

Abstract
Previous investigations suggest that DL-3-n-butylphthalide (NBP) is a promising multifaceted drug for the treatment of stroke. It is not clear whether NBP can treat traumatic brain injury (TBI) and what could be the mechanisms of therapeutic benefits. To address these issues, TBI was induced by a controlled cortical impact in adult male mice. NBP (100 mg/kg) or saline was intraperitoneally administered within 5 min after TBI. One day after TBI, apoptotic events including caspase-3/9 activation, cytochrome c release from the mitochondria, and apoptosis-inducing factor (AIF) translocation into the nucleus in the pericontusion region were attenuated in NBP-treated mice compared to TBI-saline controls. In the assessment of the nuclear factor kappa-light-chain-enhancer of activated B (NF-κB) pathway, NBP ameliorated the p65 expression and the p-IκB-α/Iκ3-α ratio, indicating reduced NF-κB activation. Consistently, NBP reduced the upregulation of proinflammatory cytokines such as tumor necrotizing factor-alpha (TNF-α) and interleukin-1beta (IL-1β) after TBI. In sub-acute treatment experiments, NBP was intranasally delivered once daily for 3 days. At 3 days after TBI, this repeated NBP treatment significantly reduced the contusion volume and cell death in the pericontusion region. In chronic experiments up to 21 days after TBI, continues daily intranasal NBP treatment increased neurogenesis, angiogenesis, and arteriogenesis in the post-TBI brain, accompanied with upregulations of regenerative genes including brain-derived neurotrophic factor (BDNF), vascular endothelial growth factor (VEGF), endothelial-derived nitric oxide synthase (eNOS), and matrix metallopeptidase 9 (MMP-9). The NBP treatment significantly improved sensorimotor functional recovery and reduced post-TBP depressive behavior. These new findings demonstrate that NBP shows multiple therapeutic benefits after TBI.
Keywords: NBP, Traumatic brain injury, Regeneration, Inflammation, Depression
Introduction
DL-3-n-butylphthalide (NBP) is a synthesized compound based on the pure component, l-3-n-butylphthalide, originally extracted from the seeds of Apium graveolens Linn (Chinese celery) (Li et al., 2010). NBP has been synthesized and approved by the State Food and Drug Administration (SFDA) of China for clinical use in patients with stroke (Huang et al., 2010). Previous studies have shown that NBP has protective effects against ischemic brain damages, resulting in reduction of the infarct volume (Li et al., 2010; Liu and Feng, 1995), attenuation of neurovascular damage (Chong and Feng, 1999; Deng and Feng, 1997; Li et al., 2010; Lin and Feng, 1996), and improvements of cognitive functions and neurological outcomes after stroke (Peng et al., 2007; Zhang and Feng, 1996). The cellular mechanisms of the protective effects may involve the improved mitochondria function and energy metabolism (Feng et al., 1995; Li et al., 2010), decreased oxidative damage and apoptosis (Chang and Wang, 2003; Dong and Feng, 2002), reduced inflammatory responses (Xu and Feng, 2000), and enhanced regional blood flow and angiogenesis (Liao et al., 2009; Yan et al., 1998). These observations were mainly from studies on ischemic stroke. It is unclear whether NBP may show neuroprotective and regenerative benefits after traumatic brain injury (TBI).
TBI is a leading cause of human death and severe disability in the United States and worldwide (Coronado et al., 2011; Runyan, 2008). Effective clinical treatments for patients with TBI have yet to be developed. Although the initial trigger of the brain damage induced by the mechanical strike in TBI is different from stroke, the pathological signaling pathways and the consequent cell death mechanisms in TBI share some similarities with ischemic strokes, and there is often a second ischemic damage after the initial TBI insult (Bramlett and Dietrich, 2004; Simard et al., 2010; Stutzmann et al., 2002). Specifically, regional ischemia is one of the main events in TBI secondary injury due to the damage to vasculatures (Simard et al., 2010; Stutzmann et al., 2002). It is also well characterized that excitotoxicity and apoptosis are mainly responsible for neuronal damage in both ischemic stroke and TBI (Liou et al., 2003). We thus hypothesized that NBP treatment would show neuroprotective effects against TBI damage. One early report showed that NBP reduced cerebral edema, disruption of the BBB, and improved memory function in the closed head injury model (Chong and Feng, 2000). Another group showed that NBP treatment ameliorated BBB damage and cerebral edema against diffuse brain injury (Zhao et al., 2013). Many patients with TBI have suffered depression due to TBI-induced severe psychological impact (Barker-Collo et al., 2013; Fann et al., 2001). Previous reports suggest that approximately 60 % of TBI patients have been diagnosed with depression within 1 year (Barker-Collo et al., 2013; Silver et al., 2011). Notably, patients with depression after TBI face poor psychosocial and functional outcomes (Barker-Collo et al., 2013). Whether NBP treatment could show an anti-depression effect is unknown. It was thus necessary and clinically essential to verify the protective effect of NBP against different TBI insults and delineate cellular and molecular mechanisms in the protection.
Endogenous neural stem/progenitor cells might play regenerative roles in brain injuries (Gage et al., 1998; Guo et al., 2012). Neurogenesis is observed in response to brain trauma or insults, suggesting that the brain has the inherent potential to restore populations of damaged or destroyed neurons. Furthermore, angiogenesis and arteriogenesis may have the potential of restoring the focal blood flow that supports tissue repair and sustained functional recovery after TBI (Carmeliet, 2000; Christoforidis et al., 2005). The effect of NBP on regenerative mechanisms has been much less investigated. In the present investigation, we evaluated the possibility that NBP may promote endogenous regenerative activities after TBI. Our present findings provide novel evidence that NBP contributes not only to acute neuroprotection against TBI, but also show other important therapeutic benefits such as enhanced regenerative activities and anti-depressive effects after TBI.
Materials and Methods
Animals and controlled cortical impact of TBI model
Adult male C57BL/6 mice (8–12 week, 22–26 g) were used in this study. Some experiments used α-smooth muscle actin (α-SMA)-GFP transgenic mice (8–12 week, 22–26 g) to examined arteriogenesis. C57BL/6 mice were purchased from Charles River Labs (Wilmington, MA), α-SMA transgenic mice were produced in the Transgenic Mouse Facility at the National Eye Institute (Maryland, MD) on a C57BL/6 strain background. These α-SMA transgenic mice express green fluorescent protein (GFP) under the control of the α-SMA promoter, resulting in the specific expression of GFP in smooth muscle cells, which facilitates the measurement of arteriogenesis (Yokota et al., 2006). Mice were housed in standard cages in 12-hr light/12-hr dark cycle and given food and water ad libitum. TBI surgery was performed as previously described (Lee et al., 2014). Mice were anesthetized with 1.5 % isoflurane and placed on a stereotaxic frame. After a midline skin incision, a 3.5 mm circular craniotomy was performed midway between lambda and bregma, 2.0 mm to the right of central suture using an electric drill. Controlled cortical impact was induced with an electric impact device using an impact tip (2.0 mm diameter with a slightly rounded edge) and the PCI3000 Precision Cortical Impactor (Hatteras Instruments, Cary, NC). The impact drop was controlled at velocity=3.0 m/s, depth=1.0 mm, and contact time=150 ms. Rectal temperature was maintained at 37.0 ± 0.5°C during surgery, using a heating pad controlled by a homeothermic blanket control unit (Harvard Apparatus, Holliston, MA). After the injury, the skin was sutured, and mice were allowed to recover in a humidity-controlled incubator (Thermocare, Incline Village, NV).
Chemicals and drug administration
Synthesized NBP was a gift from Shijiazhuang Pharmaceutical Group Ouyi Pharma Co., Ltd, (Shijiazhuang, China). We applied the intraperitoneal administration of NBP (100 mg/kg, i.p.) within 5 min after TBI to mimic an acute on-site treatment. Alternatively, we tested the intravnasal method for the non-invasive brain delivery of NBP in repeated sub-acute and chronic treatments after TBI. After the first delivery, intranasal administration of NBP (100 mg/kg/day) was performed for a total of 3 or 21 days. The dosage of NBP tested in this investigation was selected based on previous investigations on ischemic stroke and TBI (Li et al., 2010). Our own preliminary test showed that NBP at 10 mg/kg did not exhibit significant benefits in the TBI model (data not shown). At effective dosages such as 100 mg/kg/day in rodents and 200 mg/kg/day in patients, NBP barely exerts any side effects, suggesting that NBP can be a safe treatment (Li et al., 2010; Ma et al., 2009; Xiong et al., 2012).
To label proliferating cells in chronic experiments, 5-bromo-2-deoxyuridine (BrdU) (Sigma-Aldrich, St. Louis, MO) was administered to animals (50 mg/kg/day, i.p.) beginning on day 3 after TBI until sacrifice. The animal surgery and drug administration protocol was approved by the Emory University Institutional Animal Care and Use Committee (IACUC), in compliance with National Institutes of Health (NIH) guidelines.
Cresyl Violet (Nissl) staining for contusion volume measurement
Fresh-frozen brain sections of 10-μm-thick containing target lesions were collected every 200 μm and then fixed with a 1:1 solution of 10 % formalin and acetic acid for 10 min. After washing with distilled water for 5 min, slices were stained with a solution containing buffer solution (0.1 M acetate acid and 0.1 M Sodium acetate. 94:6) and Cresyl Violet acetate at a ratio of 5:1. The sections were then dehydrated in 100% ethyl alcohol and mounted.
