formation of β-glycosyl bond with neighboring group stabilization of the oxonium ion intermediate 105 and for stereochemical control of the anomeric nucleophilic substitution with an oxygen nucleophile (Figure 8).116
Figure 8.
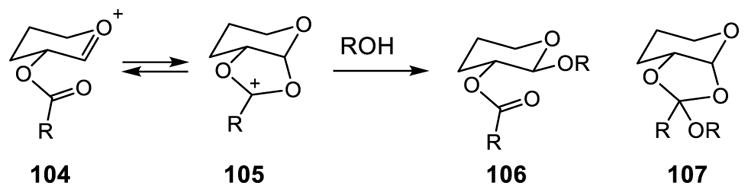
Selective β-Glycoside Formation with Neighboring Group Participation.
Initial model studies with substrates bearing conventional leaving groups such as acetate did not provide promising results. To overcome this issue, Kahne pursued the use of sulfoxide derivative 108 which could be activated under milder reaction conditions (Scheme 10).117–119 The sterically hindered base 2,3-di-tert-butyl-4-methylpyridine (DTBMP) was used to suppress formation of undesired orthoester by enhancing the nucleophilicity of the phenol.120 Addition of BF3·OEt2 minimized formation of the orthoester by promoting its rearrangement to the glycoside 110,121 and it also helped suppress Tf2O-mediated dehydration of the vancomycin-derived residue 3 carboxamide. Further improvement was made by use of a C2 azidobutyrate introduced by Kusumoto122 in place of a conventional acyl protecting group such as acetyl (56%) and pivaloyl (50%). Notably, the 4-azidobutyryl group can be selectively removed under neutral reaction conditions by treatment with PPh3 (64%), unlike acyl group removal.
Scheme 10.
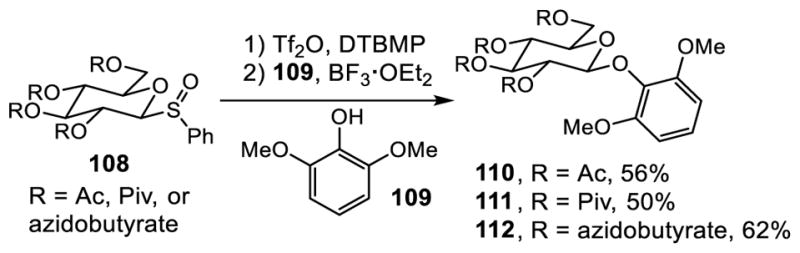
Examination of Impact of C2 Ester Functionality with Model Substrate 109.
With protocols for the coupling established, glycosylation of the vancomycin aglycon was examined (Scheme 11).103 The protected aglycon 113 was prepared from vancomycin aglycon using a 6-step protocol. This sequence includes alloc protection of the N-terminus amine (40%), temporary protection of the residue 4 phenol as a PMB ether (70%), global allylation of the C-terminus carboxylic acid and remaining three phenols (60%), acetylation of the secondary alcohols, and cleavage of the PMB ether under acidic conditions (95%). The first glycosylation with 114 afforded the intermediate compound and was followed by removal of the azidobutyrate with PPh3 to afford the protected pseudoaglycon 115 in 13% over 2 steps. Unlike the first glycosylation, the second glycosylation reaction with the vancosamine sulfoxide derivative 116 provided the disaccharide without addition of DTBMP (60%). Finally, global deprotection with removal of acetyl groups (63%) followed by the allyl and alloc groups with PdCl2(PPh3)4–Bu3SnH (78%) afforded vancomycin (1).
Scheme 11.

Kahne Synthesis of Vancomycin (1) from the Aglycon 50.
4.2.1.2 Nicolaou Synthesis
Nicolaou pursued two chemical glycosylation strategies: (1) single step direct introduction of the disaccharide 117, and (2) sequential glycosylation with two functionalized sugar coupling partners (118 and 119), as shown in Scheme 12.106,107
Scheme 12.
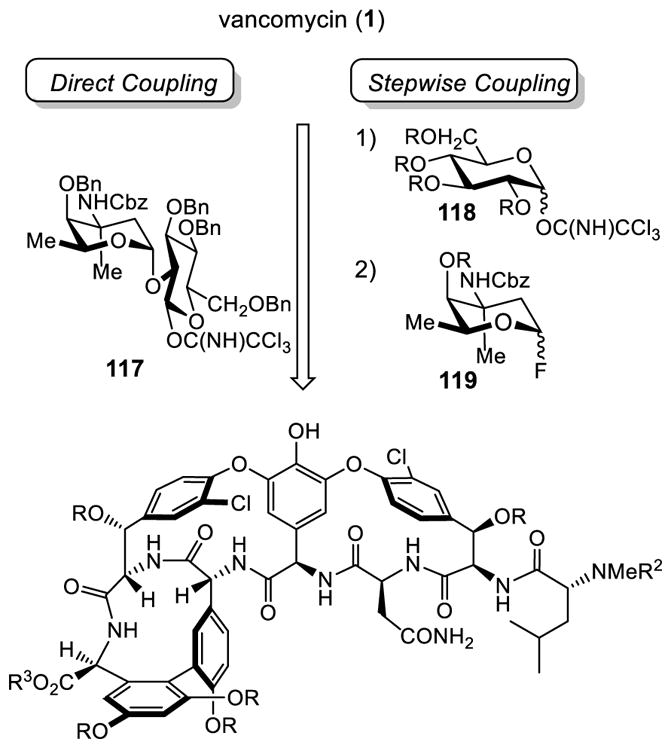
Complementary Synthetic Approaches for Chemical Glycosylation.
The protected glycosylation precursor 120 was prepared from vancomycin aglycon (50) in 4 steps (Scheme 13). The protection sequence relied on a global TBS protection with excess TBSOTf and 2,6-lutidine (72%) followed by methyl ester formation of the C-terminus carboxylic acid (92%), N-terminus free amine protection with a Cbz group (91%), and selective cleavage of the central residue TBS ether with KF–Al2O3 (60%).123 The direct coupling of the disaccharide 117 with the aglycon 120 occurred stereoselectively upon treatment with BF3·OEt2 without the addition of base to afford the fully protected vancomycin 121 solely as the β-isomer (70%). This outstanding stereoselectivity was rationalized as arising from steric interactions between the aglycon and the disaccharide, favoring equatorial disposition of the large aglycon. In the final deprotection steps of 121, E ring dechlorination occurred under the conditions required to cleave four benzyl ethers and two Cbz groups (H2, Pd/C), and was not satisfactorily avoided even after optimization of the reaction conditions or by use of other methods. In contrast, the stepwise glycosylation was successfully implemented. The first glycosylation with the alloc-protected glycosyl donor 125 proceeded smoothly affording the protected pseudoaglycon 126 in good yield (82%) accompanied by a minor side product thought to be the α-anomer.124,125 Subsequent removal of the alloc group with Pd(PPh3)4–Bu3SnH126 gave the glycosylated precursor 127 (85%). For the second glycosylation, choice of sugar protecting groups was key and had a significant impact on the glycosyl donor and acceptor reactivities. By switching the protecting group from a benzyl ether (5%) to an acetate, the protected disaccharide 129 was isolated in good yield and good stereoselectivity (84%, α/β = 8/1). Final deprotection, requiring TBS ether deprotection (80%), acetate hydrolysis (95%), N-terminus Cbz deprotection followed by hydrolysis of the methyl ester (85%, 2 steps), completed the first vancomycin total synthesis.
Scheme 13.

Direct and Sequential Glycosylations of a Protected Vancomycin Aglycon, Nicolaou Completion of the Total Synthesis of Vancomycin.
4.2.2 Enzymatic Glycosylation
In the biosynthesis of vancomycin, the carbohydrate introduction is achieved by two sequential glycosyltransferase (GtfD and GtfE) catalyzed reactions (Scheme 14).36 The latter enzyme, GtfE, is D-glucosyltransferase responsible for introduction of the first sugar on both the vancomycin and teicoplanin aglycons where TDP-glucose 133 serves as the glycosyl donor. GtfB is an analogous glycosyltransferase found in a chloroeremomycin producing strain and is able to perform the same function of installing the glucose residue on the vancomycin aglycon.
Scheme 14.

Enzymatic Carbohydrate Introduction on the Vancomycin Aglycon (50).
GtfD catalyzes the second glycosylation with the cosubstrate TDP-vancosamine 136 to afford vancomycin (Scheme 14). Early in vitro studies with the enzymes were reported by Lilly in 1997.113 The first glycosylation was achieved with a variety of NDP-sugar derivatives using Gtf’E (mutant GtfE; single amino acid mutation from serine to proline) and GtfB obtained from Amycolatopsis orientalis, which were expressed in E. coli. With use of the cell extracts containing the expressed enzymes, it was found that readily accessible and commercially available UDP-glucose (134) can be utilized as an alternative cosubstrate, displaying an indistinguishable glycosylation ability.
4.2.2.1 Walsh Synthesis
Walsh characterized the enzymatic activity of GtfB, D, and E that were obtained through subcloning from Amycolatopsis orientalis, heterologous expression and purification to homogeneity from E. coli.108,109 The GtfE reaction with vancomycin aglycon (50) and UDP-glucose 134 was carried out through incubation with 75 mM tricine (pH 9), 2.5 mM tris-(2-carboxyethyl)-phosphine (TCEP), and 1 mg/mL BSA to afford the pseudoaglycon 135 on an analytical scale in which the product formation was established and quantified by HPLC analysis.127 Consistent with previous results disclosed by Lilly with cell extracts, GtfB displayed the same level of the enzymatic reactivity in this first glycosylation. It was also demonstrated that both UDP-glucose and TDP-glucose can be used as a cosubstrate.127
The second GtfD-catalyzed glycosylation of vancomycin pseudoaglycon 135 with synthetic TDP-vancosamine 136 was carried out in 75 mM tricine (pH 9), 2.5 mM tris-(2-carboxyethyl)-phosphine (TCEP), 2 mM MgCl2, 1 mg/mL BSA to afford vancomycin (1). Unlike the first GtfE-catalyzed reaction, the second glycosylation is rapid and complete within 1.5 h.
After Walsh disclosed the enzymatic synthesis of vancomycin from the aglycon, Wong reported a one-pot glycosylation for introduction of the first sugar by using D-glucopyranosyl-1-phosphate 138 as a UDP-glucose precursor (Scheme 15).111 This method allows in situ generation of the UDP-glucose 134 from the glucose monophosphate 138. The one-pot sequence starts with coupling uridine 5′-triphosphate (UTP) and glucose phosphate 138 catalyzed by thymidyltransferase (Ep) to afford UDP-glucose 134. This UDP-glucose can be then utilized as a cosubstrate for the glycosylation of vancomycin aglycon (50) to produce the pseudoaglycon 135 with release of uridine 5′-diphosphate (UDP). Finally, the catalytic cycle is completed by transforming UDP to UTP catalyzed by pyruvate kinase (PK) in the presence of phosphoenolpyruvate (PEP).
Scheme 15.
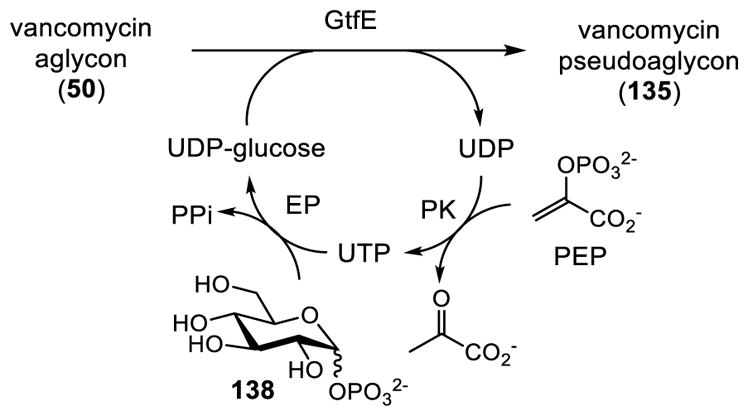
Wong One-pot Initial Glycosylation of Vancomycin Aglycon (50).
4.2.2.2 Boger Synthesis
Recently, Boger disclosed elegant total syntheses of two pocket modified vancomycin aglycons designed to address the threat of vancomycin resistance to glycopeptide antibiotics, which are detailed in Sections 7.2 and 7.3. In the course of these studies, the use of enzymatic glycosylations was pursued to install the carbohydrate on the redesigned vancomycin aglycons. Although GtfE and GtfD catalyzed glycosylations with a variety of modified sugar derivatives had been reported, there was no report at the time that utilized an aglycon containing deep-seated changes and none that used the approach on a preparative scale.
Conditions for both glycosylation reactions were established for use on a preparative scale and key elements of the aglycon scope for participation in the reactions were examined.128 The first GtfE catalyzed glycosylation was achieved by a blend of conditions detailed by Walsh and Wong and provided the pseudoaglycon 135 in good yield (92%, 2.0 mg scale); 0.5 mM aglycon, commercial 2 mM UDP-glucose, 5 μM GtfE, 75 mM tricine (pH 9.0), 2.0 mM TCEP, 1 mM MgCl2, 0.2 mg/mL BSA, glycerol (5% v/v), 37 °C, 42 h. The GtfD catalyzed glycosylation was performed with synthetic UDP-vancosamine, which was prepared by adapting a previously reported synthetic procedure disclosed by Kahne.129 This GtfD reaction did not require significant changes to the Walsh reaction conditions108,109 and afforded vancomycin (1) in good yield (87%, 1.5 mg scale); 0.5 mM pseudoaglycon, 2.0 mM UDP-vancosamine, 5.0 μM GtfD, 75 mM tricine (pH 9.0), 2.0 mM TCEP, BSA (0.2 mg/mL) 1.0 mM MgCl2, glycerol (10% v/v), 37 °C, 2 h. Importantly, although the endogenous glycosyl donors for both enzymes are the TDP-sugars, the commercially available (134) or synthetic (137) UDP-sugars displayed the same level of reactivity. The availability of the recombinant enzymes and the synthetic UDP-sugars are such that this two-step introduction of the carbohydrate can be scaled by as much as 100-fold over that reported for use in academic labs and even larger for production purposes. Its implementation by Boger completed a second total synthesis of vancomycin,128 culminating with a two-step, sequential enzymatic catalyzed glycosylation of the fully functionalized and unprotected vancomycin aglycon (Scheme 16).
Scheme 16.
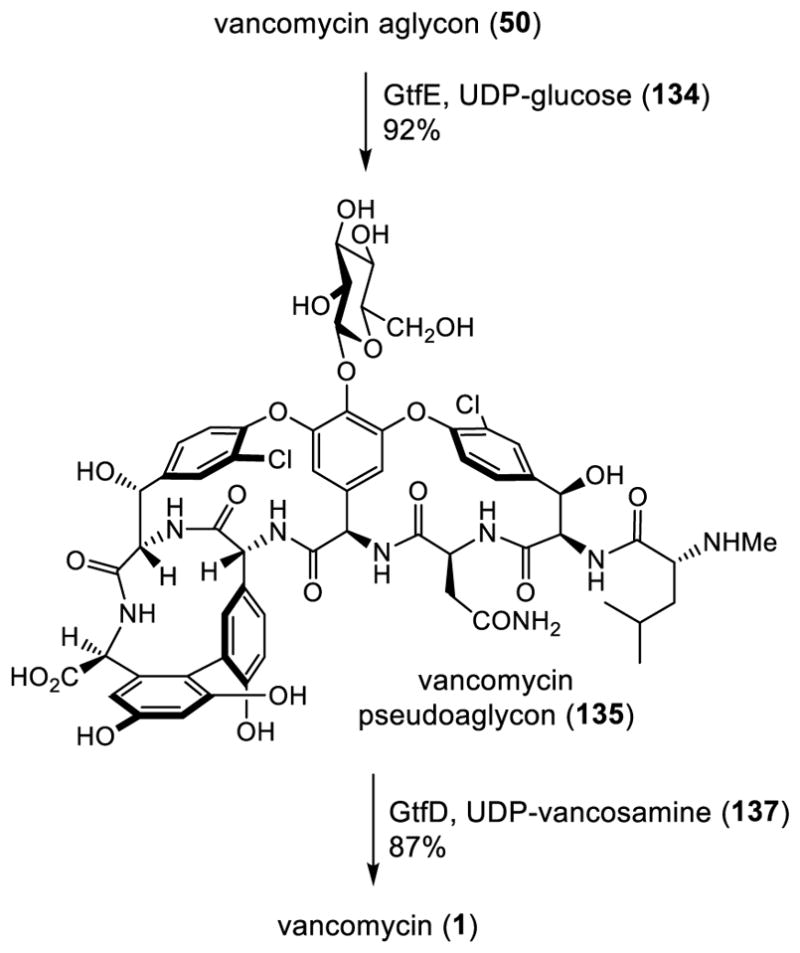
Boger Enzymatic Carbohydrate Introduction on the Vancomycin Aglycon (50), Completion of a Second Total Synthesis of Vancomycin.
5. Total Synthesis of Teicoplanin Aglycon
Isolated from Actinoplanes teicomyceticus (ATCC 31121) in 1978,130,131 teicoplanin132,133 is one of the more complex glycopeptide antibiotics related to vancomycin.3,4,29,30,134 Like vancomycin, it is a drug utilized in the clinic when resistant bacterial infections are encountered or for patients allergic to β-lactam antibiotics. Additional attributes compared to vancomycin include a 2–8 fold increased potency, a lower toxicological profile,135–137 a longer half-life in patients (40 vs 6 h),135–137 lower patient pharmacokinetic variability, and it may be administered as a single daily dose either IV or intramuscularly, making it suitable for outpatient therapy.138,139 Although teicoplanin is an approved drug in Europe, Japan, and several other countries sold under the name Targocid,140–142 it is not approved for use in the U.S..
The first total synthesis of the teicoplanin aglycon was reported in 2000 by Boger143 and shortly thereafter, a second generation strategy144 was disclosed (Figure 9). The teicoplanin ABCD ring system is identical in structure and stereochemistry to vancomycin. Additionally, the CDE atropisomer stereochemistry is identical. Key differences include the lack of a β-hydroxy group on the teicoplanin E ring phenylalanine and the presence of an additional FG ring system. As such, it represents a structurally and stereochemically more complex tetracyclic glycopeptide antibiotic aglycon than vancomycin. Both Boger and Evans disclosed total syntheses and enlisted approaches in which the teicoplanin EFG ring system was appended onto their preexisting (vancomycin) ABCD ring systems, permitting the late stage divergent total synthesis of both natural products from a common intermediate. The discussion that follows summarizes the first and second generation total synthesis reported by the Boger, followed by a summary of the later Evans total synthesis.
Figure 9.

