

Abstract
The molecular mechanisms of maladaptive response in liver tissue with respect to the acute and post-acute phase of sepsis are not yet fully understood. Long-term sepsis survivors might develop hepatocellular/hepatobiliary injury and fibrosis. Here, we demonstrate that acid sphingomyelinase, an important regulator of hepatocyte apoptosis and hepatic stellate cell (HSC) activation, is linked to the promotion of liver dysfunction in the acute phase of sepsis as well as to fibrogenesis in the long-term. In both phases, we observed a beneficial effect of partial genetic sphingomyelinase deficiency in heterozygous animals (smpd1+/−) on oxidative stress levels, hepatobiliary function, macrophage infiltration and on HSC activation. Strikingly, similar to heterozygote expression of SMPD1, either preventative (p-smpd1+/+) or therapeutic (t-smpd1+/+) pharmacological treatment strategies with desipramine – a functional inhibitor of acid sphingomyelinase (FIASMA) – significantly improved liver function and survival. The inhibition of sphingomyelinase exhibited a protective effect on liver function in the acute-phase, and the reduction of HSC activation diminished development of sepsis-associated liver fibrosis in the post-acute phase of sepsis. In summary, targeting sphingomyelinase with FDA-approved drugs is a novel promising strategy to overcome sepsis-induced liver dysfunction.
Introduction
Sepsis continues to be a serious problem worldwide with mortality rates reaching up to 18% due to consecutive organ failure and maladaptive host response1,2. The development of organ dysfunction resulting in multiple organ failure is a hallmark of the disease continuum which remains one major cause for poor outcome of these patients3. In particular, hepatic excretory dysfunction is an early and common consequence of sepsis4. In addition to challenges in managing the acute phase of the overwhelming host response to infection, long-term consequences of sepsis in survivors are still an underestimated issue compromising quality of life and resulting in an increased mortality risk following hospital discharge5. Previous studies have demonstrated that sepsis leads to increased liver-stiffness6 and prolonged sepsis-associated cholestasis with associated mortality7,8. The latter even with the risk to result in progressive sclerosing cholangitis9. These phenomena are also mimicked in a long-term murine model of sepsis where fibrosis of the liver subsequent to hepatic stellate cell (HSC) activation was shown in survivors10.
Among the plethora of pathophysiological changes in sepsis, the enhanced activity of the stress responsive enzyme sphingomyelin-phosphodiesterase 1 (SMPD1) might function as a promising target for therapeutic interventions11–13. Previous data suggest a positive association between an increased concentration as well as activity in plasma and disease severity allowing discrimination of outcome of these patients14. Furthermore, the regulation of this enzyme by a functional inhibitor improved either the survival of mice in an endotoxemia model11 or overcoming from cardiomyopathy due to polymicrobial sepsis15. SMPD1 is responsible for the fast and transient breakdown of membrane-embedded sphingomyelin to ceramide which in turn accumulates in lipid rafts of the outer layer of the cell membrane thus reorganizing protein complexes and supporting downstream signal transduction of inflammatory pathways16,17. SMPD1 is necessary for numerous signaling pathways controlling proliferation, autophagy, differentiation and apoptosis18–20. On a molecular level, there is also evidence of a direct regulation of the activation process of HSC into activated liver myofibroblasts (aLMF) by SMPD1 controlling extracellular matrix (ECM) production and deposition. The inhibition of SMPD1 abrogates the transdifferentiation of primary mouse hepatic stellate cells in vitro and reduces fibrogenesis in an in vivo model of bile-duct ligation21. Moreover, numerous studies have also provided a direct impact of SMPD1 activity on hepatocyte damage in vitro as well as in vivo 22,23. In hepatocytes of SMPD1 deficient animals, the total loss of enzyme function protects these cells from Fas- and TNF-α-mediated apoptosis24,25.
In order to characterize the effects of SMPD1 in the setting of acute- and post-acute liver dysfunction following the host response during sepsis, we opted for different inhibition strata: As a model of pharmacological inhibition, we preventively treated smpd1+/+ animals (p-smpd1+/+) for seven days prior to the induction of sepsis with desipramine, a well-known functional inhibitor of SMPD1 (FIASMA)26. Furthermore, we established a therapeutic strategy treating smpd1+/+ mice (t-smpd1+/+) with desipramine 6 hours following induction of polymicrobial sepsis, in order to evaluate a potential role of its therapeutic use. Additionally, we compared these animals with septic smpd1+/+ littermates as well as with mice partially deficient for the enzyme (smpd1+/−). At three time points representing different phases of sepsis-associated liver damage and fibrogenesis (24 hours, 3 and 28 days), we monitored the role of SMPD1 in the maintenance of hepatic (dys)function during the acute-phase and in the prevention of hepatic fibrosis during the post-acute phase of sepsis.
Results
Detrimental role of SMPD1 in liver function following polymicrobial sepsis
Previous studies have demonstrated that the increased activity of the conserved stress responsive enzyme SMPD1 is associated with impaired outcome in septic patients and animal models11,27. Measurement of the SMPD1 activity in serum samples of smpd1+/+ littermates revealed an increased activity at 24 hours and 3 days following sepsis induction, whereas at day 28 the SMPD1 activity did not differ from that in sham-treated animals. In contrast, smpd1+/− animals demonstrated significant lower activity levels at all time points as compared to smpd1+/+ mice (Fig. 1A). Furthermore, smpd1+/+ animals had depleted glutathion (GSH) levels in liver homogenates at 24 hours following sepsis induction indicating noticeable oxidative stress. The partial genetic inhibition of the enzyme significantly improved oxidative stress levels in the acute phase of sepsis (Fig. 1B). In addition, smpd1+/− animals had lower surrogates of liver dysfunction as represented by γ-GT, total-bilirubin and the transcriptional expression of Mrp2 levels at day 3 following sepsis as compared to wild-type smpd1+/+ animals (Fig. 1C–E). These results demonstrate that the stress responsive enzyme SMPD1 is an essential key player in the development of hepatic dysfunction during polymicrobial sepsis.
Figure 1.

