

Abstract
There is considerable interest in generating proteins with both high stability and high activity for biomedical and industrial purposes. One approach that has been used successfully to increase the stability of linear repeat proteins is consensus design. It is unclear the extent over which the consensus design approach can be used to produce folded and hyperstable proteins, and importantly, whether such stabilized proteins would retain function. Here we extend the consensus strategy to design a globular protein. We show that a consensus-designed homeodomain (HD) sequence adopts a cooperatively folded homeodomain structure. The unfolding free energy of the consensus-HD is 5 kcal·mol−1 higher than that of the naturally-occurring engrailed-HD from Drosophila melanogaster. Remarkably, the consensus-HD binds the engrailed-HD cognate DNA in a similar mode as the engrailed-HD with approximately 100-fold higher affinity. 15N relaxation studies show a decrease in psec-nsec backbone dynamics in the free state of consensus-HD, suggesting that increased affinity is not a result of increased plasticity. In addition to demonstrating the potential for consensus design of globular proteins with increased stability, these results demonstrate that greatly stabilized proteins can bind cognate substrates with increased affinities, showing that high stability is compatible with function.
Table of contents figure
Introduction
Designing proteins with both high stability and high activity is a significant challenge in protein engineering. Reliably achieving both these goals would be of particular benefit to industrial processes, energy technology and food science, and in particular, medicine. Protein therapeutics show great promise for treatment of a broad range of human diseases, from diabetes to cancer. Recent successes targeting cancer cells often involve using engineered antibody scaffolds to target cell surface receptors and their ligands.1 However, the high cost of these reagents limits the availability of such treatments.2 Increasing the stability, shelf life, and bioavailability of protein therapeutics while maintaining (or enhancing) biological activity would lower costs and make new treatments broadly available.
One challenge to achieving these goals is that stability and activity are often anti-correlated. It has long been recognized that active-site residues in enzymes destabilize the native fold.3,4 Moreover, high stability is often proposed to limit activity and may be necessary for function, perhaps by limiting plasticity or “dynamics.” Consistent with this proposal, homologous proteins from organisms that live at different growth temperatures (Topt) have been observed to be most active at their respective Topt, where they show similar thermostabilities.5,6
We propose that averaging sequence information over a family of proteins that have similar function may achieve these goals. Such “consensus sequences” should capture both the determinants of protein stability and activity. This approach may avoid substitutions that increase stability at the cost of activity, which are more likely to be identified by structure-based approaches.
Consensus design has been quite successful in producing linear repeat proteins with high stability, including ankryin, TPR, heat, and Arm proteins.7–12 Such repeat proteins have simplified architectures made up of 20–40 residue repeated structural elements. Since the main function of these proteins is to bind a variety of target proteins using poorly conserved surface residues, linear repeat consensus proteins do not retain biological activity, although given their high stability these proteins make excellent platforms for engineering new binding functionality.
The applicability of consensus driven design to globular proteins has not been as well explored, and has sometimes produced mixed results. Several studies have examined the effects of single point substitutions towards consensus. An early study by Steipe et al. showed that single-residue substitutions towards the consensus in a globular immunoglobulin fold often resulted in increased stability.13 Subsequent studies suggest that single-residue consensus substitutions increase stability about half the time.14
There are also a few examples of larger-scale consensus substitution in globular proteins. In an early study, a sequence corresponding to a consensus zinc finger sequence was shown to fold into a zinc finger fold upon addition of zinc.15 In another study, globular consensus phytase generated from a small sequence set with identities ranging from 60 to 80% generated proteins that display activity and, in some cases, increased thermostability.16,17 Magliery and coworkers designed several consensus β/α barrels, some of which show enzymatic activity and high thermostability, although none of these proteins demonstrate reversible folding.14,18 Most recently, the O’Neil and Buckle groups have designed consensus FN3 domains and a consensus serpin that adopt their respective folds and retain high thermostability.19–21
To further assess the consensus approach as a means to generate folded proteins with high levels of biological activity as well as high stability, we have applied the consensus approach to the homeodomain fold, a 60-residue three-helix DNA recognition motif with no cofactors or disulfide bonds. The first two N-terminal helices pack together in an anti-parallel orientation. The C-terminal helix packs perpendicular to the two N-terminal helices, and interacts extensively with the major groove of DNA. The homeodomain has high specificity for six-base duplex DNA sequences, and uses this specificity to control gene expression during development, in eukaryotes ranging from fungi to vertebrates. Additional contacts are made between the minor groove and unstructured residues at the N-terminus.22 Given its broad taxonomic distribution and high sequence representation, the homeodomain family is an excellent model system for testing the applicability of consensus sequence information on stability and recognition in a globular protein family.
In this work, we used a multiple sequence alignment to generate a consensus homedomain (consensus-HD). CD and NMR spectroscopy show that the consensus-HD adopts the homeodomain secondary and tertiary structure. Equilibrium folding studies show that the consensus-HD is significantly more stable than the naturally-occurring engrailed homeodomain from D. melanogaster (engrailed-HD). This stability gain partitions into both the folding and unfolding reaction: the consensus-HD folding kinetics are faster and unfolding kinetics are slower than the folding kinetics of engrailed-HD. Furthermore, we tested homeodomain function by measuring the affinity of the consensus-HD to duplex DNA containing the cognate engrailed-HD binding site. Surprisingly, the consensus-HD binds the engrailed-HD binding site with an affinity that is 100-fold higher than the naturally-occurring engrailed-HD.
Materials and Methods
Consensus sequence design
We used the Pfam database entry Homeobox (PF00046) to obtain a homeodomain consensus sequence. At the time the homeobox Pfam entry contained 182 homedomain sequences in the seed alignment. The average identity of the seed alignment was 37%. For our consensus-HD we used the Hidden Markov Model in Pfam to pick the amino acid at each position that had the highest probability of occurrence (Figure 1a).
Figure 1. Sequence conservation and consensus of the homeodomain family.
