

Abstract
Genotoxic carcinogens pose great hazard to human health. Uncertainty of current risk assessment strategies and long latency periods between first carcinogen exposure and diagnosis of tumors have raised interest in precictive biomarkers. Initial DNA adduct formation is a necessary step for genotoxin induced carcinogenesis. However, as DNA adducts not always translate into tumorigenesis, their predictive value is limited. Here we hypothesize that the combined analysis of pro-mutagenic DNA adducts along with time-matched gene expression changes could serve as a superior prediction tool for genotoxic carcinogenesis.
Eker rats, heterozygous for the tuberous sclerosis (Tsc2) tumor suppressor gene and thus highly susceptible towards genotoxic renal carcinogens, were continuously treated with the DNA alkylating carcinogen methylazoxymethanol acetate (MAMAc). Two weeks of MAMAc treatment resulted in a time-dependent increase of O6-methylguanine and N7-methylguanine adducts in the kidney cortex, which was however not reflected by significant expression changes of cyto-protective genes involved in DNA repair, cell cycle arrest or apoptosis. Instead, we found a transcriptional regulation of genes involved in the tumor-related MAPK, FoxO and TGF-beta pathways. Continuous MAMAc treatment for up to six months resulted in a mild but significant increase of cancerous lesions.
In summary, the combined analysis of DNA adducts and early gene expression changes could serve as a suitable predictive tool for genotoxicant-induced carcinogenesis.
Keywords: Methylazoxymethanol acetate, DNA adducts, kidney cancer, Eker rat, biomarkers of effect, biomarkers of exposure
Introduction
Humans are exposed to numerous chemicals with potential adverse health effects. Genotoxic carcinogens pose the greatest hazard as they are capable of causing cancer by directly binding and altering the genetic material of target cells. Accordingly, assessment of the genotoxic potential of chemicals is a key regulatory requirement for human safety. Hazard identification for genotoxic carcinogens mainly relies on mutagenicity studies in bacteria and mammalian cells. Additional studies in rodents are employed to confirm the in vitro findings and to establish dose-response relationships. Mathematical models are subsequently used to extrapolate risks from high dose rodent studies to putative low dose exposure effects in humans (Guerard et al. 2015; OECD 1981).
Ultrasensitive mass spectrometry methods for DNA adducts now enable direct quantitation of DNA adduct formation at their target site. This measurement of DNA adducts as “biomarkers of exposure” allows to compensate for individual, species-and tissue- specific differences in toxicokinetic factors such as carcinogen absorption, tissue distribution, metabolic activation, or cytoprotective mechanisms including phase 2 detoxification reactions, DNA repair, or programmed cell death (Swenberg et al. 2008; Swenberg et al. 2011; Jarabek et al. 2009). However, whether or not DNA adducts translate into later tumorigenesis, largely depends on the chemical stability and mutagenicity of the specific adduct types, their molecular dose at the target site and the repair capacity of the affected cell. Such complex interplay could be reflected by very specific gene expression changes, which may serve as early “effect biomarkers” in response to DNA adduct formation.
Our study hypothesized that a combined evaluation of DNA adduct formation and transcriptional activation of genes involved in cell cycle arrest, DNA repair, apoptosis or other cancer related pathways could be indicative for the effectiveness of cellular protection and therefore for the risk of DNA adducts to translate into tumor causing mutations.
Time-matched measurements of DNA adducts and transcriptional profiling after 1, 3 and 14 days of low-dose genotoxin exposure were compared with the number and incidence of pre-neoplastic lesions after 6 months of continuous exposure to unravel the predictive value of combining DNA adduct and gene expression analyses for the etiology of tumors. As a model genotoxicant we used the well-described DNA alkylating agent methylazoxymethanol acetate (MAMAc), an acetylated metabolite of the cycad plant azoglucoside cycasin (Matsumoto and Higa 1966; Matsushima et al. 1979) that causes cancer of the kidney, colon and liver at acute high doses (Laqueur et al. 1967; Notman et al. 1982; Matsubara et al. 1978). Focusing specifically on the kidney as target organ we have chosen Eker rats as highly susceptible genetic rat model of renal cancer. Eker rats carry an inherited heterozygous mutation in the tuberous sclerosis (Tsc2) tumor suppressor gene and develop renal cancer at an early age (Yeung et al. 1994). Numbers and incidence of cancerous lesions can be highly aggravated upon chemical renal carcinogen exposure (Walker et al. 1992; Wolf et al. 2000; McDorman et al. 2003; Satake et al. 2002; Morton et al. 2002; Patel et al. 2003; Stemmer et al. 2007; Stemmer et al. 2009). The quantitative nature of lesion formation in Eker rats therefore allows for the production of statistically powerful data after short-term exposures.
Collectively, short-term exposure to low dose MAMAc (250 μg/kg body weight (BW)) resulted in a time-dependent increase of DNA adducts in the Eker rat kidney, including highly stable and pro-mutagenic O6-methyguanine (O6MG) adducts. Interestingly, we found no evidence for time-matched gene expression changes involved in cellular defense mechanisms. The subsequent mild but significant increase in pre-neoplastic and neoplastic lesions in up to 6 months MAMAc treated Eker rats suggest that DNA repair was not efficient enough to protect against increasing numbers of pro-mutagenic DNA adducts. In summary, our data suggest an improved predictive value of DNA adduct quantification, if combined with time matched gene expression analysis.
Material and Methods
Methylazoxymethanol acetate (MAMAc)
Highly purified MAMAc was purchased from the Midwest Research Institute, NCI Chemical Resource Repository (64 FR 72090, 64 FR 28205).
Animals
Six to ten weeks old heterozygous Tsc2 mutant Eker rats (Tsc2+/-, Long Evans) and Tsc2+/+ wild-type littermates were purchased from the MD Anderson Cancer Center, Smithville, Texas, USA and housed at the University of Konstanz animal research facility under standard housing conditions. Prior to exposure, male and female Eker rats were randomly assigned to dose groups and allowed to acclimatize to laboratory conditions for 4 weeks. All animal experiments were approved by the State of Baden-Württemberg, Germany (Regierungspräsidium Freiburg, AZ: G-03/65).
Exposure experiments
For short-term experiments, groups of three male and female Eker rats were daily treated with MAMAc (250 μg/kg BW) by oral gavage for 1, 3, 7, and 14 days, respectively. Time-matched vehicle controls (n=3) received corresponding volumes of 0.1 M NaHCO3.
For chronic experiments, groups of 10 male and female Eker rats were treated with MAMAc (250 μg/ kg BW) or vehicle (0.1 M NaHCO3) via gavage at five days a week for 3 or 6 months.
Sample collection
At the end of each treatment period, anesthetized rats were sacrificed by exsanguination subsequent to retrograde perfusion with phosphate-buffered saline (PBS). Kidneys were collected and sectioned longitudinally into 5 mm slices. Sections were either snap frozen, fixed with RNAlater (Qiagen, Germany) or PBS buffered histology fixative buffer containing 2% paraformaldehyde and 1% glutaraldehyde for subsequent paraffin embedding and sectioning.