Increased SMPD1 activity is highly associated with liver dysfunction in polymicrobial sepsis. Polymicrobial sepsis was induced in smpd1+/+ and smpd1+/− mice by peritoneal contamination and infection to determine (A) serum SMPD1 activity (Fig. 1A; n = 4 smpd1+/+, n = 8 smpd1+/−), (B) liver glutathion (GSH) levels (Fig. 1B; n = 4 smpd1+/+, n = 4 smpd1+/−), (C) γ-GT, (D) serum total bilirubin (Fig. 1C,D; n = 4 smpd1+/+, n = 8 smpd1+/−) as well as (E) hepatic transcriptional expression of Mrp2 (Fig. 1E; n = 4 smpd1+/+, n = 8 smpd1+/−) at three different time points (24 hours, 3 and 28 days). #p < 0.05; ##p < 0.01 vs. corresponding smpd1+/+ control; *p < 0.05; **p < 0.01 vs. baseline (MWU-test). Transcriptional expression is normalized to reference gene (Actb) and shown as log2 fold changes. Cut off values were set at ± 1, representing a variation of biological significance (dotted lines).
Inhibition of SMPD1 reveals its anti-inflammatory capacity in sepsis
To further evaluate the hepatic inflammatory response, we assessed the expression of f4/80, a marker of activated macrophages, and of cytokines characteristically increased during the course of sepsis, such as Ccl2/mcp1, Tnfa as well as Il1b in liver homogenates of smpd1+/+ mice. Consistent with a detrimental role for SMPD1 in the hepatic inflammatory response during sepsis, these markers of hepatic inflammation were significantly lower in smpd1+/− animals as compared to smpd1+/+ mice (Fig. 2A–D).
Figure 2.

SMPD1 drives hepatic inflammation in polymicrobial sepsis. Hepatic gene expression of (A) f4/80, (B) Ccl2/mcp1, (C) Il1b and (D) Tnfa was determined at three different time points following polymicrobial sepsis (24 hours, 3 and 28 days; n = 4 smpd1+/+, n = 8 smpd1+/−) to evaluate the impact of SMPD1 on hepatic inflammation. Transcriptional expression is normalized to reference gene (Actb) and shown as log2 fold changes. Cut off values were set at ± 1, representing a variation of biological significance (dotted lines). #p < 0.05; ##p < 0.01 vs. corresponding smpd1+/+ control; *p < 0.05; * *p < 0.01 vs. baseline (MWU-test).
Acid sphingomyelinase controls HSC transdifferentiation and hepatic fibrosis in long-term survivors of sepsis
HSC activation, which is known to be controlled by SMPD121, ultimately resulted in ECM deposition in a long term murine sepsis model10. At day 28 following sepsis Sirius Red staining revealed fibrotic changes in the parenchyma, around the portal tract and central veins in smpd1+/+ mice, whereas smpd1+/− animals displayed less fibrotic areas in the parenchyma and around the portal tracts (Fig. 3A). Semi-quantitative scoring of ECM deposition attested a strong increase in smpd1+/+ animals, with a less pronounced collagen deposition in smpd1+/− littermates (Fig. 3B). Hepatic gene expression of α-smooth muscle actin 2 (Acta2) and cysteine and glycine rich protein 2 (Csrp2), both of which are well established measures of activated liver myofibroblasts, confirmed a significant increase in smpd1+/+ animals, which was not evident in smpd1+/− animals (Fig. 3C). The expression of Tgfb was significantly higher in smpd1+/+ as compared in smpd1+/− littermates (Fig. 3B). Consistently, hepatic expression of different collagens (Col1a1, Col3a1) was found to be increased 28 days following septic insult in smpd1+/+ but ameliorated in smpd1+/− animals (Fig. 3C). These results suggest SMPD1 as a key player in the control of post-inflammatory HSC activation and hepatic fibrogenesis as a long-term sequelae of sepsis.
Figure 3.

Reduced HSC activation and hepatic fibrosis in smpd1+/− mice after polymicrobial sepsis. In long-term survivors (28 days following sepsis induction) (A) representative Sirius Red stainings, (B) hepatic fibrosis scores and Tgfb expression (Fig. 2A/B; n = 4 smpd1+/+, n = 7 smpd1+/−) and (C) hepatic gene expression of fibrosis-associated genes (Fig. 2C; n = 4 smpd1+/+, n = 8 smpd1+/−) were determined. Transcriptional expression is normalized to reference gene (Actb) and shown as log2 fold changes. Cut off values were set at ± 1, representing a variation of biological significance (dotted lines). #p < 0.05; ##p < 0.01 vs. corresponding smpd1+/+ control; *p < 0.05; * *p < 0.01 vs. baseline (MWU-test).
Pharmacological inhibition of SMPD1 by desipramine results in improved liver function and decreased hepatic inflammatory response during polymicrobial sepsis
FIASMAs are known to have beneficial effects on numerous hepatic pathologies, such as Wilson’s disease or alcohol-induced liver cirrhosis23,28. Therefore, we tested both, a preventative and a therapeutic treatment strategy with the FIASMA desipramine, to alleviate hepatic dysfunction and liver injury after sepsis. SMPD1 was significantly diminished in smpd1+/+ animals preventatively or therapeutically treated with desipramine (Fig. 4A). Furthermore, GSH levels (Fig. 4B), and surrogates of liver dysfunction (Fig. 4C–E) were significantly improved 24 hours after sepsis induction as compared to controls. These results confirm observations from heterozygous animals where treatment with desipramine in a preventative as well as in a therapeutic fashion results in a suppression of SMPD1 activation and an improvement of liver dysfunction during systemic inflammation. Mice preventatively treated with the functional inhibitor desipramine demonstrated significantly lower expression levels of f4/80, Ccl2/mcp1, Tnfa and Il1b in liver homogenates as compared to controls (Fig. 5). Despite the normalization of functional parameters, markers of inflammation were found to be increased, but diminished compared to smpd1+/+ controls, indicating a lower extent of chronic exacerbation of inflammation. Comparable results were obtained with respect to therapeutic treatment with desipramine (Fig. 5).
Figure 4.