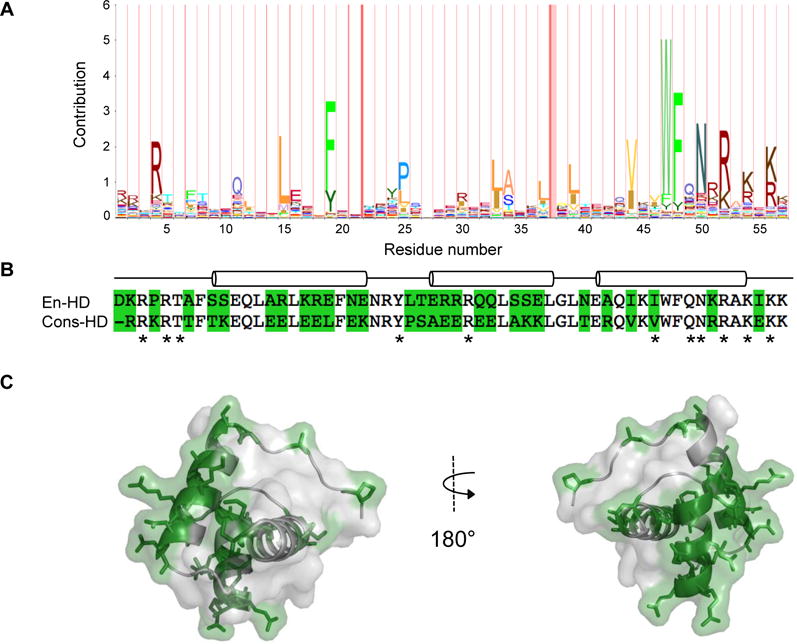
A) The sequence logo, where residue height is proportional to degree of conservation, is calculated from the Pfam version 26.0 homeodomain seed sequence from 12/21/2012. B) Sequence alignment of engrailed- and consensus-HDs. Sequence differences are shaded green. Residues that contact DNA are indicated with asterisks. The positions of the three helices in the engrailed-HD are indicated with cylinders. C) Sequence differences (shaded in green with side chains shown) mapped onto the crystal structure of engrailed-HD (PDB: 1ENH).
Subcloning, Protein expression, and purification
Genes encoding engrailed-HD and consensus-HD were synthesized by DNA 2.0. Sequences were optimized for expression in E. coli. The two genes were synthesized in tandem on a single duplex DNA, and were separated by three stop codons and a BamHI restriction site. The gene tandem was flanked by 5′ NdeI and BamHI sites and a 3′ XhoI restriction site, which were used to subclone into NdeI and XhoI restriction sites of pET24. The resulting vector expressed engrailed-HD. The expression vector for the consensus-HD was created by digesting with BamHI and ligation of the larger vector fragment, to remove the engrailed-HD gene. Expressed HD constructs include N-terminal Met-Gly-Ser tripeptide and a C-terminal His6-tag.
Constructs were expressed in E. coli (BL21 [DE3]). Cells were grown at 37°C in Luria broth with 50 μg/ml Kanamyacin. When cultures reached an OD600 of 0.6-0.8, the temperature was lowered to 20°C, and IPTG was added to a concentration of 1 mM. Induced cells were grown for 20 hours at 20°C.
The cells were pelleted and resuspended in 25 mM sodium phosphate, pH 7.0 with a cocktail of protease inhibitors (Roche Complete, EDTA-free). Cells were lysed by sonication. Lysates were cleared by centrifugation at 16,000 RPM in a Beckman JA-20 rotor. Cleared lysates were brought up to 300 mM NaCl. 250 units and 1 mg of Benzonase (Sigma) and DNaseI (Roche) were added, and were incubated at room temperature for one to two hours. Proteins were purified using Ni-NTA chromatography followed by cation exchange chromatography. Purified proteins were dialyzed into 25 mM sodium phosphate, 150 mM NaCl pH 7.0 and were frozen at −80°C.
NMR Spectroscopy
Isotopically labeled 15N- and 15N, 13C-consensus-HD proteins were expressed as described above, using M9 minimal media supplemented with 15NH4 and 13C-labeled glucose (Cambridge Isotope Laboratories). All labeled samples were purified as described above. Unless otherwise noted, NMR samples contained 100-300 μM protein, 25 mM sodium phosphate, 150 mM NaCl, and 5% D2O (pH 7.0). All experiments were performed at 20 °C, which provided optimal dispersion in the 1H- 15N HSQC spectra.
DNA for NMR experiments was purchased from IDT. The sequence 5′-GCCTAATTACCG-3′ and its sequence complement 5′-CGGTAATTAGGC-3′ were mixed in equimolar proportion and were annealed in 10 mM Tris, 50 mM NaCl, 1 mM EDTA, pH 7.5 by heating at 95 °C for 10 min and slowly cooling to room temperature. DNA solutions were dialyzed into experimental conditions overnight. DNA-bound protein samples were made with 1.25 fold molar excess of DNA to protein.
1H, 15N-HSQC experiments were performed with free consensus-HD, DNA-bound consensus-HD, and free engrailed-HD (Figure S2). Assignments were made using standard triple-resonance 3D experiments including HNCACB, CBCA(CO)NH, HBHA(CO)NH, HNCO, HN(CA)N, (H)CC(CO)NH-TOCSY, and H(CCCO)NH-TOCSY sequences. Backbone resonances were assigned for both consensus-HD and engrailed-HD. Side-chain resonance were assigned for consensus-HD. Experiments were collected on Bruker Avance and Avance II 600 MHz spectrometers equipped with cryoprobes. Backbone chemical shift data were used for secondary structure prediction of free consensus-HD using TALOS+. Chemical shift perturbations for DNA binding were determined using the following equation39:
| (1) |
3D 15N-resolved 1H{N}- 1H NOESY, 3D 13C- 15N resolved 1H{N}- 1H{C} NOESY, 3D 15N- 15N resolved 1H{N}- 1H{N} HMQC-NOESY-HSQC, and 3D methyl-selective 13C-resolved 1H{C}- 1H{C-methyl} NOESY spectra were used to identify 1H- 1H NOEs for free consensus-HD. The 3D 1H{C}- 1H {C-methyl} NOESY spectrum was collected on a consensus-HD sample that had been exchanged into D2O to suppress signal water. An NOE mixing time of 120 ms was used for all 15N-resolved experiments and a mixing time of 150 ms was used for the methyl-selective 13C-resolved experiments. All NOESY spectra were collected on a Varian 800 MHz spectrometer. All NMR data were processed using NMRPipe40 and assignments were made using Sparky.41
15N relaxation measurements were performed for consensus-HD and engrailed-HD using steady-state 1H- 15N NOE, inversion recovery for 15N R1, and CPMG sequence for 15N R2. Sample concentrations were 500 μM for consensus-HD and 1 mM for engrailed-HD. Intensities for amide 1H- 15N cross peaks were fit using Sparky. Overlapping peaks (L16, R18, E22, Q33, E37, K46 for engrailed-HD; T7, L19, E30, L34, K37, R53 for consensus-HD) and peaks with poor signal intensity (R5 and T6 for consensus-HD) were excluded from all relaxation parameter analysis. The ratio of intensities from spectra collected with and without 1H saturation was used to determine { 1H}- 15N NOE parameters. For 15N R1 and R2 measurements, spectra were recorded at six inversion recovery delays or CPMG delays respectively. Inversion recovery delays used for R1 were 100, 200, 400, 600, 800, and 900 ms. CPMG delays used for R2 were 8.4, 25.2, 42.0, 67.2, 84.0, and 100.8 ms. For both R1 and R2, spectra were duplicated at two delay time points to assess uncertainty in peak intensities. R1 and R2 parameters were determined by fitting the time series to a two-parameter single exponential decay function. Errors reported are standard errors of the fit.