Histopathology
Haematoxylin and Eosin (H&E) stained renal paraffin section were randomized and histopathological analyses were carried out by blinded microscopic examination at 40 to 400-fold magnification. Pre-neoplastic and neoplastic lesions were classified according to their histopathological phenotype as reported previously (Dietrich and Swenberg 1991). We further determined absolute numbers and incidences of pre-neoplastic and neoplastic lesions. Non-neoplastic pathologies were ranked as absent (0), mild (1), moderate (2), pronounced (3) and severe (4).
Cell proliferation analysis
Cell proliferation in paraffin embedded kidney sections of rats exposed to vehicle or MAMAc for 14 days was evaluated by immunohistochemical staining for proliferating cell nuclear antigen (PCNA) using a monoclonal primary anti-PCNA antibody (PC-10; DAKO, Germany) as described previously (Stemmer et al. 2007). Cell proliferation in 3 and 6 months MAMAc treated rats was determined by BrdU labeling. Briefly, 5 days prior to sacrifice, 5 of 10 rats per group and sex were subcutaneously implanted with ALZET® osmotic pumps (Model 2ML1, Charles River Laboratories, Germany) containing 5-bromo-2-deoxyuridine (BrdU, 20 mg/ml sterile saline, Sigma Aldrich, Germany). BrdU immunostaining was performed as described previously (Stemmer et al. 2009), using a monoclonal mouse anti-BrdU primary antibody (MU247-UC, Biogenex, USA). PCNA- and BrdU-positive S-phase nuclei were quantified on randomized sections. 20 microscopic fields (10x ocular, 40x objective) were randomly chosen within the areas of the renal cortex and the medulla /papilla. At least 1.000 nuclei were counted per kidney, distinguishing between negatively and positively stained nuclei. Nuclear labeling indices (LI %) were calculated as percentage of positive nuclei / total number of nuclei counted.
RNA isolation, microarray hybridization and gene expression profiling
RNA isolation from RNAlater fixed kidneys and Affymetrix Rat Genome RAE230A array hybridization were performed as described previously (Stemmer et al. 2007). Intensity values were extracted from digitized image files using a Mas5 condensing algorithm, which uses both perfect match and mismatch probes as published before (Ellinger-Ziegelbauer et al. 2004). Probe IDs were annotated using the information present in the R/Bioconductor package rat2302.db. Differentially expressed genes due to treatment or time were determined by the moderated t-test implemented in the R/Bioconductor package limma (Smyth 2005). P-values were corrected for multiple testing using the Benjamini-Hochberg method for controlling the false discovery rate (FDR). Differentially expressed genes with an adjusted p-value < 0.05 were considered statistically significant. ClusterProfiler (Yu et al. 2012) tested the genes of interest against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database and GO biological process terms (Ashburner et al. 2000; Gene Ontology 2015).
Real Time PCR
Isolated RNA was transcribed into cDNA by using Superscript III reverse transcriptase according to the manufacturer's instructions (Invitrogen, CA, USA). Quantitative real-time PCR (qPCR) reactions were performed in an ABI Prism 7900HT Sequence Detection System by using a TaqMan probe set for Mgmt (Rn00563462_m1, Thermo Fisher Scientific, Waltham, MA, USA). Relative expression levels were normalized to the housekeeping gene hypoxanthine guanine phosphoribosyl transferase 1 (Hprt1) using the probe set Rn01527840_m1. Expression changes were evaluated using the delta-delta CT method.
Quantification of DNA adducts
Sample preparation and quantification of O6-methyl-2′-deoxyguanosine (O6-me-dG) and N7-methylguanine (N7MG) by ultra performance liquid chromatography-tandem mass spectrometer (UPLC-MS/MS) are described in the supplemental material and methods part of the manuscript.
Statistical analysis
Statistical analyses of pre-neoplastic and neoplastic pathologies was carried out with SAS/STAT® Version 9.3. A negative binomial regression model with a log link was used to estimate the relative effects on the lesion counts of the different carcinogens in comparison to the control using SAS PROC GENMOD. The model fit used the categorical factors “treatment” (MAMAc or vehicle), “treatment duration” (3 or 6 months), sex, and interaction between treatment and time. Additional analyses were conducted using all other interaction terms.
Statistical analyses of cell proliferation data were carried out using GraphPad Prism® 6 Software. Significant differences in nuclear labeling indices (LI %) in treated and control rats were analyzed by one-way ANOVA and Tukey's multiple comparisons test.
Results
Lack of overt or fatal toxicity in response to continued low dose MAMAc treatment
Male and female Eker rats were administered with low dose MAMAc (250 μg/kg BW) as depicted in Figures 1a (acute exposure for 1, 3, 7 and 14 days) and 1b (chronic exposure for 3 or 6 months), respectively. Up to 6 months MAMAc treatment did not result in overt or fatal toxicity as indicated by 100 % survival and lack of body weight loss in males (Figure 1c) or females (Figure 1d) during the experiment. One female control rat in the 6 months treatment group died 7 weeks before the end of the experiment for unknown reasons, and was excluded from all analyses assuming no relation to treatment or study outcome.
Figure 1.
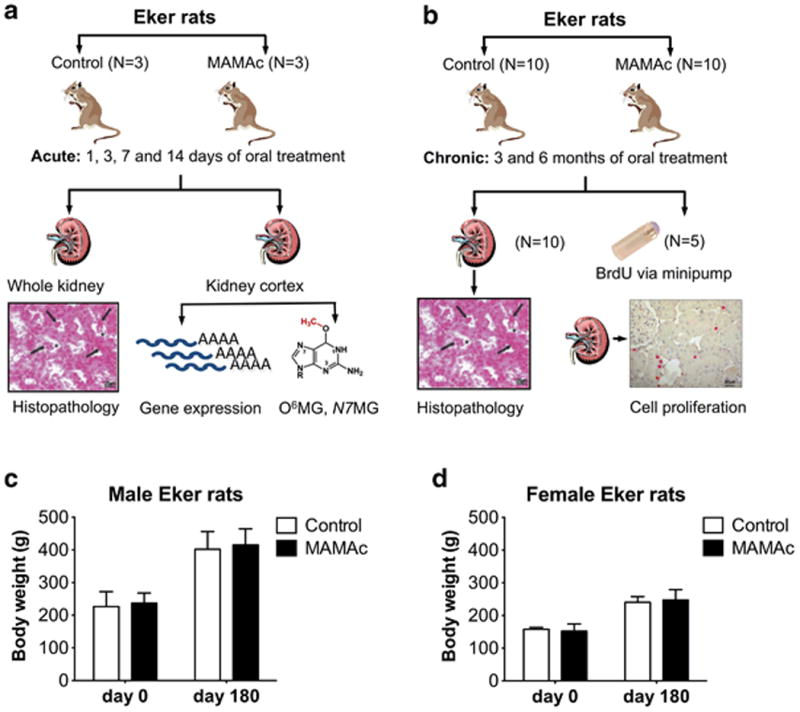
Acute (a) or chronic (b) MAMAc dosing regimen in Eker rats and follow-up analyses on gene expression, DNA adduct formation, renal pathology and cell proliferation. Body weights of male (c) and female (d) Eker rats before and after 6 months of MAMAc or vehicle treatment. Data represent means ± SEM. Student's t-tests comparing body weights at either day 0 or day 180 revealed no significant differences between MAMAc and vehicle treated groups.