After Nissl staining, the sections were digitized, and the areas of the contusion and the two hemispheres were quantified using the ImageJ software (NIH, Bethesda, MD) by an investigator blinded toward the treatment of the animals. Contusion volumes were calculated based on the contusion areas (C) obtained from 6–7 sections as follows:
C1*D+0.2(C1+C2+3+…C7), with D being the distance between two sections (0.2 mm). Additionally, to consider the effects of swelling/edema, hemispheric tissue loss was calculated as a percentage calculated by [(contralateral hemispheric volume-ipsilateral hemispheric volume)/(contralateral hemispheric volume)×100%], as previously reported (Lee et al., 2014).
Cell death assay using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)
A TUNEL assay kit was used to examine cell death by detecting fragmented DNA in 10-μm-thick coronal fresh frozen sections as described previously (Lee et al., 2014). After fixation (10% buffered formalin for 10 min and then ethanol: acetic acid (2:1) solution for 5min) and permeabilization (0.2% Triton X-100 solution), brain sections were incubated in the equilibration buffer for 10 min. Recombinant terminal deoxynucleotidyl transferase (rTdT) and nucleotide mixture were then added on the slide at 37°C for 60 min in the dark. Reactions were terminated by 2x SSC solution for 15 min. Nuclei were counterstained with Hoechst 33342 (1:20,000; Molecular Probes, Eugene, OR) for 5 min.
Isolation of subcellular fractionations
The brain pericontusion tissue was washed with ice-cold phosphate-buffered saline (PBS) and resuspended in a homogenization buffer (pH 6.7) containing 150 mM MgCl2, 10 mM KCl, 10 mM Tris, 0.25 M sucrose, and protease inhibitor cocktail (Sigma-Aldrich). For isolating the nucleus fraction, the lysate was homogenized in an NKM buffer (pH 7.4) containing 1 mM Tris-Hcl, 0.13 M NaCl, 5 mM KCl, and 7.5 mM MgCl2 with a homogenizer, centrifuged at 800g for 20 min at 4°C. The nucleus fraction was obtained by resuspending the pellet with an NKM buffer. For isolating the mitochondrial and cytosolic fractions, the supernatant was homogenized in a cytosolic buffer (pH 6.7) containing 10 mM Tris-HCl, 10 mM KCl, 0.15 mM MgCl2, 1 mM PMSF, and 1 mM DTT with a homogenizer, centrifuged at 12,000g for 15 min at 4°C. The supernatant contained cytosolic fraction and the pellet contained mitochondrial fraction. The mitochondrial fraction was further obtained by resuspending the pellet with a mitochondrial suspension buffer (pH 6.7) containing 10 mM Tris-HCl, 0.15 mM MgCl2, 0.25 M sucrose, 1 mM PMSF, and 1 mM DTT.
Western blot analysis of gene expression in brain tissues
Protein was determined with a bicinchoninic acid assay (Pierce Biotechnology, Rockford, IL). Equivalent amounts of total protein were separated by molecular weight on an SDS-polyacrylamide gradient gel, and then transferred to a PVDF membrane. The blot was incubated in 10% nonfat dry milk for 1 hr and then reacted with primary antibodies at 4°C for overnight. The primary antibodies used and the dilutions for each were rabbit anti-cleaved caspase-3 antibody (Cell Signaling, Danvers, MA) at 1:400, rabbit anti-caspase-9 antibody (Cell Signaling) at 1: 1,000, rabbit anti-cytochrome c antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at 1:200 or rabbit anti-AIF antibody (Cell Signaling) at 1:500, rabbit anti-TNF-α (Cell Signaling) at 1:1000, rabbit anti-IL-1β (Cell Signaling) at 1:1000, rabbit anti-BDNF (BD Bioscience, San Jose, CA) at 1:2,000, rabbit anti-VEGF (BD Bioscience) at 1,000, rabbit anti-GDNF (BD Bioscience) at 1:1,000, rat anti-eNOS (BD Bioscience) at 1:1,000, rabbit anti-CXCR4 (BD Bioscience) at 1:2,000, rabbit anti-p65 (Cell Signaling) at 1:1,000, mouse anti-IκB (Cell Signaling) at 1:1,000, mouse anti-p-IκB (Cell Signaling) at 1:1,000, rabbit anti-COX (Cell Signaling) at 1:2,500, mouse anti-actin (Sigma-Aldrich) at 1:5000, and rabbit anti-MMP-9 antibody (Millipore, Billerica, MA) at 1:2500. After washing with TBST, membranes were incubated with AP-conjugated or HRP-conjugated secondary antibodies (GE Healthcare, Piscataway, NJ) for 1.5 hrs at room temperature. After final washing with TBST, the signals were detected with bromochloroidolylphosphate/nitroblue tetrazolium solution (Sigma-Aldrich) or film. Signal intensity was measured by ImageJ and normalized to the actin intensity.
Immunohistochemical staining of specific cell markers
Frozen brain tissues were sliced into 10 μm-thick coronal sections using a cryostat (Leica CM 1950; Leica Microsystems, Buffalo Grove, IL). Sections were dried on the slide warmer for 30 min, fixed with 10% formalin buffer for 10 min, washed with −20°C pre-cooled ethanol: acetic acid (2:1) solution for 10 min, and finally permeabilized with 0.2% Triton-X 100 solution for 5 min. All slides were washed 3 times with PBS (5 min each) after each step. Then, tissue sections were blocked with 1% fish gelatin (Sigma-Aldrich) in PBS for 1 hr at room temperature, and subsequently incubated with the primary antibody occludin (1:200; Millipore), Glut1 (1:400; Millipore) and NeuN (1:300; Millipore) overnight at 4°C. For BrdU (1:400; AbD Serotec, Raleigh, NC) staining, slides were incubated in 2N HCl at 37°C for 40 min, followed by 0.2% Triton-X 100 incubation for 40 min. The slides were washed the next day by 3 times with PBS for 5 min each, then reacted with the secondary antibodies Alexa Fluor®488 goat anti-mouse (1:300; Life Technologies, Grand Island, NY) and Cy3-conjugated donkey anti-rabbit (1:300; Jackson ImmunoResearch Laboratories, West Grove, PA) or Cy5-conjugated donkey anti-mouse or rabbit (1:400; Jackson ImmunoResearch Laboratories) for 90 min at room temperature. After 3 washes with PBS, nuclei were stained with Hoechst 33342 (1:20,000; Molecular Probes) for 5 min as a counterstain, then mounted with Vectashield fluorescent mounting medium (Vector Laboratory, Burlingame, CA), and coverslipped for microscopy and image analysis.
Quantification of immunoreactive cells
Cell count was performed as described previously (Lee et al., 2014). Systematic random sampling was employed to ensure accurate and non-redundant cell counting. Six brain sections of 10 μm thick per animal were collected at >90 μm separation between sections for non-overlapping multistage random sampling. Six fields were chosen in each section in the pericontusion region and viewed at 20x for cell counting. ImageJ (NIH) was used to analyze each picture. All analysis was performed in a blinded fashion.
Animal behavioral tests
Adhesive dot removal test
The adhesive removal test measures sensorimotor function as previously described (Lee et al., 2014). A small adhesive dot was placed on each forepaw, and the amount of time (sec) needed to contact and remove the tape from each forepaw was recorded. Recording stopped after 6 min. Mice were trained three times before TBI surgery. Animals took more than 6 min to touch and remove the dot were removed from further test and analysis. For each trial, the test was performed three times per mouse, and the average time was used in the analysis.
Corner test
The corner test was performed 1 day before TBI and 3 and 7 days after TBI, as described previously (Choi et al., 2012). Two cardboard plates (30 × 20 × 0.3 cm) were attached at a 30° angle in a home cage. Each mouse was placed between the two plates and allowed to freely move to the corner. The number of right and left turns was counted. Twenty trials were performed for each mouse.
Cylinder test
A unilateral injury to the motor cortex results in an asymmetry in the forelimb used for support during rearing, which can be measured using the cylinder test. The mice were placed in a glass cylinder (9.5 cm diameter and 11 cm height) and the number of times each forelimb or both forelimbs were used to support the body on the wall of the cylinder was counted for 5 min. The animals were evaluated before and 3, 7 days after TBI. Two mirrors were placed behind the cylinder to view all directions. The number of impaired and non-impaired forelimb contacts was calculated as a percentage of total contacts.
Home cage behavioral test
Behavioral changes of experimental mice were monitored using the HomeCage System (Clever Sys Inc., Reston, VA) (Lee et al., 2015). The system had four cages, each was monitored by a dedicated cameras. Each cage (191 × 292 × 127 mm) contained one mouse. The behavior patterns were continuously recorded for 3 or 12 hrs during night time. After finishing the recording, the videos were analyzed by the HomeCage Software 3.0 (Clever Sys Inc.).
Forced swim test
The forced swim test (FST) was carried out to measure the effect of NBP on TBI-induced depression (Cryan et al., 2005). Mice were dropped individually into a plexiglass cylinder (height: 30 cm, diameter: 22.5 cm) filled with water to a depth of 15 cm and maintained at 23–25 °C. In this test, after initial vigorous activity of 2 min, mice acquired an immobile posture which was characterized by motionless floating in the water and made only those movements necessary to keep the head above water. The duration of immobility was recorded in seconds during the last 4 min of the 6 min test (Porsolt et al., 1977a). All mice received a 15-min training session under similar condition 24 hr before the formal test.
Open field test
The open field test (OFT) was conducted to measure baseline locomotor activity and depressive behavior as described before (Mohamad et al., 2013). In the OFT, mice were allowed to freely explore an open field container (50 × 50 × 50 cm; divided into 12 equal squares) for 60 min. Activity was recorded on a video camera. The average distance, speed, and total time spent in the central zone were recorded using TopScan CleverSys (Clever Sys, Inc.). After finishing the recording, the videos were analyzed by the TopScan Realtime Option Version 3.0 (Clever Sys Inc.).