Structure generation with CS-Rosetta
CS-Rosetta (version 3.3) was used to generate a family of structural models of the native state of consensus-HD.29,30 Backbone chemical shift data were used for dihedral restraints in fragment picking. Homologous sequences from natural-occurring homeodomains were excluded throughout the process of fragment picking. 41 unambiguous long-range NOEs identified in NOESY spectra (Table S1) were included as restraints during model generation as distance limits for proton pairs. 10,000 structural models were generated, and the 10 lowest-energy structures were selected for final analysis.
Circular dichroism spectroscopy
All CD measurements were made on an Aviv 62A DS spectropolarimeter. Far-UV measurements were collected in a 0.1 cm quartz cuvette at 20°C, with protein concentrations ranging from 8 to 20 μM. Spectra were collected with a 1 nm step-size, averaging for 5 s at each step.
Guanidine-induced unfolding was monitored by CD at 222 nm at 20°C. UltraPure guanidine hydrochloride was purchased from Invitrogen. Concentrations of guanidine hydrochloride were determined by refractometry.42 Equilibrium unfolding measurements were made using a computer controlled titrator (Hamilton). At each denaturant concentration samples were allowed to equilibrate for 5 minutes, and signal was averaged for 30 seconds. Protein concentrations were between 2-4 μM. Thermodynamic unfolding parameters were obtained by fitting a two-state model to the guanidine-induced unfolding curves.43,44
Kinetic refolding and unfolding experiments
Measurements of folding kinetics were made on an Applied Photophysics SX.18MV-R stopped-flow fluorimeter (Leatherhead, UK). Tryptophan fluorescence was detected using a 280 nm excitation and a 320 nm perpendicular cutoff filter. Proteins were diluted 11-fold to final protein concentrations of 3 to 5 μM.
Rate constants for refolding and unfolding were obtained by fitting individual progress curves using the following equation:
| (2) |
where Yobs is the observed signal, Y∞ is the signal at infinite time, ΔY is the amplitude of the decay, and kapp is the apparent rate constant. A minimum of 5 kinetic traces were obtained at each guanidine concentration. The guanidine dependence of kapp was fit according to the following equation:
| (3) |
where kf and ku are folding and unfolding rate constants; H2O subscripts indicate rate constants extrapolated to zero guanidine concentration. Values of mf and mu are independent of guanidine concentration, resulting in log-linear guanidine dependences for kf and ku. Because the unfolding arm of the consensus-HD chevron is poorly defined, we fixed the mu parameter to the fitted value for the engrailed-HD chevron.
Isothermal Titration Calorimetry
DNA oligonucleotides were purchased from IDT. The sequence 5′-GCGGCCATGTAATAACCTCCGGCG-3′ and its sequence complement 5′-CGCCGGAGGTAATTACATGGCCGC-3′ annealed as previously described. Prior to titration, protein and DNA solutions were dialyzed overnight into the same dialysate containing 25 mM HEPES and 250 mM KCl at pH 7.5. Protein and DNA concentrations were determined after dialysis using UV absorbance. All solutions were thoroughly degased under a vacuum before titration.
Titrations were carried out at 25 °C using 20 μM DNA, 200 μM protein for engrailed-HD and 5-6 μM DNA, 50-60 μM protein for consensus-HD on a VP-ITC microcalorimeter from MicroCal (Northhampton, MA). Protein solution was injected in 8 μL volumes at 300 s intervals into the DNA solution in the cell. Data were analyzed using Origin 7.0 software fitting to a single-site model. Titrations were repeated in triplicate for each protein.
Results
Consensus-HD sequence design
To determine the amino acid frequencies by position in the homeodomain family, we used the homeodomain sequence alignment from Pfam 26.0. A homeodomain consensus sequence was constructed from this alignment by selecting the most frequent amino acid at each position (Figure 1a). On average, the consensus-HD sequence is 56% identical to naturally-occurring sequences in the starting alignment. The highest sequence identity (69%) is to the short stature homeobox protein 2 from the body louse Pediculus humanus corporis. As a point of comparison, we also expressed and purified the engrailed homeodomain from Drosophila melanogaster. The stability and folding of the engrailed-HD has been studied extensively by experimental and computational methods.23,24 The engrailed-HD and consensus-HD are 59 and 58 residues long and share 50% identity (Figure 1b). In the engrailed homeodomain there are 11 residues that make direct contact with DNA.25 The consensus-HD is identical to the engrailed at 10 of 11 of these residues (Figure 1b). The single sequence difference is a conservative substitution of an isoleucine (engrailed position 47) to a valine in the consensus-HD.
Secondary structure of consensus-HD
To determine whether the consensus-HD adopts the expected α-helical structure, we obtained far-UV CD spectra of the consensus-HD. The far-UV spectrum of the consensus-HD has minima at 208 and 222 nm (Figure 2a), characteristic of proteins that have α-helical secondary structure. The far-UV spectrum of consensus-HD is similar to the engrailed-HD, indicating a similar secondary structure content.
Figure 2. Circular dichorism spectroscopy and NMR spectroscopy of consensus-HD.
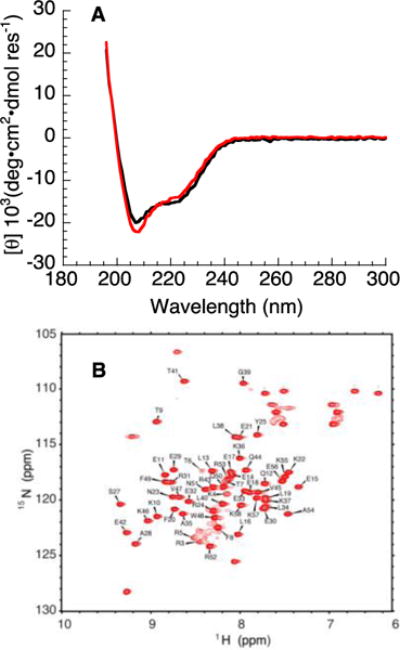
A) Far-UV CD spectra of consensus-HD (black) and engrailed-HD (red). B) 15N- 1H HSQC spectrum of uniformly 15N-labeled consensus-HD at 600 MHz. Assigned peaks are labeled. Conditions: 25 mM sodium phosphate, 150 mM NaCl, pH 7, 20 °C.
To determine whether the consensus-HD has a rigid, well-formed tertiary structure, we collected an 15N- 1H HSQC NMR spectrum of the consensus-HD. The resonances are sharp and well-dispersed (Figure 2b), indicating that the consensus-HD adopts well-defined tertiary structure in solution. There are 76 expected cross peaks for the consensus-HD: 65 non-prolyl backbone NH’s (including his6-tag), one tryptophan side chain NH, and ten asparagine and glutamine side chain NH’s. Out of the expected 76 expected cross peaks we see 69 cross peaks. To obtain higher resolution structural information, we used standard triple-resonance methods and were able to assign 96% of the backbone resonances (excluding the his6-tag and single proline residues). In addition, 88% of side chain aliphatic protons and 96% of side chain aliphatic carbons were assigned.