Time dependent accumulation of O6-methylguanine and N7-methylguanine adducts in kidneys of MAMAc treated Eker rats
We next quantified O6-methylguanine (O6MG) and N7-methylguanine (N7MG) adducts in the kidneys of MAMAc and vehicle treated Eker rats. We primarily focused on the kidney cortex as major site for MAMAc induced tumors (Gusek and Mestwerdt 1969), and pooled renal cortices from three rats per group to acquire sufficient amounts of DNA for LC-MS/MS.
MAMAc treatment induced a time dependent accumulation of O6MG in the kidney cortex of male and female Eker rats, which was measured from the extracted DNA as O6-methyl-2′-deoxyguanosine (O6-me-dG) (Figure 2a). MAMAc treated female Eker rats demonstrated slightly higher O6MG adduct levels than the corresponding treated males at all time points examined. Similarly, repeated dosing with MAMAc resulted in a time-dependent increase of N7MG adducts in male and female rats (Figure 2b). Both, O6MG and N7MG can also form via endogenous DNA alkylation processes using cellular precursors (Sharma et al. 2014). While no endogenous O6MG adduct levels could be detected in the vehicle treated controls (Figure 2a), N7MG adducts in control rats were clearly detectable, albeit with similar adduct levels at all time points investigated (Figure 2b).
Figure 2.
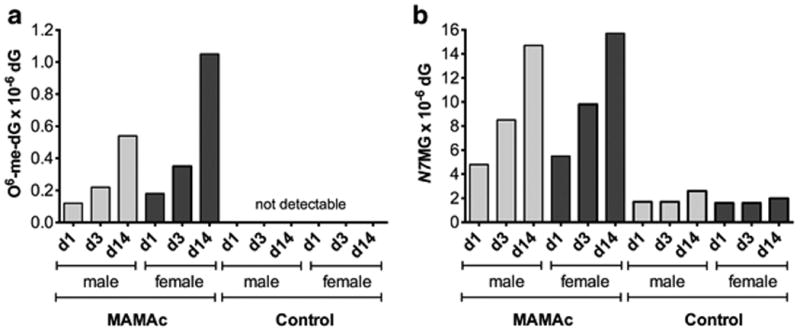
Numbers of O6-methyl-2′-deoxyguanosine (O6-me-dG) (a) and N7-methylguanine (N7MG) (b) adducts per 106 deoxyguanosine (dG), detected via LC-MS/MS in DNA of male and female Eker rats after 1, 3 and 14 days of MAMAc treatment (combined DNA pooled from renal cortices of three replicate animals).
MAMAc induced DNA adducts do not elicit a transcriptional DNA damage response in vivo
Next, we aimed to assess whether increasing amounts of DNA adducts correlate with time-matched gene expression changes in the kidney cortex. Microarray analyses of cortices from 1, 3, 7 and 14 d MAMAc treated male Eker rats revealed 87 Probe-IDs (76 could be assigned to genes) with significantly altered expression levels at least at one time point (Figure 3a). Most genes were transiently regulated, with the strongest signal after one day of exposure. KEGG pathway enrichment analyses of 76 annotated genes revealed a significant effect (adjusted p-value < 0.05) of MAMAc on 18 different pathways (Supplemental Table 1). Ten up-regulated genes could be assigned to pathways related to cancer (Tgfbr1, Tgfbr2, Smad3, Smad4, Egfr, Mapk9, Nfkb2, Gnai3, Hsp90ab1, Prkacb). Further analysis using the STRING database (Jensen et al. 2009) revealed that most of the cancer genes encode for proteins of a coherent protein-signaling network (Figure 3b), including MAPK (KEGG adj. p-value = 0.0017), FoxO (KEGG adj. p-value = 0.002) and TGF-beta pathways (KEGG adj. p-value = 0.016).
Figure 3.
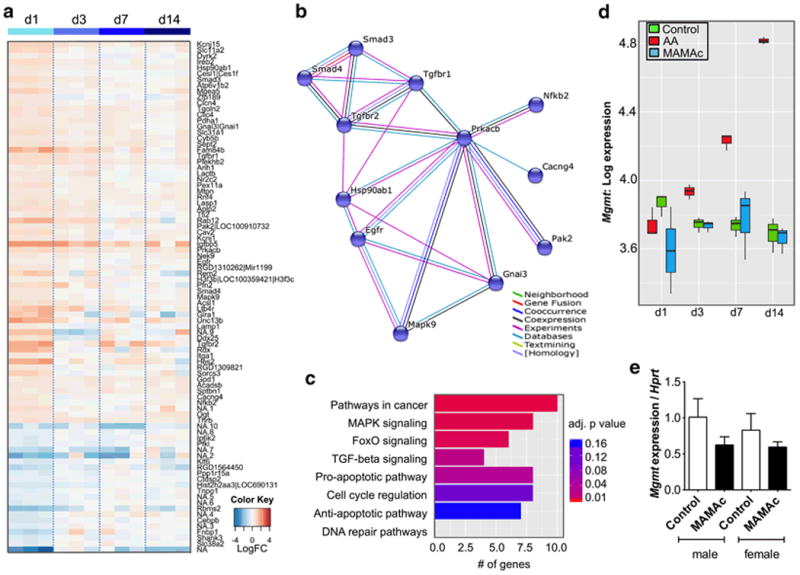
(a) Heatmap showing significant gene expression changes in the kidney cortex after 1, 3, 7 or 14 days of MAMAc exposure (n = 3 per time point). Blue: down-regulated; red: up-regulated, white: no regulation compared to the vehicle treated controls. (b) STRING interaction network of proteins encoded by genes associated with cancer-related pathways. (c) Selected KEGG pathways sorted by functional classification. Total numbers of genes per pathway are shown as horizontal bars, the significance level is indicated by the color scheme ranging from red (p < 0.01) to blue (p < 0.16). (d) Boxplots indicating microarray log expression levels of DNA repair gene Mgmt in rats treated for 1, 3, 7 and 14 days with vehicle (controls), MAMAc or the positive control aristolochic acid (AA). (e) Quantitative PCR-based Mgmt expression normalized to the housekeeping gene Hprt in male and female Eker rats treated for 14 days with MAMAc or vehicle (n = 3). Data represent means ± SEM. One-way ANOVA with Tukey's post-hoc test for multiple comparisons revealed no significant effects of MAMAc or sex on Mgmt expression levels.