Preventative and therapeutic desipramine treatment abrogates sepsis-induced liver dysfunction in smpd1+/+ mice. Smpd1+/+ mice were treated either 7 days prior (preventative, p-smpd1+/+) or 6 hours (therapeutic, t-smpd1+/+) following sepsis induction with desipramine. (A) Serum SMPD1 activity (Fig. 4A; n = 4 smpd1+/+, n ≥ 6 p-smpd1+/+, t-smpd1+/+), (B) liver glutathion (GSH) levels (n = 4 smpd1+/+, n = 4 p-smpd1+/+, t-smpd1+/+), (C) γ-GT, (D) total serum bilirubin (n = 4 smpd1+/+, n ≥ 6 p-smpd1+/+, t-smpd1+/+) and (E) hepatic gene expression of Mrp2 (Fig. 4E; n = 4 smpd1+/+, n ≥ 5 p-smpd1+/+, t-smpd1+/+) are shown. Transcriptional expression is normalized to reference gene (Actb) and shown as log2 fold changes. Cut off values were set at ± 1, representing a variation of biological significance (dotted lines). #p < 0.05; ##p < 0.01 vs. corresponding smpd1+/+ control; *p < 0.05; * *p < 0.01 vs. baseline (MWU-test).
Figure 5.

Preventative treatment strategy as well as therapeutic treatment diminishes hepatic inflammatory response during polymicrobial sepsis. Hepatic gene expression of (A) f4/80, (B) Ccl2/mcp1, (C) Il1b and (D) Tnfa was determined at three different time points following polymicrobial sepsis (24 hours, 3 and 28 days; n = 4 smpd1+/+, n = 8 p-smpd1+/+, t-smpd1+/+) to evaluate the impact of SMPD1 on hepatic inflammation following either preventative (p-smpd1+/+) or therapeutic (t-smpd1+/+) treatment with desipramine during sepsis. Transcriptional expression is normalized to reference gene (Actb) and shown as log2 fold changes. Cut off values were set at ± 1, representing a variation of biological significance (dotted lines). #p < 0.05; ##p < 0.01 vs. corresponding smpd1+/+ control; *p < 0.05; **p < 0.01 vs. baseline (MWU-test).
Pharmacological inhibition of SMPD1 reduced hepatic fibrosis after sepsis
Since it is known that functional inhibitors of SMPD1, such as imipramine, are capable of reducing hepatic fibrosis, we were interested whether desipramine, also a functional inhibitor, is capable to reduce hepatic fibrosis occurring in sepsis survivors as a long-term sequela. Collagen deposition was reduced in desipramine-treated animals 28 days after the onset of sepsis according to Sirius Red staining (Fig. 6A,B). Consistently, the hepatic expression of Acta2, Csrp2, Col1a1 and Col3a1 was improved by both treatment strategies indicating reduced activation of liver myofibroblasts and reduced expression of extracellular matrix (Fig. 6C). To confirm a protective effect of desipramine on HSC activation in vitro, we treated LX-2 cells with desipramine in different concentrations for 24 hours. The stimulation with desipramine resulted in a downregulation of ACTA2 as well as COL1A1 expression in a dose-dependent manner (Fig. 6D).
Figure 6.

Pharmacological inhibition of SMPD1 results in reduced HSC activation and reduced hepatic fibrosis. In long-term survivors (28 days following sepsis induction) (A) representative Sirius Red stainings, (B) hepatic fibrosis scores and Tgfb expression (Fig. 6A/B; n = 4 smpd1+/+, n ≥ 5 p-smpd1+/+, t-smpd1+/+) and (C) hepatic gene expression of fibrosis-associated genes (Fig. 6C; n = 4 smpd1+/+, n = 8 p-smpd1+/+, t-smpd1+/+) were determined. Transcriptional expression is normalized to reference gene (Actb) and shown as log2 fold changes. (D) Furthermore, stimulation for 24 hours with different desipramine concentrations (5 µM, 10 µM, 20 µM) of LX-2 cells were performed presenting dose dependent inhibition of ACTA2 as well as COL1A1 expression rate (Fig. 6D; n = 4). Transcriptional expression is normalized to reference genes (GAPDH) and presented in log2 fold changes. Cut off values were set at ± 1, representing a variation of biological significance (dotted lines). #p < 0.05; ##p < 0.01 vs. corresponding smpd1+/+ control; *p < 0.05; * *p < 0.01 vs. baseline (MWU-test).
SMPD1 inhibition improves survival following polymicrobial sepsis
As a previous study has already demonstrated a beneficial role of SMPD1 inhibition for survival in a murine model of sterile endotoxemia11, we tested the hypothesis if this holds true in a polymicrobial sepsis model of peritoneal contamination and infection (PCI) with subsequent antibiotic rescue. As shown in Fig. 7, the overall survival revealed a rate of 50% in smpd1+/+ animals in an observation period of 96 hours. In contrast, smpd1+/− animals displayed a significant reduction of mortality following polymicrobial sepsis with similar findings for preventative treatment with desipramine 7 days prior to sepsis induction as well as therapeutic treatment with desipramine 6 hours following sepsis induction.
Figure 7.
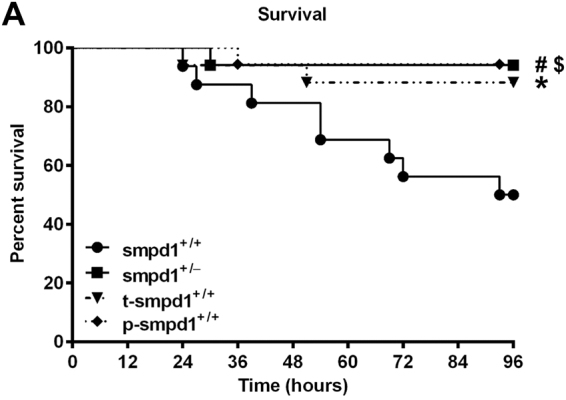
Partial genetic and pharmacological inhibition of SMPD1 improves survival in polymicrobial sepsis. Kaplan-Meier survival analysis [96 hours] with respect to SMPD1 controlled either by genotype (smpd1+/−) or by treatment (p-smpd1+/+, t-smpd1+/+) following polymicrobial sepsis. Randomly selected smpd1+/+ mice were pretreated 7 days prior or 6 hours following sepsis induction with desipramine and compared to smpd1+/+, smpd1+/− littermates. Data were obtained from at least n ≥ 15 of each stratum. Statistical analyses were performed using log-rank test for trend (p = 0.0152); subsequently performed log-rank test attested significant differences between smpd1+/+ and smpd1+/− (#p = 0.0057); smpd1+/+ and p-smpd1+/+ ($p = 0.0042); smpd1+/+ and t-smpd1+/+ (*p = 0.0245).