Boger First and Second Generation Retrosynthetic Analysis of Teicoplanin Aglycon.
5.1 Boger Synthesis
The first and second generation total synthesis disclosed by Boger both enlisted synthesis of the same E, F and G ring subunits. The absolute stereochemistry found in the E ring (139) subunit was obtained through use of a cuprate-mediated Schöllkopf asymmetric alkylation145 of the respective 4-fluoro-3-nitrobenzyl bromide. For rings F (140) and G (141), the absolute stereochemistry of the phenylglycines was introduced through Sharpless asymmetric aminohydroxylation of their respective styrene precursors.146 The distinction in the two approaches was the order and timing of the closure of the DE and FG ring systems. Because of the facile residue 3 (F ring) phenylglycine epimerization observed within the confines of the teicoplanin FG ring system that provides the unnatural diastereomer (1% aq. NaHCO3, 80 °C; >95:5 unnatural R:S),147 the more conservative first generation total synthesis closed this ring system last. In addition, the FG diaryl ether was formed with acyclic phenylglycinol substrates incapable of epimerization. Thus, the F and G rings were coupled via a room temperature intermolecular SNAr reaction to provide 142 (70%), and was followed by conversion of the activating nitro group to the requisite aryl methyl ether 145 (Scheme 17). After benzyl ether protection of the primary alcohol (91%), MEM ether cleavage, and hydrolysis of the trifluoroacetamide (96%), the linear EFG fragment 147 was obtained by peptide coupling of 141 and 146 (89%). Sequential Dess–Martin and Pinnick oxidation of the primary alcohol yielded 148 (86%), completing the synthesis of the precursor EFG fragment and allowing attachment of the free carboxylic acid to the ABCD ring system.
Scheme 17.

Boger First Generation Synthesis of the Acyclic EFG Tripeptide.
The coupling of the ABCD ring system 96 and EFG tripeptide precursor 148 was effected by DEPBT148 (83%) without competitive epimerization (Scheme 18). In contrast to DEPBT, typical coupling reagents provided near 1:1 mixtures of epimers in this especially challenging coupling reaction. Room temperature macrocyclization upon treatment with CsF (DMSO) provided 149 in excellent yield (80%) and good atropodiastereoselecitivity (3:1), favoring the natural P-atropisomer. The second generation total synthesis improved this atropodiastereoselectivity (18:1).
Scheme 18.

Final Stages of the Boger First Generation Total Synthesis of Teicoplanin Aglycon.
Concurrent with cyclization, the N-terminus Teoc group was also cleaved. Subsequent TBS ether protection of the secondary alcohol (89%) followed by Troc protection of the primary amine (93%) proceeded smoothly to afford 151. Single step, O-debenzylation and reduction of the aryl nitro group to the aniline was affected by treatment with H2 and Pd/C. This was conducted under specially designed conditions (1% Cl3CCO2H–MeOH) that accelerated the sluggish benzyl ether hydrogenolysis and avoided competitive Troc dechlorination. Aniline diazotization and Sandmeyer substitution with CuCl2–CuCl yielded the fully functionalized E ring and set the stage for FG ring closure. Stepwise oxidation of the primary alcohol 152 to the carboxylic acid 153 (74%), N-Troc deprotection (89%), and macrolactamization with PyBop provided 155 (66%) with closure of the 14-membered FG ring system. Lastly, the C-terminus was converted to the free carboxylic acid, and a global deprotection (48%) effected by treatment of 157 with AlBr3–EtSH served to cleave the six methyl ethers, the TBS ether, and the N-Boc group and completed the first total synthesis of the teicoplanin aglycon (158).
Shortly following the initial disclosure, a second generation total synthesis of teicoplanin aglycon was published by Boger144 that proved to be more convergent, eliminating 4 steps from the longest linear sequence, and proceeded with a much higher kinetic atropodiastereoselectivity (18:1 vs 3:1) for closure of the DE ring system. This was accomplished by altering the order of the ring closures such that the FG macrolactamization (95%) preceded coupling of EFG tripeptide to the ABCD ring system and subsequent DE ring closure. This latter SNAr macrocyclization with diaryl ether formation proceed with high diastereoselection (18:1, 76%) without competitive racemization, provided tempered reaction conditions were used.
From intermediate 145, PyBop promoted coupling of the E ring subunit 139 with 159 after deprotection of the trifluoroacetamide provided 160 (Scheme 19). Stepwise oxidation of the primary alcohol to the carboxylic acid 161 (81%) followed by Teoc deprotection afforded 162 (87%). Macrolactamization of the 14-membered FG ring system with PyBop afforded 163 in excellent yield (95%). In preparation for the attachment to the ABCD ring system, MEM ether cleavage, followed by stepwise oxidation of the released primary alcohol afforded the necessary carboxylic acid 165 of the EFG fragment.
Scheme 19.

Boger Second Generation Synthesis of the Teicoplanin Aglycon FG Ring System.
With the two major subunits of the structural framework of teicoplanin in hand, amide bond coupling promoted by DEPBT148 afforded the heptapeptide backbone of teicoplanin in 72% yield without competitive epimerization even for this challenging reaction (Scheme 20). This set the stage for the key SNAr reaction with formation of the last macrocycle. While the first generation total synthesis of teicoplanin aglycon was designed to avoid epimerization by not incorporating a rigid FG macrocycle prior to formation of the DE ring system, suppression of competitive epimerization proved possible. Although CsF-promoted macrocyclization under conventional reaction conditions (25 °C, DMSO) was not especially productive, affording 160 in low yields (23–37%) along with numerous epimeric or degradation products, the use of tempered reaction conditions (10 °C, DMF) provided 160 in high yield (76%) as essentially a single diastereomer (>10:1, ca. 18:1) with little or no competitive epimerization. The key macrocyclization proceeded with greater ease and with a much higher atropodiastereoselectivity with the intact FG ring system installed in the cyclization substrate. This contrasts with the more modest diastereoselectivity observed with 149, bearing the intact ABCD ring system without the intact FG ring system (3:1) and the nonselective closure of the isolated teicoplanin DE ring system (1:1–3). Conversion of the nitro group to the corresponding chloride by reduction, aniline diazotization, and Sandmeyer substitution, followed by protection of the secondary alcohol as its TBS ether provided 168, junctioning with a late stage intermediate in the first generation total synthesis. During these studies, the fully functionalized monocyclic FG and DEF ring systems and the bicyclic DEFG ring systems were prepared.143,144,149 The isolated FG ring systems was found to adopt a single rigid solution conformation consistent with that found in teicoplanin. Atropisomerization of the teicoplanin DE ring system proved nearly identical to that of the vancomycin DE ring system, proceeding at comparable rates and with no thermodynamic atropisomer preference (P vs M). Most remarkable was the observation that the 16-membered vancomycin and teicoplanin DE ring systems isomerize at similar rates regardless of the presence of the FG ring system. Both equilibrate much more readily than the 16-membered CD ring system even in the absence of the confines of the AB ring system and both can be preferentially equilibrated in the presence of the ABCD ring system.96,97,150
Scheme 20.

Final Stages of the Boger Second Generation Total Synthesis of Teicoplanin Aglycon.
5.2 Evans Synthesis
Shortly after the disclosure of both the first and second generation total synthesis of teicoplanin aglycon by Boger, Evans disclosed a route similar to the Boger second generation total synthesis with notable distinctions (Figure 10).151 The EFG subunit outfitted with an E ring 4-fluoro-3-nitrophenylalanine for SNAr DE ring closure and complete with the macrocyclic FG ring system intact also served as the key advanced intermediate. In the Evans approach, the FG biaryl ether was installed by a Cu(II)-mediated phenol coupling with an aryl boronic acid152,153 and the macrolactamization with closure of the FG ring system was conducted at a different amide site. An additional important difference was that the amino acid subunits were incorporated at the correct carboxylic acid oxidation state during the entirety of the synthesis of the EFG fragment, enlisting N-methylamide protection from competitive epimerization that was subject to selective cleavage to the corresponding carboxylic acid when and as needed.
Figure 10.
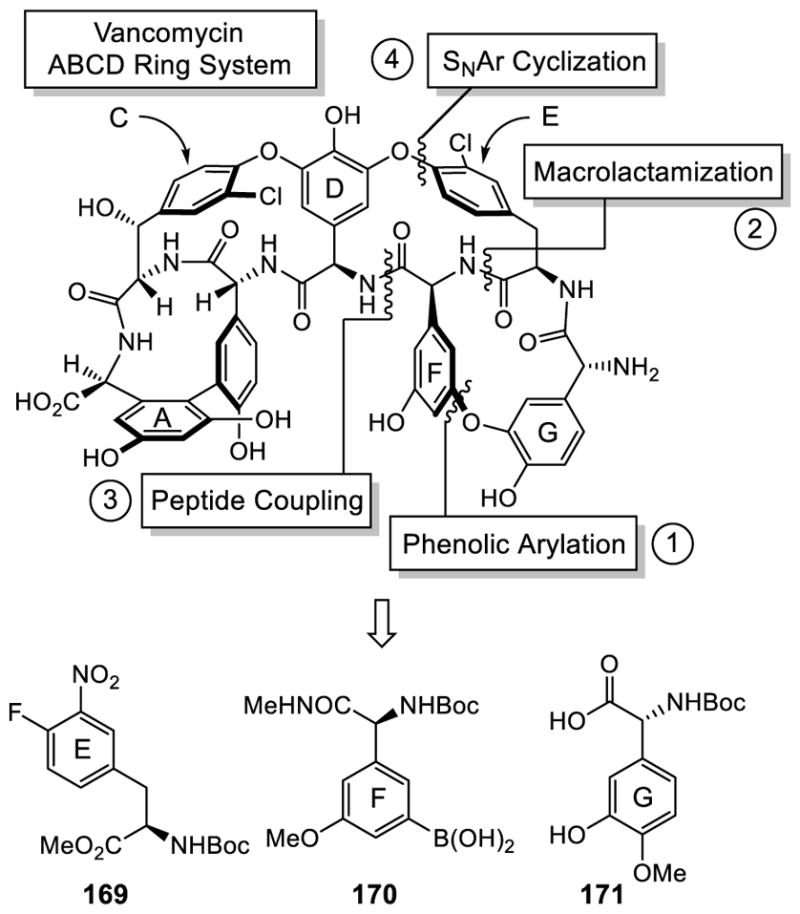
Evans Retrosynthetic Analysis of Teicoplanin Aglycon.
The construction of the E ring 169 was achieved through an asymmetric rhodium-catalyzed hydrogenation of methyl (Z)-2-acetamido-3-(4-fluoro-3-nitrophenyl) acrylate,154 whereas the F ring 170 was prepared as an aryl boronic acid from the respective aryl bromide.
The appropriately protected E and G amino acid subunits were coupled with EDCI. After amine protecting group exchange (N-Boc to trifluoroacetamide),155 the FG diaryl ether was formed through a Cu(OAc)2-mediated coupling of phenol 175 with the arylboronic acid 170, affording 176 in 80% yield (Scheme 21). The 14-membered macrocycle was formed upon macrolactamization with HATU after hydrolysis of the methyl ester and Boc removal. Lastly, the C-terminus N-methylamide was hydrolyzed to the corresponding carboxylic acid 180 upon selective nitrosation (N2O4, 0 °C) and mild nitrosamide hydrolysis (2:1 DMF–H2O, 60 °C) in the absence of added base.
Scheme 21.

Evans Synthesis of the Teicoplanin EFG Fragment.
With the EFG ring system in hand, this fragment was coupled to the ABCD ring system with adoption of the DEPBT-mediated reaction conditions143,144 used by Boger to provide the product in good yield without epimerization (Scheme 22). Closure of the DE ring system with 16-membered diaryl ether formation was effected by treatment with CsF to promote the intramolecular nucleophilic aromatic substitution reaction, using the tempered reaction conditions (DMF, 10 °C) disclosed by Boger and providing 182 in 75% yield with high (>15:1) atropodiastereoselectivity. Reduction of the E ring nitro group to the aniline, diazotization, and Sandmeyer reaction with CuCl and CuCl2 installed the E ring chloro substituent (183). Lastly, hydrolysis of the C-terminus N-methylamide upon successive nitrosation (N2O4, 0 °C) and a pH neutral hydrolysis, followed by global demethylation and trifluoroacetamide cleavage provided teicoplanin aglycon (158).
Scheme 22.

Final Stages of the Evans Total Synthesis of Teicoplanin Aglycon.
6. Total Synthesis of Ristocetin A Aglycon
A ristocetin complex, Spontin containing both ristocetin A and B, was reported in 1956. It was isolated from Amycolatopsis orientalis subs. Lurida, collected in 1951 in Colorado Springs, CO, by Abbott Laboratories.156 Shortly following this initial disclosure, the complex was introduced into the clinic in 1957. Reports of incidents of patient mortality forced this antibiotic to be pulled from the market after two years of clinical use.157,158 A common link was identified where patients missing a platelet factor, those with von Willebrand’s disease, suffered platelet aggregation, attributing to the deaths.159–161 As a result, ristocetin A aggregation of patient platelet in drawn blood samples is used today to diagnosis this disease and to detect abnormalities in this protein.162–164 Later, studies identified an aspect of the ristocetin structure that could be easily removed through selective enzymatic cleavage of rhamnose, which eliminated the induced platelet aggregation in vivo.165
Nearly 30 years after its isolation and based largely on the spectroscopic studies of Williams, the structure of ristocetin A (4) was elucidated by Harris in 1982 and confirmed by others.166–168 Among the family of vancomycin related glycopeptides, ristocetin A uniquely contains a tetrasaccharide bound to the central aryl subunit that is composed of arabinose, mannose, glucose, and rhamnose. Due to the presence of this tetrasaccharide, ristocetin A has the weakest dimerization constant compared to other glycopeptides when bound to a cell wall precursor substrate.169 Like vancomycin, ristocetin A also was the only other glycopeptide within this family to receive FDA approval in the U.S. without further modification of the natural product. Since then, and with the clinical withdrawal of ristocetin A, focus shifted to semisynthetic means for its antimicrobial improvement. Notably, ristocetin A aglycon is slightly more active than ristocetin A itself, free of platelet aggregation activity, and emerged as an entry point for such semisynthetic antibiotic development.
6.1 Boger Synthesis
Given the importance of the glycopeptides, the total synthesis of ristocetin A aglycon was undertaken by Boger and reported in 2004.100 The total synthesis of the ristocetin A aglycon, which constitutes a tetracyclic aglycon similar to teicoplanin, was heavily influenced by the second generation teicoplanin total synthesis previously discussed (Scheme 17–20).144 In contrast to teicoplanin, (1) both the CD and DE rings of ristocetin no longer possess elements of planar chirality, lacking the aryl chlorides and simplifying the synthetic challenges. Ristocetin also incorporates (2) a β-hydroxy group on the E ring phenylalanine, (3) a methyl substituent on the F ring, and (4) a C-terminus capped as the methyl ester in place of a free carboxylic acid. Thus, the aglycon was prepared in a highly convergent approach from 185 and 186 representing the intact ABCD ring system and the EFG subunit complete with the preformed FG ring system (Figure 11). DE ring closure by a nucleophilic aromatic substitution reaction would not only introduce the diaryl ether linkage but also complete the assemblage of the tetracyclic ring system. The key DE ring closure was anticipated to benefit from preorganization of the substrate, resulting in closure under conditions much milder than those required of vancomycin and with a higher atropodiastereoselectivity. Offsetting this advantage is the propensity for epimerization under even mildly basic conditions which might have precluded implementation of this approach.
Figure 11.

Boger Retrosynthetic Analysis of Ristocetin A Aglycon.
In turn, the ABCD ring system was prepared through sequential CD and AB ring closures analogous to the efforts on vancomycin. Control of the CD atropisomer stereochemistry is not an issue with ristocetin by virtue of its lack of a C ring aryl chloride rendering the diastereoselectivity of the diaryl ether macrocyclization unimportant. Thus, the stereochemical issue associated with this approach simplified to the control of the AB atropisomer stereochemistry. This could be effectively addressed with an anticipated thermodynamic preference for the natural stereochemistry (ca. 3:1) and easily adjusted on the biaryl precursor preceding AB macrolactamization.
Given the similarity between the total synthesis of ristocetin aglycon and the second generation total synthesis of teicoplanin aglycon, the reader can refer to both Section 5.1 and the disclosed report by Boger for full details.100 The diaryl ether that links the FG ring of the EFG macrocycle was prepared from the appropriately substituted F ring 187 and G ring 141 through an intermolecular SNAr reaction, using NaH and affording 188 in 69% yield (Scheme 23). Although this substitution reaction could be effected with K2CO3 in the total synthesis of teicoplanin, its use resulted in low yields for 188 (20–30%) due to the increased steric hindrance of the F ring methyl substituent. The nitro group on the G ring was transformed to the corresponding methoxy group, yielding 191. After deprotection of the amine, the peptide bond between the FG fragment and the E ring was formed with HATU, which suppressed epimerization observed with other peptide coupling reagents. Notably, the synthesis of the E ring subunit was improved relative to that originally introduced with vancomycin through use of a diastereoselective anti-aldol reaction (94% de) of a glycine imine bearing an α-hydroxypinanone chiral auxiliary (94% ee).170 Stepwise oxidation (Dess–Martin and Pinnick) of the primary alcohol and Teoc deprotection primed intermediate 196 for subsequent macrolactamization with PyBop, closing the 14-membered EFG ring system and providing 197 in superb yield (92%). Silyl ether protection of the β-hydroxy group (96%), followed by MEM ether cleavage and reintroduction of the N-Boc group (86%), and finally Jones oxidation of the primary alcohol (79%) completed synthesis of the EFG ring fragment 186.
Scheme 23.

Boger Synthesis of the Ristocetin A EFG Fragment.
The cyclic tripeptide 86, representing the CD ring system, was synthesized as previously discussed in Section 4.1.3. However, it is worth highlighting that the preparation of the C ring subunit was improved through use of a Schöllkopf171 aldol-type addition of the transmetalated Zr anion,95 leading to excellent control of not only the α-amino acid stereochemistry (>99:1), but also the β-hydroxy diastereoselectivity (5:1). Removal of the nitro group over two steps afforded 200 (77%), and Suzuki–Miyaura cross-coupling with the A ring provided 201 with nonselective formation of the biaryl axial chirality (Scheme 24). Thermal equilibration of the post-coupling mixture afforded a 3:1 (P:M) ratio in favor of the natural atropisomer (P)-201. The minor unnatural atropisomer was recycled by subjection to thermal equilibration in o-dichlorobenzene at 130 °C, regenerating the 3:1 (P:M) mixture and funneling all material into the synthesis. TBS deprotection of the natural atropisomer (P)-201 with Bu4NF (95%), methyl ester hydrolysis with LiOH (96%), and Cbz hydrogenolysis with H2–Pd/C yielded 204 (98%). Macrolactamization with EDCI closed the 12-membered AB macrocycle in 51% yield. Lastly, removal of the Boc group without affecting the MEM ether afforded the ristocetin A ABCD ring system 206.
Scheme 24.