Enrichment analyses that only used KEGG pathways and GO Biological Processes of interest (Figure 3c) showed that 8 out of 76 genes with significant expression changes after MAMAc treatment were involved in pro-apoptotic pathways (GO adj. p-value = 0.028; up-regulated: Tgfbr, Smad3, Itga1, Slc11a2, Mapk9, Pak2; down-regulated: Ip6k2, Ppp1r15a). In contrast, no pathway enrichment was detected for 7 significantly regulated anti-apoptotic genes (GO adj. p-value = 0.16; up-regulated: Egfr, Kcnj1, Smad3, Tgfbr1, Hsp90ab1; down-regulated: Cebpb) or genes involved in cell cycle (GO adj. p-value = 0.1; up-regulated: Prkacb, Rdx, Egfr, Smad3, Cav2, Rnf4; down-regulated: Ctdsp2).
Despite the formation of DNA adducts in MAMAc treated rats, not a single gene involved in DNA repair was significantly up- or down-regulated (Figure 3c). The expression of the primary repair enzyme for O6MG, O6-Methylguanin-DNA-Methyltransferase (Mgmt) remained unchanged after MAMAc exposure (Figure 3d). This was in contrast to the strong and time dependent transcriptional induction of Mgmt in the kidney cortex of male Eker rats following treatment with the positive control genotoxicant aristolochic acid (AA) (Stemmer et al. 2007) (Figure 3d). Mgmt microarray expression data were further verified by real-time PCR at the 14 days time-point (Figure 3e).
Despite the early transcriptional changes of genes involved in cancer relevant pathways or apoptosis, histopathological analyses of the kidneys from the same animals did not reveal any signs of increased apoptotic cell death or mitotic events in MAMAc treated rats (Supplemental Table 2).
Similar lack of DNA damage response in Eker and wild type rats
Recent evidence revealed that the complete loss of TSC2 enhances etoposide-induced DNA damage and apoptosis in mouse embryonic fibroblasts and Eker rat uterine leiomyoma cells (Wang et al. 2013). In addition, partial loss of TSC2 in kidneys of Eker rats resulted in markedly reduced expression levels of the DNA repair enzyme 8-oxoG-DNA glycosylase (Ogg1) compared to wild type rats (Habib 2010). We therefore aimed to address whether DNA repair pathways, chronically reduced by genetic haploinsufficiency of TSC2 in our Eker rat model, could explain the observed lack of transcriptional regulation after MAMAc treatment in Eker rats.
Expression levels of 169 known genes involved in DNA replication, mismatch repair, excision repair, homologous recombination (HR), direct reversal, base excision repair, non-homologous end joining and the Fanconi anemia (FA) pathway were compared in in vehicle- and MAMAc-treated Eker rats and in wild type littermates. Similar to Eker rats, wild type littermates failed to show a significant induction of DNA repair genes following up to 14 days of low dose MAMAc treatment (data not shown). When comparing both genotypes, we only detected 6 genes with significantly altered expression levels in Eker vs. wild type rats (Table 1), indicating that a partial loss of TSC2 only marginally affects the overall transcription levels of DNA repair genes.
Table 1. Differentially expressed DNA repair genes in Eker and wild type rats.
| Gene | KEGG pathway | Direction of regulation | Adjusted p-value |
|---|---|---|---|
| Ssbp1 | DNA replication, Homologous recombination, Mismatch repair | Up | 5.772972e-05 |
| Stra13 | Fanconi anemia pathway | Up | 0.0001458302 |
| Lig3 | Base excision repair | Down | 0.0000180946 |
| Brcc | Homologous recombination | Up | 0.0001258071 |
| Rnaseh2b | DNA replication | Down | 1.007141e-05 |
| Pole4 | DNA replication, Nucleotide excision repair, Base excision repair | Down | 0.000120004 |
| Bre | Homologous recombination | Down | 9.037353e-06 |
| Lig3 | Base excision repair | Up | 0.0008834112 |
DNA repair genes from selected KEGG pathways with differential expression in vehicle treated Eker rats vs. wild type littermates. P-values were obtained using Limma's moderated F-statistic, which tests the overall significance for each gene and adjusted for multiple testing applying the Benjamini and Hochberg method.
Increased numbers and incidences of pre-neoplastic and neoplastic lesions in Eker rats following 3 and 6 months of MAMAc treatment
Next, we aimed to investigate if the increased DNA adduct formation in MAMAc treated rats in absence of a transcriptional induction of cytoprotective genes translates into tumors of the kidney. Male and female Eker rats were continuously dosed with MAMAc (250 μg/kg BW) for 3 and 6 months. Histopathological analyses of renal sections demonstrated the presence of pre-neoplastic and neoplastic lesions of different progressional stages, i.e. basophilic atypical tubules (bATs) (Figure 4a), basophilic hyperplasia (bAHs) (Figure 4b), encapsulated adenomas and invasive carcinomas (Figure 4c) of a basophilic phenotype in all groups examined (Table 2).
Figure 4.
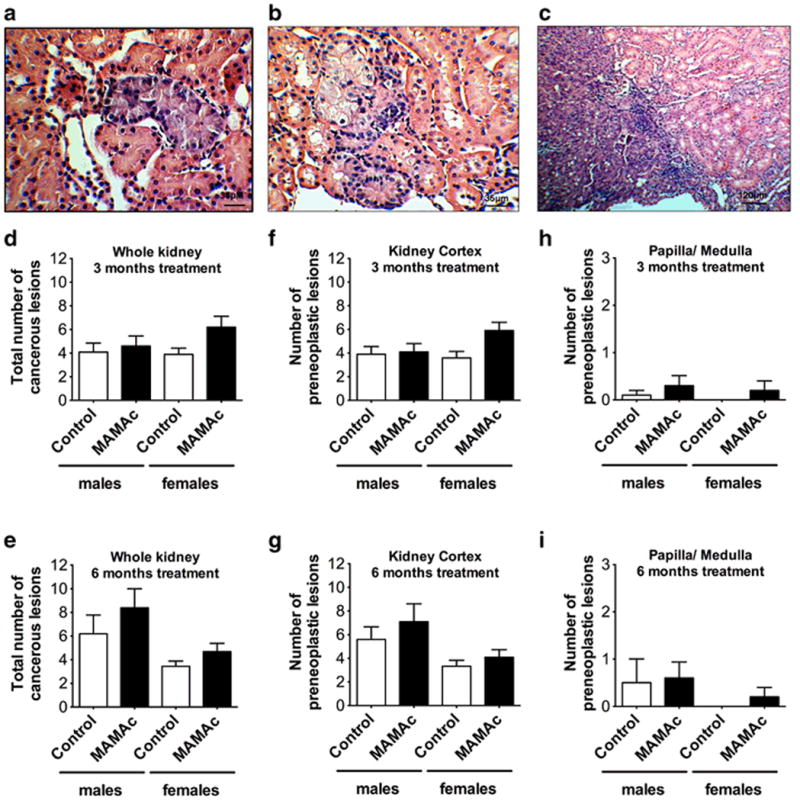
(a) Basophilic atypical tubule (bAT), (b) basophilic atypical hyperplasia (bAH) and (c) invasive carcinoma in H&E stained renal sections of Eker rats. Total numbers of all pre-cancerous and cancerous lesions in male and female Eker rats after 3 months (d) or 6 months (e) of MAMAc exposure. Zonal occurrence of pre-neoplastic lesions (bATs and bAHs) in the renal cortex (f, g) or medulla / papilla (h, i) of male and female Eker rats following 3 months (f, h) or 6 months (g, i) of MAMAc exposure. Data represent means ± SEM. N = 10 per group in 3 and 6 months MAMAc and 3 months vehicle treated control rats. N = 9 for the female 6 months treated control group.