Statistical analysis
GraphPad Prism 6 (GraphPad Software, San Diego, CA) was used for statistical analysis and graphic presentation. Student’s two-tailed t-test was used for comparison of 2 experimental groups, and one-way ANOVA followed by Bonferroni correction was used for multiple-group comparisons. Two-way ANOVA followed by Bonferroni correction was used for repeated measurements. In the statistical analysis of the Western blot data, we assumed the distribution of protein expression followed normal distribution patterns. Significant differences between groups were identified by a P value of <0.05. All data are presented as Mean±SEM.
Results
NBP treatment attenuated contusion volume and cell death 3 days after TBI
The closed impact TBI insult induced a focal and significant contusion in the right sensorimotor cortex of the adult mouse (Lee et al., 2014). In an acute “on-site” treatment setting, NBP (100 mg/kg, i.p.) or saline control was administered within 5 min after the traumatic blow. We assessed the contusion volume at 3 days after TBI to allow sufficient time for contusion development and there was no mortality after the cortical TBI insult. Early time points such as 1 day after TBI was not used because the contusion volume in this model showed large variations at early stages (data not shown). In the sub-acute experiments, we tested intranasal delivery of NBP (100 mg/kg daily) for acute and sub-acute treatment of consecutive 3 days. Animals were sacrificed for brain damage assessments at 3 days after TBI. The intranasal route was selected because it is an established method of bypassing the blood brain barrier (BBB) for non-invasive brain delivery of therapeutics including proteins, peptides and even stem cells (Djupesland et al.; Lee et al.). The contusion volume was measured in brain sections with Nissl staining, showing 7.32±0.57 mm3 in TBI-saline controls and decreased to 5.83±0.69 mm3 in the NBP treatment group (P<0.05, n=12 and 13, respectively) (Fig. 1A–1C). TBI mice with NBP treatment had significantly fewer TUNEL-positive cells than TBI-saline controls at 1 and 3 days after TBI (Fig. 1D and 1E).
Figure 1. Tissue and cellular protective effects of NBP treatments after TBI.
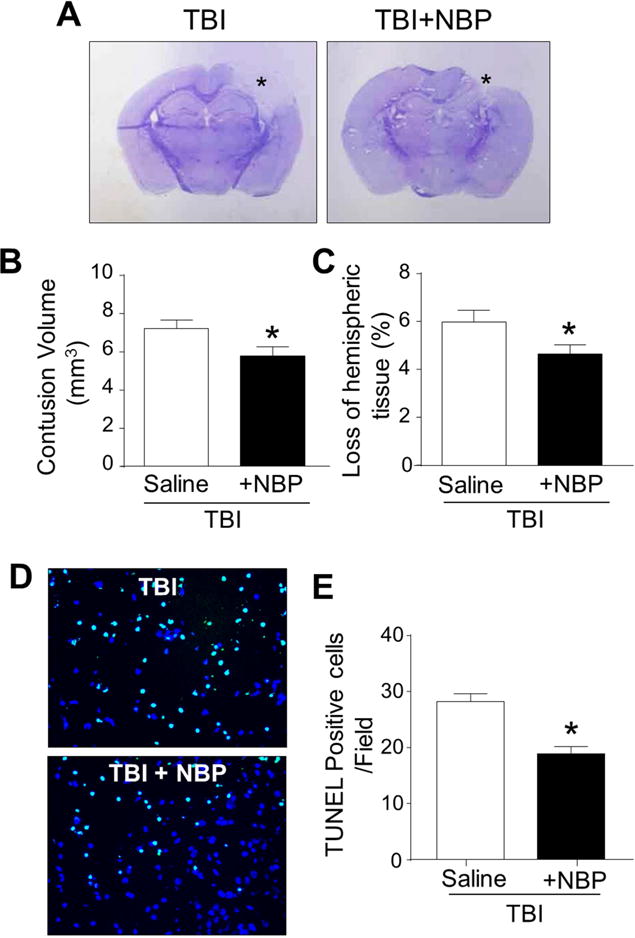
TBI mice received repeated NBP treatments 5 min after TBI followed by 2 daily intranasal administrations. TBI-induced contusion volume and cell death were measured 3 days after the insult. A. Nissl staining shows brain sections from a TBI-saline control mouse and a TBI mouse received NBP treatments. The NBP treatment resulted in smaller contusion area (*). B and C. Bar graphs summarizes the measurements on contusion volume and the loss of hemispheric tissues in TBI-saline group and TBI plus NBP group. * P<0.05 versus TBI-saline controls by Student t test; N=12–13 per group. D. TUNEL staining revealed DNA damages and cell death 3 days after TBI. Total cells were visualized with Hoechst 33342 staining (blue). Massive TUNEL-positive cells (green) were observed in the TBI injured cortical region. The NBP treatment reduced TUNEL-positive cells. Scale bar = 50 μm. E. The percentage of TUNEL positive cells among total cells in the pericontusion area at 1 and 3 days after TBI. The bar graph summarized that NBP treatment after TBI significantly decreased death. * P<0.05 versus TBI-saline control group by Student t test; N=7 per group.
NBP treatment reduced apoptotic cell death acutely after TBI
Western blot analysis was performed to investigate apoptotic signals at 24 hrs after TBI. In the pericontusion region, TBI-saline control mice exhibited significant increases of cleaved caspase-3 and caspase-9 compared to sham mice. This caspase activation was substantially suppressed by the acute NBP treatment (Fig. 2A).
Figure 2. Acute NBP treatment prevented both caspase-dependent and -independent apoptosis after TBI.
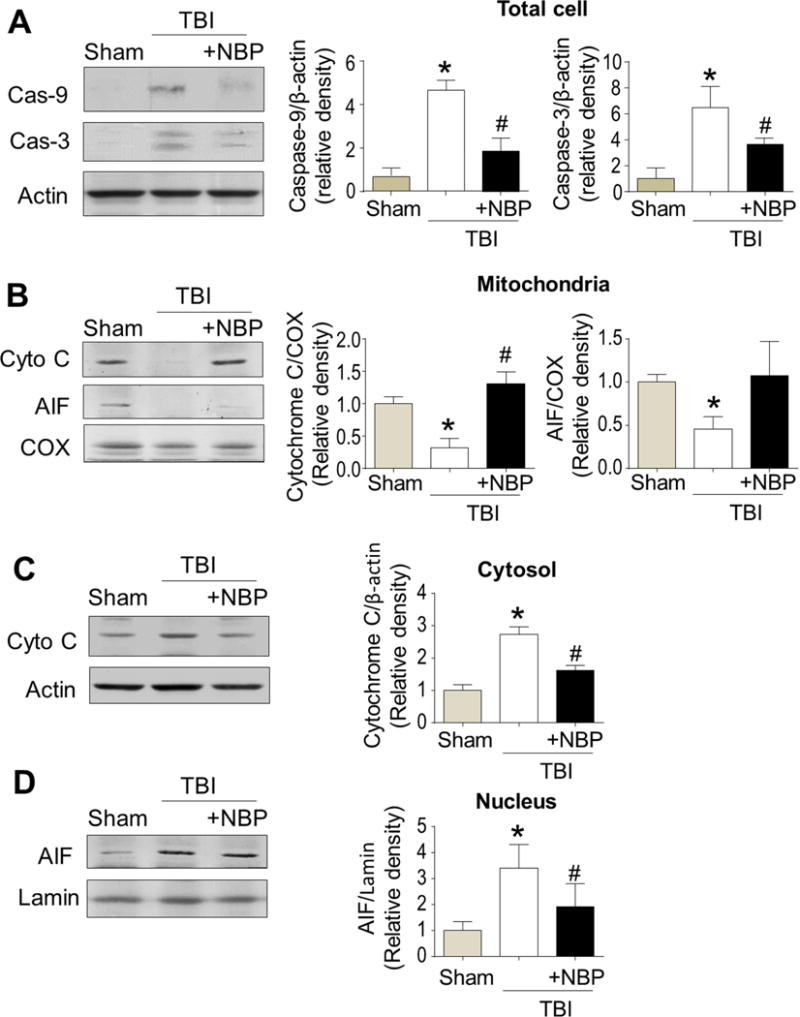
NBP (100 mg/kg, i.p.) was administrated within 5 min after TBI. Expressions or activation of key apoptotic molecules in the peri-contusion tissue was measured using Western blot analysis 24 hrs after TBI. A. TBI significantly enhanced the expression of cleaved caspase-3 and caspase-9. The NBP treatment largely reduced the caspase activation. N=3 for sham control, n=5 for Caspase-3 and -9 groups. B–D. Expressions of cytochrome c and AIF in mitochondrial (B), cytosolic (C), and nucleic (D) fractions after TBI. TBI reduced the expressions of cytochrome c and AIF in the mitochondrial fractionation, while the NBP treatment reversed these changes (B). Cytochrome c oxidase (COX) was measured as an internal control for mitochondrial fractionation. N=5 per group except n=3 for sham control. In panel C, the Western blotting and bar graph show increased cytochrome c in the cytosolic fractionation, correlating to its decreased level in the mitochondria. The NBP treatment prevented the cytosolic increase of cytochrome c. N=5 per group except n=3 for sham control. Increased AIF was observed in the nucleus, whereas NBP reduced this AIF translocation (D). Lamin was used as an internal control for nucleus fractionation. N=5 per group except n=3 for sham control. * P<0.05 versus TBI saline group by one-way ANOVA followed by Bonferroni correction.