Boger Synthesis of the Ristocetin A ABCD Ring System.
The EFG and ABCD ring systems were coupled with DEPBT148 (Scheme 25), which after extensive exploration was determined to be the reagent of choice for minimizing epimerization, affording the product in excellent diastereoselectivity (>10:1). The DE ring closure was promoted by CsF in DMF, yielding the desired product 207 in exceptional yield (>95%) and excellent atropodiastereoselectivity (>15:1). Additionally, like teicoplanin, no epimerization was observed. The remaining steps and conditions are nearly identical to those developed in the second generation total synthesis of teicoplanin. Reduction of the nitro group, aniline diazotization, and in situ reduction provided 208 (79%). Protection of the secondary alcohols, MEM deprotection, and oxidation of the released C-terminus alcohol to the carboxylic acid followed by esterification afforded 211. Next, global deprotection with AlBr3–EtSH with removal of the six methyl ethers and cleavage of the methyl ester, two TBS ethers, and the terminal N-Boc group (10 protecting groups) provided 212 in remarkably high yield (78%). Finally, the Boc group was reintroduced on the N-terminus (91%), and was followed by selective methylation of the carboxylic acid (93%), and acid-catalyzed Boc removal (98%) to afford ristocetin A aglycon (215).
Scheme 25.

Final Stages of the Boger Total Synthesis of Ristocetin A Aglycon.
7. Total Syntheses of Residue 4 Modified Vancomycins and Key Derivatives
7.1 [Φ[CH2NH]Tpg4]Vancomycin Aglycon
Boger extended his group’s efforts to the total synthesis of novel redesigned vancomycin analogues with the ambitious goal of addressing vancomycin resistant bacterial infections. The elaborate mechanism of resistance orchestrates a simple one atom change in bacteria cell wall precursors that significantly reduces both the binding affinity of vancomycin for its target and its antimicrobial activity 1000-fold.48 Boger began the studies by defining the origin of this loss in binding affinity by partitioning it into: (1) the loss of a H-bond, and (2) introduction of a destabilizing lone pair/lone pair repulsion between the vancomycin residue 4 amide carbonyl oxygen and the lactate ester oxygen (Figure 12).172
Figure 12.

Evaluation of Binding Affinity of Vancomycin with Model Ligands.
The binding studies were carried out with vancomycin and the model ligands 216–218. This included the ketone ligand 217, containing a linking methylene which lacks a lone pair and is incapable of H-bonding. These studies revealed that it is the destabilizing electrostatic repulsion (100-fold), more so than the H-bond (10-fold), that is responsible for the 1000-fold loss in binding affinity. This has significant ramifications on the redesign of vancomycin for treatment of vancomycin resistant bacteria, indicating that simply removing the destabilizing lone pair/lone pair interaction without reengineering a reverse H-bond could improve affinity and activity by as much as 100-fold. As a result and in initial studies, a binding pocket modification in the vancomycin core that replaced the residue 4/5 amide carbonyl with an aminomethylene linkage was targeted to remove the destabilizing lone pair interactions.173 More subtly, such a modification provides the antibiotic with balanced dual ligand binding capabilities needed for vancomycin resistant organisms (D-Ala-D-Ala and D-Ala-D-Lac), while maintaining its ability to bind D-Ala-D-Ala required for vancomycin sensitive bacteria.
The plan for the synthesis the aminomethylene analogue 219 was based largely on the route implemented in the vancomycin aglycon total synthesis,90 albeit with improvements as shown in Figure 13.173 Key elements of the approach include synthesis of the modified vancomycin ABCD ring system featuring a reductive amination for installation of the amide modification, the first of two diaryl ether closures for formation of the modified CD ring system (76%, 2.5–3.1 kinetic atropodiastereoselectivity), a Suzuki coupling for installation of the hindered AB biaryl bond (90%) on which the atropisomer stereochemistry could be thermally adjusted, and a macrolactamization closure of the AB ring system (70%). Subsequent DE ring system introduction enlisted a room temperature aromatic nucleophilic substitution reaction for formation of the remaining diaryl ether (86%, 6–7:1 kinetic atropodiastereoselectivity), completing the carbon skeleton of 219. The methyl carbamate was selected as the protecting group for the aminomethylene group, which was tolerant of all chemical transformations throughout the total synthesis, yet capable of removal in the final global deprotection step with AlBr3–EtSH. Moreover, the relatively small carbamate protecting group minimized any undesired steric features that might have effected the CD and AB ring closures. Finally, and despite the apparent flexibility introduced into the CD ring system by removal of the amide, the recognition that the rigid AB ring system complete with its cis amide controls the conformation of the surrounding CD ring system insured that the ABCD ring system as well as the final analogue 219 would adopt an overall conformation analogous to that of vancomycin.
Figure 13.

Boger Retrosynthetic Analysis of [Φ[CH2NH]Tpg4]Vancomycin Aglycon.
The synthesis started with a reductive amination of aldehyde 222 with free amine 221 to produce 223 in good yield (75%) and in excellent diastereoselectivity (12:1) in spite of ease of epimerization of 222 or the intermediate imine (Scheme 26). Subsequent methyl carbamate protection of the secondary amine (85%), followed by benzyl ether deprotection upon treatment with Raney Ni (98%), and hydrolysis of the methyl ester afforded 224 (>99%). Peptide coupling of 76 with 224 effected by DEPBT148 (70%, dr = 14:1) preceded a base-mediated (K2CO3–CaCO3) SNAr cyclization to provide the CD ring system 226 in good yield (76%) and in good kinetic atropodiastereoselectivity (2.5–3:1, P:M). Reduction of the nitro group followed by aniline diazotization and Sandmeyer substitution afforded the aryl chloride 227 (70%). The same key aryl chloride 227 was also prepared in later efforts from thioamide 246, which was synthesized in studies culminating in the total synthesis of the residue 4 thioamide of vancomycin aglycon (see Section 7.2). Reduction of the thioamide with H2, Ra–Ni in the presence of formamidine acetate gave 228 while suppressing aryl dechlorination (60%). Methyl carbamate protection of the amine (91%) and selective cleavage of the phenol TBS ether also gave 227 in good yield (95%).
Scheme 26.

Synthesis of the Modified CD Ring System 227.
Suzuki coupling of 227 with the boronic acid 79 provided the biaryl atropisomers in a separable 1:1.3 ratio (90%) (Scheme 27). Thermal re-equilibration of the unnatural M-atropisomer upon simple heating provided a 1:1.1 separable atropisomer mixture, permitting the recycling of all material into the synthesis. TBS ether deprotection (80%), Cbz deprotection (95%), followed by methyl ester saponification (96%), and macrolactamization closure of the AB ring system with PyBop (70%) completed the synthesis of the modified ABCD ring system. Deprotection of the N-terminus Boc group by treatment with HCO2H provided the free amine 235 in good yield (84%).
Scheme 27.

Synthesis of Modified ABCD Ring System 235.
The final peptide coupling of the ABCD ring system 235 and the E ring tripeptide 96 was carried out with DEPBT148 to afford the heptapeptide in good yield (73%) with excellent diastereoselectivity (12:1), suffering little competitive racemization (Scheme 28). Subsequent ring closure of the DE ring system was achieved by SNAr cyclization that proceeded under milder reaction conditions (CsF–CaCO3, 25 °C, DMF) than those disclosed for vancomycin and provided 236 in good yield (74%) and good atropodiastereoselectivity (6–7:1). To further improve the efficiency of the approach, focused efforts were made to optimize the E ring chloride introduction and the final global deprotection steps. Reduction of the aryl nitro group with zinc nanoparticles resulted in a rapid conversion (30 min) to the aniline compared to the previously reported conditions (H2, Pd/C, 12 h). The subsequent diazonium salt formation and Sandmeyer substitution reaction for chloride introduction were carried out under improved conditions minimizing reduction (dechlorination) by changing the solvent ratio (CH3CN/H2O) and lowering the reaction temperature (–35 °C) to afford the aryl chloride 237. Next, 237 was subjected to TBS ether protection of both secondary alcohols with CF3CONMeTBS to produce the TBS protected aryl chloride 238 in improved overall conversions (55–60% over 3 steps). MEM ether deprotection (80%) and a two-step oxidation of the released primary alcohol provided the C-terminus carboxylic acid (80%), and was followed by hydration of the nitrile to afford the primary carboxamide 241 (87%). The final global deprotection, promoting the removal of four aryl methyl ethers, two TBS ethers, the N-Boc group, and the methyl carbamate, was carried out with AlBr3–EtSH at a defined reaction concentration (0.1 M) to produce [Φ[CH2NH]Tpg4]vancomycin aglycon (242) in excellent yield (80%). When the global deprotection was conducted at more dilute reaction concentrations (0.01 M vs 0.1 M), only partial cleavage of the E ring TBS ether was observed.
Scheme 28.

Completion of the Boger Total Synthesis of [Φ[CH2NH]Tpg4]Vancomycin Aglycon.
7.2 [Φ[C(=S)NH]Tpg4]Vancomycin and [Φ[C(=NH)NH]Tpg4] Vancomycin Aglycons
Based on the initial success of a binding pocket modification of vancomycin aglycon, Boger explored additional vancomycin analogues with the potential of displaying greater activity by introduction of a residue 4 amidine (Figure 14).174,175 Importantly, the amidine 243 was found to display dual binding affinity toward both the wild type dipeptide (D-Ala-D-Ala) and the modified resistant dipeptide (D-Ala-D-Lac), where the amidine serves as both a H-bond donor to reinstate the lost H-bond with the altered ligand, while maintaining the ability to serve as a H-bond acceptor toward the wild type cell wall peptidoglycan dipeptide.
Figure 14.
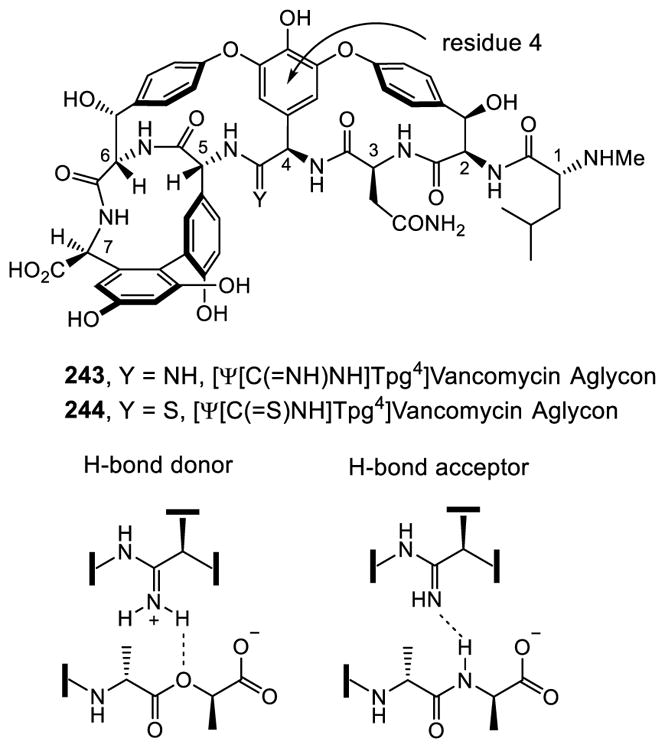
Structures of [Φ[C(=NH)NH]Tpg4]Vancomycin Aglycon (243), [Φ[C(=S)NH]Tpg4] Vancomycin Aglycon (244), and the Dual Binding Behavior of the Amidine in 243.
At the time of the work, there was no precedent on which to base the expected results and, even today, reports of the use of an amidine as an amide isostere in cyclic peptides are limited.176–178 Thus, the binding behavior of the amidine in 243 was not clear and it was not evident whether the ester oxygen of D-Ala-D-Lac could serve as a H-bond acceptor, or whether the amidine,179 typically protonated under physiological conditions, could act as the H-bond acceptor for binding D-Ala-D-Ala. As a result, and even though it was perhaps the most important modification to examine, it was not the first binding pocket modified vancomycin analogue pursued by Boger.
The overall synthetic strategy envisioned for 243 relied on the synthetic route developed for the total synthesis of vancomycin aglycon (50)90 and subsequently adopted for the aminomethylene vancomycin aglycon 242 (Figure 15).173 Key to its synthesis, a residue 4 thioamide was chosen to serve as a masked precursor to be converted to the amidine in a final step and conducted on a fully functionalized and deprotected vancomycin aglycon. By design, this approach permits the divergent synthesis of additional pocket modified vancomycin analogs from a common late stage intermediate, including substituted amidines180 and the aminomethylene derivative 242. Its implementation required development of a new reaction for such a direct single step amidine formation on substrates only soluble in protic solvents. At the time of the work, it was established empirically that introduction of the residue 4 thioamide could only be introduced effectively on the early stage synthetic intermediate 88, introducing a host of challenges for the ensuing total synthesis and requiring notable alterations in the approach.
Figure 15.

Boger Retrosynthetic Analysis of [Φ[C(=NH)NH]Tpg4]Vancomycin Aglycon.
The total synthesis started with preparation of cyclic tetrapeptide 88 as shown in Scheme 29. TBS protection of the phenol in 88 afforded 245 (100%), preventing competitive reaction with a thionation reagent. Subsequent thionation with Lawesson’s reagent selectively generated the thioamide 246 in excellent yield (92%). Although the thioamide could be subjected to Suzuki coupling with the Ring A boronic acid 79, the reaction was not dependably reproducible. Even after careful removal of sulfur byproducts from the thionation reaction, which were thought to be poisoning the reaction, the coupling reaction remained capricious. It was suspected that the residue 4 thioamide itself might be affecting the coupling reaction. Thus, the residue 4 thioamide was masked as its less nucleophilic methyl thioimidate 247 by treatment of 246 with MeI/K2CO3 (92%). The thioimidate proved to be an effective and reproducible coupling partner for the Suzuki reaction with 79 and afforded the corresponding biaryl product as a separable mixture of atropisomers (1:1.2) in good yield (65–80%), displaying a reactivity similar to the corresponding amide substrate. Reintroduction of the thioamide upon treatment of the methyl thioimidate with H2S in the presence of collidine provided the thioamide (P)-249 and its separable unnatural isomer (M)-249 in good yield (95%). Recycling the unnatural atropisomer (M)-249 was carried out by thermal equilibration, affording a 2.6–1.8:1 (P:M) separable mixture of atropisomers with 83–99% overall recovery. Completion of the synthesis of the modified ABCD ring system required alteration of the order of deprotections. Removal of Cbz group under ionic hydrogenolysis conditions with Pd(OAc)2–Et3SiH (97%)181,182 to avoid competitive thioamide reduction was carried out prior to TBS deprotection (95%). Hydrolysis of the methyl ester (95%), followed by macrolactamization of the AB ring system with DEPBT afforded thioamide 253 in good yield (71%, dr 7:1). Finally, Boc deprotection by treatment with HCO2H completed the synthesis of the modified ABCD ring system 254 in good yield (98%).
Scheme 29.

Synthesis of the ABCD Ring System Bearing a Residue 4 Thioamide.
Introduction of the E ring tripeptide was effected by coupling 254 with 96 mediated by DEPBT or T3P to afford the product (56%) as shown in Scheme 30. The final DE diaryl ether macrocyclization was achieved upon treatment with Cs2CO3, promoting a room temperature SNAr cyclization to produce 255 in superb yield (74–78%) and good atropodiastereoselectivity (5–8:1), favoring the natural P-atropisomer. The minor unnatural isomer was recycled and funneled into the total synthesis by selective thermal atropisomer equilibration, generating additional amounts of the natural isomer as a separable mixture (1:1). Subsequent conversion of the E ring aryl nitro group to the chloride and the final global deprotection was further refined. In addition to adopting the Zn nanoparticle conditions for the nitro group reduction, a decrease in amounts of the diazotization reagent (1.0 vs 1.1 equiv of HBF4/t-BuONO) in the chloride substitution process minimized competitive oxidative formation of the residue 4 amide. TBS protection of both secondary alcohols with CF3CONMeTBS afforded 257 (50–59%, over 3 steps). Subsequent MEM deprotection upon treatment with BCB followed by Boc reprotection of the N-terminus amine provided the primary alcohol 258 in good yield (75%). Complicated by the presence of the oxidation prone thioamide, the C-terminus oxidation with Dess–Martin periodinane, Swern oxidation, SO3–Pyr, Bu4NRuO4 (TPAP), pyridinium dichromate (PDC),183 pyridinium chlorochromate (PCC),184,185 or 2-iodoxybenzoic acid (IBX) resulted in little formation of the desired carboxylic acid or resulted in a complex mixture. Remarkably, it was found that Jones oxidation (CrO3, aq. H2SO4–acetone)186,187 selectively oxidized the primary alcohol directly to the carboxylic acid (70%) in single step while generating the amide only as a minor byproduct (>10:1). It was additionally demonstrated that formation of the amide could be further minimized by conversion of an unusually stable contaminant thioamide S-oxide (<10%) back to the thioamide in the subsequent global deprotection steps. After Jones oxidation, a purified mixture of the thioamide and the minor S-oxide (<10%) was treated with TFA to convert the nitrile to the primary carboxamide,188 followed by global deprotection using AlBr3–EtSH to afford the [Φ[C(=S)NH]Tpg4]vancomycin aglycon (244, 44–52%, over 3 steps). Remarkably, the minor thioamide S-oxide tolerated the mild TFA-mediated hydration step without change, and was converted back to the thioamide in the global deprotection step. Finally, the residue 4 thioamide was transformed directly to the amidine 243 (50%) through use of a newly developed method (AgOAc–NH3, MeOH) for the direct conversion. This reaction could be conducted on the fully deprotected and fully functionalized aglycon 244, bearing both the C-terminus carboxylic acid and N-terminus free amine, in protic solvents where such substrates are soluble.
Scheme 30.