Table 2. Numbers and incidence of pre-neoplastic and neoplastic lesions.
| 3 months treatment | 6 months treatment | |||||||
|---|---|---|---|---|---|---|---|---|
| Male | Female | Male | Female | |||||
| Vehicle | MAMAc | Vehicle | MAMAc | Vehicle | MAMAc | Vehicle | MAMAc | |
| N = 10 | N = 10 | N = 10 | N = 10 | N = 10 | N = 10 | N = 9 | N = 10 | |
| Cortex | ||||||||
| bAT | 34 | 35 | 35 | 53 | 45 | 60 | 28 | 36 |
| bAH | 5 | 6 | 1 | 6 | 11 | 11 | 2 | 5 |
| Medulla | ||||||||
| bAT | 1 | 3 | 0 | 2 | 5 | 5 | 0 | 0 |
| bAH | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 |
| Papilla | ||||||||
| bAT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| bAH | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 2 |
| Total Kidney | ||||||||
| Total # bAT | 35 | 38 | 35 | 55 | 50 | 65 | 28 | 36 |
| Total # bAH | 5 | 6 | 1 | 6 | 11 | 12 | 2 | 7 |
| Total # other ATs | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 1 |
| Total # adenoma | 0 | 1 | 0 | 1 | 0 | 2 | 0 | 1 |
| Total # carcinoma | 1 | 1 | 3 | 0 | 1 | 3 | 1 | 2 |
| Total # all lesions | 41 | 46 | 39 | 62 | 62 | 84 | 31 | 47 |
| Incidence pre-neoplastic lesions | 9 / 10 | 10 / 10 | 10 / 10 | 10 / 10 | 10 / 10 | 10 / 10 | 9 / 9 | 10 / 10 |
| Incidence neoplastic lesions | 1 / 10 | 2 / 10 | 3 / 10 | 1 / 10 | 1 / 10 | 5 / 10 | 1 / 9 | 3 / 10 |
| Incidence all lesions | 9 / 10 | 10 / 10 | 10 / 10 | 10 / 10 | 10 / 10 | 10 / 10 | 9 / 9 | 10 / 10 |
Total numbers of pre-neoplastic and neoplastic lesions in 3 and 6 months MAMAc or vehicle treated male and female Eker rats. bAT: basophilic atypical tubule, bAH: basophilic atypical hyperplasia. Other ATs include atypical tubules with either oncocytic, eosinophilic or chromophobe phenotype. A detailed summary of the statistical analysis is given in the text.
The proportion of rats with renal pre-neoplastic or neoplastic lesions was 100 % in all groups (10/10), except for the three months male vehicle treated control group, which displayed an incidence of 90 % (9/10) (Table 2). Most kidneys displayed multiple lesions, which occurred primarily in the renal cortex (Table 2).
Mean total numbers of pre-neoplastic and neoplastic lesions on whole kidney sections in male and female Eker rats after 3 and 6 months treatment, respectively, are displayed in Figures 4d and 4e. To assess statistically significant differences in total numbers of pre-neoplastic and neoplastic lesions between groups, we applied a negative binomial regression with factors carcinogen, treatment duration, sex, and interaction between time and carcinogen. This model accounts for an increased variance with increasing counts, evaluates relative (percent) changes instead of absolute changes, and compared to an ANOVA allows for a more comprehensive and sensitive statistical analysis of groups with limited sample size. MAMAc treated Eker rats showed an increased lesion rate by a factor of 1.36, i.e. an increase of +36 % compared to the vehicle treated rats (95 % confidence interval (CI): 7 % - 73 %). The interaction between treatment duration and treatment in MAMAc treated rats was negligible, which indicates an unchanged carcinogenic effect size between rats treated for 3 or 6 months (-2 % with 95 % CI: -40 % - +58 %). Further differentiation between the kidney cortex (Figure 4f, g) and the medulla/papilla zone (Figure 4h, i) of the kidney revealed the cortex as major target site of spontaneous and MAMAc induced lesion development.
Positive correlation between increased cell proliferation and lesion numbers
Proliferating cells are often presumed to have a higher susceptibility for mutations than quiescent cells because they have less time to repair DNA damage before DNA replication (Bielas and Heddle 2000). We therefore aimed to investigate if increased cell proliferation in MAMAc treated rats could have turned the potentially unrepaired DNA damage into manifest mutations and thus increased lesion numbers.
In our experimental setup, PCNA staining of S-phase nuclei in kidneys from 14 days MAMAc or vehicle treated Eker rats revealed no significant difference in proliferation (Figure 5a). Similarly, continuous MAMAc exposure over 3 and 6 months (Figure 5b, c) had no impact on cell proliferation when analyzed by BrdU staining. Both techniques have been successfully applied for the detection of cell proliferating differences in carcinogen or vehicle treated Eker rats (Stemmer et al. 2009), indicating adequate sensitivity of both staining techniques. Assessing non-neoplastic pathologies in 3 and 6 months treated rats further revealed the absence of MAMAc induced cell death (Supplemental Table 3), which is typically followed by regenerative cell proliferation. Interestingly, when comparing total numbers of pre-neoplastic lesions with cell proliferation data, we found the highest (1.59-fold) increase in total lesions in the 3 months MAMAc treated group, which also displayed the highest (1.55-fold) increase in cell proliferation (Figure 4d, 4f and 5b).
Figure 5.
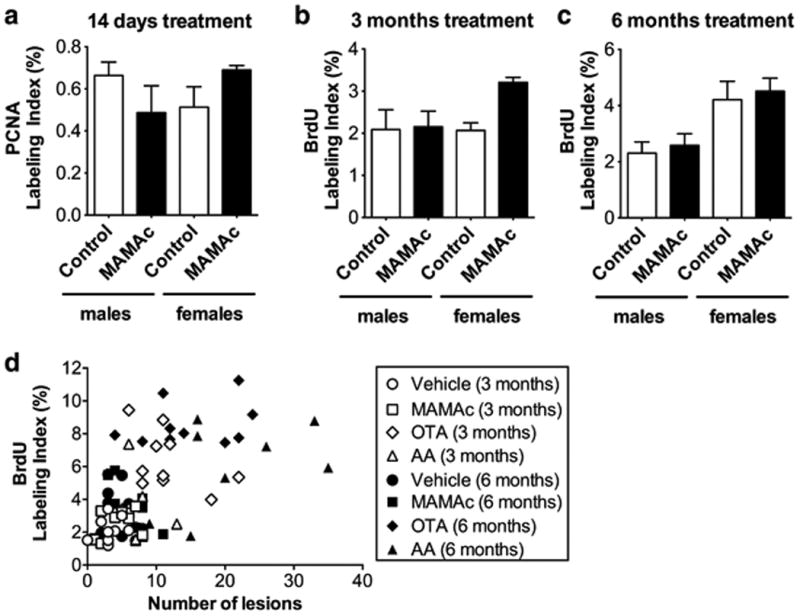
(a-c) Assessment of MAMAc effects on proliferation via (a) proliferating-cell-nuclear-antigen (PCNA) S-phase labeling indices (LI %) in the renal cortex of 14-days MAMAc treated male and female Eker rats (n=3), or via (b, c) BrdU S-phase labeling indices of male and female Eker rats treated with MAMAc for 3 (b) and 6 months (c), respectively (n=5). (d) Pearson correlation analysis of BrdU labeling indices versus the total number of renal lesions determined by renal histopathology in rats (N = 5 per group), either 3 months (white) or 6 months (black) treated with vehicle (circles), MAMAc (squares), ochratoxin A (OTA, diamonds), and aristolochic acid (AA, triangles). Data represent means ± SEM. One-way ANOVA with Tukey's post-hoc test (a-c) was used to test for statistical significance.