Consistent with the upregulation of caspase-9, we observed cytochrome c release from mitochondria to cytosol 24 hr after TBI (Fig. 2B and 2C). TBI mice received NBP treatment had lower levels of cytochrome c in the cytosol and more cytochrome c remained in the mitochondria compared to TBI-saline controls (Fig. 2B and 2C). For caspase-independent apoptosis, we examined the levels of apoptosis-inducing factor (AIF) in the mitochondria and nucleus. In TBI-saline control mice, reduced mitochondrial AIF expression was observed, accompanied with increased AIF in the nuclear, suggesting a pro-apoptotic translocation of AIF (Fig. 2B and 2D). NBP treatment retained more AIF protein in the mitochondria and less AIF in nucleus compared to TBI-saline controls (Fig. 2B and 2D). These data suggested that both caspase-dependent and -independent apoptotic events developed in the TBI brain while the NBP acute treatment was able to suppress these two apoptotic pathways during the early stage after TBP.
NBP treatment reduced NF-κB activation and early inflammatory response after TBI
In Western blot analysis, the level of NF-κB p65 in nuclear protein extracts of pericontusion region was significantly increased 24 hr after TBI compared to sham mice. The TBI-induced increase of nuclear NF-κB p65 was significantly decreased by NBP treatment (Fig. 3A). The level of NF-κB p65 in cytosolic protein fractions of pericontusion region decreased after TBI compared to sham mice. Accordingly, the ratio of nuclear p65 against cytosolic p65 increased in the TBI brain was substantially restored by the NBP treatment (Fig. 3A).
Figure 3. The anti-inflammatory effects of acute NBP treatment in the post-TBI brain.
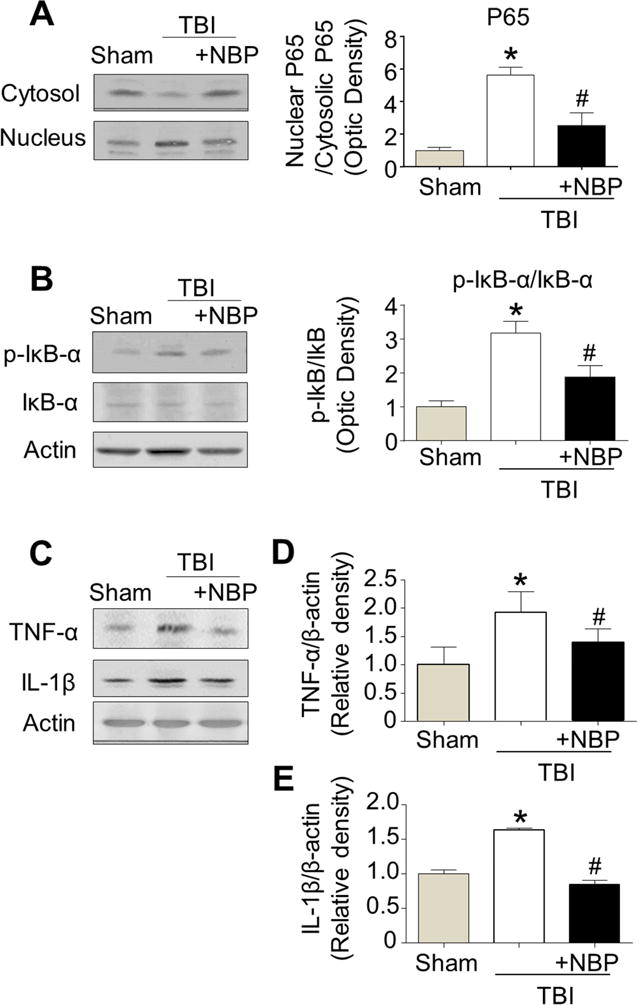
TBI-induced NF-κB activation and inflammatory response were measured using Western blot in the pericontusion region 24 hr after the TBI insult. A. TBI increased translocation of NF-κB p65 from cytosol to nucleus; the increase was inhibited by the acute NBP treatment (100 mg/kg, i.p.). Bar graphs summarize the ratios of nucleic p65/cytosolic p65 in these groups. * P<0.05 versus sham group; # P<0.05 versus TBI-saline control (symbols show the same comparisons in A to E). One-way ANOVA and Bonferroni correction were used in the comparisons. N=3 in sham group, n=5 in TBI-saline control and TBI with NBP treatment, respectively. B. Western blot analysis of p-IκB-α and IκB-α showed the increased ratio of p-IκB-α/IκB-α after TBI while the NBP acute treatment significantly prevented the increase. N=6 in TBI-saline control and TBI plus NBP, respectively. C. Western blot analysis of inflammatory cytokines. D and E. Summarized Western blot assays. TBI increased proinflammatory cytokines including TNF-α (D) and IL-1β (E); The NBP treatment largely prevented these cytokines increases. n=3–5 per group.
In addition, the cytoplasmic level of IκB-α protein was examined by Western blot analysis (Fig. 3B). Results are expressed as the ratio of p-IκB-α vs. total IκB-α, which was significantly elevated in TBI-saline control mice compared to sham mice. However, TBI mice received NBP treatment showed a significantly reduced p-IκB-α/total IκB-α ratio compared to TBI-saline controls (Fig. 3B).
Next we assessed the effect of NBP on the expression of inflammatory mediators. Western blot analysis revealed increases in the expression of tumor necrotizing factor-alpha (TNF-α) and interleukin-1beta (IL-1β) in the TBI brain (Fig. 3C–3E). The expression ratios of TNF-α and IL-1β in NBP-treated TBI mice were significantly decreased compared to TBI-saline controls (Fig. 3D and 3E).
Effects of NBP on delayed expressions of regenerative-related genes after TBI
We were interested to learn whether a NBP chronic treatment could enhance long-term regeneration. After the TBI insult and the acute NBP treatment (100 mg/kg), animals received intranasal NBP treatment (100 mg/kg/day) for 20 days. Western blot analysis was performed at 21 days after TBI to detect the delayed expression of regenerative-related genes including BDNF, VEGF, GDNF, eNOS, CXCR4, and MMP-9 in the pericontusion regions (Fig. 4A). Compared to TBI-saline control mice, the levels of neurotrophin BDNF and pro-angiogenic factors VEGF, eNOS, MMP-9 were significantly increased in the NBP-treated brain (Fig. 4A and 4B). The expression of CXCR4 showed an increase tendency, but did not reach statistical difference (Fig. 4B). There was no difference on GDNF expression among groups.
Figure 4. Chronic NBP treatment increased the expression of BDNF, VEGF, eNOS, and MMP-9 in the post-TBI brain.
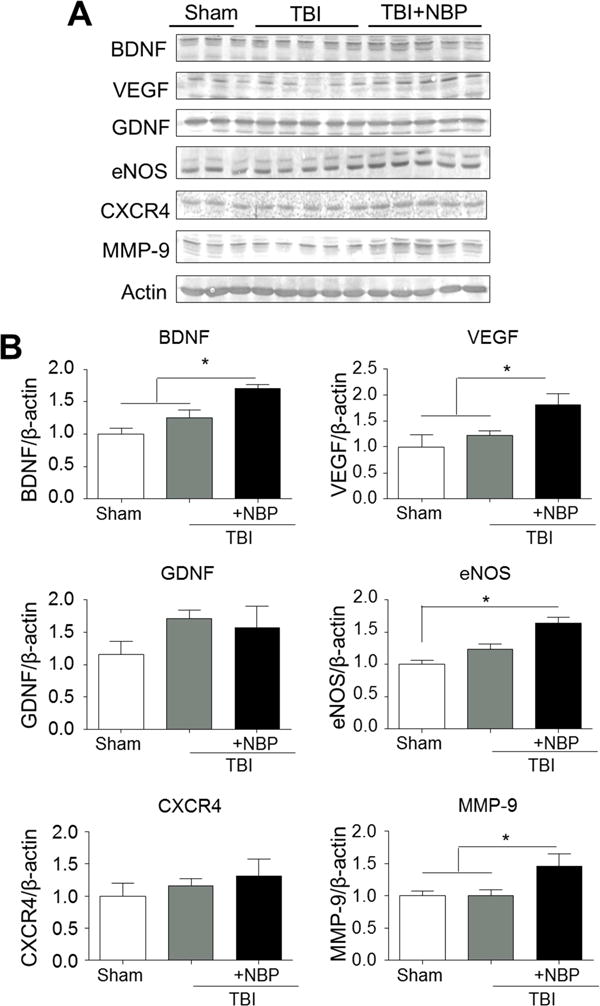
TBI mice received acute as well as daily TBI intranasal treatments until sacrifice. Expressions of regenerative factors were measured using Western blot 21 days after TBI. A. The protein levels of BDNF, VEGF, GDNF, eNOS, CXCR4, and MMP-9. B. Quantified data from A. NBP treatments significantly enhanced the expression of BDNF, VEGF, eNOS, and MMP-9 compared to the control group. There was no significant change in GDNF and CXCR4 expressions among groups. * P<0.05 versus sham group; # P<0.05 versus TBI-saline control group by one-way ANOVA followed by Bonferroni correction; n=5 per group.
NBP treatment increased long-term regenerative activities after TBI
Based on increased regenerative factors after repeated NBP treatments, we evaluated neurogenesis and angiogenesis in the post-TBI brain. Immunohistochemistry analysis of the pericontusion region was performed to detect cells co-stained with NeuN and BrdU (NeuN+/BrdU+) 21 days after TBI. In TBI mice with NBP treatment, the number of NeuN+/BrdU+ cells was significantly greater than that in the TBI control brain (Fig. 5A and 5B). To examine angiogenesis, we stained for Glut-1, a specific marker for glucose transporter in brain vascular endothelial cells (Fig. 5A and 5C). The number of Glut-1+ microvessels co-labeled with BrdU in the pericontusion region was significantly larger compared to that in TBI control mice.