Completion of the Boger Total Synthesis of [Φ[C(=S)NH]Tpg4]Vancomycin Aglycon and [Φ[C(=NH)NH]Tpg4]Vancomycin Aglycon.
Full details of the development of this reaction and its use in the divergent synthesis of substituted amidine derivatives (=NMe and =NCN vs =NH) that clarified the protonation state of the amidine upon binding D-Ala-D-Ala and D-Ala-D-Lac were disclosed in a separate account.180
The three residue 4 modified vancomycin aglycons were examined for their ability to bind model D-Ala-D-Ala and D-Ala-D-Lac ligands and for their in vitro antimicrobial activity against vancomycin resistant enterococci (VanA VRE, Figure 16).
Figure 16.
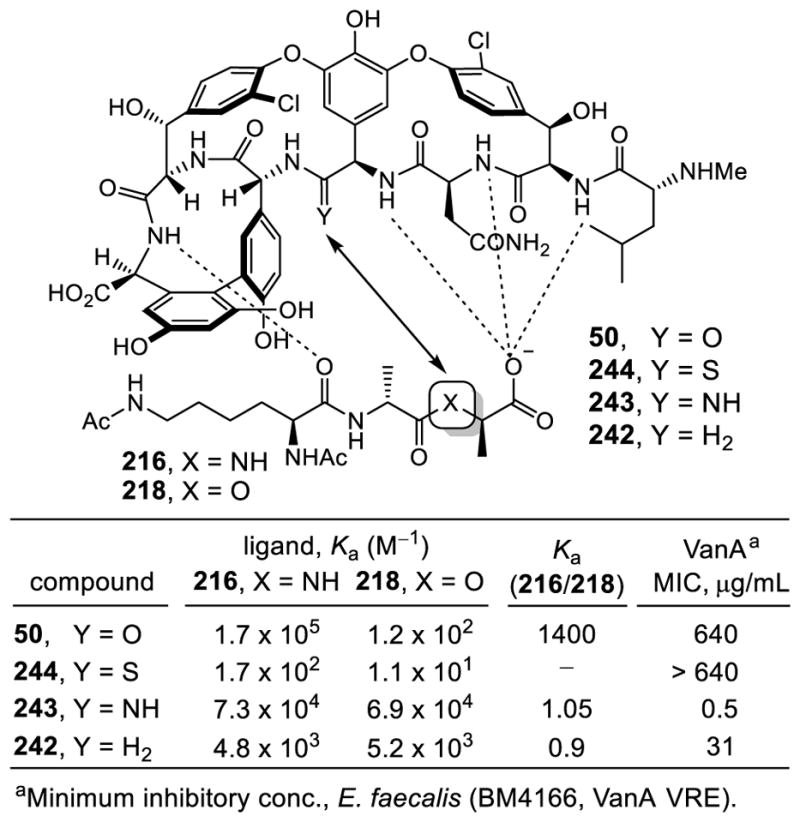
Biological Evaluation of Residue 4 Modified Vancomycin Aglycons.
As designed, both 243 and 242 were found to exhibit dual ligand binding properties, each displaying equal affinities for D-Ala-D-Ala or D-Ala-D-Lac. The amidine exhibited the greater affinity for both ligands, establishing its productive behavior even though its success was not obvious at the onset of the studies. Moreover, the amidine 243 bound both ligands with an affinity nearly equivalent to that found for vancomycin and its binding to D-Ala-D-Ala, effectively mimicking this latter behavior. However, the amidine 243 also binds the D-Ala-D-Lac ligand 600-fold better than vancomycin. Beautifully, both 243 and 242 exhibited activity against VanA VRE, displaying potencies that perfectly mirror their relative binding affinities. Most notable, amidine 243 was found to be equipotent to vancomycin but also now displays this activity against vancomycin resistant organisms. Just as remarkable and initially surprising, the thioamide 244 failed to bind either D-Ala-D-Ala or D-Ala-D-Lac and displayed no antimicrobial activity against either vancomycin sensitive or resistant bacteria. This was attributed to the increased size of a sulfur atom and the longer thiocarbonyl bond length that serve to displace the ligand from the binding pocket. This behavior, which contrasts that of both 243 and 242, highlight just how remarkable the successful behaviors of 243 and 242 are. It also provides an attractive target for preparation by coersed biosynthesis, providing a vancomycin analogue capable of semisynthetic conversion to the active pocket modified vancomycins that will not impede the growth of producing organisms.
7.3 [Φ[C(=S)NH]Tpg4]Vancomycin, [Φ [C(=NH)NH]Tpg4]Vancomycin, and [Φ [CH2NH]Tpg4]Vancomycin and Their (4-Chlorobiphenyl)methyl Derivatives
With the stunning results of the pocket modified vancomycin aglycon analogues in hand, Boger initiated efforts to install the disaccharide in preparation for their more comprehensive examination.189 The results of these studies, enlisting two enzyme-catalyzed glycosylation reactions on the fully deprotected aglycon and culminating in the completion of a total synthesis of vancomycin,128 are summarized in part in Section 4.2.2. The use of this methodology for the total synthesis of three pocket modified analogues, each bearing a single atom change in the structure of vancomycin, is summarized below. With the residue 4 modified vancomycins in hand, Boger additionally pursued the development of analogues that would display even greater potency. The approach focused on the combined use of a binding pocket modification and an added peripheral modification. One of the most widely recognized peripheral modifications of glycopeptide antibiotics is the hydrophobic substitution of the vancosamine free amine first discovered at Eli Lilly and inspired by the lipophilic acyl groups found in teicoplanin.190–192 To date, three semi-synthetic analogues; oritavancin (259, 2014, The Medicines Company),193 telavancin (260, 2009, Theravance),194,195 and dalbavancin (261, 2014, Vicuron Pharmaceuticals Inc.)196–198 that bear such substitution have been approved for clinical use (Figure 17). Importantly, the hydrophobic substituent enhances the antibiotic potency against both vancomycin sensitive and resistant pathogens.199–203 Boger examined the (4-chlorobiphenyl)methyl (CBP) group substitution of vancosamine on the residue 4 modified vancomycin derivatives (Figure 18).189,204 The synthetic approach relied on a 3- or 4-step route from the fully functionalized and protecting group free pocket modified vancomycin aglycons. This sequence involved two sequential enzymatic glycosylations, primary amine N-alkylation via a reductive amination, and, in the case of the amidine, AgOAc–NH3 promoted amidine formation from the residue 4 thioamide. By following the glycosylation procedure established for vancomycin (Section 4.2.2.2), the first GtfE catalyzed glycosylation of the residue 4 modified aglycons was examined by reverse phase HPLC, following product formation (Figure 18).
Figure 17.

Clinically Approved Semisynthetic Glycopeptide Antibiotics.
Figure 18.

Comparison of the Relative Rates and Efficiency of the GtfE-catalyzed Glycosylation Reaction of 50, and 242–244.
The thioamide (244) and aminomethylene vancomycin aglycons (242) displayed sufficient reactivity under the glycosylation conditions to preparatively provide the pseudoaglycons 263 (75%, HPLC conversion 86–92%), 264 (72%, HPLC conversion 85–95%), with only slightly reduced isolated yields compared to the parent vancomycin aglycon (135, 92%, HPLC conversion 100%). In contrast, the amidine substituted vancomycin aglycon (243) was not successfully glycosylated and only a trace amount of product 262 was detected.
The GtfD catalyzed second glycosylation of the pseudoaglycons with UDP-vancosamine was rapid and complete within 2 h to afford the disaccharides 265 (87%, HPLC conversion >95%) and 267 (76%, HPLC conversion >95%) in good yields (Scheme 31). Finally, the residue 4 thioamide was converted to 266 via AgOAc-promoted amidine formation (10 equiv of AgOAc, NH3/MeOH, 25 °C, 6 h) without competitive deglycosylation.
Scheme 31.

Synthesis of Residue 4 Modified Vancomycins and their CBP Derivatives.
Subsequent introduction of the (4-chlorobiphenyl)methyl group via reductive amination, using reported reaction conditions for the synthesis of CBP-vancomycin (268, 61–74%) disclosed by Kahne205,206 (1.5 equiv of 4-(4-chlorophenyl)benzaldehyde, 5 equiv of i-Pr2NEt, DMF, 50–70 °C; 100 equiv of NaCNBH3, 70 °C, 5 h), provided the chlorobiphenyl derivatives 269 (57%) and 271 (41%). The reductive amination was conducted without competing alkylation of the N-terminus or residue 4 secondary amine. Finally, conversion of the residue 4 thioamide to the amidine was carried out with use of the AgOAc–NH3 promoted amidine formation (10 equiv of AgOAc, NH3/MeOH, 25 °C, 6 h) to afford 270 in 45% yield.
A summary of the in vitro antimicrobial activity of the pocket modified vancomycin analogues is presented in Figure 19. As expected, introduction of the disaccharide on the residue 4 modified vancomycin aglycons did not alter their antimicrobial potency. Like the aglycons, sequential improvements in activity against vancomycin resistant bacteria (VanA VRE) were observed with 267 and 266 that mirrored their dual ligand binding affinities and where the activity of amidine 266 matched the potency of vancomycin observed against sensitive bacteria. The CBP introduction on vancomycin enhanced the potency against both vancomycin sensitive and resistant strains (VSSA and MRSA) by 20-fold and 100-fold, respectively, but 268 still remains nearly 100-fold less active against vancomycin resistant bacteria. Both CBP-amidine 270 and CBP-aminomethylene 271 displayed 100-fold increases in antibiotic potency against VRE (VanA) and exhibited this same level of potency against vancomycin sensitive bacteria (e.g. VSSA and MRSA), reinstating potent antimicrobial activity against vancomycin resistant bacteria. Importantly, introduction of the CBP substituent on the thioamide vancomycin, lacking the ability to bind D-Ala-D-Ala/D-Ala-D-Lac, provided an analogue 269 that displayed less active, but equally potent activity against vancomycin sensitive and resistant strains. This reflects antibacterial activity derived from a second mechanism of action independent of D-Ala-D-Ala/D-Ala-D-Lac binding, one that is responsible for the improved activity of all CBP analogues in Figure 19. This second mechanism of action entails direct inhibition of transglycosylase also resulting in inhibition of bacterial cell wall biosynthesis.199–203
Figure 19.

In vitro Antimicrobial Activity.
7.4 Development of Durable Antibiotics by Design
In a remarkable extension of these latter studies, Boger incorporated a second peripheral modification into the pocket modified vancomycin analogues that provided a different second mechanism of action independent of D-Ala-D-Ala/D-Ala-D-Lac binding and demonstrated that the antimicrobial activity similarly increased 200-fold against VanA VRE.207 Further, this C-terminus modification could be combined with the peripheral CBP modification to provide even more potent antimicrobial agents whose activity can be attributed to three independent mechanisms of action, only one of which requires D-Ala-D-Ala/D-Ala-D-Lac binding. Importantly, such peripherally and binding pocket modified vancomycins display little propensity for acquired resistance and they display a durability as well as increased potency that follow now predictable trends: 3 > 2 > 1 mechanisms of action.
In these studies, an additional peripheral modification on the C-terminus was explored, introducing an amide bearing a permanent cationic charge.207 A variety of commercially available quaternary ammonium salts are used as antimicrobial agents and many cationic naturally occurring antimicrobial peptides are known,208–210 which serve to disrupt the cell membrane integrity and increase cell wall permeability.
Recently, Haldar disclosed that such functionalization of vancomycin provided compounds active against vancomycin resistant bacteria,211,212 albeit without inhibiting the bacteria cell wall biosynthesis and displaying an independent mode of action. Thus, Boger envisioned modifications first focused on combining the binding pocket modification and C-terminus quaternary ammonium salt functionalization. This study was conducted with [Φ[CH2NH]Tpg4]vancomycin (267), the most readily available pocket modified vancomycin analogue available through total synthesis (Figure 20).173 It also represents the least potent of the two pocket modified analogues, permitting the most accurate assessment of the impact of sequential peripheral modifications. Several amines bearing a variety of quaternary ammonium salts were attached to the C-terminus carboxylic acid of vancomycin under modified reaction conditions with HBTU and NMM to afford the coupling products 272–276 in good yield (58–68%). Subsequent antimicrobial assessments indicated that the tetradecyl substituted ammonium salt was the most potent derivative, enhancing the antibacterial activity 125-fold against VanA VRE. Thus, the tetradecyl ammonium salt was introduced to the aminomethylene vancomycin 267 under the same coupling conditions to afford 276 in 64% yield. This peripherally modified pocket analogue 276 displayed more potent antimicrobial activity, further increasing the activity 200-fold, and it inhibited VanA VRE bacterial cell growth by two independent mechanisms of action, only one of which relied on dual D-Ala-D-Ala/D-Ala-D-Lac binding.
Figure 20.

Peripheral C-terminus Modifications of Vancomycin and the Binding Pocket Modified Vancomycin Analogue 267.
Next, the combination of the C-terminus quaternary ammonium salt modification with the vancomycin analogue that contained both a binding pocket modification and the peripheral CBP substituent was examined (Figure 21). Coupling CBP-vancomycin 268 with the same set of functionalized amines generated the products 277–281 in good yield (55–76%). Interestingly, the introduction a C14 ammonium salt did not alter the antibiotic activity against vancomycin resistant bacteria, but the simpler trimethyl ammonium salt proved enhance the potency against VRE 10-fold. When combined with CBP-[Φ[CH2NH]Tpg4]vancomycin, this C-terminus peripheral modification similarly improved the MIC values to the remarkable levels of 0.01–0.005 μg/mL. This molecule was shown to exert its impressive potency through three independent mechanisms of action, only one of which is dependent upon the dual D-Ala-D-Ala/D-Ala-D-Lac binding. The pocket modification reinstates inhibition of transpeptidase via D-Ala-D-Ala/D-Ala-D-Lac binding, the CBP modification results in direct inhibition of transglycosylase, and the C-terminus trimethyl ammonium salt induces cell wall permeability. Even more importantly, it was demonstrated that such peripherally and binding pocket modified vancomycins display little propensity for acquired resistance by VRE and that their durability against such challenges as well as their antimicrobial potency follow now predictable trends (3 > 2 > 1 mechanisms of action).
Figure 21.

Combined CBP and C-terminus Peripheral Modifications of Vancomycin and the Binding Pocket Modified Vancomycin Analogue 267.
8. Total Synthesis of Chloropeptins and Complestatins
Complestatin (282, chloropeptin II) was first disclosed as an inhibitor of an alternate pathway of human complement in 1980 (Figure 22).213–215 Its original isolation216 from Streptomyces lavendulae and partial structure elucidation217 were reported in 1989. Four years later, Omura reported the isolation218–220 of chloropeptin I (283) and chloropeptin II (282, completstatin) from Streptomyces sp. along with details of their structure elucidation221 and disclosed their biological activity against HIV infectivity and their cytopathic effect.222 Because they inhibit HIV through two independent mechanisms of action, it is expected that resistance would develop only slowly. Singh and Patane later demonstrated that complestatin (chloropeptin II) (282) can be converted to the less strained chloropeptin I (283) through a remarkable TFA-catalyzed rearrangement that proceeds with retention of the atropisomer stereochemistry.223 Singh also isolated and characterized isocomplestatin, bearing the unnatural (S)-indole tryptophan-derived biaryl ether linkage.222 Later in 2006, Hoveyda reported that the compound thought to be isocomplestatin was in fact complestatin (282).224 The related complestatin A (284) and B (285), bearing a 2-oxindole or a 3-hydroxy-2-oxiindole, were isolated by Merck from Streptomyces sp. MA7234.222 At the same time, these compounds were independently disclosed as neuroprotection A and B and were isolated from Streptomyces sp. Q27107.225,226 Although they are not strictly members of the glycopeptide antibiotics, the natural products in this group contain structural features that place them in discussions of the class.
Figure 22.

Structure of Chloropeptins and Complestatins (Neuroprotectins).
Although the complestatins and chloropeptins contain many of the same structural features and the overall complexity of glycopeptide antibiotics, the significant difference is the replacement of a central diaryl ether linkage with a tryptophan indole embedded in the macrocyclic core as part of a biaryl linkage that adopts a single (R)-atropisomer configuration incapable of thermal interconversion.
8.1. Snapper–Hoveyda Total Synthesis of Chloropeptin I
Snapper and Hoveyda reported the first total synthesis of chloropeptin I (283), the least strained of the two chloropeptins.227 Their synthetic strategy enlisted a late stage intramolecular Stille coupling for biaryl formation on a substrate that contains the left-hand macrocycle and exclusively afforded the natural (R)-atropisomer (38–42%, 3 steps) (Figure 23). They also examined the extension of this strategy for the preparation of complestatin (282, chloropeptin II). It was found that the Suzuki coupling reaction exclusively provided isocomplestatin, bearing the unnatural biaryl (S)-atropisomer in 63% yield, whereas closure of the isolated ring system proceeded in a non-atropodiastereoselective manner (52%, 1:1 R:S).224 The atropodiastereoselectivity of these macrocyclization reactions was also later confirmed by Zhu.228 The first ring closure was carried out with use of a Cu(II)-mediated coupling reaction of a phenol with an aryl boronic acid for diaryl ether formation analogous to the reaction employed in the total synthesis of teicoplanin disclosed by Evans.151
Figure 23.

Snapper and Hoveyda Retrosynthetic Analysis of Chloropeptin I.
8.2 Boger Total Synthesis of Chloropeptin I and II, and Complestatins A and B
By virtue of single step acid-catalyzed conversion of chloropeptin II (282) to chloropeptin I (283), Boger envisioned a total synthesis of both natural products by first preparing the more strained chloropeptin II and conducting its subsequent single step acid catalyzed rearrangement to chloropeptin I (283).229–231 The key elements of their synthetic strategy were an intramolecular SNAr cyclization and Larock indole annulation for the ring closure of the BCD and DEF ring systems, respectively. The latter intramolecular Larock indole synthesis232,233 utilized a 2-bromoaniline234 and its reaction with an alkyne that incorporates a removable large terminal substituent (−SiEt3) to sterically dictate the indole cyclization regioselectivity.235,236 Importantly, the aniline acetamide not only served to deactivate the strained indole toward subsequent electrophilic reactions, but also favorably influenced the cyclization atropodiastereoselectivity.237 They pursued two synthetic approaches, which are distinct in the order and timing of the key ring closures. In the first generation total synthesis, the DEF ring system macrocyclization was first conducted via the Larock annulation to afford the functionalized right hand subunit in superb isolated yield (89%) and in good atropodiastereoselectivity (4:1). This represented the first reported example of an intramolecular Larock macrocyclization strategy (Figure 24). Later studies further improved on this atropodiastereoselectivity (20:1) by subtle alteration of the aniline acyl group (eq. 1).229–231,237 The second macrocyclization via SNAr diaryl ether formation with closure of the BCD ring system afforded the cyclized compound in good yield (81%) and in superb atropodiastereoselectivity (>10:1). The first generation approach provided the first total synthesis of complestatin (chloropeptin II) and completion of an approach in which both chloropeptin I and II were prepared in a common route.
Figure 24.

Boger First Generation Total Synthesis of Complestatin (Chloropeptin II) and Chloropeptin I.
 |
(1) |
The second generation synthesis was conducted by reversing the order of the macrocyclization reactions (Figure 25). Notably, the late stage Larock annulation provided the macrocycle exclusively as a single atropisomer (>20:1), possessing the natural (R)-configuration. Thus, the reverse macrocyclization order increased the Larock indole annulation atropodiastereoselectivity, and further improved on their first generation approach. Most notable, it uniquely gives the (R)-atropisomer in high atropodiastereoselectivity, and represents a complete reversal of the atropodiastereoselectivity observed by Hoveyda224 and Zhu228 with the similar macrocyclization performed using a Suzuki–Miyaura coupling.
Figure 25.