To further investigate a positive correlation between increased cell proliferation and lesion numbers, we included data sets from a previously published paralleled study, where male and female Eker rats were treated with the genotoxin aristolochic acid (AA) or the mitogenic carcinogen ochratoxin A (OTA) for 3 or 6 months (Stemmer et al. 2009). Both groups shared the same controls as the MAMAc treated groups. Indeed, the combined analysis revealed a significant correlation between the number of total lesions and the percentage of positively stained S-phase nuclei after 3 (p = 0.0027, r = 0.4621) and 6 months (p = 0.0004, r = 0.5432) of exposure (Figure 5d). These data provide additional evidence that the combination of DNA damage and increased cell proliferation is crucial for the development of cancerous lesions. Increased cell proliferation, as detected for AA and OTA after up to 6 months of treatment, was already reflected in early expression changes of genes involved in cell cycle regulation (Supplemental Figure 1).
Discussion
DNA adducts are widely accepted as biomarkers of exposure, representing the end-product of a process involving carcinogen absorption, distribution, metabolic activation, detoxication, and DNA repair. An increasing amount of studies describe the formation of DNA adducts in response to genotoxin exposure and associated DNA adduct levels with an increased risk of cancer.
The genotoxin used in our studies, MAMAc, shows a similar association between DNA adducts formation and tumor risk. A single high dose of MAMAc (20 mg/kg BW, ip.) was found to cause O6MG and N7MG adducts primarily in the liver, kidneys (Sohn et al. 2001) and the brain (Kisby et al. 2011). Other studies reported MAMAc-induced tumors of the kidney, colon and liver when applied at acute high doses of 1 mg/g diet for 14 days (Laqueur et al. 1967), 35 mg/kg BW given as single intraveneous (i.v.) bolus (Notman et al. 1982), or 25 mg/kg BW applied once weekly via i.v. injection for 10 weeks (Matsubara et al. 1978).
We applied an accumulated dose of 30 mg/kg body weight of MAMAc over 6 months (5 days per week, i.e. 120 oral applications of 250 μg/kg BW) to better reflect typical exposures of humans over a prolonged time but at low doses. We also assessed early effects of the single daily low dose exposure of 250 μg/kg BW of MAMAc on DNA adduct formation, and found O6MG and N7MG adducts in the kidney cortex already after one day of exposure. Both adduct types further accumulated over 14 days of continuous treatment indicating that adduct formation exceeded the rate of adduct loss.
While N7MG is chemically unstable with a half-life ranging from 2 h to 150 h (Boysen et al. 2009), O6MG are highly persistent and are frequently linked to cancer (Margison et al. 2002; Swann 1990; Beranek 1990). The pro-mutagenic character of O6MG is primarily due to its tendency of inducing a miss-pair with thymine during DNA replication (Coulondre and Miller 1977). The DNA repair enzyme MGMT protects cells against these lesions by transferring the alkyl group to its own cysteine, leading to the targeted degradation of the enzyme (Srivenugopal et al. 1996). Thus, the number of O6MG adducts is directly related to the intracellular MGMT concentration and the rates of its de novo synthesis. The relevance of MGMT mediated removal of O6MG in the protection against tumor causing mutations by alkylating genotoxicants has been largely confirmed in studies using MGMT deficient mice (Sakumi et al. 1997; Fahrer et al. 2015; Fu et al. 2012). In addition, an important role of MGMT in the repair of MAMAc induced DNA damage has been demonstrated previously (Kisby et al. 2011).
Surprisingly, in our study the profound formation of O6MG following 14 days of MAMAc treatment failed to induce a transcriptional activation of Mgmt or any other enzymes involved in DNA damage repair. The lack of an early transcriptional induction was in contrast to our previous study, where 1 - 3 days exposure to different genotoxins were sufficient enough to detect transcriptional changes in the DNA repair machinery in the liver (Ellinger-Ziegelbauer et al. 2004; Ellinger-Ziegelbauer et al. 2005) and the kidney (Stemmer et al. 2007). Together these findings suggest a model whereby a threshold level of DNA adducts has to be reached to activate a detectable transcriptional activation of DNA damage response genes. Supporting evidence is provided by a recent in vitro study, where exposure to the three genotoxic agents etoposide, quercetin, and methyl methanesulphonate failed to induce changes in P53 and downstream DNA repair genes, but caused a significant increases in micronuclei formation (Clewell et al. 2014). The latter is indicative for the potential of a chemical to cause clastogenic effects.
Interestingly, instead of the expected transcriptional induction of DNA repair genes, up to 14 days of MAMAc treatment caused significant changes of cancer related genes, which could be assigned to a coherent network including TGF-β, MAPK and FOXO pathways. It remains to be determined whether these gene regulations are indeed a consequence of pro-mutagenic DNA adduct formation. Nevertheless, they may serve as predictive tool by reflecting a possible translation of MAMAc-induced DNA adducts into tumor causing mutations.
We next aimed to address the question if potentially unrepaired pro-mutagenic DNA adducts may further translate into cancerous lesions. Continuous treatment of male and female Eker rats with 250 μg/kg BW MAMAc for up to 6 months resulted in a significant increase of pre-neoplastic and neoplastic renal lesions. However, the increase in tumor incidence after MAMAc treatment was small compared to vehicle-treated rats, especially when put in relation to the increased sensitivity of Eker rats towards other genotoxic carcinogens such as dimethylnitrosamine (Walker et al. 1992) or aristolochic acid (Stemmer et al. 2007; Stemmer et al. 2009). Accordingly, our data suggests that only few adducts have manifested in tumor causing mutations. This low manifestation of MAMAc-induced tumors may be explained by the lack of MAMAc to increase cell proliferation. Increased cell proliferation is a critical event in carcinogenesis and necessary to convert DNA damage into heritable mutations (Cohen and Ellwein 1990; Preston-Martin et al. 1990). For instance, regenerative proliferation induced by cytotoxicity has been shown to decrease the latency period for genotoxic carcinogens (Williams et al. 1996). An extended correlation analysis including data from this and previous exposure studies (Stemmer et al. 2009) confirmed this association between toxin-induced cell proliferation and renal (pre)-neoplasia incidence in Eker rats. Moreover, increased cell proliferation after 6 months of OTA and AA treatment (Stemmer et al. 2009) was predicted by early expression changes of genes involved in cell cycle regulation following up to 14 days of AA and OTA treatment (Stemmer et al. 2007). Low doses of MAMAc used in this study caused negligible expression changes of genes involved in cell cycle regulation when applied for up to 14 days of treatment. Further, when applied chronically we found no increase in cell proliferation, which may explain why the effect of MAMAc on pre-neoplastic lesion development was only very mild.