Figure 5. Chronic NBP treatment enhanced regenerative activities in the post-TBI brain.
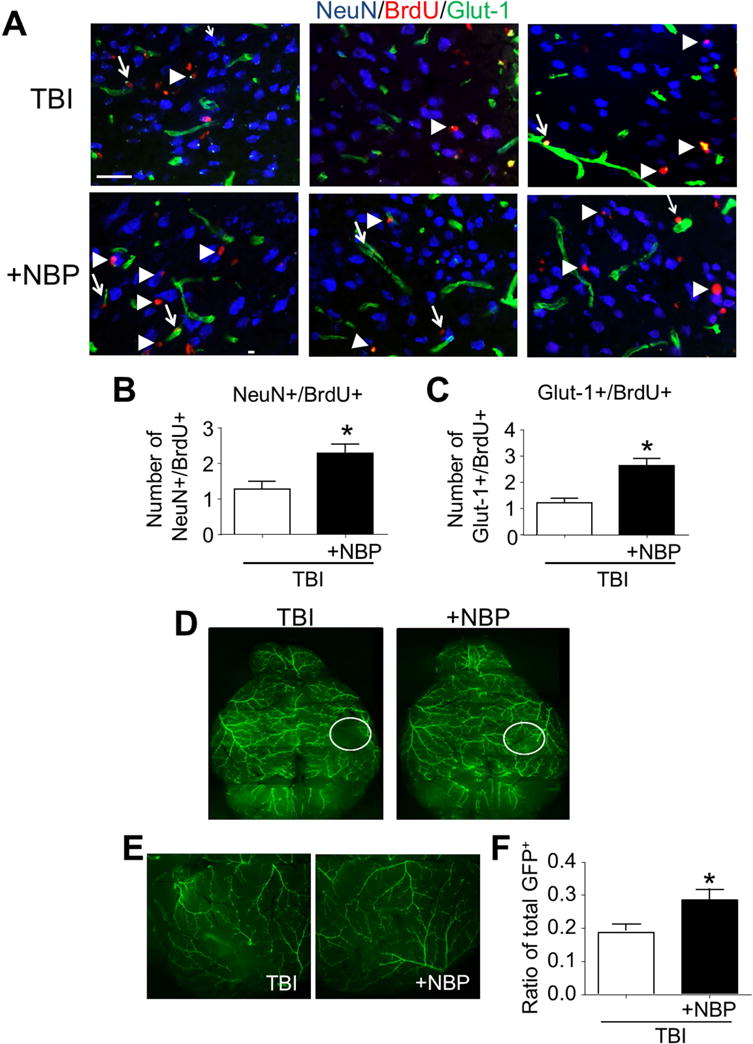
In TBI mice received and acute and chronic NBP treatments, neurogenesis and angiogenesis were analyzed using immunohistochemical staining 21 days after TBI. To reveal arteriogenesis, α-SMA-GFP transgenic mice was tested in this test. A. Neurogenesis and angiogenesis in the pericontusion region were identified by the colocalization of the proliferation marker BrdU (red) with the neuronal marker NeuN (blue) or the vascular endothelial cell marker Glut-1 (green). Scale bar = 50 μm. B and C. Quantified data of cell counts show increased numbers of NeuN/BrdU-positive cells (B) and Glut-1/BrdU-positive cells (C) in the NBP treatment group compared with TBI-saline controls. * P<0.05 versus TBI group; n=6 per group. D and E. Arteries were visualized by the green color of smooth muscle actin in the α-SMA-GFP transgenic brain. Circles in D images illustrate the contusion region. Enlarged images from different animals in E show increased vasculature in the contusion core of the TBI plus NBP brain. Scale bar = 50 μm. F. The bar graph summarizes the ratio of total GFP+ arteries, which is significantly higher than that in TBI-saline control group. * P<0.05 versus TBI-saline control group by Student t test; n=5 per group.
We next examined whether the chronic NBP treatment could influence arteriogenesis. Alpha-smooth muscle actin (α-SMA)-GFP transgenic mice were used to reveal the arteries in the brain. Twenty-one days after TBI, TBI mice received NBP treatment exhibited a higher density of GFP+ arteries around TBI contusion regions compared to TBI control mice (Fig. 5D and 5E). The ratio of total GFP+ arteries in TBI mice with NBP treatment was significantly higher than that of TBI-saline control mice (Fig. 5F).
NBP treatment improves functional outcome after TBI
To examine functional outcomes after TBI, adhesive dot removal, corner, and cylinder tests were performed. In the adhesive dot removal test, the latency times to sense and remove the adhesive from the right and left forelimbs were recorded before, 3 days after, and 7 days after TBI (Fig. 6A). Before TBI, the contact and removal times were similar among groups. After TBI damage to the right side of the sensorimotor cortex, injured animals showed prolonged times in response to the sticky dot attached to their left paws, indicating impaired sensorimotor function on the right side of the brain. TBI mice with the daily repeated NBP treatment showed significantly improved performance 3 days after TBI on both time to detect and time to remove the adhesive (Fig. 6A). In the corner test, the TBI-saline control mice exhibited an abnormal corner turn score (Fig. 6B). TBI mice with NBP treatment exhibited better scores in this test. In addition, TBI mice with NBP treatment performed significantly better in the cylinder test (Fig. 6C). After the TBI injury, the sensorimotor function showed a spontaneous recovery alone time. By day 7 post TBI, there was no more difference between TBI-saline control mice and NBP-treated TBI mice (Fig. 6A–6C). Even delayed tests were thus not performed.
Figure 6. Chronic NBP treatment improved sensorimotor functional recovery after TBI.
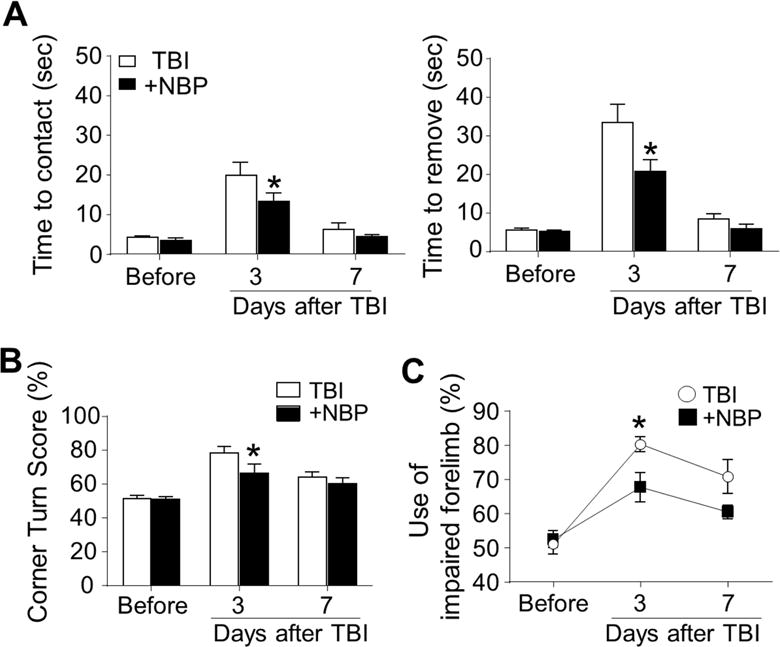
In the long-term experiments of chronic NBP treatment, the adhesive dot removal test, corner test, and cylinder test were used to evaluate the sensorimotor functional recovery different days after TBI. A. The latency to recognize the sticky dot and removal time for the right and left forelimb was recorded at 3 and 7 days after TBI. After TBI damage to the right sensorimotor cortex, both time to contact and time to remove the sticky dot were increased. NBP treatment showed significantly improved performance. * P<0.05 versus TBI-saline control group by two-way ANOVA followed by Bonferroni correction; n=6–7 per group. B. In the corner test, normal mice make right and left turns equally (50% value in the figure) while unilateral TBI damage results uneven turns to one direction. NBP treatment significantly corrected the functional deficit at 3 days after TBI. At day 7, no significant difference was seen because the spontaneous recovery in TBI-saline control mice. * P<0.05 versus TBI-saline control group by two-way ANOVA followed by Bonferroni correction; n=6–7 per group. C. Cylinder test showed that forelimb activities of TBI mice were impaired during the first few days after TBI. NBP treatment group performed significantly better than TBI-saline control group. * P<0.05 versus TBI-saline control group by two-way ANOVA followed by Bonferroni correction; n=6–7 per group.
NBP treatment antagonized the development of depression and anxiety after TBI
Depression-like behavior is a common syndrome after TBI. We investigated the effect of NBP treatments in depression tests different days after TBI. In the forced swim test (FST), the immobility of mice placed in water is reversed by antidepressant treatments (Porsolt et al., 1977b). In our study, immobility during the FST was tested at 3, 14, and 21 days after TBI with saline or repetitive daily NBP treatments. In TBI-saline control mice, we observed increased immobility time at sub-acute and chronic time points after TBI compared to sham mice, indicating the development of depression-like behavior (Fig. 7A). NBP-treated mice had significantly decreased immobility when compared to TBI-saline controls. In the open field test (OFT), TBI-saline control mice spent significantly less time in the center and fewer entrances into the center area measured at 21 days after TBI (Fig. 7B and 7C). These reduced activities suggestive of anxiety were largely corrected in NBP-treated TBI mice (Fig. 7B and 7C). During the OFT test, the locomotor activity such as total distance and velocity were examined to determine whether impaired locomotion after TBI could affect these behavior tests. Although there was a tendency for TBI-saline mice to travel less and slower, the differences were not significant compared either to sham or TBI-NBP groups (Fig. 7D and 7E).