Boger Second Generation Total Synthesis of Complestatin (Chloropeptin II) and Chloropeptin I.
With the total synthesis of the chloropeptins completed, Boger also achieved the first divergent total syntheses of complestatin A (284) and B (285) (neuroprotectin A and B)229–231 (Scheme 32). Mild reaction conditions were developed for two unique indole oxidations that take advantage of the strain in 16-membered macrocycle. The reactions selectively provide either a 2-oxindole or 3-hydroxy-2-oxindole by late stage treatment of 282 with concentrated aqueous HCl in DMSO238,239 or stoichiometric NBS or NCS in aqueous THF,240–246 respectively.
Scheme 32.

Boger Synthesis of Complestatin A and B (Neuroprotectin A and B).
9. Conclusions
A series of total syntheses of members of the vancomycin related glycopeptide antibiotics, their aglycons, and key analogues have been completed as summarized herein. Key to their success was the development or advancement of new synthetic methodology that was enlisted for the preparation of the unnatural amino acid subunits. Powerful macrocyclization methodology was developed or adopted for closure of the imbedded biaryl or diaryl ether cross-linked 12-membered, 14-membered, and 16-membered ring systems, using kinetic, thermodynamic, or ordered adjustments for introduction of the non-conventional elements of axial or planar chirality (atropisomers) found in the individual ring systems. Finally, chemical or enzymatic methods were developed for the carbohydrate introductions. Beyond the total synthesis of the natural products themselves, this has provided a foundation on which previously inaccessible but important synthetic analogues that contain deep-seated modifications may be prepared. Already such efforts have not only provided key, rationally designed analogues of vancomycin that address the underlying molecular basis of vancomycin resistance, but they have even provided further modified derivatives that display remarkable antimicrobial potencies, multiple independent mechanisms of action, and antimicrobial durabilities that extend well beyond those of the natural products.
Finally, the studies detailed herein complement those conducted on other glycopeptide antibiotic classes including ramoplanin,247–252 the total synthesis of other classes of naturally occurring antibiotics253,254 and reviews that chronicle the role of organic synthesis in the exploration and development of antibiotics.255
Figure 5.