In summary, our data support the current concept that the quantification of DNA adducts can only serve as biomarker for internal exposure, rather than as marker for cancer-relevant DNA damage or tumorigenesis. Although DNA adduct formation is considered to be necessary for genotoxin induced carcinogenesis, other events such as mutagenesis in critical genes and cell proliferation are necessary for the translation of DNA adducts into cancer. Thus, a more reliable prediction of genotoxic carcinogenesis should be based on the concomitant detection of distinct biomarkers that reflect the multistage mechanisms of carcinogenesis. Here, we provide evidence that the accumulation of pro-mutagenic DNA adducts in MAMAc treated Eker rats, in absence of a cyto-protective or mitogenic gene expression response, could be indicative for an increased risk of DNA adducts manifesting into tumor-causing mutations. Our data suggests an improved predictive value of DNA adduct quantification for chemical carcinogenesis when combined with time matched gene expression analyses.
Supplementary Material
Acknowledgments
The authors thank Tanja Lampertsdoerfer, Gudrun von Scheven, Evelyn O'Brien and Alexandra Heussner for their skillful assistance during the animal experiment and Paul Pfluger for critically reading the manuscript. This work was supported by the Federal Ministry of Education and Research (BMBF: 0313024). The UNC Biomarker Mass Spectrometry Facility is partially supported by NIEHS grant P30-ES010126.
References
- Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231(1):11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- Bielas JH, Heddle JA. Proliferation is necessary for both repair and mutation in transgenic mouse cells. Proc Natl Acad Sci U S A. 2000;97(21):11391–6. doi: 10.1073/pnas.190330997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boysen G, Pachkowski BF, Nakamura J, Swenberg JA. The formation and biological significance of N7-guanine adducts. Mutat Res. 2009;678(2):76–94. doi: 10.1016/j.mrgentox.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clewell RA, Sun B, Adeleye Y, et al. Profiling dose-dependent activation of p53-mediated signaling pathways by chemicals with distinct mechanisms of DNA damage. Toxicol Sci. 2014;142(1):56–73. doi: 10.1093/toxsci/kfu153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SM, Ellwein LB. Cell proliferation in carcinogenesis. Science. 1990;249(4972):1007–11. doi: 10.1126/science.2204108. [DOI] [PubMed] [Google Scholar]
- Coulondre C, Miller JH. Genetic studies of the lac repressor. IV. Mutagenic specificity in the lacI gene of Escherichia coli. J Mol Biol. 1977;117(3):577–606. doi: 10.1016/0022-2836(77)90059-6. [DOI] [PubMed] [Google Scholar]
- Dietrich DR, Swenberg JA. Preneoplastic lesions in rodent kidney induced spontaneously or by non-genotoxic agents: predictive nature and comparison to lesions induced by genotoxic carcinogens. Mutat Res. 1991;248(2):239–60. doi: 10.1016/0027-5107(91)90060-2. [DOI] [PubMed] [Google Scholar]
- Ellinger-Ziegelbauer H, Stuart B, Wahle B, Bomann W, Ahr HJ. Characteristic expression profiles induced by genotoxic carcinogens in rat liver. Toxicol Sci. 2004;77(1):19–34. doi: 10.1093/toxsci/kfh016. [DOI] [PubMed] [Google Scholar]
- Ellinger-Ziegelbauer H, Stuart B, Wahle B, Bomann W, Ahr HJ. Comparison of the expression profiles induced by genotoxic and nongenotoxic carcinogens in rat liver. Mutat Res. 2005;575(1-2):61–84. doi: 10.1016/j.mrfmmm.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Fahrer J, Frisch J, Nagel G, et al. DNA repair by MGMT, but not AAG, causes a threshold in alkylation-induced colorectal carcinogenesis. Carcinogenesis. 2015;36(10):1235–44. doi: 10.1093/carcin/bgv114. [DOI] [PubMed] [Google Scholar]
- Fu D, Calvo JA, Samson LD. Balancing repair and tolerance of DNA damage caused by alkylating agents. Nat Rev Cancer. 2012;12(2):104–20. doi: 10.1038/nrc3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gene Ontology C. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015;43(Database issue):D1049–56. doi: 10.1093/nar/gku1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerard M, Baum M, Bitsch A, et al. Assessment of mechanisms driving non-linear dose-response relationships in genotoxicity testing. Mutat Res Rev Mutat Res. 2015;763:181–201. doi: 10.1016/j.mrrev.2014.11.001. [DOI] [PubMed] [Google Scholar]
- Gusek W, Mestwerdt W. [Cycasin-induced renal tumors in the Wistar rat with special consideration of the adenoma] Beitr Pathol Anat. 1969;139(2):199–218. [PubMed] [Google Scholar]
- Habib SL. Tuberous sclerosis complex and DNA repair. Adv Exp Med Biol. 2010;685:84–94. doi: 10.1007/978-1-4419-6448-9_8. [DOI] [PubMed] [Google Scholar]
- Jarabek AM, Pottenger LH, Andrews LS, et al. Creating context for the use of DNA adduct data in cancer risk assessment: I. Data organization. Crit Rev Toxicol. 2009;39(8):659–78. doi: 10.1080/10408440903164155. [DOI] [PubMed] [Google Scholar]
- Jensen LJ, Kuhn M, Stark M, et al. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009;37(Database issue):D412–6. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisby GE, Fry RC, Lasarev MR, et al. The cycad genotoxin MAM modulates brain cellular pathways involved in neurodegenerative disease and cancer in a DNA damage-linked manner. PLoS One. 2011;6(6):e20911. doi: 10.1371/journal.pone.0020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laqueur GL, McDaniel EG, Matsumoto H. Tumor induction in germfree rats with methylazoxymethanol (MAM) and synthetic MAM acetate. J Natl Cancer Inst. 1967;39(2):355–71. [PubMed] [Google Scholar]
- Margison GP, Santibanez Koref MF, Povey AC. Mechanisms of carcinogenicity/chemotherapy by O6-methylguanine. Mutagenesis. 2002;17(6):483–7. doi: 10.1093/mutage/17.6.483. [DOI] [PubMed] [Google Scholar]
- Matsubara N, Mori H, Hirono I. Effect of colostomy on intestinal carcinogenesis by methylazoxymethanol acetate in rats. J Natl Cancer Inst. 1978;61(4):1161–4. [PubMed] [Google Scholar]