Discussion
This study provides first evidence of a detrimental role of the conserved stress responsive enzyme SMPD1 in the development of sepsis-induced liver dysfunction and fibrosis, which both affect the outcome from sepsis and the quality of life in sepsis survivors. We show that reducing SMPD1 activity, either by partial genetic deficiency or mostly important by preventative or therapeutic pharmacological inhibition, is beneficial for liver function in the acute phase of sepsis and for preventing inflammation, HSC activation and hepatic fibrogenesis as a prolonged consequence of sepsis.
Altered SMPD1 activity has been linked to the pathogenesis of numerous disease entities such as alcoholic cirrhosis, non-alcoholic steatohepatitis, as well as cachexia and mortality in chronic heart failure20,23,29. Protective effects due to deficiency could be reversed by treatment with exogenously added ceramide30,31. In addition, in vitro treatment of rat hepatocytes with human placenta SMPD1 induced oxidative stress and initiated apoptosis32.
Moreover, an increased activity of secreted SMPD1 in plasma samples of septic patients correlates with disease severity and poor outcome, and its inhibition improved survival rate in a murine model of severe endotoxemia11, in a Staphylococcus aureus or polymicrobial sepsis model15,27. In order to clarify the role of SMPD1 as a stress enzyme in sepsis-induced liver dysfunction, we used mice, which are partially deficient in the enzyme. It is controversially discussed whether it is appropriate to use this strain of genetically deficient mice in order to prove pathophysiological effects and cellular mechanisms based on the fact that mice compensate their total loss of enzyme function33. The complete knock-out of SMPD1 gene results in Niemann-Pick disease which is characterized by rapid central nervous degeneration alongside an impairment of liver function34. However, minimal residual enzymatic activity is sufficient to prevent Niemann-Pick disease and its neurological symptoms35. In line with these studies, we observed increased oxidative stress as well as elevated cytokine responses in knock-out animals36, which encouraged us to investigate mice heterozygous for SMPD1 (smpd1+/−) presenting with decreased but still residual SMPD1 activity levels. In addition to the beneficial effect of partial deficiency of SMPD1 in a bile duct ligation21 as well as in a Fas-mediated apoptosis model25, we infer from our sepsis model that residual activity of SMPD1 in smpd1+/− mice is sufficient to prevent maladaptive effects caused by the overwhelming activity in smpd1+/+ animals.
In this study smpd1+/− mice had a favorable phenotype resulting in improved liver function and reduced hepatic oxidative stress in the course of sepsis. In the case of sepsis-induced liver dysfunction there is still a broad discussion regarding its etiology and underlying molecular mechanism. One of these concepts denotes that immune cells, such as liver invading monocytes, contribute to the pathogenesis of sepsis-induced liver failure due to the overwhelming production of cytokines37. Consistent with the fact that SMPD1 is a key player in the regulation of macrophage differentiation and cytokine secretion, we demonstrate that activation of these cells and further cytokine expression is a function of SMPD1 activity. Furthermore, reduced hepatic fibrogenesis as a long-term sequela of sepsis, was proven by the expression profile of Acta238 and Csrp239, which are well established markers for activated liver myofibroblasts. Our data revealed that in smpd1+/− mice the number of aLMF was reduced which might be mirrored in a diminished deposition of ECM in the liver. In fact, the expression profile of two components of ECM (Col1a1, Col3a1) was found reduced on both, the molecular and the histological level. Our data are in line with a previous study from Moles et al. identifying SMPD1 as a key player HSC activation and hepatic fibrogenesis21.
In our study we elaborated causes and consequences of sepsis-induced fibrogenesis and its modulation by approved drugs. For that issue we analyzed samples from tissue homogenates and sections from representative locations. To solve the more specific question, whether there is not only a change in (trans-)activation state of hepatic stellate cells but also a variation in number (i.e. proliferation) or even a phenotypic dissociation of stellate cells affected, one might consider the temporal-spatial conditions of liver tissue undergoing adaptive response, but also the presence of granulomatous inflammation as a response to persistent inflammation/infection10 due to inflammasome activation in stellate cells40. Then, the generation and distribution of tissue abundant abscesses and the status of inflammasome activation might be correlated with a putative phenotypic dissociation of stellate cells.
Our data demonstrate that desipramine, a functional inhibitor of this stress enzyme, is capable of alleviating elevated enzyme activity during the host response towards in order to reduce sepsis-induced hepatic injury and fibrogenesis. This observation is of major interest since the used inhibitor is a member of a class of drugs (FIASMA) all of which regulate the activity of SMPD1 and have been used in daily clinical care for decades in treatment of neurological disorders41,42. A series of studies have provided evidence that the functional inhibition of SMPD1 improves cellular stress responses, reduces oxidative stress and improves viability in vivo 15 or in vitro 30,32,43,44. In our study, we expand the knowledge of a beneficial effect of SMPD1 inhibition by treating animals with desipramine 7 days prior and 6 hours following to severe infection, resulting in decreased SMPD1 activity thus followed by improved sepsis-induced oxidative stress. It is known that GSH belongs to intracellular antioxidant defense strategies and plays an important role in the balance of oxidants and antioxidants45. Interestingly, desipramine was demonstrated to restore glutathione levels46 and to further possess anti-inflammatory capacity47. These data are supported by the fact that p-smpd1+/+ as well as t-smpd1+/+ animals revealed less pronounced markers of inflammation as well as liver dysfunction such as total-Bilirubin and γ-GT as well as oxidative stress compared to smpd1+/+ animals. Comparison of smpd1+/− mice to p-smpd1+/+ and t-smpd1+/+ animals indicates similarities or minimal differences in all parameters, especially with respect to chronic exacerbation of inflammation. Of note, in our experimental setting, no immune-suppressive effect of desipramine treatment was observed48.
In addition, we show an improved survival rate after polymicrobial sepsis in desipramine-treated smpd1+/+ mice as well as in smpd1+/− mice.