To confirm the location of helical elements within the consensus-HD sequence, we obtained backbone chemical shift-based secondary structure predictions using TALOS+.26 This analysis identifies three regions within the consensus-HD sequence showing high probability of α-helical structure (Figure 3a). These regions of predicted α-helical structure in the consensus-HD align perfectly in sequence with the three α-helices seen in engrailed-HD crystal structure27, based on the sequence alignment in Figure 1.
Figure 3. Secondary-structure of consensus-HD determined by NMR spectroscopy.
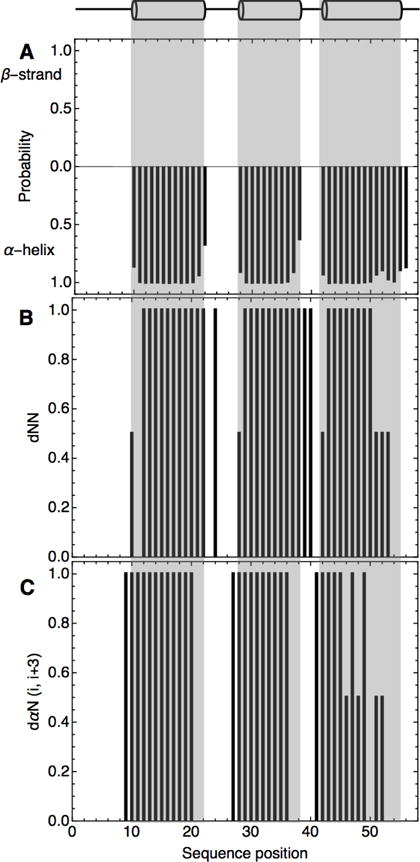
A) Plot of chemical shift-based secondary structure predictions by TALOS+29 B) 1HNi- 1HNi+1 and C) 1Hαi- 1HNi+3 NOEs. Strong NOEs were given value of one. Weak but detectable NOEs were given a value of 0.5. Positions with no bar showed no detectable NOE. Helix boundaries (top) are based on the engrailed-HD structure. Sequence positions are as in alignment in Figure 1B where position 1 corresponds to the N-terminal D in engrailed-HD and position 2 corresponds to the N-terminal R in consensus-HD. Conditions: 25 mM sodium phosphate, 150 mM NaCl, pH 7, 20 °C.
We further confirmed the secondary structure using 1H- 1H NOE connectivities. NOE connectivities between sequential backbone amide protons ( 1HNi- 1HNi±1) and between alpha protons and the amide proton three residues forward in sequence ( 1Hαi- 1HNi+3) are characteristic of α-helical secondary structure elements.28 The short-range NOEs observed in the consensus-HD are consistent with the α-helical structure predicted by TALOS+ and the homeodomain fold (Figures 3b and 3c).
Tertiary structure of the consensus-HD
To obtain information on the tertiary structure of the consensus-HD, we measured 1H- 1H NOEs using both 13C-edited and 15N-edited NOE experiments. In the 13C-edited experiment, a methyl-selective pulse sequence was used to identify NOEs between side chain methyl protons and aliphatic protons. In the 15N-edited experiment, NOEs were identified between amide backbone protons and side chain aliphatic protons. From all NOESY spectra, 41 unambiguous long-range NOEs (|i - j| > 4 residues) were identified (Table S1).
These long-range NOEs were used along with chemical shifts to generate structural models of the consensus-HD native state using restrained CS-Rosetta.29,30 During the fragment generation stage, we excluded fragments derived from homeodomain structures. We generated 10,000 Rosetta structures, and found a modestly funneled distribution of energy scores versus Cα RMSD (from the lowest scoring structure), with a weak, positive correlation (Figure S1). We examined the ten lowest energy structures, which all adopt a three-helix homeodomain fold (Figure 4). When superposed onto engrailed-HD, the lowest energy structure has a Cα RMSD of 2.46 Å over all residues. Excluding the relatively unrestrained N-terminus (residues 1-9 of consensus-HD) gives an RMSD of 2.14 Å. When the ten lowest energy structures are compared to each other, an average Cα RMSD of 2.16Å is obtained (excluding the N-terminus, Figure 4B).
Figure 4. CS-Rosetta generated model of consensus-HD.
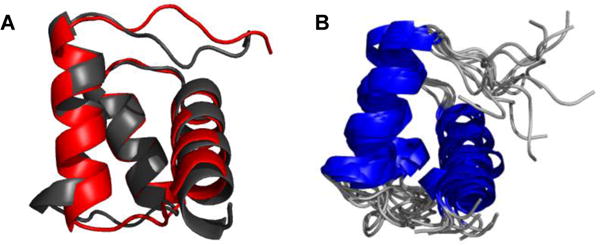
A) Alignment of the backbone atoms of engrailed-HD (red, 1ENH.pdb) and the lowest energy CS-Rosetta structure of consensus-HD (black). B) Overlay of ten lowest energy structures from CS-Rosetta model generation.
Equilibrium stability of the engrailed- and consensus-HD folds
To compare the stabilities of the consensus-HD and engrailed-HD, guanidine-induced unfolding transitions were measured using CD spectroscopy (Figure 5a). Both constructs unfold in single cooperative transitions, indicative of two-state folding. Remarkably, the midpoint of the consensus-HD unfolding transition is approximately 3 M guanidine higher than that of the engrailed-HD (Figure 5a, Table 1). The fitted free energy of unfolding of the consensus-HD is 8.1 kcal·mol−1, approximately 5 kcal·mol−1 higher than that of engrailed-HD. The fitted m-value of the consensus-HD is similar to that of the engrailed-HD, indicating that the high degree of cooperativity seen for the engrailed-HD is maintained in the consensus design.
Figure 5. Stability and folding kinetics of consensus-HD compared to engrailed-HD.
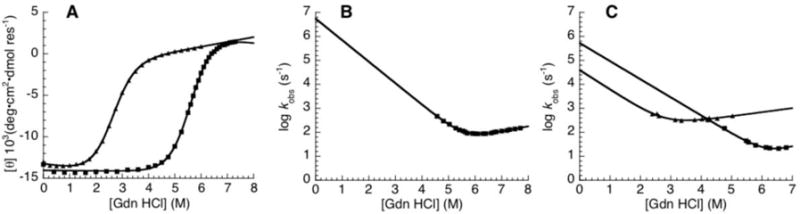
A) Guanidine-induced unfolding of consensus-HD (black) and engrailed-HD (red) at 20 °C. B) Guanidine dependence of the refolding and unfolding rate constant of consensus-HD at 20 °C. C) Guanidine dependence of the refolding and unfolding rate constants of consensus-HD (black) and engrailed-HD (red) at 10 °C. Solid lines are obtained from fitting a kinetic two-state model (equation 3) to the data. Conditions: 25 mM sodium phosphate, 150 mM NaCl, pH 7, temperature noted.