- Matsumoto H, Higa HH. Studies on methylazoxymethanol, the aglycone of cycasin: methylation of nucleic acids in vitro. Biochem J. 1966;98(2):20C–22C. doi: 10.1042/bj0980020c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima T, Matsumoto H, Shirai A, Sawamura M, Sugimura T. Mutagenicity of the naturally occurring carcinogen cycasin and synthetic methylazoxymethanol conjugates in Salmonella typhimurium. Cancer Res. 1979;39(9):3780–2. [PubMed] [Google Scholar]
- McDorman KS, Hooth MJ, Starr TB, Wolf DC. Analysis of preneoplastic and neoplastic renal lesions in Tsc2 mutant Long-Evans (Eker) rats following exposure to a mixture of drinking water disinfection by-products. Toxicology. 2003;187(1):1–12. doi: 10.1016/s0300-483x(03)00004-0. [DOI] [PubMed] [Google Scholar]
- Morton LD, Youssef AF, Lloyd E, Kiorpes AL, Goldsworthy TL, Fort FL. Evaluation of carcinogenic responses in the Eker rat following short-term exposure to selected nephrotoxins and carcinogens. Toxicol Pathol. 2002;30(5):559–64. doi: 10.1080/01926230290105794. [DOI] [PubMed] [Google Scholar]
- Notman J, Tan QH, Zedeck MS. Inhibition of methylazoxymethanol-induced intestinal tumors in the rat by pyrazole with paradoxical effects on skin and kidney. Cancer Res. 1982;42(5):1774–80. [PubMed] [Google Scholar]
- OECD. OECD Guidelines for the Testing of Chemicals. Section 4. Health effects 1981 [Google Scholar]
- Patel SK, Ma N, Monks TJ, Lau SS. Changes in gene expression during chemical-induced nephrocarcinogenicity in the Eker rat. Mol Carcinog. 2003;38(3):141–54. doi: 10.1002/mc.10153. [DOI] [PubMed] [Google Scholar]
- Preston-Martin S, Pike MC, Ross RK, Jones PA, Henderson BE. Increased cell division as a cause of human cancer. Cancer Res. 1990;50(23):7415–21. [PubMed] [Google Scholar]
- Sakumi K, Shiraishi A, Shimizu S, Tsuzuki T, Ishikawa T, Sekiguchi M. Methylnitrosourea-induced tumorigenesis in MGMT gene knockout mice. Cancer Res. 1997;57(12):2415–8. [PubMed] [Google Scholar]
- Satake N, Miyagawa M, Sakurai J, et al. N-ethyl-N-hydroxyethylnitrosamine (EHEN)-induced renal and hepatocarcinogenesis in the tumor suppressor Tsc2 transgenic rat. Cancer Lett. 2002;184(2):157–63. doi: 10.1016/s0304-3835(02)00209-4. [DOI] [PubMed] [Google Scholar]
- Sharma V, Collins LB, Clement JM, Zhang Z, Nakamura J, Swenberg JA. Molecular dosimetry of endogenous and exogenous O(6)-methyl-dG and N7-methyl-G adducts following low dose [D3]-methylnitrosourea exposures in cultured human cells. Chem Res Toxicol. 2014;27(4):480–2. doi: 10.1021/tx5000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK. bioinformatics and computational biology solutions using R and bioconductor. New York: Springer; 2005. Limma: linear models for microarray data. [Google Scholar]
- Sohn OS, Fiala ES, Requeijo SP, Weisburger JH, Gonzalez FJ. Differential effects of CYP2E1 status on the metabolic activation of the colon carcinogens azoxymethane and methylazoxymethanol. Cancer Res. 2001;61(23):8435–40. [PubMed] [Google Scholar]
- Srivenugopal KS, Yuan XH, Friedman HS, Ali-Osman F. Ubiquitination-dependent proteolysis of O6-methylguanine-DNA methyltransferase in human and murine tumor cells following inactivation with O6-benzylguanine or 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochemistry. 1996;35(4):1328–34. doi: 10.1021/bi9518205. [DOI] [PubMed] [Google Scholar]
- Stemmer K, Ellinger-Ziegelbauer H, Ahr HJ, Dietrich DR. Carcinogen-specific gene expression profiles in short-term treated Eker and wild-type rats indicative of pathways involved in renal tumorigenesis. Cancer Res. 2007;67(9):4052–68. doi: 10.1158/0008-5472.CAN-06-3587. [DOI] [PubMed] [Google Scholar]
- Stemmer K, Ellinger-Ziegelbauer H, Ahr HJ, Dietrich DR. Molecular characterization of preneoplastic lesions provides insight on the development of renal tumors. Am J Pathol. 2009;175(4):1686–98. doi: 10.2353/ajpath.2009.081071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann PF. Why do O6-alkylguanine and O4-alkylthymine miscode? The relationship between the structure of DNA containing O6-alkylguanine and O4-alkylthymine and the mutagenic properties of these bases. Mutat Res. 1990;233(1-2):81–94. doi: 10.1016/0027-5107(90)90153-u. [DOI] [PubMed] [Google Scholar]
- Swenberg JA, Fryar-Tita E, Jeong YC, et al. Biomarkers in toxicology and risk assessment: informing critical dose-response relationships. Chem Res Toxicol. 2008;21(1):253–65. doi: 10.1021/tx700408t. [DOI] [PubMed] [Google Scholar]
- Swenberg JA, Lu K, Moeller BC, et al. Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol Sci. 2011;120(Suppl 1):S130–45. doi: 10.1093/toxsci/kfq371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker C, Goldsworthy TL, Wolf DC, Everitt J. Predisposition to renal cell carcinoma due to alteration of a cancer susceptibility gene. Science. 1992;255(5052):1693–5. doi: 10.1126/science.1553556. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hu Z, Liu Z, et al. MTOR inhibition attenuates DNA damage and apoptosis through autophagy-mediated suppression of CREB1. Autophagy. 2013;9(12):2069–86. doi: 10.4161/auto.26447. [DOI] [PubMed] [Google Scholar]
- Williams GM, Iatropoulos MJ, Wang CX, et al. Diethylnitrosamine exposure-responses for DNA damage, centrilobular cytotoxicity, cell proliferation and carcinogenesis in rat liver exhibit some non-linearities. Carcinogenesis. 1996;17(10):2253–8. doi: 10.1093/carcin/17.10.2253. [DOI] [PubMed] [Google Scholar]
- Wolf DC, Goldsworthy TL, Janszen DB, et al. Promotion by sodium barbital induces early development but does not increase the multiplicity of hereditary renal tumors in Eker rats. Carcinogenesis. 2000;21(8):1553–8. [PubMed] [Google Scholar]
- Yeung RS, Xiao GH, Jin F, Lee WC, Testa JR, Knudson AG. Predisposition to renal carcinoma in the Eker rat is determined by germ-line mutation of the tuberous sclerosis 2 (TSC2) gene. Proc Natl Acad Sci U S A. 1994;91(24):11413–6. doi: 10.1073/pnas.91.24.11413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.