With the aim of fostering the excretory capacity also the functionality of hepatocytes and cholangiocytes as well as the restitution of excretory function in presence of inhibitors of sphingomyelinase needs enhancing perception for upcoming experimental work. Primary hepatocytes for ex-vivo experimentation or animal experiments with distinct (mixed) genotypes might function as a reliable tool to get further insights into the complex regulatory mechanisms of membrane integration of MDR2, its cytoskeletal anchoring (phosphorylation/ dephosphorylation), or ultimately reorganization upon inflammation in membranous subdomains characterized by a specific lipid composition4. Since the lipid mediator ceramide is known to affect and also to control mostly all of these key events, the exploration of the (local) kinetic) and dynamic of ceramide generation and its accumulation is of great interest, especially with respect to MDR2-function.
Overall, results from our study support the notion that an increased activity level of SMPD1 displays a detrimental role with respect to regulation of liver function in both the acute and post-acute phase of sepsis. The decreased, but residual SMPD1 activity in smpd1+/−, p-smpd1+/+ and t-smpd1+/+ mice are even sufficient for regulation of adaptive mechanisms in the liver during host response. Finally, our data highlights the favorable effect of functional SMPD1 inhibition, i.e. by desipramine, in order to protect the liver from hepatocyte damage in vivo which is underlined by obtained data from smpd1+/− animals. Furthermore, the pharmacological inhibitor diminished the activation of HSC and prevented fibrogenesis in survivors as a prolonged consequence of severe infection. We are proposing a concept of a causative therapeutic strategy by evaluation of a novel target. As these SMPD1 inhibiting drugs are FDA-approved drug, which are frequently prescribed for neurological disorders, observational studies in well-defined patients could unravel, whether FIASMA use is associated with reduced liver injury in patients with sepsis.
Material and Methods
Cell culture experiments
An immortalized hepatic stellate cell line (LX-2)49 of human origin, was cultured under standard conditions (1% heat inactivated fetal calf serum [FCS] in Dulbecco’s Modified Eagle Medium (DMEM) media, 5% CO2). Cells were treated 24 hours with different concentrations of desipramine (5 µM, 10 µM, 20 µM) prior to stimulation with cytokine mix (TNF-α, IFN-γ, IL-1β [each: 10 ng/mL] and 100 ng/mL of endotoxin) for 24 hours as described previously50,51.
Animals
All experiments were performed in accordance with the German legislation on protection of animals and with approval of the local animal welfare committee (Thueringer Landesamt fuer Lebensmittelsicherheit und Verbraucherschutz, 02-009/12). We compared smpd1+/− animals with a partial deficiency in SMPD1 function to smpd1+/+ littermates52. For each experiment, similar proportions of male and female gender were randomly selected at the age of 8–12 weeks (mean B.W. 22.2 g). Animals were housed under controlled/standardized day-night conditions (12 h/12 h) at room temperature (23 ± 1 °C, 30%–60% environmental humidity), received a standard diet and water ad libitum and were allowed to adapt to laboratory conditions for at least 2 days prior to the start of the experiments.
Desipramine treatment and sepsis mode
Smpd1+/+ animals were treated 7 days prior or 6 hours following sepsis induction with desipramine-hydrochloride (isotonic saline solution, 20 mg/kg B.W.) every 24 hours subcutaneously which was continued daily up to euthanasia. Sepsis was induced by the standardized and established peritoneal contamination and infection approach53. Briefly, intraperitoneal injection of fecal slurry (diluted 1:4 in saline solution) was performed (3.0 µl/g B.W.) into the right lower quadrant of the abdomen with a 21-gauge cannula. Without supportive treatment, severity of the insult results in an approximately 100% mortality rate within 48 hours (data not shown). However, administration of antibiotics and volume resuscitation rescued animals and adjusted survival rate to 50% (20 mg/kg B.W. of meropenem every 24 hours s.c. and 25 µl/g B.W. of physiological saline solution s.c. twice daily over 4 days, starting 6 hours following sepsis induction). In the post-acute phase (day 3) treatment was restrained to desipramine-HCl/vehicle administration once daily (isotonic saline solution, 20 mg/kg B.W.). Disease progression and outcome of mice during host response was continuously assessed using the clinical severity scores (CSS) (data not shown) as previously described53. Animals were deeply anesthetized before any procedure by isoflurane (2%) which was followed by harvesting tissues and whole blood (right-ventricular heart puncture).
SMPD1 activity
Serum was collected from each stratum (smpd1+/+, smpd1+/−, p-smpd1+/+, t-smpd1+/+) at baseline, 24 h, 3 and 28 days following the insult. SMPD1 activity was determined by the hydrolysis of fluorescently labeled sphingomyelin (NBD-SM; Molecular Probes, Eugene, OR) as a substrate, chromatographic product separation, and image analysis as described previously54,55. Serum samples were diluted 1:10 with the incubation buffer (sodium acetate, pH 5.0) before analysis. Finally, the reaction mixture was composed from 20 μl diluted serum and the extraction was carried out using a SpeedVac concentrator Plus (Eppendorf, Hamburg, Germany).
GSH
Glutathione in its reduced form (GSH) was determined according to Ellman56. Briefly, fresh frozen liver samples were homogenized with 11 volumes of 0.2 M sodium phosphate buffer (5 mM EDTA; pH 8.0) and 4 volumes of 25% metaphosphoric acid. Following centrifugation (12000 x g; 4 °C; 30 min), GSH was measured in the supernatants photometrically. For analysis, data were obtained from n = 4 for baseline and n = 4 animals following polymicrobial sepsis from each stratum (smpd1+/+, smpd1+/−, p-smpd1+/+, t-smpd1+/+).
Laboratory markers
Serum from each stratum (smpd1+/+, smpd1+/−, p-smpd1+/+, t-smpd1+/+) at baseline, 24 hours, day 3 and 28 following sepsis induction was used for analysis of clinically used laboratory parameters of hepatobiliary damage and dysfunction (total-bilirubin T-Bil and γ-glutamyl-transferase γ-GT) using the clinical chemistry analyzer Fuji Dri-Chem 3500i (Sysmex, Leipzig, Germany) according to manufacturer’s instructions.