Nicolaou Retrosynthetic Analysis of Vancomycin Aglycon.
Acknowledgments
The authors gratefully acknowledge the financial support of the National Institutes of Health (CA041101, DLB) and the award of a National Institutes of Health postdoctoral fellowship (F32 GM114948, NAI).
Abbreviations
- BCB
B-bromocatecholborane
- Boc
tert-butyloxycarbonyl
- Bop
(1-benzotriazolyloxy)tris(dimethylamino) phosphonium hexafluoride
- BSA
bovine serum albumin
- dba
dibenzylideneacetone
- DEAD
diethyl azodicarboxylate
- DEPBT
(3-(diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one)
- Ddm
4,4′-dimethoxydiphenylmethyl
- DPPA
diphenyl phosphorazidate
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- DTBMP
2,6-di-tert-butyl-4-methylpyridine
- EDCI
1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide
- FDPP
pentafluorophenyl diphenylphosphinate
- HATU
2-(1H-7-azabenzotriazol)-1-yl-1,1,3,3-tetramethyluronium hexafluorophosphate
- HBTU
N,N,N′,N′-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate
- HOAt
1-hydroxy-7-azabenzotriazole
- HOBt
1-hydroxybenzotriazole
- MEM
2-methoxyethoxymethyl ether
- NIS
N-iodosuccinimide
- NMM
N-methylmorpholine
- NMP
N-methyl-2-pyrrolidone
- Piv
pivalate
- PMP
p-methoxybenzyl
- PyBop
benztriazol-1-yl-oxy-tris(pyrrolidino)phosphonium hexafluorophosphate
- PyBrop
bromo-tris(pyrrolidino)phosphonium hexafluorophosphate
- MRSA
methicillin-resistant Staphylococcus aureus
- Ms
methanesulfonyl
- T3P
propylphosphonic anhydride
- TBS
tert-butyldimethylsilyl
- TCEP
tris(2-carboxyethyl)phosphine
- Teoc
2-trimethylsilylethoxycarbonyl
- Tf
trifluoromethylsulfonyl
- TFA
trifluoroacetic acid
- TFAA
trifluoroacetic anhydride
- TFFH
fluoro-N,N,N′,N′-tetramethylformamidinium hexafluorophosphate
- TMS
trimethylsilyl
- TMSE
2-(trimethylsilyl)ethyl
- Troc
2,2,2-trichloroethyl carbonyl
- VRE
vancomycin resistant enterococcus
- VRSA
vancomycin resistant Staphylococcus aureus
Biographies
Dale L. Boger is the Richard & Alice Cramer Chair in Chemistry at The Scripps Research Institute (1990–present) where he serves as Chairman (2012–present) of the Department of Chemistry. He received his B.S. in Chemistry from the University of Kansas (1975) and his Ph.D. in Chemistry from Harvard University (1980). He was on the faculty at the University of Kansas and Purdue University before joining TRSI in 1990.
Akinori Okano was born in 1981 in Hyogo, Japan, and received his B.S. degree in Pharmaceutical Sciences from Osaka University of Pharmaceutical Sciences in 2005. He moved to Osaka University and obtained his M.S. degree in Pharmaceutical Sciences from Osaka University under the supervision of Professor Hiroaki Ohno and Professor Tetsuaki Tanaka in 2007. He moved to Kyoto University to continue his Ph.D. research under the supervision of Professor Hiroaki Ohno and Professor Nobutaka Fujii (2009–2010; as a Japan Society for the Promotion of Science [JSPS] Research Fellow). After completion of his Ph.D. in 2010, he joined Professor Boger’s group at The Scripps Research Institute as a post-doctoral research associate (2010–2011; as a JSPS Research Fellow). In 2013, he was appointed to research Assistant Professor in the Boger group.
Nicholas A. Isley received his B.S. from Western Washington University (2010). After being exposed to organometallic research as an undergraduate, he decided to obtain a Ph.D. at the University of California Santa Barbara (2015). Working with Bruce H. Lipshutz, he developed organic and organometallic reactions in water via micellar catalysis with benign “designer” surfactants. In 2015, he moved to San Diego for postdoctoral studies with Professor Boger at The Scripps Research Institute.
Footnotes
The authors declare no competing financial interest.
References
- 1.Malabarba A, Nicas TI, Thompson RC. Structural modifications of glycopeptide antibiotics. Med Res Rev. 1997;17:69–137. doi: 10.1002/(sici)1098-1128(199701)17:1<69::aid-med3>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 2.Ashford PA, Bew SP. Recent advances in the synthesis of new glycopeptide antibiotics. Chem Soc Rev. 2012;41:957–978. doi: 10.1039/c1cs15125h. [DOI] [PubMed] [Google Scholar]
- 3.McCormick MH, Stark WM, Pittenger GE, Pittenger RC, McGuire JM. Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot Annu. 1955–1956:606–611. [PubMed] [Google Scholar]
- 4.Cooper GL, Given DB. Vancomycin, A Comprehensive Review of 30 Years of Clinical Experience; Park Row Publications; Indianapolis, IN: 1986. The development of vancomycin; pp. 1–5. [Google Scholar]
- 5.Walsh CT, Fischbach MA. New ways to squash superbugs. Sci Am. 2009;301:44–51. doi: 10.1038/scientificamerican0709-44. [DOI] [PubMed] [Google Scholar]
- 6.Laxminarayan R, Duse A, Wattal C, Zaidi AKM, Wertheim HFL, Sumpradit N, Vlieghe E, Hara GL, Gould IM, Goossens H, et al. Antibiotic resistance – the need for global solutions. Lancet Infect Dis. 2013;13:1057–1098. doi: 10.1016/S1473-3099(13)70318-9. [DOI] [PubMed] [Google Scholar]
- 7. [accessed Nov 30 2016];National Strategy for Combating Antibiotic Resistant Bacteria. 2014 Sep; http://www.cdc.gov/drugresistance/pdf/carb_national_strategy.pdf.
- 8.Nagarajan R, editor. Glycopeptide Antibiotics. Marcel Dekker Inc; New York: 1994. [Google Scholar]
- 9.James RC, Pierce JG, Okano A, Xie J, Boger DL. Redesign of glycopeptide antibiotics: back to the future. ACS Chem Biol. 2012;7:797–804. doi: 10.1021/cb300007j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leclercq R, Derlot E, Duval J, Courvalin P. Plasmid-mediated resistance to vancomycin and teicoplanin in Enterococcus faecium. N Engl J Med. 1988;319:157–161. doi: 10.1056/NEJM198807213190307. [DOI] [PubMed] [Google Scholar]
- 11.Weigel LM, Clewell DB, Gill SR, Clark NC, McDougal LK, Flannagan SE, Kolonay JF, Shetty J, Killgore GE, Tenover FC. Genetic analysis of a high-level vancomycin-resistant isolate of Staphylococcus aureus. Science. 2003;302:1569–1571. doi: 10.1126/science.1090956. [DOI] [PubMed] [Google Scholar]
- 12.Howden BP, Davies JK, Johnson PD, Stinear TP, Grayson ML. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev. 2010;23:99–139. doi: 10.1128/CMR.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Walsh TR, Howe RA. The prevalence and mechanisms of vancomycin resistance in Staphylococcus aureus. Ann Rev Microbiol. 2002;56:657–675. doi: 10.1146/annurev.micro.56.012302.160806. [DOI] [PubMed] [Google Scholar]
- 14.Perichon B, Courvalin P. VanA-type vancomycin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2009;53:4580–4587. doi: 10.1128/AAC.00346-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woodford N, Johnson AP, Morrison D, Speller DCE. Current perspectives on glycopeptide resistance. Clin Microbiol Rev. 1995;8:585–615. doi: 10.1128/cmr.8.4.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murray BE. Vancomycin-resistant enterococci. Am J Med. 1997;102:284–293. doi: 10.1016/S0002-9343(99)80270-8. [DOI] [PubMed] [Google Scholar]
- 17.Levy SB. The challenge of antibiotic resistance. Sci Am. 1998;3:46–53. doi: 10.1038/scientificamerican0398-46. [DOI] [PubMed] [Google Scholar]
- 18.Brown ED, Wright GD. Antibacterial drug discovery in the resistance era. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- 19.Levine D. Vancomycin: a history. Clin Infect Dis. 2006;42:5–12. doi: 10.1086/491709. [DOI] [PubMed] [Google Scholar]
- 20.Marshall FJ. Structural studies on vancomycin. J Med Chem. 1965;8:18–22. doi: 10.1021/jm00325a004. [DOI] [PubMed] [Google Scholar]
- 21.Smith KA, Williams DH, Smith GA. Structure studies on the antibiotic vancomycin; the nature of the aromatic rings. J Chem Soc Perkin Trans 1. 1974:2369–2376. [PubMed] [Google Scholar]
- 22.Smith GA, Smith KA, Williams DH. Structural studies on the antibiotic vancomycin: evidence for the presence of modified phenylgylcine and β-hydroxytyrosine units. J Chem Soc Perkin Trans 1. 1975:2108–2115. doi: 10.1039/p19750002108. [DOI] [PubMed] [Google Scholar]
- 23.Williams DH, Kalman JR. Structural and mode of action studies on the antibiotic vancomycin. Evidence from 270-MHz proton magnetic resonance. J Am Chem Soc. 1977;99:2768–2774. doi: 10.1021/ja00450a058. [DOI] [PubMed] [Google Scholar]
- 24.Sheldrick GM, Jones PG, Kennard O, Williams DH, Smith GA. Structure of vancomycin and its complex with acetyl-D-alanyl-D-alanine. Nature. 1978;271:223–225. doi: 10.1038/271223a0. [DOI] [PubMed] [Google Scholar]
- 25.Williamson MP, Williams DH. Structure revision of the antibiotic vancomycin. Use of nuclear Overhauser effect difference spectroscopy. J Am Chem Soc. 1981;103:6580–6585. [Google Scholar]
- 26.Harris CM, Harris TM. Structure of the glycopeptide antibiotic vancomycin. Evidence for an asparagine residue in the peptide. J Am Chem Soc. 1982;104:4293–4295. [Google Scholar]
- 27.Harris CM, Kopecka H, Harris TM. Vancomycin: structure and transformation to CDP-1. J Am Chem Soc. 1983;105:6915–6922. [Google Scholar]
- 28.Perkins HR. Vancomycin and related antibiotics. Pharmacol Ther. 1982;16:181–197. doi: 10.1016/0163-7258(82)90053-5. [DOI] [PubMed] [Google Scholar]
- 29.Williams DH. The glycopeptide story – how to kill the dealy ‘superbugs’. Nat Prod Rep. 1996;13:469–477. doi: 10.1039/np9961300469. [DOI] [PubMed] [Google Scholar]
- 30.Williams DH, Bardsley B. The vancomycin group of antibiotics and the fight against resistant bacteria. Angew Chem, Int Ed. 1999;38:1172–1193. doi: 10.1002/(SICI)1521-3773(19990503)38:9<1172::AID-ANIE1172>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 31.Nicolaou KC, Boddy CNC, Bräse S, Winssinger N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew Chem, Int Ed. 1999;38:2096–2152. doi: 10.1002/(sici)1521-3773(19990802)38:15<2096::aid-anie2096>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 32.Courvalin P. Resistance of enterococci to glycopeptides. Antimicrob Agents Chemother. 1990;34:2291–2296. doi: 10.1128/aac.34.12.2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Courvalin P. Vancomycin resistance in Gram-positive cocci. Clin Infect Dis. 2006;42:25–34. doi: 10.1086/491711. [DOI] [PubMed] [Google Scholar]
- 34.Walsh CT. Vancomycin resistance: decoding the molecular logic. Science. 1993;261:308–309. doi: 10.1126/science.8392747. [DOI] [PubMed] [Google Scholar]
- 35.Hubbard BK, Walsh CT. Vancomycin assembly: nature’s way. Angew Chem, Int Ed. 2003;42:730–735. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- 36.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 37.Binda E, Marinelli F, Marcone GL. Old and new glycopeptide antibiotics: action and resistance. Antibiotics. 2014;3:572–594. doi: 10.3390/antibiotics3040572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butler MS, Hansford KA, Blaskovich MAT, Halai R, Cooper MA. Glycopeptide antibiotics: back to the future. J Antibiot. 2014;67:631–644. doi: 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- 39.Boneca IG, Chiosis G. Vancomycin resistance: occurrence, mechanisms and strategies to combat it. Expert Opin Ther Targets. 2003;7:311–328. doi: 10.1517/14728222.7.3.311. [DOI] [PubMed] [Google Scholar]
- 40.Anderson JS, Matsuhashi M, Haskin MA, Strominger JL. Lipid-phosphoacetylmuramyl-pentapeptide and lipid-phosphodisaccharide-pentapeptide: presumed membrane transport intermediates in cell wall synthesis. Proc Natl Acad Sci US A. 1965;53:881–889. doi: 10.1073/pnas.53.4.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perkins HR. Specificity of combination between mucopeptide precursors and vancomycin or ristocetin. Biochem J. 1969;111:195–205. doi: 10.1042/bj1110195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nieto M, Perkins HR. Modifications of the acyl-D-alanyl-D-alanine terminus affecting complex-formation with vancomycin. Biochem J. 1971;123:789–803. doi: 10.1042/bj1230789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams DH, Williamson MP, Butcher DW, Hammond SJ. Detailed binding sites of the antibiotics vancomycin and ristocetin A: determination of intermolecular distances in antibiotic/substrate complexes by use of the time-dependent NOE. J Am Chem Soc. 1983;105:1332–1339. [Google Scholar]
- 44.Schäfer M, Schneider TR, Sheldrick GM. Crystal structure of vancomycin. Structure. 1996;4:1509–1515. doi: 10.1016/s0969-2126(96)00156-6. [DOI] [PubMed] [Google Scholar]
- 45.Bugg TD, Wright GD, Dutka-Malen S, Arthur M, Courvalin P, Walsh CT. Molecular basis for vancomycin resistance in Enterococcus faecium BM4147: biosynthesis of a depsipeptide peptidoglycan precursor by vancomycin resistance proteins VanH and VanA. Biochemistry. 1991;30:10408–10415. doi: 10.1021/bi00107a007. [DOI] [PubMed] [Google Scholar]
- 46.Marshall CG, Lessard IAD, Park IS, Wright GD. Glycopeptide antibiotic resistance genes in glycopeptide-producing organisms. Antimicrob Agents Chemother. 1998;42:2215–2220. doi: 10.1128/aac.42.9.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Healy VL, Lessard IA, Roper DI, Knox JR, Walsh CT. Vancomycin resistance in enterococci: reprogramming of the D-Ala-D-Ala ligases in bacterial peptidoglycan biosynthesis. Chem Biol. 2000;7:109–119. doi: 10.1016/s1074-5521(00)00116-2. [DOI] [PubMed] [Google Scholar]
- 48.Walsh CT, Fisher SL, Park IS, Prahalad M, Wu Z. Bacterial resistance to vancomycin: five genes and one missing hydrogen bond tell the story. Chem Biol. 1996;3:21–28. doi: 10.1016/s1074-5521(96)90079-4. [DOI] [PubMed] [Google Scholar]
- 49.Evans DA, Dinsmore CJ, Ratz AM, Evrar DA, Barrow JC. Synthesis and conformational properties of the M(4–6)(5–7) bicyclic tetrapeptide common to the vancomycin antibiotics. J Am Chem Soc. 1997;119:3417–3418. [Google Scholar]
- 50.Evans DA, Barrow JC, Watson PS, Ratz AM, Dinsmore CJ, Evrard DA, DeVries KM, Ellman JA, Rychovsky SD, Lacour J. Approaches to the synthesis of the vancomycin antibiotics. Synthesis of orienticin C (bis-dechlorovancomycin) aglycon. J Am Chem Soc. 1997;119:3419–3420. [Google Scholar]
- 51.Tsuji N, Kobayashi M, Kamigauchi T, Yoshimura Y, Terui Y. New glycopeptide antibiotics. I. The structures of orienticins. J Antibiot. 1988;41:819–822. doi: 10.7164/antibiotics.41.819. [DOI] [PubMed] [Google Scholar]
- 52.Evans DA, Ellman JA, DeVries KM. The oxidative macrocyclization of phenolic peptides. A biomimetic approach to the synthesis of the vancomycin family of antibiotics. J Am Chem Soc. 1989;111:8912–8914. [Google Scholar]
- 53.Evans DA, Dinsmore CJ, Evrard DA, DeVries KM. Oxidative coupling of arylglycine-containing peptides. A biomimetic approach to the synthesis of the macrocyclic actinoidinic-containing vancomycin subunit. J Am Chem Soc. 1993;115:6426–6427. [Google Scholar]
- 54.Evans DA, Dinsmore CJ. Kinetic and thermodynamic atropdiastereoselection in the synthesis of the M(5–7) tripeptide portion of vancomycin. Tetrahedron Lett. 1993;34:6029–6032. [Google Scholar]
- 55.Nishiyama S, Nakamura K, Suzuki Y, Yamamura S. Synthesis of piperazinomycin, a novel antifungal antibiotic. Tetrahedron Lett. 1986;27:4481–4484. [Google Scholar]
- 56.Suzuki Y, Nishiyama S, Yamamura S. Synthetic study vancomycin: synthesis of a macrocyclic tetrapeptide as a plausible active center in vancomycin. Tetrahedron Lett. 1989;30:6043–6046. [Google Scholar]
- 57.Suzuki Y, Nishiyama S, Yamamura S. Synthesis of a bicyclic hexapeptide as a plausible active center in vancomycin. Tetrahedron Lett. 1990;31:4053–4056. [Google Scholar]
- 58.Nakamura K, Nishiyama S, Yamamura S. Synthetic studies on vancomycin: synthesis of seco-aglucovancomycins. Tetrahedron Lett. 1995;36:8621–8624. [Google Scholar]
- 59.Evans DA, Watson PS. Synthesis of the orienticin C M(2–4) macrocycle utilizing a nucleophilic aromatic substitution strategy. Tetrahedron Lett. 1996;37:3251–3254. [Google Scholar]
- 60.Boger DL, Borzilleri RM, Nukui S. Synthesis of appropriately functionalized vancomycin CD and DE ring systems. Bioorg Med Chem Lett. 1995;5:3091–3096. [Google Scholar]
- 61.Evans DA, Weber AE. Synthesis of the cyclic hexapeptide echinocandin D. New approaches to the asymmetric synthesis of β-hydroxy α-amino acids. J Am Chem Soc. 1987;109:7151–7157. [Google Scholar]
- 62.Evans DA, Weber AE. Asymmetric glycine enolate aldol reactions: synthesis of cyclosporin’s unusual amino acid, MeBmt. J Am Chem Soc. 1986;108:6757–6761. [Google Scholar]
- 63.Evans DA, Britton TC, Ellman JA, Dorow RL. The asymmetric synthesis of α-amino acids. Electrophilic azidation of chiral imide enolates, a practical approach to the synthesis of (R)- and (S)-α-azido carboxylic acids. J Am Chem Soc. 1990;112:4011–4031. [Google Scholar]
- 64.Evans DA, Evrard DA, Rychnovsky SD, Fruh T, Whittingham WG, DeVries KM. A general approach to the asymmetric synthesis of vancomycin-related arylglycines by enolate azidation. Tetrahedron Lett. 1992;33:1189–1192. [Google Scholar]
- 65.Cacci S, Ciattini PG, Morera E, Ortar G. Palladium-catalyzed triethylammonium formate reduction of aryl triflates. A selective method for the deoxygenation of phenols. Tetrahedron Lett. 1986;27:5541–5544. [Google Scholar]
- 66.Shine HJ, Rhee ES. Heavy-atom kinetic isotope effects and mechanism of the acid-catalyzed o-semidine and p-semidine rearrangements and disproportionation of 4,4′-dichlorohydrazobenzene. J Am Chem Soc. 1986;108:1000–1006. [Google Scholar]
- 67.Bois-Choussy M, Beugelmans R, Bouillon JP, Zhu J. Synthesis of a modified carboxylate-binding pocket of vancomycin. Tetrahedron Lett. 1995;36:4781–4784. [Google Scholar]
- 68.Evans DA, Carter PH, Dinsmore CJ, Barrow JC, Katz JL, Kung DW. Mild nitrosation and hydrolysis of polyfunctional amides. Tetrahedron Lett. 1997;38:4535–4538. [Google Scholar]
- 69.Garcia J, Gonzales J, Segura R, Vilarrasa J. Nitrosation of peptide bonds. Cleavage of nitrosated peptides by pyrrolidine and α-amino esters. Tetrahedron. 1984;40:3121–3127. [Google Scholar]
- 70.Evans DA, Wood MR, Trotter BW, Richardson TI, Barrow JC, Katz JL. Total syntheses of vancomycin and eremomycin aglycons. Angew Chem, Int Ed. 1998;37:2700–2704. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2700::AID-ANIE2700>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 71.Evans DA, Dinsmore CJ, Watson PS, Wood MR, Richardson TI, Trotter BW, Katz JL. Nonconventional stereochemical issues in the design of the synthesis of the vancomycin antibiotics: challenges imposed by axial and nonplanar chiral elements in the heptapeptide aglycons. Angew Chem, Int Ed. 1998;37:2704–2708. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2704::AID-ANIE2704>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 72.Boger DL, Borzilleri RM, Nukui S, Beresis RT. Synthesis of the vancomycin CD and DE ring systems. J Org Chem. 1997;62:4721–4736. [Google Scholar]
- 73.Boger DL, Yohannes D. Total synthesis of deoxybouvardin and RA-VII: macrocyclization via an intramolecular Ullman reaction. J Am Chem Soc. 1991;113:1427–1429. [Google Scholar]
- 74.Boger DL, Yohannes D. Total synthesis of L,L-isodityrosine and isodityrosine-derived agents: K-13, OF4949-III, and OF4949-IV. J Org Chem. 1999;55:6000–6017. [Google Scholar]
- 75.Felix AM, Heimer EP, Lambros TJ, Tzougraki C, Meienhofer J. Rapid removal of protecting groups from peptides by catalytic transfer hydrogenation with 1,4-cyclohexadiene. J Org Chem. 1978;43:4194–4196. [Google Scholar]
- 76.Nicolaou KC, Natarajan S, Li H, Jain NF, Hughes R, Solomon ME, Ramanjulu JM, Boddy CNC, Takayanagi M. Total synthesis of vancomycin aglycon—part 1: synthesis of amino acids 4–7 and construction of the AB–COD ring skeleton. Angew Chem, Int Ed. 1998;37:2708–2714. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2708::AID-ANIE2708>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 77.Nicolaou KC, Jain NF, Natarajan S, Hughes R, Solomon ME, Li H, Ramanjulu JM, Takayanagi M, Koumbis AE, Bando T. Total synthesis of vancomycin aglycon—part 2: synthesis of amino acids 1–3 and construction of the AB–COD–DOE ring skeleton. Angew Chem, Int Ed. 1998;37:2714–2716. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2714::AID-ANIE2714>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 78.Nicolaou KC, Takayanagi M, Jain NF, Natarajan S, Koumbis AE, Bando T, Ramanjulu JM. Total synthesis of vancomycin aglycon—part 3: final stages. Angew Chem, Int Ed. 1998;37:2717–2719. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2717::AID-ANIE2717>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 79.Nicolaou KC, Boddy CNC, Natarajan S, Yue TY, Li H, Bräse JM, Ramanjulu JM. New synthetic technology for the synthesis of aryl ethers: construction of C–O–D and D–O–E ring model systems of vancomycin. J Am Chem Soc. 1997;119:3421–3422. [Google Scholar]