Table 1.
Thermodynamic and kinetic parameters for engrailed-HD and consensus-HD.
| Folding thermodynamics | Folding kinetics | Binding thermodynamics | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||
| ΔG°(H2O)(kcal·mol−1) | m-value (kcal·mol−1·M−1) | (s−1) | mf (Μ−1) | (s−1) | mu (Μ−1) | n | Kd (nM) | ΔΗ° (kcal·mol−1) | TΔS° (kcal·mol−1) | |
|
|
||||||||||
| Engrailed-HD | 3.62 ± 0.03 | 1.39 ±0.01 | 3.9 × 104 | −0.81 | 67.1 | 0.17 | 0.98 ± 0.01 | 737 ± 38 | −9.2 ±0.1 | −0.8 ± 0.1 |
| Consensus-HD | 8.05 ±0.10 | 1.45 ± 0.02 | 1.1 × 10e | −0.83 | 1.4 | NAa | 1.00 ±0.01 | 8.1 ±1.6 | −16 ±0.2 | −5.0 ± 0.3 |
| Consensus-HD (10°C) | 8.51 ± 0.27 | 1.35 ± 0.02 | 5.50 × 10 | −0.90 | 3.24 | 0.23 | ND | ND | ND | ND |
Folding thermodynamic: parameters were obtained by fitting gaunidine-induced denaturation curves. Uncertainies are standard errors on the mean of three titrations. Conditions: 25 mM NaPO4 (pH 7.0), 150 mM NaCI, 20°C
Folding kinetics: Parameters were obtained by fitting equation (2) to the guanidine dependence of the rate constants for the refolding and unfolding 1 phases (Figure 5B, C).
For consensus-HD, the mu parameter was fixed to the fitted value for the engrailed-HD (0.17 M−1). Conditions: 25 mM NaPO4, 150 mM NaCI, temperatures as indicated.
Binding thermodynamics: Data were anaylzed using Origin 7.0 software fitting to a single site model. Uncertainies are standard errors on the mean of three independent experiments Conditions: 25 mM HEPES (pH 7.5), 250 mM KCI, 25°C.
Kinetic characterization of the engrailed- and consensus-HD folding
To determine how the stability differences between the consensus-HD and engrailed-HD relate to the folding kinetics, we measured rates of refolding and unfolding using stopped-flow fluorescence. For the consensus-HD, we were able to measure the folding and unfolding kinetics at 20 °C, over a range of guanidine concentrations (Figure 5b). Both refolding and unfolding kinetic traces fit well to a single exponential (data not shown). Over the guanidine concentrations that rates were experimentally accessible, the chevron has linear refolding and unfolding arms. The free energy of unfolding determined from the extrapolated rate constants is 8.07 kcal·mol−1, which is within error of the value (8.51 kcal·mol−1) measured in our equilibrium experiments.
Unfortunately, the folding and unfolding rates of engrailed-HD at 20 °C are too fast to measure reliably in a standard stopped-flow apparatus.23,24 To compare the folding kinetics of engrailed-HD and consensus-HD, we collected kinetic traces of both proteins at 10 °C (Figure 5c). At this temperature, reproducible kinetic traces could be resolved for both constructs. Again, the unfolding and refolding kinetic traces of both constructs were fit well by a single exponential (data not shown), and show approximately linear refolding arms. Comparing the two chevron plots, it is clear that the consensus HD folds faster, and unfolds more slowly than the natural engrailed-HD.
Because we were limited by both protein concentrations and guanidine solubility, the unfolding arm of the consensus-HD chevron could not be adequately defined at 10 °C. To fit the chevron plot of the consensus-HD we fixed the mu to that from the fit of the engrailed-HD chevron. Extrapolating fitted rate constants indicates that the kinetic partitioning of consensus-HD stabilization is maintained in the absence of denaturant. Thus, the transition state for folding is also stabilized by consensus substitutions, albeit to a smaller extent than the native state.
Structural characterization of DNA binding
To determine whether consensus-HD retains DNA binding function, we collected a 1H- 15N HSQC spectrum of 15N-labelled consensus-HD in the presence of an unlabeled DNA duplex containing the six base-pair cognate binding site for engrailed-HD. The spectrum of consensus-HD in the presence of DNA contains significant changes from that of the free protein, indicating that the consensus-HD binds the engrailed-HD cognate sequence (Figure 6a). At substoichiometric DNA concentrations we see sharp resonances for free and bound consensus-HD, demonstrating slow exchange (not shown). To determine which residues are involved in the interaction with DNA, we collected standard triple-resonance NMR experiments to successfully assign 95% of the backbone amide resonances. We then determined chemical shift perturbations between bound and free spectra using equation 1. Significant perturbations are seen for the flexible N-terminal arm and the C-terminal recognition helix (Figure 6b). This is consistent with DNA binding architecture of the homeodomain family, where the C-terminal helix makes specific contacts with DNA bases in the major groove and the N-terminal arm fits into the minor grove (Figure 6c).22,25
Figure 6. DNA-binding architecture of consensus-HD confirmed by NMR spectroscopy.
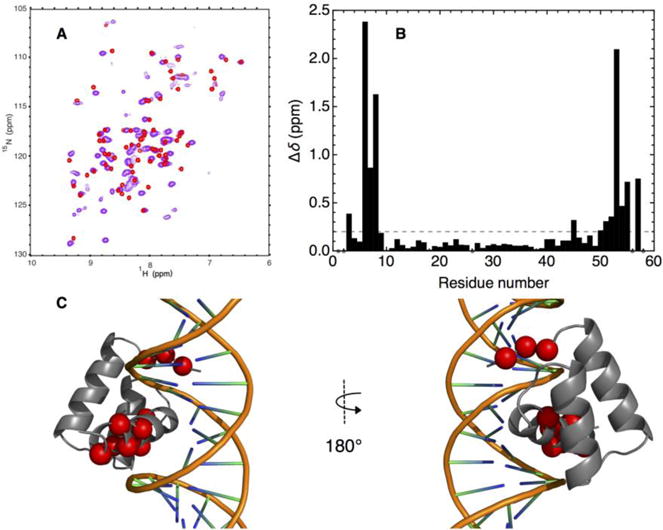
A) Overlay of 15N- 1H HSQC spectra of uniformly 15N labeled free consensus-HD (red) and DNA-bound consensus-HD (purple, 1.25-fold molar excess of duplex DNA) at 600 MHz. Conditions: 25 mM sodium phosphate, 150 mM NaCl, pH 7, 20 °C. B) Plot of weighted chemical shift perturbations for DNA binding to consensus-HD, calculated using equation 1, versus residue number. Dots at zero shift perturbation correspond to unassigned residues. Sequence numbering as in Figure 3. C) Residues with large chemical shift perturbations mapped on to the homeodomain structure (engrailed-HD:DNA cocrystal structure, PDB: 3HDD). Residues with chemical shift perturbations larger than 0.2 ppm (dashed line, Fig. 7b) are highlighted in red spheres centered at Cα.