Real-time PCR
Fresh frozen liver tissue samples (15–20 mg) were homogenized in 600 µL lysis buffer (RLT lysis puffer, supplemented with 1% β-mercaptoethanol; Qiagen, Hilden, Germany). RNA was isolated using RNeasy Mini Kit according to manufacturer’s instructions (Qiagen, Hilden, Germany). RNA concentration and integrity were analyzed using NanodropTM spectrophometer and QIAxcel microcapillar electrophoresis apparatus and were followed by reverse transcription of 1.0 µg mRNA using standard conditions (Thermo Scientific, Germany)57. The expression profile of selected mRNA was measured using Rotor-Gene Q 2plex system. The following primer sequences were used:
ACTA2fw: tgatcaccatcggaaatgaa, rv: agaggtccttcctgatgtcaa
COL1A1 fw: caagaaccccaaggacaaga, rv: aggaaggtcagctggatgg
GAPDH fw: ctctgctcctcctgttcgac, rv: caatacgaccaaatccgttgac
Acta2fw: ccgagatctcaccgactacc, rv: tccagagcgacatagcacag
f4/80 fw: tttcctcgcctgcttcttc, rv: ccccgtctctgtattcaacc
Ccl2/mcp1 fw: aggtgtcccaaagaagctgtag, rv: aatgtatgtctggacccattcc
Tgfb fw: tatagcaacaattcctggcg, rv: tgctgtcacaagagcagtg
Tnfa fw: gtctactgaacttcggggtgat, rv: atgatctgagtgtgagggtctg
Il1b fw: gaagagcccatcctctgtga, rv: ttcatctcggagcctgtagtg
Csrp2 fw: gctacggaaagaagtatggacc, rv: ctcagtcagagttgtagactcc
Col1a1 fw: caaggtccttctggatcaagtg, rv: cctttatgcctctgtcaccttg
Col3a1 fw: acgtagatgaattgggatgcag, rv: gggttggggcagtctagtg
Mrp2 fw: aacttgggttgctccatga, rv: caggaccaggattttggattt
Actb fw: gctcttttccagccttcctt, rv: cggatgtcaacgtcacactt
Histology
Representative liver samples from smpd1+/+, smpd1+/−, p-smpd1+/+, t-smpd1+/+ were fixed in neutral buffered formaldehyde 4% for at least 24 hours directly following harvesting. Samples were dehydrated in rising alcohol concentration and then embedded in paraffin. Livers were sliced into 3 µm sections and deparaffinized through xylene and reversed graded alcohol series. At least seven organs of each stratum were stained in Sirius Red, which was used to detect and to visualize extracellular matrix deposition. Two experienced researchers independently scored collagen deposition using a semi-quantitative scoring system in 20 randomly sampled high-power fields per animal semi-quantitatively in a blinded fashion as 0 (absent), 1 (mild), 2 (moderate), 3 (strong). Scores were accumulated for each animal.
Statistics
For survival analysis, log rank test was used. Unpaired Mann-Whitney-U test (MWU) was performed to determine statistical differences between groups and over time. A level of p ≤ 0.05 was considered to be statistically significant. With respect to analyses of expression profile values log2 fold variations at least ± 1 were considered to be of biological significance.
Acknowledgements
This work was supported by the Center for Sepsis Control and Care (Integriertes Forschungs- und Behandlungszentrum Sepsis und Sepsisfolgen – Center for Sepsis Control and Care, sponsored by German Federal Ministry of Education and Research, BMBF FKZ 01EO1002; M.D. fellowships to HYC and BG) and by a grant of German research foundation (DFG; CL-173/3-2; SPP 1267: Sphingolipids - Signal and Disease). We also thank Edith Walter, Danny Himsel, Ulrike Vetterling, Brigitte Specht and Sascha Grossmann for excellent technical assistance.
Author Contributions
H.Y.C. designed and performed research, analysed and interpreted data incl. statistical analysis, and wrote the first draft of the manuscript. C.J.W. performed all experiments regarding to therapeutic treatment strategy. N.J. substantially contributed to acquisition of data, analysis as well as interpretation of data together with R.A.C. J.H.-O. and B.G. contributed to experiments and interpreted data. A.L. and M.H.G. performed measures of oxidative stress and sphingolipids, respectively. F.A.G., T.B. and A.S. substantially contributed to study design and interpretation of clinical data. R.A.C. supervised the study, drafted and revised the final version of the manuscript. All authors approved the final version of the draft.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Engel C, et al. Epidemiology of sepsis in Germany: results from a national prospective multicenter study. Intensive care medicine. 2007;33:606–618. doi: 10.1007/s00134-006-0517-7. [DOI] [PubMed] [Google Scholar]
- 2.Gaieski DF, Edwards JM, Kallan MJ, Carr BG. Benchmarking the incidence and mortality of severe sepsis in the United States. Critical care medicine. 2013;41:1167–1174. doi: 10.1097/CCM.0b013e31827c09f8. [DOI] [PubMed] [Google Scholar]
- 3.Gustot T. Multiple organ failure in sepsis: prognosis and role of systemic inflammatory response. Current opinion in critical care. 2011;17:153–159. doi: 10.1097/MCC.0b013e328344b446. [DOI] [PubMed] [Google Scholar]
- 4.Recknagel P, et al. Liver dysfunction and phosphatidylinositol-3-kinase signalling in early sepsis: experimental studies in rodent models of peritonitis. PLoS medicine. 2012;9:e1001338. doi: 10.1371/journal.pmed.1001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winters BD, et al. Long-term mortality and quality of life in sepsis: a systematic review. Critical care medicine. 2010;38:1276–1283. doi: 10.1097/CCM.0b013e3181d8cc1d. [DOI] [PubMed] [Google Scholar]
- 6.Koch A, et al. Increased liver stiffness denotes hepatic dysfunction and mortality risk in critically ill non-cirrhotic patients at a medical ICU. Crit Care. 2011;15:R266. doi: 10.1186/cc10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhai R, et al. Serum bilirubin levels on ICU admission are associated with ARDS development and mortality in sepsis. Thorax. 2009;64:784–790. doi: 10.1136/thx.2009.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nesseler N, et al. Clinical review: The liver in sepsis. Crit Care. 2012;16:235. doi: 10.1186/cc11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulaksiz H, Heuberger D, Engler S, Stiehl A. Poor outcome in progressive sclerosing cholangitis after septic shock. Endoscopy. 2008;40:214–218. doi: 10.1055/s-2007-967024. [DOI] [PubMed] [Google Scholar]