- 80.Kolb HC, Van Nieuwenhze MS, Sharpless KB. Catalytic asymmetric dihydroxylation. Chem Rev. 1994;94:2483–2547. [Google Scholar]
- 81.Tao B, Schlingloff G, Sharpless KB. Reversal of regioselection in the asymmetric aminohydroxylation of cinnamates. Tetrahedron Lett. 1998;39:2507. [Google Scholar]
- 82.Nicolaou KC, Li H, Boddy CNC, Ramanjulu JM, Yue TY, Natarajan S, Chu XJ, Bräse S, Rübsam F. Total synthesis of vancomycin—part 1: design and development of methodology. Chem Eur J. 1999;5:2584–2601. [Google Scholar]
- 83.Nicolaou KC, Boddy CNC, Li H, Koumbis AE, Hughes R, Natarajan S, Jain NF, Ramanjulu JM, Bräse S, Solomon ME. Total synthesis of vancomycin—part 2: retrosynthetic analysis, synthesis of amino acid building blocks and strategy evaluations. Chem Eur J. 1999;5:2602–2621. [Google Scholar]
- 84.Nicolaou KC, Koumbis AE, Takayanagi M, Natarajan S, Jain NF, Bando T, Li H, Hughes R. Total synthesis of vancomycin—part 3: synthesis of the aglycon. Chem Eur J. 1999;5:2622–2647. [Google Scholar]
- 85.Nicolaou KC, Ramanjulu JM, Natarajan S, Bräse S, Li H, Boddy CNC, Rübsam F. A Suzuki coupling–macrolactamization approach to the AB–COD bicyclic system of vancomycin. Chem Commun. 1997:1899–1900. [Google Scholar]
- 86.Chen S, Xu J. Pentafluorophenyl diphenylphosphinate a new efficient coupling reagent in peptide chemistry. Tetrahedron Lett. 1991;32:6711–6714. [Google Scholar]
- 87.Boger DL, Miyazaki S, Kim SH, Wu JH, Loiseleur O, Castle SL. Diastereoselective total synthesis of the vancomycin aglycon with ordered atropisomer equilibrations. J Am Chem Soc. 1999;121:3226–3227. [Google Scholar]
- 88.Dess DB, Martin JC. A useful 12-I-5 triacetoxyperiodinane (the Dess–Martin periodinane) for the selective oxidation of primary or secondary alcohols and a variety of related 12-I-5 species. J Am Chem Soc. 1991;113:7277–7287. [Google Scholar]
- 89.Abiko A, Roberts JC, Takemasa T, Masamune TS. KMnO4 revisited: oxidation of aldehydes to carboxylic acids in the tert-butyl alcohol–aqueous NaH2PO4 system. Tetrahedron Lett. 1986;27:4537–4540. [Google Scholar]
- 90.Boger DL, Miyazaki S, Kim SH, Wu JH, Castle SL, Loiseleur O, Jin Q. Total synthesis of the vancomycin aglycon. J Am Chem Soc. 1999;121:10004–10011. [Google Scholar]
- 91.Boger DL, Nomoto Y, Teegarden BR. Vancomycin and ristocetin models: synthesis via the Ullmann macrocyclization reaction. J Org Chem. 1993;58:1425–1433. [Google Scholar]
- 92.Boger DL, Borzilleri RM. An unusually facile SNAr 14-membered biaryl ether macrocyclization reaction suitable for preparation of the cycloisodityrosine subunit of bouvardin, deoxybouvardin and related agents. Bioorg Med Chem Lett. 1995;5:1187–1190. [Google Scholar]
- 93.Boger DL, Zhou J, Borzilleri RM, Nukui S. Synthesis of (9S,12S)-cycloisodityrosine and its unnatural (9R,12S)-diastereomer. Bioorg Med Chem Lett. 1996;6:1089–1092. [Google Scholar]
- 94.Boger DL, Zhou J, Borzilleri RM, Nukui S, Castle SL. Synthesis of (9R,12S)- and (9S,12S)-cycloisodityrosine and their N-methyl derivatives. J Org Chem. 1997;62:2054–2069. doi: 10.1021/jo961346o. [DOI] [PubMed] [Google Scholar]
- 95.Boger DL, Castle SL, Miyazaki S, Wu JH, Beresis RT, Loiseleur O. Vancomycin CD and DE macrocyclization and atropisomerism studies. J Org Chem. 1999;64:70–80. doi: 10.1021/jo980880o. [DOI] [PubMed] [Google Scholar]
- 96.Boger DL, Loiseleur O, Castle SL, Beresis RT, Wu JH. Thermal atropismerism of fully functionalized vancomycin CD, DE, and CDE ring systems. Bioorg Med Chem Lett. 1997;7:3199–3202. doi: 10.1016/s0960-894x(98)00110-3. [DOI] [PubMed] [Google Scholar]
- 97.Boger DL, Beresis RT, Loiseleur O, Wu JH, Castle SL. Synthesis of the vancomycin CDE ring system. Bioorg Med Chem Lett. 1998;8:721–724. doi: 10.1016/s0960-894x(98)00110-3. [DOI] [PubMed] [Google Scholar]
- 98.Boger DL, Miyazaki S, Loiseleur O, Beresis RT, Castle SL, Wu JH, Jin Q. Thermal atropisomerism of aglucovancomycin derivatives: preparation of (M,M,M)- and (P,M,M)-aglucovancomycins. J Am Chem Soc. 1998;120:8920–8926. [Google Scholar]
- 99.Boger DL, Borzilleri RM, Nukui S. Synthesis of (R)-(4-methoxy-3,5-dihydroxyphenyl)glycine derivatives: the central amino acid of vancomycin and related agents. J Org Chem. 1996;61:3561–3565. [Google Scholar]
- 100.Crowley BM, Mori Y, McComas CC, Tang D, Boger DL. Total synthesis of the ristocetin aglycon. J Am Chem Soc. 2004;126:4310–4317. doi: 10.1021/ja039795a. [DOI] [PubMed] [Google Scholar]
- 101.Hartwig JF, Paul F. Oxidative addition of aryl bromide after dissociation of phosphine from a two-coordinate palladium(0) complex, bis(tri-o-tolylphosphine)palladium(0) J Am Chem Soc. 1995;117:5373–5374. [Google Scholar]
- 102.Ge M, Thompson C, Kahne D. Reconstruction of vancomycin by chemical glycosylation of the pseudoaglycon. J Am Chem Soc. 1998;120:11014–11015. [Google Scholar]
- 103.Thompson C, Ge M, Kahne D. Synthesis of vancomycin from the aglycon. J Am Chem Soc. 1999;121:1237–1244. [Google Scholar]
- 104.Leimkuhler C, Chen Z, Kruger RG, Oberthur M, Lu W, Walsh CT, Kahne D. Glycosylation of glycopeptides: a comparison of chemoenzymatic and chemical methods. Tetrahedron: Asymmetry. 2005;16:599–603. [Google Scholar]
- 105.Ritter TK, Mong KKT, Liu T, Nakatani T, Wong CH. A programmable one-pot oligosaccharide synthesis for diversifying the sugar domains of natural products: a case study of vancomycin. Angew Chem, Int Ed. 2003;42:4657. doi: 10.1002/anie.200351534. [DOI] [PubMed] [Google Scholar]
- 106.Nicolaou KC, Mitchell HJ, Jain NF, Winssinger N, Hughes R, Bando T. Total synthesis of vancomycin. Angew Chem, Int Ed. 1999;38:240–244. [Google Scholar]
- 107.Nicolaou KC, Mitchell HJ, Jain NF, Bando T, Hughes R, Winssinger N, Natarajan S, Koumbis AE. Total synthesis of vancomycin–part 4: attachment of the sugar moieties and completion of the synthesis. Chem Eur J. 1999;5:2648–2667. [Google Scholar]
- 108.Losey HC, Peczuh MW, Chen Z, Eggert US, Dong SD, Pelczer I, Kahne D, Walsh CT. Tandem action of glycosyltransferases in the maturation of vancomycin and teicoplanin aglycones: novel glycopeptides. Biochemistry. 2001;40:4745–4755. doi: 10.1021/bi010050w. [DOI] [PubMed] [Google Scholar]
- 109.Losey HC, Jiang J, Biggins JB, Oberthur M, Ye X, Dong SD, Kahne D, Thorson JS, Walsh CT. Incorporation of glucose analogs by GtfE and GtfD from the vancomycin biosynthetic pathway to generate variant glycopeptides. Chem Biol. 2002;9:1305–1309. doi: 10.1016/s1074-5521(02)00270-3. [DOI] [PubMed] [Google Scholar]
- 110.Oberthur M, Leimkuhler C, Kruger RG, Lu W, Walsh CT, Kahne D. A systematic investigation of the synthetic utility of glycopeptide glycosyltransferases. J Am Chem Soc. 2005;127:10747–10752. doi: 10.1021/ja052945s. [DOI] [PubMed] [Google Scholar]
- 111.Thayer DA, Wong CH. Vancomycin analogues containing monosaccharides exhibit improved antibiotic activity: a combined one-pot enzymatic glycosylation and chemical diversification strategy. Chem Asian J. 2006;1:445–452. doi: 10.1002/asia.200600084. [DOI] [PubMed] [Google Scholar]
- 112.Thayer DA, Yu HN, Galan MC, Wong CH. A general strategy toward S-linked glycopeptides. Angew Chem Int Ed. 2005;44:4596–4599. doi: 10.1002/anie.200500090. [DOI] [PubMed] [Google Scholar]
- 113.Solenberg PJ, Matsushima P, Stack DR, Wilkie SC, Thompson RC, Baltz RH. Production of hybrid glycopeptide antibiotics in vitro and in Streptomyces toyocaensis. Chem Biol. 1997;4:195–202. doi: 10.1016/s1074-5521(97)90288-x. [DOI] [PubMed] [Google Scholar]
- 114.Fu X, Albermann C, Zhang CS, Thorson JS. Diversifying vancomycin via chemoenzymatic strategies. Org Lett. 2005;7:1513–1515. doi: 10.1021/ol0501626. [DOI] [PubMed] [Google Scholar]
- 115.Fu X, Albermann C, Jiang JQ, Liao JC, Zhang CS, Thorson JS. Antibiotic optimization via in vitro glycorandomization. Nat Biotechnol. 2003;21:1467–1469. doi: 10.1038/nbt909. [DOI] [PubMed] [Google Scholar]
- 116.Toshima K, Tatsuta K. Recent progress in O-glycosylation methods and its application to natural products synthesis. Chem Rev. 1993;93:1503–1531. [Google Scholar]
- 117.Kim SH, Augeri D, Yang D, Kahne D. Concise synthesis of the calicheamicin oligosaccharide using the sulfoxide glycosylation method. J Am Chem Soc. 1994;116:1766–1775. [Google Scholar]
- 118.Raghavan S, Kahne D. A one-step synthesis of the ciclamycin trisaccharide. J Am Chem Soc. 1993;115:1580–1581. [Google Scholar]
- 119.Gildersleeve J, Pascal RA, Kahne D. Sulfenate intermediates in the sulfoxide glycosylation reaction. J Am Chem Soc. 1998;120:5961–5969. [Google Scholar]
- 120.Brown HC, Kanner B. 2,6-Di-butylpyridine–an unusual pyridine base. J Am Chem Soc. 1953;75:3865. [Google Scholar]
- 121.Wang W, Kong F. New synthetic methodology for regio- and stereoselective synthesis of oligosaccharides via sugar ortho ester intermediates. J Org Chem. 1998;63:5744–5745. doi: 10.1021/jo981135e. [DOI] [PubMed] [Google Scholar]
- 122.Kusumoto S, Sakai K, Shiba T. 4-Azidobutyryl group for temporary protection of hydroxyl functions. Bull Chem Soc Jpn. 1986;59:1296–1298. [Google Scholar]
- 123.Schmittling EA, Sawyer JS. Selective desilylation of tert-butyldimethylsilyl ethers of phenols using potassium fluoride–alumina and ultrasound. Tetrahedron Lett. 1991;32:7207–7210. [Google Scholar]
- 124.Mukaiyama T, Murai Y, Shoda S. An efficient method for glucosylation of hydroxy compounds using glucopyranosyl fluoride. Chem Lett. 1981:431–432. [Google Scholar]
- 125.Nicolaou KC, Chucholowski A, Dolle RE, Randall JL. Reactions of glycosyl fluorides. Synthesis of O-, S-, and N-glycosides. J Chem Soc Chem Commun. 1984:1155–1156. [Google Scholar]
- 126.Guibe F, Saint M’leux Y. The allyloxycarbonyl group for alcohol protection: quantitative removal or transformation into allyl protecting group via π-allyl complexes of palladium. Tetrahedron Lett. 1981;22:3591–3594. [Google Scholar]
- 127.Dong SD, Oberthur M, Losey HC, Anderson JW, Eggert US, Peczuh MW, Walsh CT, Kahne D. The structural basis for induction of VanB resistance. J Am Chem Soc. 2002;124:9064–9065. doi: 10.1021/ja026342h. [DOI] [PubMed] [Google Scholar]
- 128.Nakayama A, Okano A, Feng Y, Collins JC, Collins KC, Walsh CT, Boger DL. Enzymatic glycosylation of vancomycin aglycon: completion of a total synthesis of vancomycin and N- and C-terminus substituent effects of the aglycon substrate. Org Lett. 2014;16:3572–3575. doi: 10.1021/ol501568t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Oberthur M, Leimkuhler C, Kahne D. A practical method for the stereoselective generation of β-2-deoxyglycosyl phosphates. Org Lett. 2004;6:2873–2876. doi: 10.1021/ol049187f. [DOI] [PubMed] [Google Scholar]
- 130.Bardone MR, Paternoster M, Coronelli C. Teichomycins, new antibiotics from Actinoplanes teichomyceticus nov. sp. II. Extraction and chemical characterization. J Antibiot. 1978;31:170–177. doi: 10.7164/antibiotics.31.170. [DOI] [PubMed] [Google Scholar]
- 131.Parenti F, Beretta G, Berti M, Arioli V. Teichomycins, new antibiotics from Actinoplanes teichomyceticus Nov. Sp. I. Description of the producer strain, fermentation studies and biological properties. J Antibiot. 1978;31:276–283. doi: 10.7164/antibiotics.31.276. [DOI] [PubMed] [Google Scholar]
- 132.Hunt AH, Molloy RM, Occolowitz JL, Marconi GG, Debono M. Structure of the major glycopeptide of the teicoplanin complex. J Am Chem Soc. 1984;106:4891–4895. [Google Scholar]
- 133.Barna JCJ, Williams DH, Stone DJM, Leung TWC, Doddrell DM. Structure elucidation of the teicoplanin antibiotics. J Am Chem Soc. 1984;106:4895–4902. [Google Scholar]
- 134.Wiedemann B, Grimm H. In: Antibiotics in Laboratory Medicine. Lorian V, editor. Williams and Wilkins; Baltimore: 1996. pp. 900–1168. [Google Scholar]
- 135.De Lalla F, Tramarin A. A risk-benefit assessment of teicoplanin in the treatment of infections. Drug Saf. 1995;13:317–328. doi: 10.2165/00002018-199513050-00005. [DOI] [PubMed] [Google Scholar]
- 136.Greenwood D. Microbiological properties of teicoplanin. J Antimicrob Chemother. 1988;21(Suppl A):1–13. doi: 10.1093/jac/21.suppl_a.1. [DOI] [PubMed] [Google Scholar]
- 137.Cavalcanti AB, Goncalves AR, Almeida CS, Bugano DD, Silva E. Teicoplanin versus vancomycin for proven or suspected infection. Cochrane Database Syst Rev. 2010:1–53. doi: 10.1002/14651858.CD007022.pub2. [DOI] [PubMed] [Google Scholar]
- 138.Brogden RN, Peters DH. Teicoplanin. A reappraisal of its antimicrobial activity, pharmacokinetic properties and therapeutic efficacy. Drugs. 1994;47:823–854. doi: 10.2165/00003495-199447050-00008. [DOI] [PubMed] [Google Scholar]
- 139.Pea F, Brollo L, Viale P, Pavan F, Furlanut M. Teicoplanin therapeutic drug monitoring in critically ill patients: a retrospective study emphasizing the importance of a loading dose. J Antimicrob Chemother. 2003;51:971–975. doi: 10.1093/jac/dkg147. [DOI] [PubMed] [Google Scholar]
- 140.Jovetic S, Zhu Y, Marcone GL, Marinelli F, Tramper J. β-Lactam and glycopeptide antibiotics: first and last line of defense? Trends Biotechnol. 2010;28:596–604. doi: 10.1016/j.tibtech.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 141.Rossolini GM, Arena F, Pollini S. Novel infectious diseases and emerging Gram-positive multiresistant pathogens in hospital and community acquired infections. In: Marinelli F, Genilloud O, editors. Antimicrobials. Springer Verlag; Berlin Heidelberg, Germany: 2014. [Google Scholar]
- 142.Drugs.com website. [accessed Nov 15 2016];Targocid. http://www.drugs.com/international/targocid.html.
- 143.Boger DL, Kim SH, Miyazaki S, Strittmatter H, Weng JH, Mori Y, Rogel O, Castle SL, McAtee JJ. Total Synthesis of the teicoplanin aglycon. J Am Chem Soc. 2000;122:7416–7417. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 144.Boger DL, Kim SH, Mori Y, Weng JH, Rogel O, Castle SL, McAtee JJ. First and second generation total synthesis of the teicoplanin aglycon. J Am Chem Soc. 2001;123:1862–1871. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- 145.Bois-Choussy M, Neuville L, Beugelmans R, Zhu J. Synthesis of modified carboxyl binding pockets of vancomycin and teicoplanin. J Org Chem. 1996;61:9309–9322. [Google Scholar]
- 146.Reddy KL, Sharpless KB. From styrenes to enantiopure α-arylglycines in two steps. J Am Chem Soc. 1998;120:1207–1217. [Google Scholar]
- 147.Barna JCJ, Williams DH, Strazzolini P, Malabarba A, Leung TWC. Structure and conformation of epimers derived from the antibiotic teicoplanin. J Antibiot. 1984;37:1204–1208. doi: 10.7164/antibiotics.37.1204. [DOI] [PubMed] [Google Scholar]
- 148.Li H, Jiang X, Ye YH, Fan C, Romoff T, Goodman M. 3-(Diethoxyphosphoryloxy)-1,2,3-benzotriazin-4(3H)-one (DEPBT): a new coupling reagent with remarkable resistance to racemization. Org Lett. 1999;1:91–94. doi: 10.1021/ol990573k. [DOI] [PubMed] [Google Scholar]
- 149.Mori Y, McAtee JJ, Rogel O, Boger DL. Alternative synthesis and thermal atropisomerism of a fully functionalized DEFG ring system of teicoplanin. Tetrahedron Lett. 2001;42:6061–6064. [Google Scholar]
- 150.Boger DL, Weng JH, Miyazaki S, McAtee JJ, Castle SL, Kim SH, Mori Y, Rogel O, Strittmatter H, Jin Q. Thermal atropisomerism of teicoplanin aglycon derivatives: preparation of the P,P,P and M,P,P atropisomers of the teicoplanin aglycon via selective equilibration of the DE ring system. J Am Chem Soc. 2001;122:10047–10055. [Google Scholar]
- 151.Evans DA, Katz JL, Peterson GS, Hintermann T. Total synthesis of teicoplanin aglycon. J Am Chem Soc. 2001;123:12411–12413. doi: 10.1021/ja011943e. [DOI] [PubMed] [Google Scholar]
- 152.Evans DA, Kart JL, West TR. Synthesis of diaryl ethers through the copper-promoted arylation of phenols with arylboronic acids. An expedient synthesis of thyroxine. Tetrahedron Lett. 1998;39:2937–2340. [Google Scholar]
- 153.Chan AMT, Monaco KL, Wang RP, Winters MP. New N-and O-arylations with phenylboronic acids and copper acetate. Tetrahedron Lett. 1998;39:2933–2936. [Google Scholar]
- 154.Evans DA, Campos KR, Tedrow JS, Michael FE, Gagné MR. Application of chiral mixed phosphorus/sulfur ligands to palladium-catalyzed allylic substitutions. J Am Chem Soc. 2000;122:7905–7920. [Google Scholar]
- 155.Burk MJ, Allen JG. A mild amide to carbamate transformation. J Org Chem. 1997;62:7054–7057. [Google Scholar]
- 156.Philip JE, Schenck JR, Hargie MP. Ristocetins A and B, two new antibiotics; isolation and properties. Antibiot Annu. 1956–1957:699–705. [PubMed] [Google Scholar]
- 157.Dries CP, Koch R. Ristocetin, a laboratory and clinical evaluation in children. J Pediatr. 1960;56:498–504. doi: 10.1016/s0022-3476(60)80363-0. [DOI] [PubMed] [Google Scholar]
- 158.Herting RL, Lees B, Zimmerman AJ, Berryman GH. Ristocetin; a statistical review of three hundred thirty-three cases. J Am Med Assoc. 1959;170:176–178. doi: 10.1001/jama.1959.03010020034010. [DOI] [PubMed] [Google Scholar]
- 159.Jawetz E. Polymyxins, colistin, bacitracin, ristocetin and vancomycin. Pediatr Clin North Am. 1968;15:85–94. doi: 10.1016/s0031-3955(16)32090-9. [DOI] [PubMed] [Google Scholar]
- 160.Howard MA, Firkin BG. Ristocetin–a new tool in the investigation of platelet aggregation. Thromb Diath Haemorrh. 1971;26:362–369. [PubMed] [Google Scholar]
- 161.Jenkins CSP, Meyer D, Dreyfus MD, Larreu MJ. Willebrand factor and ristocetin I. Mechanism of ristocetin-induced platelet aggregation. Brit J Haematol. 1974;28:561–578. doi: 10.1111/j.1365-2141.1974.tb06675.x. [DOI] [PubMed] [Google Scholar]
- 162.Macfarlane DE, Stibbe J, Kirby EP, Zucker MB, Grant RA, McPherson J. Letter: A method for assaying von Willebrand factor (ristocetin cofactor) Thromb Diath Haemorrh. 1975;34:306–308. [PubMed] [Google Scholar]
- 163.Weiss HJ, Rogers J, Brand H. Defective ristocetin-induced platelet aggregation in von Willebrand’s disease and its correction by Factor VIII. J Clin Invest. 1973;52:2697–2707. doi: 10.1172/JCI107464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Othman M, Kaur H, Emsley J. Platelet-type von Willebrand disease: new insights into the molecular pathophysiology of a unique platelet defect. Semin Thromb Hemost. 2013;39:663–673. doi: 10.1055/s-0033-1353442. [DOI] [PubMed] [Google Scholar]
- 165.Kuwahara S, Chambers WT. Destruction of the platelet aggregating activity of ristocetin A. Experientia. 1978;34:532–534. doi: 10.1007/BF01935974. [DOI] [PubMed] [Google Scholar]
- 166.Harris CM, Harris TM. Structure of ristocetin A: configurational studies of the peptide. J Am Chem Soc. 1982;104:363–365. [Google Scholar]
- 167.Nahoum V, Spector S, Loll PJ. Structure of ristocetin A in complex with a bacterial cell-wall mimetic. Acta Crystallogr Sect D: Biol Crystallogr. 2009;65:832–838. doi: 10.1107/S0907444909018344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Nahoum V, Spector S, Loll PJ. Structure of ristocetin A in complex with a bacterial cell-wall mimetic. Corrigendum. Acta Crystallogr D Biol Crystallogr. 2011;67:832–838. doi: 10.1107/S0907444909018344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mackay JP, Gerhard U, Beauregard DA, Westwell MS, Searle MS, Williams DH. Glycopeptide antibiotic activity and the possible role of dimerization: a model for biological signaling. J Am Chem Soc. 1994;116:4581–4590. [Google Scholar]
- 170.Solladié-Cavallo A, Nsenda T. A four-step synthesis of erythro-m-chloro-3-hydroxy tyrosine ethyl ester enantiomerically pure. Tetrahedron Lett. 1998;39:2191–2194. [Google Scholar]
- 171.Schöllkopf U, Nozulak J, Grauert M. Asymmetric synthesis via heterocyclic intermediates; XXIV1. Asymmetric synthesis of enantiomerically and diastereomerically pure D-threonine by the bis-lactim ether method. Synthesis. 1985:55–56. [Google Scholar]
- 172.McComas CC, Crowley BM, Boger DL. Partitioning the loss in vancomycin binding affinity for D-Ala-D-Lac into lost H-bond and repulsive lone pair contributions. J Am Chem Soc. 2003;125:9314–9315. doi: 10.1021/ja035901x. [DOI] [PubMed] [Google Scholar]