Characterization of DNA binding thermodynamics
To investigate how consensus substitution affects DNA affinity, we used isothermal calorimetry (ITC) to monitor the binding of the engrailed- and consensus-HDs to DNA duplex containing the six base engrailed recognition sequence (5′-TAATTA-3′). At 150 mM KCl, the consensus-HD shows saturation binding (data not shown). Although saturation binding demonstrates that binding is tight, it precludes accurate determination of binding affinity. To quantify binding affinity we increased salt concentration in an attempt to weaken the consensus-HD DNA complex. At 250 mM KCl, we obtained enough curvature in the thermogram to accurately determine affinity (Figure 7). Under these conditions the consensus-HD binds the cognate DNA with high affinity (Kd = 8.1 nM) with a 1:1 stoichiometry. This high DNA binding affinity is achieved through a very favorable binding enthalpy that is slightly compensated by a smaller unfavorable binding entropy (Table 1). Surprisingly, consensus-HD binds the cognate DNA with an affinity rougly two orders of magnitude tighter than engrailed-HD (Kd = 737.4 nM; Figure 6d, Table 1). The higher affinity of consensus-HD is the result of a significantly more favorable enthalpy of binding, which is partly offset by a more unfavorable entropy penalty.
Figure 7. Isothermal titration calorimetry of consensus- and engrailed-HDs binding to DNA.
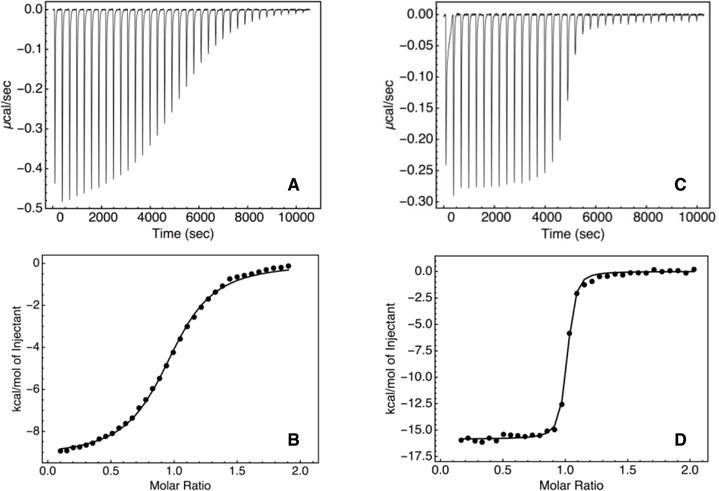
A) Differential power and B) integrated heat peaks resulting from titration of the engrailed-HD into DNA. C) Differential power and D) integrated heat peaks resulting from titration of the consensus-HD into DNA. The black lines show a least-squares fit using a single-site model. Conditions: 25 mM HEPES, 250 mM KCl pH 7.5, 25 °C.
Dynamics of the consensus-HD on the psec-nsec timescale
To examine whether the increased stability and/or DNA binding affinity of the consensus-HD correlate with changes in protein dynamics, we measured backbone 15N-relaxation parameters ({ 1H}- 15N NOE, 15N R1, and 15N R2) for consensus and engrailed-HD (Figure 8 and Table S2). We see similar overall profiles for each of these parameters, however, consensus-HD appears to have a decreased amplitude of dynamic motions in the N- and C-termini, as well as in the loops between the three helices. The high R2 values in the loop between helices 1 and 2 of engrailed-HD suggests are additional μsec-msec motions, as previously observed31, that are not observed in consensus-HD. These changes may result directly from the two sequence substitutions in this loop, or from a combination of the other 26 differences between engrailed- and consensus-HD.
Figure 8. 15N relaxation parameters for consensus-HD and engrailed-HD.
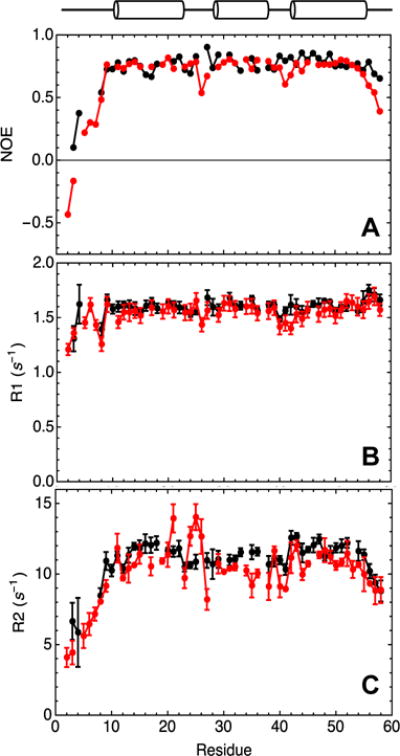
A) Heteronuclear NOE, B) R1, and C) R2 parameters for consensus-HD (black) and engrailed-HD (red) shown as a function of sequence position. Errors shown for R1 and R2 parameters for each residue are standard errors of fits. Overlapping peaks (L16, R18, E22, Q33, E37, K46 for engrailed-HD; T7, L19, E30, L34, K37, R53 for consensus-HD) and peaks with low signal intensity (R5 and T6 for consensus-HD) were excluded from analysis. Helix positions are based on engrailed-HD crystal structure. Sequence numbering as in Figure 3. Conditions: 25 mM sodium phosphate, 150 mM NaCl, pH 7.0, 20°C.
Discussion
The residues that stabilize protein folds are often different from those that contribute to activity, and in many cases have been shown to be conflicting. Using sequence averages (that is, consensus sequences) from large groups of proteins may provide a route to optimize stability while preserving (or enhancing) activity, as long as high stability itself does not interfere with activity. This approach has been used to design linear repeat protein with high stability, and more recently, globular proteins. In some cases, partial biological function is maintained, but in other cases it is lost. Here, we find a stabilized consensus version of the homeodomain to bind DNA significantly tighter than a naturally-occurring homeodomain, demonstrating that function is encoded in the consensus, and is not antagonized by high stability.