- 10.Gonnert FA, et al. Hepatic Fibrosis in a Long-term Murine Model of Sepsis. Shock. 2012;37:399–407. doi: 10.1097/SHK.0b013e31824a670b. [DOI] [PubMed] [Google Scholar]
- 11.Claus RA, et al. Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:1719–1721. doi: 10.1096/fj.04-2842fje. [DOI] [PubMed] [Google Scholar]
- 12.Kott M, et al. Acid sphingomyelinase serum activity predicts mortality in intensive care unit patients after systemic inflammation: a prospective cohort study. PloS one. 2014;9:e112323. doi: 10.1371/journal.pone.0112323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claus R, Russwurm S, Meisner M, Kinscherf R, Deigner HP. Modulation of the ceramide level, a novel therapeutic concept? Current drug targets. 2000;1:185–205. doi: 10.2174/1389450003349272. [DOI] [PubMed] [Google Scholar]
- 14.Chung, H. Y. et al. Acid sphingomyelinase promotes endothelial stress response in systemic inflammation and sepsis. Mol Med22, doi:10.2119/molmed.2016.00140 (2016). [DOI] [PMC free article] [PubMed]
- 15.Chung HY, et al. Adjustment of Dysregulated Ceramide Metabolism in a Murine Model of Sepsis-Induced Cardiac Dysfunction. Int. J. Mol. Sci. 2017;18:839. doi: 10.3390/ijms18040839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bollinger CR, Teichgraber V, Gulbins E. Ceramide-enriched membrane domains. Biochimica et biophysica acta. 2005;1746:284–294. doi: 10.1016/j.bbamcr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Kornhuber J, Rhein C, Muller CP, Muhle C. Secretory sphingomyelinase in health and disease. Biological chemistry. 2015;396:707–736. doi: 10.1515/hsz-2015-0109. [DOI] [PubMed] [Google Scholar]
- 18.Morales A, Lee H, Goni FM, Kolesnick R, Fernandez-Checa JC. Sphingolipids and cell death. Apoptosis: an international journal on programmed cell death. 2007;12:923–939. doi: 10.1007/s10495-007-0721-0. [DOI] [PubMed] [Google Scholar]
- 19.Langmann T, et al. Transcription factors Sp1 and AP-2 mediate induction of acid sphingomyelinase during monocytic differentiation. Journal of lipid research. 1999;40:870–880. [PubMed] [Google Scholar]
- 20.Fucho, R. et al. Asmase Regulates Autophagy and Lysosomal Membrane Permeabilization and its Inhibition Prevents Early Stage Nonalcoholic Steatohepatitis. Journal of hepatology doi:10.1016/j.jhep.2014.06.009 (2014). [DOI] [PMC free article] [PubMed]
- 21.Moles A, et al. Acidic sphingomyelinase controls hepatic stellate cell activation and in vivo liver fibrogenesis. The American journal of pathology. 2010;177:1214–1224. doi: 10.2353/ajpath.2010.091257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Llacuna L, Mari M, Garcia-Ruiz C, Fernandez-Checa JC, Morales A. Critical role of acidic sphingomyelinase in murine hepatic ischemia-reperfusion injury. Hepatology. 2006;44:561–572. doi: 10.1002/hep.21285. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez A, et al. ASMase is required for chronic alcohol induced hepatic endoplasmic reticulum stress and mitochondrial cholesterol loading. Journal of hepatology. 2013;59:805–813. doi: 10.1016/j.jhep.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirschnek S, et al. CD95-mediated apoptosis in vivo involves acid sphingomyelinase. The Journal of biological chemistry. 2000;275:27316–27323. doi: 10.1074/jbc.M002957200. [DOI] [PubMed] [Google Scholar]
- 25.Lin T, et al. Role of acidic sphingomyelinase in Fas/CD95-mediated cell death. J Biol Chem. 2000;275:8657–8663. doi: 10.1074/jbc.275.12.8657. [DOI] [PubMed] [Google Scholar]
- 26.Kornhuber J, et al. Identification of novel functional inhibitors of acid sphingomyelinase. PloS one. 2011;6:e23852. doi: 10.1371/journal.pone.0023852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peng H, et al. Acid sphingomyelinase inhibition protects mice from lung edema and lethal Staphylococcus aureus sepsis. Journal of molecular medicine. 2015;93:675–689. doi: 10.1007/s00109-014-1246-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lang PA, et al. Liver cell death and anemia in Wilson disease involve acid sphingomyelinase and ceramide. Nature medicine. 2007;13:164–170. doi: 10.1038/nm1539. [DOI] [PubMed] [Google Scholar]
- 29.Doehner W, et al. Secretory sphingomyelinase is upregulated in chronic heart failure: a second messenger system of immune activation relates to body composition, muscular functional capacity, and peripheral blood flow. European heart journal. 2007;28:821–828. doi: 10.1093/eurheartj/ehl541. [DOI] [PubMed] [Google Scholar]
- 30.Garcia-Ruiz C, et al. Defective TNF-alpha-mediated hepatocellular apoptosis and liver damage in acidic sphingomyelinase knockout mice. The Journal of clinical investigation. 2003;111:197–208. doi: 10.1172/JCI16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paris F, et al. Natural ceramide reverses Fas resistance of acid sphingomyelinase(-/-) hepatocytes. The Journal of biological chemistry. 2001;276:8297–8305. doi: 10.1074/jbc.M008732200. [DOI] [PubMed] [Google Scholar]
- 32.Garcia-Ruiz C, et al. Human placenta sphingomyelinase, an exogenous acidic pH-optimum sphingomyelinase, induces oxidative stress, glutathione depletion, and apoptosis in rat hepatocytes. Hepatology. 2000;32:56–65. doi: 10.1053/jhep.2000.8267. [DOI] [PubMed] [Google Scholar]
- 33.Moles A, Tarrats N, Fernandez-Checa JC, Mari M. Cathepsin B overexpression due to acid sphingomyelinase ablation promotes liver fibrosis in Niemann-Pick disease. The Journal of biological chemistry. 2012;287:1178–1188. doi: 10.1074/jbc.M111.272393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lidove, O., Sedel, F., Charlotte, F., Froissart, R. & Vanier, M. T. Cirrhosis and Liver Failure: Expanding Phenotype of Acid Sphingomyelinase-Deficient Niemann-Pick Disease in Adulthood. JIMD reports, doi:10.1007/8904_2014_306 (2014). [DOI] [PMC free article] [PubMed]