Figure 7. Chronic NBP treatment attenuated post-TBI depressive behaviors.
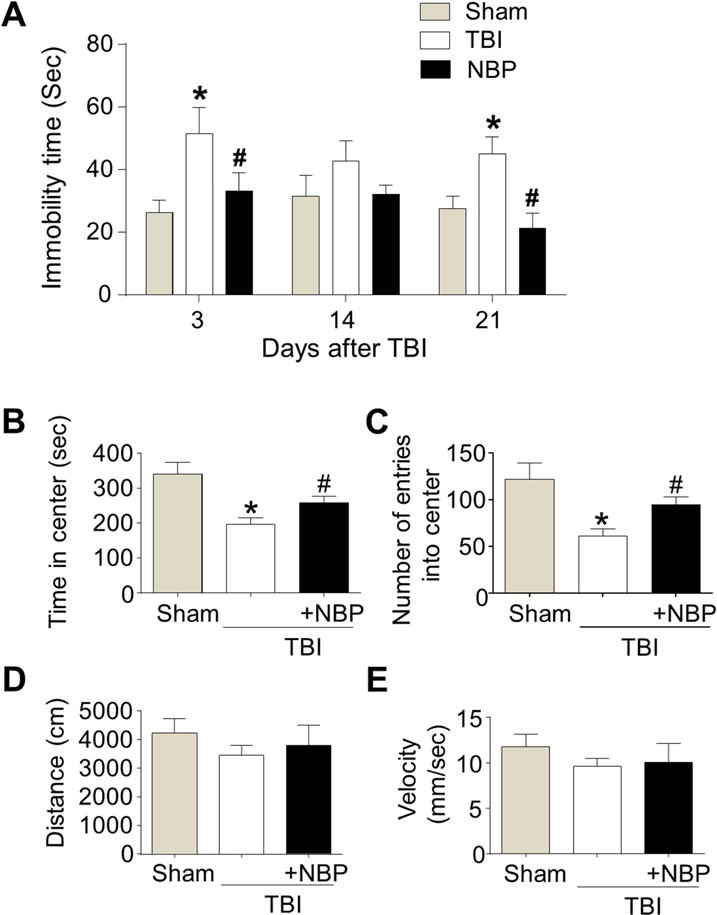
In TBI mice received repeated NBP treatments, forced swim test (FST) and open field test (OFT) were performed to examine the effect of NBP on TBI-induced psychological outcomes. A. In the FST, TBI increased immobility time at 3, 14, and 21 days after TBI. NBP treatment significantly reduced the abnormal behavior. * P<0.05 versus sham group by two-way ANOVA followed by Bonferroni correction; # P<0.05 versus TBI group; n=4–7 per group. B and C. In the OFT, the TBI group showed less time in the center and fewer entries into the center area 21 days after TBI. NBP treatment reversed the behavior change. * P<0.05 versus sham group by two-way ANOVA followed by Bonferroni correction; # P<0.05 versus TBI group; n=4–7 per group. D and E. Summarized data of the distance animals travelled and the velocity during the OFT. There was no significant difference among groups. * P<0.05 versus sham group; # P<0.05 versus TBI-saline controls by two-way ANOVA followed by Bonferroni correction; n=4–7 per group.
Discussion
In the present study, an acute treatment soon after TBI and a combination of acute and chronic treatments of the synthesized neuroprotective compound NBP were tested for the potential brain protective and regenerative benefits in a TBI model of mice. The NBP effect was tested by an acute treatment and a repeated daily treatment of intranasal administration in clinical relevant settings. Intranasal drug administration is a non-invasive method for brain specific drug delivery suitable for chronic treatments. Our findings provide new evidence that, in addition to its neuroprotection against stroke as shown before, NBP is also protective against TBI-induced brain damage. We demonstrate that the NBP neuroprotection was mediated by inhibiting apoptotic cell death and NF-kB-mediated inflammation in the TBI brain. Moreover, we show novel evidence that NBP treatment enhanced important regenerative growth/trophic factors including BDNF, VEGF, and others chronically after the TBI insult. The neuroprotective action and the regenerative effects on neurogenesis, angiogenesis and arteriogenesis are likely the mechanisms that promote long-term functional recovery after TBI. In previous investigations, the neuroprotective effect of NBP were mostly demonstrated after ischemic strokes. So far, there have been only a few reports about the effect of NBP on TBI damages, showing that NBP reduced the size of brain edema, protect the blood brain barrier (BBB) and improved behavioral functions after TBI (Chong and Feng, 2000; Zhao et al., 2013). The underlying mechanism of the NBP effects, however, has not been well defined. In the present study, NBP markedly reduced the brain contusion volume and apoptotic cell death, indicating its strong protective effects at cellular and tissue levels against TBI damages. Our previous findings in the ischemic stroke model provided the evidence that NBP attenuated both cytochrome c release and AIF translocation (Li et al., 2010). Consistently, NBP attenuated the release of cytochrome c and the translocation of AIF after TBI. It is known that cytochrome c and AIF lead to caspase-dependent and caspase-independent apoptotic cascades, respectively (Zhang et al., 2002). Therefore, these findings suggest that NBP could have cellular protective effects through inhibiting not only caspase-dependent but also caspase-independent apoptosis.
Accumulating data from preclinical and clinical studies suggest that inflammatory response can be a key mechanism in the development of TBI damage leading to neural death and increased brain edema (Clark et al., 1994; Corps et al., 2015; Scheff et al., 2013). Post-TBI inflammation is characterized by the production of pro- and anti-inflammatory cytokines, the accumulation of neutrophils, and the activation of microglia in the injured brain (Clark et al., 1994; Morganti-Kossmann et al., 2002). Activation of NF-κB signaling is considered a primary regulator of inflammatory responses (O’Neill and Kaltschmidt, 1997). After cellular damage, NF-κB releases from IκB-α, enters the nucleus and promotes the transcription of its target genes including inflammatory-related genes, which leads to the expression of a large variety of proinflammatory molecules that can eventually contribute to cell death after TBI (O’Neill and Kaltschmidt, 1997). In line with these findings, our previous study showed that TBI induced inflammatory response including not only microglial activation but also increases in pro-inflammatory cytokines (TNF-α and IL-1β) (Lee et al., 2014). In the present study, TBI damage increased both IκB-α activation and NF-κB nuclear translocation and enhanced the expressions of proinflammatory cytokines such as TNF-α and IL-1β These observations agree with the idea that the NF-κB signaling pathway participates in the TBI-induced inflammatory response. Importantly, NBP treatment suppressed not only IκB-α activation and NF-κB nuclear translocation but also proinflammatory cytokines induction. Furthermore, previous findings revealed that inflammation impairs neurogenesis levels after injury through increasing activated microglia and secretion of numerous proinflammatory cytokines including TNF-α and IL-1β (Ben-Hur et al., 2003; Curtis et al., 2007; Jakubs et al., 2008). This idea was supported when minocycline, an anti-inflammatory drug, alleviated reduction of new neurons after lipopolysaccharide exposure (Ekdahl et al., 2003). Taken together, it is likely that NBP may attenuate TBI-induced cell death through the inhibitory action on inflammation including blocking of the NF-κB activation.
Neurogenesis has been observed in response to trauma and other insults, suggesting that the brain has the inherent potential to restore populations of damaged or destroyed neurons (Rolfe and Sun, 2015). Therefore, therapeutic treatments and approaches with neurogenic capacity could be applicable therapies for treating patients with stroke and TBI. In this study, immunohistochemical results showed increased NeuN/BrdU-positive cells in the NBP treatment group, consistent with enhanced expression of BDNF. BDNF increases neuronal differentiation, neurite outgrowth, and cell survival of neural progenitor cells and neurons during development and in the adult brain (Malberg et al., 2000; Palmer et al., 1997; Takahashi et al., 1999). Therefore, it is suggested that NBP treatment may enhance neurogenesis through regulating BDNF; further study will be needed to reveal the mediator role of BDNF in NBP induced effects.
The enhanced growth of functional blood vessels is essential for the restoration of blood flow to decrease brain damage and improve long-term functional outcome (Liu et al., 2008; Tobin et al., 2014). Angiogenesis and arteriogenesis support restored perfusion in the brain injury and promoted long-term outcome in stroke and TBI patients (Christoforidis et al., 2005; Wei et al., 2001). Therefore, enhancement of angiogenesis and arteriogenesis in the brain injury can be an attractive therapy for stroke and TBI (Carmeliet, 2000; Erdo and Buschmann, 2007; Semenza, 2007). We provided new findings that NBP treatment enhances angiogenesis and arteriogenesis after TBI. Also, we observed that VEGF and eNOS were upregulated 21 days after TBI. VEGF is known as an important factor in the process of angiogenesis and arteriogenesis (Carmeliet, 2000). The binding of VEGF to its receptor, VEGFR-2, on the surface of endothelial cells activates intracellular tyrosine kinases, triggering multiple downstream signals that promote angiogenesis and arteriogenesis (Greenberg and Jin, 2013). eNOS also plays an important role in the revascularization process including endothelial cell proliferation and migration, smooth muscle cell differentiation, angiogenic processes, and arterial-venous differentiation (Gertz et al., 2006; Luque Contreras et al., 2006). In our study, NBP treatment increased MMP-9 expression 21 days after TBI. MMP-9 has been shown to be essential for capillary branching, invasion and tube formation of endothelial cells, and enhanced angiogenesis (Collen et al., 2003; Johnson et al., 2004).