Consensus-HD adopts a cooperatively folded homeodomain structure with very high stability
CD and NMR spectroscopy show that consensus-HD is folded and adopts the expected α-helical homedomain fold (Figures 2–4). Like the engrailed-HD, the consensus-HD has a single cooperative unfolding transition that is well described by a two-state model. The fitted m-value of the consensus-HD is 1.45 kcal·mol−1·M−1, similar to that of engrailed-HD (Table 1) and is consistent with an empirical prediction based on chain-length.32 Thus, for the homeodomain, consensus design captures the high degree of cooperativity seen in the folding of globular proteins.
The free energy of unfolding of the consensus-HD is approximately 5 kcal·mol−1 greater than that of engrailed-HD. When assessed by thermal denaturation the Tm of the consensus-HD is 23 °C higher than Tm of engrailed-HD, although the thermal denaturation of consensus-HD is not fully reversible (data not shown). Such a drastic increase in stability is surprising given the high sequence similarity between consensus-HD and engrailed-HD and the small size of the two domains. However, this is consistent with the large stability enhancement observed for consensus repeat proteins.8–10
The high stability of consensus-HD results from surface substitutions
The increased stability of the consensus-HD must result from some subset of the 30 sequence substitutions (~50% difference) from the engrailed HD. Not surprisingly, the positions that have the highest conservation within the homeodomain family are identical in the engrailed- and consensus-HDs. These conserved positions are nearly all core residues. The 30 sequence substitutions are nearly all surface residues (Figure 1c). Given the importance of core residues in protein stability, it is somewhat surprising that this large stability increment is due to differences at surface positions. Although we did not intentionally target surface residues in our design, the success of the consensus-HD construct suggests a strategy for designing increased stability that limits consensus substitution to surface residues.
Stability enhancement through surface substitution may have a significant electrostatic component. The consensus-HD has considerably more charged residues (29/57), but lower net charge (+5) than the engrailed-HD (23/58, +9). Balanced surface charge, even at high charge density, has been shown to be an effective means to stabilize small globular proteins33,34, and is consistent with the observation that proteins from thermostable organisms have a higher fraction of charged residues than mesophilic homologues.35
Stability increase is partitioned into both the folding and unfolding kinetics
Increased stability implies faster folding, slower unfolding, or both. We find that the increase in stability of the consensus-HD partitions into both faster folding kinetics and slower unfolding kinetics (Table 1). The extrapolated folding rate constant increases by around 25-fold for consensus-HD, whereas the unfolding rate constant decreases by about 50-fold. This partitioning indicates that the stabilizing consensus interactions are partly (but incompletely) developed in the transition state ensemble. The folding rate constant for consensus-HD in water is estimated to be approximately 106 sec−1. Although such a rate constant is near the estimated speed limit for folding,36 it is clear that considerable folding rate enhancements can be obtained by consensus stabilization. Likewise, consensus stabilization appears to provide a means to significantly enhance kinetic stability against unfolding.
Consensus-HD has high binding affinity for DNA
Although the consensus homeodomain sequence gives significant enhancement of stability, it is unclear how this increased stability should affect function. The common observation that proteins are marginally stable has been interpreted by some to suggest that high stability would impair function, particularly if conformational heterogeneity (often referred to as “dynamics”) is important for function.35,37 Our finding that consensus-HD binds DNA with native-like architecture and a hundredfold increase in affinity is inconsistent with interpretation that stability and function are in opposition. Rather, our measurements of backbone dynamics demonstrate that increased affinity correlates with decrease backbone motion (Figure 8). Thus, whereas consensus stabilization may lead to a more rigid native state, it does not appear to compromise binding energy.
The enhanced affinity is somewhat surprising given the decreased positive charge of the consensus-HD. Although the direct DNA contact residues from the engrailed-HD are conserved in the consensus-HD, the more numerous, distant sequence substitutions have a rather profound effect on affinity. This result is consistent with an alanine substitution study by Abate and coworkers on the Msx homeodomain, which showed that positions with relatively low conservation (particularly in the N-terminal arm) were required for high affinity DNA binding.38 The enhancement in consensus-HD binding is enthalpically driven, but is partly offset by an entropy penalty. These changes may reflect increased favorable bonding interactions for the consensus-HD:DNA complex, which may have increased rigidity. Alternatively, these changes may be related to changes in salt ion displacement or hydration. Regardless, the increase in affinity suggests that consensus sequences also encode functional properties. The extent to which function- and stability-enhancing substitutions overlap remains to be determined.
Conclusions
Our work here demonstrates the design of a functional globular α-helical protein using consensus information. The consensus-HD adopts a homeodomain fold, has a cooperative unfolding transition and is dramatically stabilized over the naturally occurring engrailed-HD. The consensus designed homedomain captures the native DNA-binding function of the homeodomain, with an affinity that is significantly tighter than the engrailed-HD. This work suggests that using consensus information, especially at surface positions, may provide a route to stabilize target proteins that retain function. How this finding holds true for larger helical domains and folds that have β-sheet secondary structure remains to be determined.
Acknowledgments
We thank the Johns Hopkins University Biomolecular NMR Center and the Center for Molecular Biophysics for providing facilities and resources. We thank Jeliazko Jeliazkov for help with restrained CS-Rosetta modeling. Chemical shifts have been deposited in the BMRB (accession code 26995). This work was supported by National Insitutes of Health grants GM068462 to D.B and by an NIH Training grant T32 GM008403.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Details include observed long-range NOEs (Table S1), 15N relxation parameters (Table S2), CS-Rosetta structural analysis data (Figure S1) and engrailed-HD HSQC spectrum (Figure S2).
Notes
The authors declare no competing financial interest.