- 35.Marathe S, et al. Creation of a mouse model for non-neurological (type B) Niemann-Pick disease by stable, low level expression of lysosomal sphingomyelinase in the absence of secretory sphingomyelinase: relationship between brain intra-lysosomal enzyme activity and central nervous system function. Human molecular genetics. 2000;9:1967–1976. doi: 10.1093/hmg/9.13.1967. [DOI] [PubMed] [Google Scholar]
- 36.Jbeily N, et al. Hyperresponsiveness of mice deficient in plasma-secreted sphingomyelinase reveals its pivotal role in early phase of host response. Journal of lipid research. 2013;54:410–424. doi: 10.1194/jlr.M031625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nature reviews. Immunology. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hautekeete ML, Geerts A. The hepatic stellate (Ito) cell: its role in human liver disease. Virchows Archiv: an international journal of pathology. 1997;430:195–207. doi: 10.1007/BF01324802. [DOI] [PubMed] [Google Scholar]
- 39.Herrmann J, et al. The expression of CSRP2 encoding the LIM domain protein CRP2 is mediated by TGF-beta in smooth muscle and hepatic stellate cells. Biochemical and biophysical research communications. 2006;345:1526–1535. doi: 10.1016/j.bbrc.2006.05.076. [DOI] [PubMed] [Google Scholar]
- 40.Meng N, et al. Activation of NLRP3 inflammasomes in mouse hepatic stellate cells during Schistosoma J. infection. Oncotarget. 2016;7:39316–39331. doi: 10.18632/oncotarget.10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kornhuber, J., Tripal, P., Gulbins, E. & Muehlbacher, M. Functional inhibitors of acid sphingomyelinase (FIASMAs). Handbook of experimental pharmacology 169–186, doi:10.1007/978-3-7091-1368-4_9 (2013). [DOI] [PubMed]
- 42.Olfson M, Druss BG, Marcus SC. Trends in mental health care among children and adolescents. N Engl J Med. 2015;372:2029–2038. doi: 10.1056/NEJMsa1413512. [DOI] [PubMed] [Google Scholar]
- 43.Mari M, et al. Acidic sphingomyelinase downregulates the liver-specific methionine adenosyltransferase 1A, contributing to tumor necrosis factor-induced lethal hepatitis. The Journal of clinical investigation. 2004;113:895–904. doi: 10.1172/JCI200419852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Claus RA, Dorer MJ, Bunck AC, Deigner HP. Inhibition of sphingomyelin hydrolysis: targeting the lipid mediator ceramide as a key regulator of cellular fate. Current medicinal chemistry. 2009;16:1978–2000. doi: 10.2174/092986709788682182. [DOI] [PubMed] [Google Scholar]
- 45.Macdonald J, Galley HF, Webster NR. Oxidative stress and gene expression in sepsis. British journal of anaesthesia. 2003;90:221–232. doi: 10.1093/bja/aeg034. [DOI] [PubMed] [Google Scholar]
- 46.Guemei AA, El Din NM, Baraka AM, El Said Darwish I. Do desipramine [10,11-dihydro-5-[3-(methylamino) propyl]−5H-dibenz[b,f]azepine monohydrochloride] and fluoxetine [N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]-propan-1-amine] ameliorate the extent of colonic damage induced by acetic acid in rats? The Journal of pharmacology and experimental therapeutics. 2008;327:846–850. doi: 10.1124/jpet.108.141259. [DOI] [PubMed] [Google Scholar]
- 47.Roumestan C, et al. Anti-inflammatory properties of desipramine and fluoxetine. Respiratory research. 2007;8:35. doi: 10.1186/1465-9921-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Johnson, B. L., 3rd et al. Amitriptyline Usage Exacerbates the Immune Suppression Following Burn Injury. Shock, doi:10.1097/SHK.0000000000000648 (2016). [DOI] [PMC free article] [PubMed]
- 49.Weiskirchen R, et al. Genetic characteristics of the human hepatic stellate cell line LX-2. PloS one. 2013;8:e75692. doi: 10.1371/journal.pone.0075692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ceppi ED, Smith FS, Titheradge MA. Effect of multiple cytokines plus bacterial endotoxin on glucose and nitric oxide production by cultured hepatocytes. The Biochemical journal. 1996;317(Pt 2):503–507. doi: 10.1042/bj3170503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ekaney ML, et al. Preserved Expression of mRNA Coding von Willebrand Factor-Cleaving Protease ADAMTS13 by Selenite and Activated Protein C. Molecular medicine. 2015;21:355–363. doi: 10.2119/molmed.2014.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Horinouchi K, et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nature genetics. 1995;10:288–293. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 53.Gonnert FA, et al. Characteristics of clinical sepsis reflected in a reliable and reproducible rodent sepsis model. The Journal of surgical research. 2011;170:e123–134. doi: 10.1016/j.jss.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 54.Loidl A, Claus R, Deigner HP, Hermetter A. High-precision fluorescence assay for sphingomyelinase activity of isolated enzymes and cell lysates. Journal of lipid research. 2002;43:815–823. [PubMed] [Google Scholar]
- 55.Loidl A, Claus R, Ingolic E, Deigner HP, Hermetter A. Role of ceramide in activation of stress-associated MAP kinases by minimally modified LDL in vascular smooth muscle cells. Biochimica et biophysica acta. 2004;1690:150–158. doi: 10.1016/j.bbadis.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 56.Ellman GL. Tissue sulfhydryl groups. Archives of biochemistry and biophysics. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 57.Sieber MW, et al. Substantial performance discrepancies among commercially available kits for reverse transcription quantitative polymerase chain reaction: a systematic comparative investigator-driven approach. Analytical biochemistry. 2010;401:303–311. doi: 10.1016/j.ab.2010.03.007. [DOI] [PubMed] [Google Scholar]