Depression is a commonly reported symptom in patients with TBI (Barker-Collo et al., 2013; Fann et al., 2001). Although the exact etiological factors leading to the development of depression after TBI still remain unclear, accumulating data suggest that a complex interaction of neurological, psychological, and social factors after TBI contribute to the development of depression (Anderson et al., 2004; Rosenberg et al., 2015; Williams and Evans, 2003). Importantly, previous studies have shown that depression after TBI is strongly linked to poor psychosocial and functional outcomes (Barker-Collo et al., 2013). In the present study, NBP treatment effectively suppressed depressive-like symptoms. A previous study revealed that chronic antidepressant treatment increases neurogenesis in the adult rat hippocampus (Malberg et al., 2000). Anti-depressants increase the expression of BDNF in the hippocampus and prefrontal cortex (Castren et al., 2007; Nibuya et al., 1995). We show here that NBP has an anti-depressive effect after TBI in two psychiatric behavior tests. As reduced neurogenesis has been thought to be involved in the neuropathology of depression, the NBP’s antidepressant action may be due to its ability to promote neurogenesis and the inhibitory action on inflammation. Although the underlying molecular and cellular mechanisms of NBP’s anti-depressive effect remain to be further investigated, these findings raise the possibility that NBP treatment might be applied for patients with post-TBI depression.
Highlights.
DL-3-n-butylphthalide (NBP) treatment reduced contusion volume after TBI.
NBP treatment blocked apoptotic neuronal cell death.
NBP treatment suppressed inflammation.
Chronic intranasal NBP delivery increased regeneration in the post-TBI brain.
NBP promoted functional recovery and prevented post-TBI depression.
Acknowledgments
This study was supported by NS075338 (LW), NS085568 (LW/SPY), NS091585 (LW), AHA Postdoctoral Fellowship 15POST25680013 (JHL), and a VA Merit grant RX000666 (SPY).
Abbreviations
- NBP
DL-3-n-butylphthalide
- TBI
Traumatic brain injury
- CCI
Controlled cortical impact
- TUNEL
Terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling
- AIF
Apoptosis-inducing factor
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- IκB
Inhibitor of NF-κB
- p-IκB
Phosphorylation of IκB
- TNF-α
Tumor necrotizing factor-alpha
- IL-1 β
interleukin-1beta
- BDNF
Brain-derived neurotrophic factor
- VEGF
Vascular endothelial growth factor
- GDNF
Glial cell-derived neurotrophic factor
- eNOS
Endothelial-derived nitric oxide synthase
- MMP-9
Matrix metallopeptidase 9
- α-SMA
Alpha-smooth muscle actin
- GFP
Green fluorescent protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Disclosure Statement
NBP was a gift from Shijiazhuang Pharmaceutical Group Ouyi Pharma Co., Ltd, (Shijiazhuang, China). No competing financial interests exist.
References
- Anderson VA, Morse SA, Catroppa C, Haritou F, Rosenfeld JV. Thirty month outcome from early childhood head injury: a prospective analysis of neurobehavioural recovery. Brain. 2004;127:2608–2620. doi: 10.1093/brain/awh320. [DOI] [PubMed] [Google Scholar]
- Barker-Collo S, Starkey N, Theadom A. Treatment for depression following mild traumatic brain injury in adults: a meta-analysis. Brain Inj. 2013;27:1124–1133. doi: 10.3109/02699052.2013.801513. [DOI] [PubMed] [Google Scholar]
- Ben-Hur T, Ben-Menachem O, Furer V, Einstein O, Mizrachi-Kol R, Grigoriadis N. Effects of proinflammatory cytokines on the growth, fate, and motility of multipotential neural precursor cells. Mol Cell Neurosci. 2003;24:623–631. doi: 10.1016/s1044-7431(03)00218-5. [DOI] [PubMed] [Google Scholar]
- Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: similarities and differences. J Cereb Blood Flow Metab. 2004;24:133–150. doi: 10.1097/01.WCB.0000111614.19196.04. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–395. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- Castren E, Voikar V, Rantamaki T. Role of neurotrophic factors in depression. Curr Opin Pharmacol. 2007;7:18–21. doi: 10.1016/j.coph.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Chang Q, Wang XL. Effects of chiral 3-n-butylphthalide on apoptosis induced by transient focal cerebral ischemia in rats. Acta Pharmacol Sin. 2003;24:796–804. [PubMed] [Google Scholar]
- Choi KE, Hall CL, Sun JM, Wei L, Mohamad O, Dix TA, Yu SP. A novel stroke therapy of pharmacologically induced hypothermia after focal cerebral ischemia in mice. FASEB J. 2012;26:2799–2810. doi: 10.1096/fj.11-201822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Z, Feng Y. dl-3-n-butylphthalide reduces brain damage in mice with closed head injury. Chin Med J (Engl) 2000;113:613–616. [PubMed] [Google Scholar]
- Chong ZZ, Feng YP. dl-3-n-butylphthalide attenuates reperfusion-induced blood-brain barrier damage after focal cerebral ischemia in rats. Zhongguo Yao Li Xue Bao. 1999;20:696–700. [PubMed] [Google Scholar]
- Christoforidis GA, Mohammad Y, Kehagias D, Avutu B, Slivka AP. Angiographic assessment of pial collaterals as a prognostic indicator following intra-arterial thrombolysis for acute ischemic stroke. AJNR Am J Neuroradiol. 2005;26:1789–1797. [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Schiding JK, Kaczorowski SL, Marion DW, Kochanek PM. Neutrophil accumulation after traumatic brain injury in rats: comparison of weight drop and controlled cortical impact models. J Neurotrauma. 1994;11:499–506. doi: 10.1089/neu.1994.11.499. [DOI] [PubMed] [Google Scholar]
- Collen A, Hanemaaijer R, Lupu F, Quax PH, van Lent N, Grimbergen J, Peters E, Koolwijk P, van Hinsbergh VW. Membrane-type matrix metalloproteinase-mediated angiogenesis in a fibrin-collagen matrix. Blood. 2003;101:1810–1817. doi: 10.1182/blood-2002-05-1593. [DOI] [PubMed] [Google Scholar]
- Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, Guzman BR, Hemphill JD. Surveillance for traumatic brain injury-related deaths—United States, 1997–2007. MMWR Surveill Summ. 2011;60:1–32. [PubMed] [Google Scholar]
- Corps KN, Roth TL, McGavern DB. Inflammation and Neuroprotection in Traumatic Brain Injury. JAMA Neurol. 2015 doi: 10.1001/jamaneurol.2014.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Page ME, Lucki I. Differential behavioral effects of the antidepressants reboxetine, fluoxetine, and moclobemide in a modified forced swim test following chronic treatment. Psychopharmacology (Berl) 2005;182:335–344. doi: 10.1007/s00213-005-0093-5. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Kam M, Nannmark U, Anderson MF, Axell MZ, Wikkelso C, Holtas S, van Roon-Mom WM, Bjork-Eriksson T, Nordborg C, Frisen J, Dragunow M, Faull RL, Eriksson PS. Human neuroblasts migrate to the olfactory bulb via a lateral ventricular extension. Science. 2007;315:1243–1249. doi: 10.1126/science.1136281. [DOI] [PubMed] [Google Scholar]
- Deng W, Feng Y. Effect of dl-3-n-butylphthalide on brain edema in rats subjected to focal cerebral ischemia. Chin Med Sci J. 1997;12:102–106. [PubMed] [Google Scholar]
- Djupesland PG, Messina JC, Mahmoud RA. The nasal approach to delivering treatment for brain diseases: an anatomic, physiologic, and delivery technology overview. Ther Deliv. 5:709–733. doi: 10.4155/tde.14.41. [DOI] [PubMed] [Google Scholar]
- Dong GX, Feng YP. Effects of NBP on ATPase and anti-oxidant enzymes activities and lipid peroxidation in transient focal cerebral ischemic rats. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. 2002;24:93–97. [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdo F, Buschmann IR. Arteriogenesis: a new strategy of therapeutic intervention in chronic arterial disorders. Cellular mechanism and experimental models. Orv Hetil. 2007;148:633–642. doi: 10.1556/OH.2007.27916. [DOI] [PubMed] [Google Scholar]
- Fann JR, Uomoto JM, Katon WJ. Cognitive improvement with treatment of depression following mild traumatic brain injury. Psychosomatics. 2001;42:48–54. doi: 10.1176/appi.psy.42.1.48. [DOI] [PubMed] [Google Scholar]
- Feng YP, Hu D, Zhang LY. Effect of DL-butylphthalide (NBP) on mouse brain energy metabolism in complete brain ischemia induced by decapitation. Yao Xue Xue Bao. 1995;30:741–744. [PubMed] [Google Scholar]
- Gage FH, Kempermann G, Palmer TD, Peterson DA, Ray J. Multipotent progenitor cells in the adult dentate gyrus. J Neurobiol. 1998;36:249–266. doi: 10.1002/(sici)1097-4695(199808)36:2<249::aid-neu11>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Gertz K, Priller J, Kronenberg G, Fink KB, Winter B, Schrock H, Ji S, Milosevic M, Harms C, Bohm M, Dirnagl U, Laufs U, Endres M. Physical activity improves long-term stroke outcome via endothelial nitric oxide synthase-dependent augmentation of neovascularization and cerebral blood flow. Circ Res. 2006;99:1132–1140. doi: 10.1161/01.RES.0000250175.14861.77. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci. 2013;70:1753–1761. doi: 10.1007/s00018-013-1282-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc. 2012;7:2005–2012. doi: 10.1038/nprot.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JZ, Chen YZ, Su M, Zheng HF, Yang YP, Chen J, Liu CF. dl-3-n-Butylphthalide prevents oxidative damage and reduces mitochondrial dysfunction in an MPP(+)-induced cellular model of Parkinson’s disease. Neurosci Lett. 2010;475:89–94. doi: 10.1016/j.neulet.2010.03.053. [DOI] [PubMed] [Google Scholar]