- 173.Crowley BM, Boger DL. Total synthesis and evaluation of [Φ[CH2NH]Tpg4]vancomycin aglycon: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2006;128:2885–2892. doi: 10.1021/ja0572912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Xie J, Pierce JG, James RC, Okano A, Boger DL. A redesigned vancomycin engineered for dual D-Ala-D-Ala and D-Ala-D-Ala binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J Am Chem Soc. 2011;133:13946–13949. doi: 10.1021/ja207142h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Xie J, Okano A, Pierce JG, James RC, Stamm S, Crane CM, Boger DL. Total synthesis of [Φ[C(=S)NH]Tpg4]vancomycin aglycon, [Φ[C(=NH)NH]Tpg4]vancomycin aglycon, and related key compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2012;134:1284–1297. doi: 10.1021/ja209937s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Kamenecka TM, Park YJ, Lin LS, Laszlo S, McCauley ED, Riper GV, Egger L, Kidambi U, Mumford RA, Sharon Tong S, et al. Amidines as amide bond replacements in VLA-4 antagonists. Bioorg Med Chem Lett. 2004;14:2323–2330. doi: 10.1016/j.bmcl.2004.01.100. [DOI] [PubMed] [Google Scholar]
- 177.Inokuchi E, Oishi S, Kubo T, Ohno H, Shimura K, Matsuoka M, Fujii N. Potent CXCR4 antagonists containing amidine type peptide bond isosteres. ACS Med Chem Lett. 2011;2:477–480. doi: 10.1021/ml200047e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Inokuchi E, Yamada A, Hozumi K, Tomita K, Oishi S, Ohno H, Nomizu M, Fujii N. Design and synthesis of amidine-type peptide bond isosteres: application of nitrile oxide derivatives as active ester equivalents in peptide and peptidomimetics synthesis. Org Biomol Chem. 2011;9:3421–3427. doi: 10.1039/c0ob01193b. [DOI] [PubMed] [Google Scholar]
- 179.Molcanov K, Kojic-Prodic B, Raos N. Analysis of the less common hydrogen bonds involving ester oxygen sp3 atoms as acceptors in the crystal structures of small organic molecules. Acta Crystallogr. 2004;B60:424–432. doi: 10.1107/S0108768104014442. [DOI] [PubMed] [Google Scholar]
- 180.Okano A, James RC, Pierce J, Xie J, Boger DL. Silver(I)-promoted conversion of thioamides to amidines: divergent synthesis of a key series of vancomycin aglycon residue 4 amidines that clarify binding behavior to model ligands. J Am Chem Soc. 2012;134:8790–8793. doi: 10.1021/ja302808p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Coleman RS, Shah JA. Chemoselective cleavage of benzyl ethers, esters, and carbamates in the presence of other easily reducible groups. Synthesis. 1999:1399–1400. [Google Scholar]
- 182.Sakaitani M, Ohfune Y. Syntheses and reactions of silyl carbamates. 1. Chemoselective transformation of amino protecting groups via tert-butyldimethylsilyl carbamates. J Org Chem. 1990;55:870–876. [Google Scholar]
- 183.Corey EJ, Schmidt G. Useful procedures for the oxidation of alcohols involving pyridinium dichromate in aprotic media. Tetrahedron Lett. 1979;20:399–402. [Google Scholar]
- 184.Corey EJ, Suggs JW. Pyridinium chlorochromate. An efficient reagent for oxidation of primary and secondary alcohols to carbonyl compounds. Tetrahedron Lett. 1975;16:2647–2650. [Google Scholar]
- 185.Corey EJ, Boger DL. Oxidative cationic cyclization reactions effected by pyridinium chlorochromate. Tetrahedron Lett. 1978;19:2461–2464. [Google Scholar]
- 186.Bowden K, Heilbron IM, Jones ERH, Weedon BC. Researches on acetylenic compounds. Part I. The preparation of acetylenic ketones by oxidation of acetylenic carbinols and glycols. J Chem Soc. 1946:39–45. [Google Scholar]
- 187.Bowers A, Halsall TG, Jones ERH, Lemin AJ. The chemistry of the triterpenes and related compounds. Part XVIII. Elucidation of the structure of polyporenic acid C. J Chem Soc. 1953:2548–2560. [Google Scholar]
- 188.Moorthy JN, Singhal N. Facile and highly selective conversion of nitriles to amides via indirect acid-catalyzed hydration using TFA or AcOH–H2SO4. J Org Chem. 2005;70:1926–1929. doi: 10.1021/jo048240a. [DOI] [PubMed] [Google Scholar]
- 189.Okano A, Nakayama A, Schammel AW, Boger DL. Total synthesis of [Φ[C(=NH)NH]Tpg4]vancomycin and its (4-chlorobiphenyl)methyl derivative: impact of peripheral modifications on vancomycin analogues redesigned for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J Am Chem Soc. 2014;136:13522–13525. doi: 10.1021/ja507009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Nicas TI, Mullen DL, Flokowitsch JE, Preston DA, Snyder NJ, Zweifel MJ, Wilkie SC, Rodriguez MJ, Thompson RC, Cooper RD. Semisynthetic glycopeptide antibiotics derived from LY264826 active against vancomycin-resistant enterococci. Antimicrob Agents Chemother. 1996;40:2194–2199. doi: 10.1128/aac.40.9.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cooper RDG, Snyder NJ, Zweifel MJ, Staszak MA, Wilkie SC, Nicas TI, Mullen DL, Butler TF, Rodriguez MJ, Huff BE, Thompson RC. Reductive alkylation of glycopeptide antibiotics: synthesis and antibacterial activity. J Antibiot. 1996;49:575–581. doi: 10.7164/antibiotics.49.575. [DOI] [PubMed] [Google Scholar]
- 192.Nagarajan R, Schabel AA, Occolowitz JL, Counter FT, Ott JL, Felty–Duckworth AM. Synthesis and antibacterial evaluation of N-alkyl vancomycins. J Antibiot. 1989;42:63–72. doi: 10.7164/antibiotics.42.63. [DOI] [PubMed] [Google Scholar]
- 193.Markham A. Oritavancin: first global approval. Drugs. 2014;74:1823–1828. doi: 10.1007/s40265-014-0295-4. [DOI] [PubMed] [Google Scholar]
- 194.Candiani G, Abbondi M, Borgonovi M, Romano G, Parenti F. In vitro and in vivo antibacterial activity of BI 397, a new semi-synthetic glycopeptide antibiotic. J Antimicrob Chemother. 1999;44:179–192. doi: 10.1093/jac/44.2.179. [DOI] [PubMed] [Google Scholar]
- 195.Anderson VR, Keating GM. Dalbavancin. Drugs. 2008;68:639–648. doi: 10.2165/00003495-200868050-00006. [DOI] [PubMed] [Google Scholar]
- 196.Judice JK, Pace JL. Semi-synthetic glycopeptide antibacterials. Bioorg Med Chem Lett. 2003;13:4165–4168. doi: 10.1016/j.bmcl.2003.08.067. [DOI] [PubMed] [Google Scholar]
- 197.Leadbetter MR, Adams SM, Bazzini B, Fatheree PR, Karr DE, Krause KM, Lam BMT, Linsell MS, Nodwell MB, Pace JL, et al. Hydrophobic vancomycin derivatives with improved ADME properties: discovery of telavancin (TD-6424) J Antibiot. 2004;57:326–336. doi: 10.7164/antibiotics.57.326. [DOI] [PubMed] [Google Scholar]
- 198.Corey GR, Stryjewski ME, Weyenberg W, Yasothan U, Kirkpatrick P. Telavancin. Nat Rev Drug Discovery. 2009;8:929–930. doi: 10.1038/nrd3051. [DOI] [PubMed] [Google Scholar]
- 199.Allen NE, Hobbs JN, Jr, Nicas TI. Inhibition of peptidoglycan biosynthesis in vancomycin-susceptible and -resistant bacteria by a semisynthetic glycopeptide antibiotic. Antimicrob Agents Chemother. 1996;40:2356–2362. doi: 10.1128/aac.40.10.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Ge M, Chen Z, Onishi HR, Kohler J, Silver LL, Kerns R, Fukuzawa S, Thompson C, Kahne D. Vancomycin derivatives that inhibit peptidoglycan biosynthesis without binding D-Ala-D-Ala. Science. 1999;284:507–511. doi: 10.1126/science.284.5413.507. [DOI] [PubMed] [Google Scholar]
- 201.Chen L, Walker D, Sun B, Hu Y, Walker S, Kahne D. Vancomycin analogues active against VanA-resistant strains inhibit bacterial transglycosylase without binding substrate. Proc Natl Acad Sci US A. 2003;100:5658–5663. doi: 10.1073/pnas.0931492100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Goldman RC, Baizman ER, Longley CB, Branstrom AA. Chlorobiphenyl-desleucyl-vancomycin inhibits the transglycosylation process required for peptidoglycan synthesis in bacteria in the absence of dipeptide binding. FEMS Microbiol Lett. 2000;183:209–214. doi: 10.1111/j.1574-6968.2000.tb08959.x. [DOI] [PubMed] [Google Scholar]
- 203.Allen NE, Nicas TI. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol Rev. 2003;26:511–532. doi: 10.1111/j.1574-6976.2003.tb00628.x. [DOI] [PubMed] [Google Scholar]
- 204.Okano A, Nakayama A, Wu K, Lindsey EA, Schammel AW, Feng Y, Collins KC, Boger DL. Total synthesis and initial examination of [Φ[C(=S)NH]Tpg4]vancomycin, [Φ[C(=NH)NH]Tpg4]vancomycin, [Φ[CH2NH]Tpg4]vancomycin and their (4-chlorobiphenyl)methyl derivatives: synergistic binding pocket and peripheral modifications for the glycopeptide antibiotics. J Am Chem Soc. 2015;137:3693–3704. doi: 10.1021/jacs.5b01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chen Z, Eggert US, Dong SD, Shaw SJ, Sun B, LaTour JV, Kahne D. Structural requirements for VanA activity of vancomycin analogues. Tetrahedron. 2002;58:6585–6594. [Google Scholar]
- 206.Kerns R, Dong SD, Fukuzawa S, Carbeck J, Kohler J, Silver L, Kahne D. The role of hydrophobic substituents in the biological activity of glycopeptide antibiotics. J Am Chem Soc. 2000;122:12608–12609. [Google Scholar]
- 207.Okano A, Isley NA, Boger DL. Durable antibiotics by design: installation of peripheral modifications and additional mechanisms of action on pocket redesigned vancomycin analogs synergistically improves both antimicrobial potency and durability. J Am Chem Soc. 2016 submitted. [Google Scholar]
- 208.Jennings MC, Minbiole KPC, Wuest WM. Quaternary ammonium compounds: an antimicrobial mainstay and platform for innovation to address bacterial resistance. ACS Infect Dis. 2015;1:288–303. doi: 10.1021/acsinfecdis.5b00047. [DOI] [PubMed] [Google Scholar]
- 209.Mitchell MM, Iannetta AA, Jennings MC, Fletcher MH, Wuest WM, Minbiole KPC. Scaffold-hopping of multicationic amphiphiles yields three new classes of antimicrobials. ChemBioChem. 2015;16:2299–2303. doi: 10.1002/cbic.201500381. [DOI] [PubMed] [Google Scholar]
- 210.Zasloff M. Antimicrobial peptide of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 211.Yarlagadda V, Akkapeddi P, Manjunath GB, Haldar J. Membrane active vancomycin analogues: a strategy to combat bacterial resistance. J Med Chem. 2014;57:4558–4568. doi: 10.1021/jm500270w. [DOI] [PubMed] [Google Scholar]
- 212.Yarlagadda V, Samaddar S, Paramanandham K, Shome BR, Haldar J. Membrane disruption and enhanced inhibition of cell wall biosynthesis: a synergistic approach to tackle vancomycin-resistant bacteria. Angew Chem, Int Ed. 2015;54:13644–13649. doi: 10.1002/anie.201507567. [DOI] [PubMed] [Google Scholar]
- 213.Kaneko I, Fearon DT, Austen KF. Inhibition of the alternative pathway of human complement in vitro by a natural mirobial product, complestatin. J Immunol. 1980;124:1194–1198. [PubMed] [Google Scholar]
- 214.Tachikawa K, Hasumi K, Endo A. Enhancement of plasminogen binding to U937 cells and fibrin by complestatin. Thromb Haemost. 1997;77:137–142. [PubMed] [Google Scholar]
- 215.Tachikawa K, Hasumi K, Endo A. Enhancement of plasminogen binding and fibrinolysis by chloropeptin. Thrombosis Res. 1997;87:571–576. doi: 10.1016/s0049-3848(97)00186-2. [DOI] [PubMed] [Google Scholar]
- 216.Kaneko I, Kamoshida K, Takahashi S. Complestatin, a potent anti-complement substance produced by Streptomyces lavendulae. J Antibiot. 1989;42:236–241. doi: 10.7164/antibiotics.42.236. [DOI] [PubMed] [Google Scholar]
- 217.Seto H, Fujioka T, Furihata K, Kaneko I, Takahashi S. Structure of complestatin, a very strong inhibitor of protease activity of complement in the human complement system. Tetrahedron Lett. 1989;30:4987–4990. [Google Scholar]
- 218.Matsuzaki K, Ikeda H, Ogino T, Matsumoto A, Woodruff HB, Tanaka H, Omura S. Loropeptins I and II, novel inhibitors against gp120-CD4 binding from Streptomyces sp. J Antibiot. 1994;47:1173–1174. doi: 10.7164/antibiotics.47.1173. [DOI] [PubMed] [Google Scholar]
- 219.Tanaka H, Matsuzaki K, Nakashima H, Ogino T, Matsumoto A, Ikeda H, Woodruff HB, Omura S. Chloropeptins, new anti-HIV antibiotics inhibiting gp120-CD4 binding from Streptomyces sp. I. Taxonomy, fermentation, isolation, and physico-chemical properties and biological activities. J Antibiot. 1997;50:58–65. doi: 10.7164/antibiotics.50.58. [DOI] [PubMed] [Google Scholar]
- 220.Matsuzaki K, Ogino T, Sunazuka T, Tanaka H, Omura S. Chloropeptins, a new anti-HIV antibiotics inhibiting gp120-CD4 binding from Streptomyces sp. II. Structure elucidation of chloropeptin I. J Antibiot. 1997;50:66–69. doi: 10.7164/antibiotics.50.66. [DOI] [PubMed] [Google Scholar]
- 221.Gouda H, Matsuzaki K, Tanaka H, Hirono S, Omura S, McCauley JA, Sprengeler PA, Furst GT, Smith AB. Stereostructure of (–)-chloropeptin I, a novel inhibitor of gp120-CD4 binding, via high temperature molecular dynamics, monte carlo conformational searching, and NMR spectroscopy. J Am Chem Soc. 1996;118:13087–13088. [Google Scholar]
- 222.Singh SB, Jayasuriya H, Salituro GM, Zink DL, Shafiee A, Heimbuch B, Silverman KC, Lingham RB, Genilloud O, Teran A, Vilella D, Felock P, Hazuda D. The complestatins as HIV-1 integrase inhibitors. Efficient isolation, structure elucidation, and inhibitory activities of isocomplestatin, chloropeptin I, new complestatins, A and B, and acid-hydrolysis products of chloropeptin I. J Nat Prod. 2001;64:874–882. doi: 10.1021/np000632z. [DOI] [PubMed] [Google Scholar]
- 223.Jayasuriya H, Salituro GM, Smith SK, Heck JV, Gould SJ, Singh SB, Homnick CF, Holloway MK, Pitzenberger SM, Patane MA. Complestatin to chloropeptin I via a quantitative acid catalyzed rearrangement. Absolute stereochemical determination of complestatin. Tetrahedron Lett. 1998;39:2247–2248. [Google Scholar]
- 224.Shinohara T, Deng H, Snapper ML, Hoveyda AH. Isocomplestatin: total synthesis and stereochemical revision. J Am Chem Soc. 2005;127:7334–7336. doi: 10.1021/ja051790l. [DOI] [PubMed] [Google Scholar]
- 225.Kobayashi H, Shin-Ya K, Furhata K, Nagai K, Suzuki KI, Hayakawa Y, Seto H, Yun BS, Ryoo IJ, Kim JS, et al. Neuroprotectins A and B, dicyclohexapeptides protecting chick telencephalic neurons from excitotoxicity II. Structure determination. J Antibiot. 2001;54:1019–1024. doi: 10.7164/antibiotics.54.1019. [DOI] [PubMed] [Google Scholar]
- 226.Kobayashi H, Shin-Ya K, Nagai K, Suzuki KI, Hayakawa Y, Seto H, Yun BS, Ryoo IJ, Kim JS, Kim CJ, et al. Neuroprotectins A and B, bicyclohexapeptides protecting chick telencephalic neuronal cells from excitotoxicity I. Fermentation, isolation, physico-chemical properties and biological activity. J Antibiot. 2001;54:1013–1018. doi: 10.7164/antibiotics.54.1013. [DOI] [PubMed] [Google Scholar]
- 227.Deng H, Jung JK, Liu T, Kuntz KW, Snapper ML, Hoveyda AH. Total synthesis of anti-HIV agent chloropeptin I. J Am Chem Soc. 2003;125:9032–9034. doi: 10.1021/ja030249r. [DOI] [PubMed] [Google Scholar]
- 228.Jia Y, Bois-Choussy M, Zhu J. Synthesis of diastereomers of complestatin and chloropeptin I: substrate-dependent atropstereoselectivity of the intramolecular Suzuki–Miyaura reaction. Angew Chem, Int Ed. 2008;47:4167–4172. doi: 10.1002/anie.200800599. [DOI] [PubMed] [Google Scholar]
- 229.Garfunkle J, Kimball FS, Trzupek JD, Takazawa S, Shimamura H, Tomishima M, Boger DL. Total synthesis of chloropeptin II (complestatin) and chloropeptin I. J Am Chem Soc. 2009;131:16036–16038. doi: 10.1021/ja907193b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Shimamura H, Breazzano SP, Garfunkle J, Kimball FS, Trzupek JD, Boger DL. Total synthesis of complestatin: development of a Pd(0)-mediated indole annulation for macrocyclization. J Am Chem Soc. 2010;132:7776–7783. doi: 10.1021/ja102304p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Breazzano SP, Boger DL. Synthesis and stereochemical determination of complestatin A and B (neuroprotection A and B) J Am Chem Soc. 2011;133:18495–18502. doi: 10.1021/ja208570q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Larock RC, Yum EK. Synthesis of indoles via palladium-catalyzed heteroannulation of internal alkynes. J Am Chem Soc. 1991;113:6689–6690. [Google Scholar]
- 233.Larock RC, Yum EK, Refvik MD. Synthesis of 2,3-disubstituted indoles via palladium-catalyzed annulation of internal alkynes. J Org Chem. 1998;63:7652–7662. [Google Scholar]
- 234.Shen M, Li G, Lu BZ, Hossain A, Roschangar F, Farina V, Senanayaka CH. The first regioselective palladium-catalyzed indolization of 2-bromo- or 2-chloroanilines with internal alkynes: a new approach to 2,3-disubstituted indoles. Org Lett. 2004;6:4129–4132. doi: 10.1021/ol048114t. [DOI] [PubMed] [Google Scholar]
- 235.Ma C, Liu X, Li X, Flippen-Anderson J, Yu S, Cook JM. Efficient asymmetric synthesis of biologically important tryptophan analogues via a palladium-mediated heteroannulation reaction. J Org Chem. 2001;66:4525–4542. doi: 10.1021/jo001679s. [DOI] [PubMed] [Google Scholar]
- 236.Jeschke T, Wensbo D, Annby U, Gronowitz S, Cohen LA. A novel approach to Bz-substituted tryptophans via Pd-catalysed coupling/annulation. Tetrahedron Lett. 1993;34:6471–6474. [Google Scholar]
- 237.Breazzano SP, Poudel YB, Boger DL. A Pd(0)-mediated indole (macro)cyclization reaction. J Am Chem Soc. 2013;135:1600–1606. doi: 10.1021/ja3121394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Savige WE, Fontana A. New procedure for oxidation of 3-substituted indoles to oxindoles: modification of tryptophan residues in peptides and proteins. J Chem Soc, Chem Commun. 1976:599–600. [Google Scholar]
- 239.Szabo-Pustay K, Szabo L. A simple general method for the oxidation of indoles to oxindoles. Synthesis. 1979:276–277. [Google Scholar]
- 240.Hinman RL, Bauman CP. Reactions of N-bromosuccinimide and indoles. A simple synthesis of 3-bromooxindoles. J Org Chem. 1964;29:1206–1215. [Google Scholar]
- 241.Lawson WB, Witkop R. A simple method for the preparation of oxindoleacetic and propionic acids from the parent indoles. J Org Chem. 1961;26:263–264. [Google Scholar]
- 242.Lawson WB, Patchornik A, Witkop R. Substitution, oxidation and group participation in the bromination of indoles. J Am Chem Soc. 1960;82:5918–5923. [Google Scholar]
- 243.Fuchs JR, Funk RL. Total synthesis of (±)-perophoramidine. J Am Chem Soc. 2004;126:5068–5069. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]
- 244.Fuchs JR, Funk RL. Indol-2-one intermediates: mechanistic evidence and synthetic utility. Total syntheses of (±)-flustramines A and C. Org Lett. 2005;7:677–680. doi: 10.1021/ol047532v. [DOI] [PubMed] [Google Scholar]
- 245.Hino T, Tonozuka M, Nakagawa M. Bromination of 3-phenylindoles. Tetrahedron. 1974;30:2123–2133. [Google Scholar]
- 246.Hino T, Nakamura T, Nakagawa M. Halogenation of 1-substituted skatoles. Preparation of 3-bromomethylindoles. Chem Pharm Bull Jpn. 1975;23:2990–2997. [Google Scholar]
- 247.Walker S, Chen L, Hu Y, Rew Y, Shin D, Boger DL. Chemistry and biology of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. Chem Rev. 2005;105:449–476. doi: 10.1021/cr030106n. [DOI] [PubMed] [Google Scholar]
- 248.Jiang W, Wanner J, Lee RJ, Bounaud PY, Boger DL. Total synthesis of the ramoplanin A2 and ramoplanose aglycon. J Am Chem Soc. 2003;125:1877–1887. doi: 10.1021/ja0212314. [DOI] [PubMed] [Google Scholar]
- 249.Rew Y, Shin D, Hwang I, Boger DL. Total synthesis and examination of three key analogues of ramoplanin: a lipoglycodepsipeptide with potent antibiotic activity. J Am Chem Soc. 2004;126:1041–1043. doi: 10.1021/ja039671y. [DOI] [PubMed] [Google Scholar]
- 250.Shin D, Rew Y, Boger DL. Total synthesis and structure of the ramoplanin A1 and A3 aglycons: two minor components of the ramoplanin complex. Proc Natl Acad Sci US A. 2004;101:11977–11979. doi: 10.1073/pnas.0401419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Nam J, Shin D, Rew Y, Boger DL. Alanine scan of [L-dap2]ramoplanin A2 aglycon: assessment of the importance of each residue. J Am Chem Soc. 2007;124:8747–8755. doi: 10.1021/ja068573k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Boger DL. Vancomycin, teicoplanin, and ramoplanin: synthetic and mechanistic studies. Med Res Rev. 2001;21:356–381. doi: 10.1002/med.1014. [DOI] [PubMed] [Google Scholar]
- 253.Seiple IB, Zhang ZY, Jakubec P, Langlois-Mercier A, Wright PM, Hog DT, Yabu K, Allu SR, Fukuzaki T, Carlsen PN, et al. A platform for the discovery of new macroclide antibiotics. Nature. 2016;533:338–345. doi: 10.1038/nature17967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG. A convergent enantioselective route to structurally diverse 6-deoxytetracycline antibiotics. Science. 2005;308:395–398. doi: 10.1126/science.1109755. [DOI] [PubMed] [Google Scholar]
- 255.Wright PM, Seiple IB, Myers AG. The evolving role of chemical synthesis in antibacterial drug discovery. Angew Chem, Int Ed. 2014;53:8840–8869. doi: 10.1002/anie.201310843. [DOI] [PMC free article] [PubMed] [Google Scholar]