References
- 1.Fesnak AD, June CH, Levine BL. Nat Rev Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Farid SS. Polyclonal Monoclon Antib Prod Purif Process Prod Anal. 2007;848:8–18. [Google Scholar]
- 3.Shoichet BK, Baase WA, Kuroki R, Matthews BW. Proc Natl Acad Sci U S A. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beadle BM, Shoichet BK. J Mol Biol. 2002;321:285–296. doi: 10.1016/s0022-2836(02)00599-5. [DOI] [PubMed] [Google Scholar]
- 5.Wolf-Watz M, Thai V, Henzler-Wildman K, Hadjipavlou G, Eisenmesser EZ, Kern D. Nat Struct Mol Biol. 2004;11:945–949. doi: 10.1038/nsmb821. [DOI] [PubMed] [Google Scholar]
- 6.Holland LZ, McFall-Ngai M, Somero GN. Biochemistry (Mosc) 1997;36:3207–3215. doi: 10.1021/bi962664k. [DOI] [PubMed] [Google Scholar]
- 7.Mosavi LK, Minor DL, Peng Z. Proc Natl Acad Sci. 2002;99:16029–16034. doi: 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aksel T, Majumdar A, Barrick D. Structure. 2011;19:349–360. doi: 10.1016/j.str.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devi VS, Binz HK, Stumpp MT, Plückthun A, Bosshard HR, Jelesarov I. Protein Sci. 2004;13:2864–2870. doi: 10.1110/ps.04935704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Main ERG, Xiong Y, Cocco MJ, D’Andrea L, Regan L. Structure. 2003;11:497–508. doi: 10.1016/s0969-2126(03)00076-5. [DOI] [PubMed] [Google Scholar]
- 11.Parmeggiani F, Pellarin R, Larsen AP, Varadamsetty G, Stumpp MT, Zerbe O, Caflisch A, Plückthun A. J Mol Biol. 2008;376:1282–1304. doi: 10.1016/j.jmb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 12.Urvoas A, Guellouz A, Valerio-Lepiniec M, Graille M, Durand D, Desravines DC, van Tilbeurgh H, Desmadril M, Minard P. J Mol Biol. 2010;404:307–327. doi: 10.1016/j.jmb.2010.09.048. [DOI] [PubMed] [Google Scholar]
- 13.Steipe B, Schiller B, Plückthun A, Steinbacher S. J Mol Biol. 1994;240:188–192. doi: 10.1006/jmbi.1994.1434. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan BJ, Nguyen T, Durani V, Mathur D, Rojas S, Thomas M, Syu T, Magliery TJ. J Mol Biol. 2012;420:384–399. doi: 10.1016/j.jmb.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krizek BA, Amann BT, Kilfoil VJ, Merkle DL, Berg JM. J Am Chem Soc. 1991;113:4518–4523. [Google Scholar]
- 16.Lehmann M, Kostrewa D, Wyss M, Brugger R, D’Arcy A, Pasamontes L, van Loon APGM. Protein Eng. 2000;13:49–57. doi: 10.1093/protein/13.1.49. [DOI] [PubMed] [Google Scholar]
- 17.Lehmann M, Loch C, Middendorf A, Studer D, Lassen SF, Pasamontes L, van Loon APGM, Wyss M. Protein Eng. 2002;15:403–411. doi: 10.1093/protein/15.5.403. [DOI] [PubMed] [Google Scholar]
- 18.Sullivan BJ, Durani V, Magliery TJ. J Mol Biol. 2011;413:195–208. doi: 10.1016/j.jmb.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Jacobs SA, Diem MD, Luo J, Teplyakov A, Obmolova G, Malia T, Gilliland GL, O’Neil KT. Protein Eng Des Sel PEDS. 2012;25:107–117. doi: 10.1093/protein/gzr064. [DOI] [PubMed] [Google Scholar]
- 20.Porebski BT, Nickson AA, Hoke DE, Hunter MR, Zhu L, McGowan S, Webb GI, Buckle AM. Protein Eng Des Sel PEDS. 2015;28:67–78. doi: 10.1093/protein/gzv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porebski BT, Keleher S, Hollins JJ, Nickson AA, Marijanovic EM, Borg NA, Costa MGS, Pearce MA, Dai W, Zhu L, Irving JA, Hoke DE, Kass I, Whisstock JC, Bottomley SP, Webb GI, McGowan S, Buckle AM. Sci Rep. 2016;6:33958. doi: 10.1038/srep33958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolberger C, Vershon AK, Liu B, Johnson AD, Pabo CO. Cell. 1991;67:517–528. doi: 10.1016/0092-8674(91)90526-5. [DOI] [PubMed] [Google Scholar]
- 23.Mayor U, Guydosh NR, Johnson CM, Grossmann JG, Sato S, Jas GS, Freund SMV, Alonso DOV, Daggett V, Fersht AR. Nature. 2003;421:863–867. doi: 10.1038/nature01428. [DOI] [PubMed] [Google Scholar]
- 24.Religa TL, Markson JS, Mayor U, Freund SMV, Fersht AR. Nature. 2005;437:1053–1056. doi: 10.1038/nature04054. [DOI] [PubMed] [Google Scholar]
- 25.Kissinger CR, Liu BS, Martin-Blanco E, Kornberg TB, Pabo CO. Cell. 1990;63:579–590. doi: 10.1016/0092-8674(90)90453-l. [DOI] [PubMed] [Google Scholar]
- 26.Shen Y, Delaglio F, Cornilescu G, Bax A. J Biomol NMR. 2009;44:213–223. doi: 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clarke ND, Kissinger CR, Desjarlais J, Gilliland GL, Pabo CO. Protein Sci. 1994;3:1779–1787. doi: 10.1002/pro.5560031018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wemmer DE, Reid BR. Annu Rev Phys Chem. 1985;36:105–137. [Google Scholar]
- 29.Shen Y, Lange O, Delaglio F, Rossi P, Aramini JM, Liu G, Eletsky A, Wu Y, Singarapu KK, Lemak A, Ignatchenko A, Arrowsmith CH, Szyperski T, Montelione GT, Baker D, Bax A. Proc Natl Acad Sci U S A. 2008;105:4685–4690. doi: 10.1073/pnas.0800256105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowers PM, Strauss CE, Baker D. J Biomol NMR. 2000;18:311–318. doi: 10.1023/a:1026744431105. [DOI] [PubMed] [Google Scholar]
- 31.Stollar EJ, Mayor U, Lovell SC, Federici L, Freund SMV, Fersht AR, Luisi BF. J Biol Chem. 2003;278:43699–43708. doi: 10.1074/jbc.M308029200. [DOI] [PubMed] [Google Scholar]
- 32.Myers JK, Nick Pace C, Martin Scholtz J. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schweiker KL, Zarrine-Afsar A, Davidson AR, Makhatadze GI. Protein Sci Publ Protein Soc. 2007;16:2694–2702. doi: 10.1110/ps.073091607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gribenko AV, Patel MM, Liu J, McCallum SA, Wang C, Makhatadze GI. Proc Natl Acad Sci U S A. 2009;106:2601–2606. doi: 10.1073/pnas.0808220106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das R, Gerstein M. Funct Integr Genomics. 2000;1:76–88. doi: 10.1007/s101420000003. [DOI] [PubMed] [Google Scholar]
- 36.Kubelka J, Hofrichter J, Eaton WA. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 37.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 38.Shang Z, Isaac VE, Li H, Patel L, Catron KM, Curran T, Montelione GT, Abate C. Proc Natl Acad Sci U S A. 1994;91:8373–8377. doi: 10.1073/pnas.91.18.8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Williamson MP. Prog Nucl Magn Reson Spectrosc. 2013;73:1–16. doi: 10.1016/j.pnmrs.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 40.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 41.Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: [Google Scholar]
- 42.Nozaki Y. Methods Enzymol. 1972;26:43–50. doi: 10.1016/s0076-6879(72)26005-0. [DOI] [PubMed] [Google Scholar]
- 43.Pace CN. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 44.Street TO, Courtemanche N, Barrick D. Methods Cell Biol. 2008;84:295–325. doi: 10.1016/S0091-679X(07)84011-8. [DOI] [PubMed] [Google Scholar]