- Jakubs K, Bonde S, Iosif RE, Ekdahl CT, Kokaia Z, Kokaia M, Lindvall O. Inflammation regulates functional integration of neurons born in adult brain. J Neurosci. 2008;28:12477–12488. doi: 10.1523/JNEUROSCI.3240-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C, Sung HJ, Lessner SM, Fini ME, Galis ZS. Matrix metalloproteinase-9 is required for adequate angiogenic revascularization of ischemic tissues: potential role in capillary branching. Circ Res. 2004;94:262–268. doi: 10.1161/01.RES.0000111527.42357.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Wei L, Deveau TC, Gu X, Yu SP. Expression of the NMDA receptor subunit GluN3A (NR3A) in the olfactory system and its regulatory role on olfaction in the adult mouse. Brain Struct Funct. 2015 doi: 10.1007/s00429-015-1099-3. [DOI] [PubMed] [Google Scholar]
- Lee JH, Wei L, Gu X, Wei Z, Dix TA, Yu SP. Therapeutic effects of pharmacologically induced hypothermia against traumatic brain injury in mice. J Neurotrauma. 2014;31:1417–1430. doi: 10.1089/neu.2013.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Li Y, Ogle M, Zhou X, Song M, Yu SP, Wei L. DL-3-n-butylphthalide prevents neuronal cell death after focal cerebral ischemia in mice via the JNK pathway. Brain Res. 2010;1359:216–226. doi: 10.1016/j.brainres.2010.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao SJ, Lin JW, Pei Z, Liu CL, Zeng JS, Huang RX. Enhanced angiogenesis with dl-3n-butylphthalide treatment after focal cerebral ischemia in RHRSP. Brain Res. 2009;1289:69–78. doi: 10.1016/j.brainres.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Lin JF, Feng YP. Effect of dl-3-n-butylphthalide on delayed neuronal damage after focal cerebral ischemia and intrasynaptosomes calcium in rats. Yao Xue Xue Bao. 1996;31:166–170. [PubMed] [Google Scholar]
- Liou AK, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog Neurobiol. 2003;69:103–142. doi: 10.1016/s0301-0082(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Liu XB, Wang JA, Yu SP, Keogh CL, Wei L. Therapeutic strategy of erythropoietin in neurological disorders. CNS Neurol Disord Drug Targets. 2008;7:227–234. doi: 10.2174/187152708784936617. [DOI] [PubMed] [Google Scholar]
- Liu XG, Feng YP. Protective effect of dl-3-n-butylphthalide on ischemic neurological damage and abnormal behavior in rats subjected to focal ischemia. Yao Xue Xue Bao. 1995;30:896–903. [PubMed] [Google Scholar]
- Luque Contreras D, Vargas Robles H, Romo E, Rios A, Escalante B. The role of nitric oxide in the post-ischemic revascularization process. Pharmacol Ther. 2006;112:553–563. doi: 10.1016/j.pharmthera.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Ma S, Xu S, Liu B, Li J, Feng N, Wang L, Wang X. Long-term treatment of l-3-n-butylphthalide attenuated neurodegenerative changes in aged rats. Naunyn Schmiedebergs Arch Pharmacol. 2009;379:565–574. doi: 10.1007/s00210-009-0398-8. [DOI] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamad O, Song M, Wei L, Yu SP. Regulatory roles of the NMDA receptor GluN3A subunit in locomotion, pain perception and cognitive functions in adult mice. J Physiol. 2013;591:149–168. doi: 10.1113/jphysiol.2012.239251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr Opin Crit Care. 2002;8:101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Palmer TD, Takahashi J, Gage FH. The adult rat hippocampus contains primordial neural stem cells. Mol Cell Neurosci. 1997;8:389–404. doi: 10.1006/mcne.1996.0595. [DOI] [PubMed] [Google Scholar]
- Peng Y, Xu S, Chen G, Wang L, Feng Y, Wang X. l-3-n-Butylphthalide improves cognitive impairment induced by chronic cerebral hypoperfusion in rats. J Pharmacol Exp Ther. 2007;321:902–910. doi: 10.1124/jpet.106.118760. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Bertin A, Jalfre M. Behavioral despair in mice: a primary screening test for antidepressants. Arch Int Pharmacodyn Ther. 1977a;229:327–336. [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977b;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Rolfe A, Sun D. Stem Cell Therapy in Brain Trauma: Implications for Repair and Regeneration of Injured Brain in Experimental TBI Models. In: Kobeissy FH, editor. Brain Neurotrauma. Molecular, Neuropsychological, and Rehabilitation Aspects; Boca Raton (FL): 2015. [PubMed] [Google Scholar]
- Rosenberg H, Dethier M, Kessels RP, Westbrook RF, McDonald S. Emotion Perception After Moderate-Severe Traumatic Brain Injury: The Valence Effect and the Role of Working Memory, Processing Speed, and Nonverbal Reasoning. Neuropsychology. 2015 doi: 10.1037/neu0000171. [DOI] [PubMed] [Google Scholar]
- Runyan DK. The challenges of assessing the incidence of inflicted traumatic brain injury: a world perspective. Am J Prev Med. 2008;34:S112–115. doi: 10.1016/j.amepre.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Scheff SW, Ansari MA, Roberts KN. Neuroprotective effect of Pycnogenol(R) following traumatic brain injury. Exp Neurol. 2013;239:183–191. doi: 10.1016/j.expneurol.2012.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Vasculogenesis, angiogenesis, and arteriogenesis: mechanisms of blood vessel formation and remodeling. J Cell Biochem. 2007;102:840–847. doi: 10.1002/jcb.21523. [DOI] [PubMed] [Google Scholar]
- Silver JM, McAllister TW, Yudofsky SC. Textbook of traumatic brain injury. American Psychiatric Pub; Washington, DC: 2011. [Google Scholar]
- Simard JM, Kahle KT, Gerzanich V. Molecular mechanisms of microvascular failure in central nervous system injury–synergistic roles of NKCC1 and SUR1/TRPM4. J Neurosurg. 2010;113:622–629. doi: 10.3171/2009.11.JNS081052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutzmann JM, Mary V, Wahl F, Grosjean-Piot O, Uzan A, Pratt J. Neuroprotective profile of enoxaparin, a low molecular weight heparin, in in vivo models of cerebral ischemia or traumatic brain injury in rats: a review. CNS Drug Rev. 2002;8:1–30. doi: 10.1111/j.1527-3458.2002.tb00213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi J, Palmer TD, Gage FH. Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J Neurobiol. 1999;38:65–81. [PubMed] [Google Scholar]
- Tobin MK, Bonds JA, Minshall RD, Pelligrino DA, Testai FD, Lazarov O. Neurogenesis and inflammation after ischemic stroke: what is known and where we go from here. J Cereb Blood Flow Metab. 2014;34:1573–1584. doi: 10.1038/jcbfm.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L, Erinjeri JP, Rovainen CM, Woolsey TA. Collateral growth and angiogenesis around cortical stroke. Stroke. 2001;32:2179–2184. doi: 10.1161/hs0901.094282. [DOI] [PubMed] [Google Scholar]
- Williams WH, Evans JJ. Brain injury and emotion: An overview to a special issue on biopsychosocial approaches in neurorehabilitation. Neuropsychol Rehabil. 2003;13:1–11. doi: 10.1016/S0960-9822(02)01374-X. [DOI] [PubMed] [Google Scholar]
- Xiong N, Huang J, Chen C, Zhao Y, Zhang Z, Jia M, Zhang Z, Hou L, Yang H, Cao X, Liang Z, Zhang Y, Sun S, Lin Z, Wang T. Dl-3-n-butylphthalide, a natural antioxidant, protects dopamine neurons in rotenone models for Parkinson’s disease. Neurobiol Aging. 2012;33:1777–1791. doi: 10.1016/j.neurobiolaging.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Xu HL, Feng YP. Inhibitory effects of chiral 3-n-butylphthalide on inflammation following focal ischemic brain injury in rats. Acta Pharmacol Sin. 2000;21:433–438. [PubMed] [Google Scholar]
- Yan CH, Feng YP, Zhang JT. Effects of dl-3-n-butylphthalide on regional cerebral blood flow in right middle cerebral artery occlusion rats. Zhongguo Yao Li Xue Bao. 1998;19:117–120. [PubMed] [Google Scholar]
- Yokota T, Kawakami Y, Nagai Y, Ma JX, Tsai JY, Kincade PW, Sato S. Bone marrow lacks a transplantable progenitor for smooth muscle type alpha-actin-expressing cells. Stem Cells. 2006;24:13–22. doi: 10.1634/stemcells.2004-0346. [DOI] [PubMed] [Google Scholar]
- Zhang LY, Feng YP. Effect of dl-3-n-butylphthalide (NBP) on life span and neurological deficit in SHRsp rats. Yao Xue Xue Bao. 1996;31:18–23. [PubMed] [Google Scholar]
- Zhang X, Chen J, Graham SH, Du L, Kochanek PM, Draviam R, Guo F, Nathaniel PD, Szabo C, Watkins SC, Clark RS. Intranuclear localization of apoptosis-inducing factor (AIF) and large scale DNA fragmentation after traumatic brain injury in rats and in neuronal cultures exposed to peroxynitrite. J Neurochem. 2002;82:181–191. doi: 10.1046/j.1471-4159.2002.00975.x. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Li J, Zhang P, Chen C, Li S. Protective effects of dl-3n-butylphthalide against diffuse brain injury. Neural Regen Res. 2013;8:2615–2624. doi: 10.3969/j.issn.1673-5374.2013.28.003. [DOI] [PMC free article] [PubMed] [Google Scholar]