

Abstract
Spider venom toxins, such as Protoxin-II (ProTx-II), have recently received much attention as selective Nav1.7 channel blockers, with potential to be developed as leads for the treatment of chronic nocioceptive pain. ProTx-II is a 30-amino acid peptide with three disulfide bonds that has been reported to adopt a well-defined inhibitory cystine knot (ICK) scaffold structure. Potential drawbacks with such peptides include poor pharmacodynamics and potential scrambling of the disulfide bonds in vivo. In order to address these issues, in the present study we report the solid-phase synthesis of lanthionine-bridged analogues of ProTx-II, in which one of the three disulfide bridges is replaced with a thioether linkage, and evaluate the biological properties of these analogues. We have also investigated the folding and disulfide bridging patterns arising from different methods of oxidation of the linear peptide precursor. Finally, we report the X-ray crystal structure of ProTx-II to atomic resolution; to our knowledge this is the first crystal structure of an ICK spider venom peptide not bound to a substrate.
Introduction
Peptide toxins hold considerable promise as novel therapeutics due to the potency and specificity with which they interact with their biological targets, low cost of synthesis, and largely well-understood metabolic pathways.1 These peptides are often cysteine-rich and contain intricate disulfide bonding patterns that are crucial for their biological activity.2 Despite these encouraging properties, however, to date only one peptide toxin-based therapeutic (Ziconitide, marketed as Prialt) has been approved for clinical use.3 This is largely due to commonly encountered problems in the in vivo stability of peptide toxins. Poor transport across the intestinal wall and susceptibility to enzymatic degradation mean that peptide toxins are not often suitable for oral administration and must be administered by intrathecal injection.3 Poor pharmacodynamics profiles are also an issue: none of these peptide toxins can cross the blood-brain or blood-nerve barriers easily.4 In addition, many toxins still show a lack of receptor subtype selectivity, leading to undesired side effects.5 Finally, the disulfide bonds in these toxins are susceptible to reduction in extracellular environments, by glutathione and disulfide bond isomerases, leading to chain unfolding and oxidative refolding with consequent loss of biological activity.6 Considerable efforts have therefore been directed toward developing replacements for the disulfide bonds in peptide toxins that are not susceptible to reduction or oxidative refolding. One of the earliest methods employed was the use of diselenide bonds, which have only slight steric differences to the native disulfide bond.6 Further studies showed that the diselenide analogues did not undergo reduction in blood plasma and actually showed slightly improved potency over the native structure.7 Both unsaturated8,9 and saturated10 dicarba-bridges have also been used as disulfide bond replacements in cystine-rich toxins, using either ring-closing metathesis8,9,11 or solid-phase peptide synthesis (SPPS) using differentially protected diamino diacids10 as the synthetic strategy. These disulfide bond replacements resulted in peptide toxin analogues of comparable potency,8 and in some cases replacing different cystine bridges can enable the resulting toxin analogue to be tuned to one or other of two possible receptor targets.9 Lactam bridges12 and cystathione analogues13 have also been employed to stabilize the structure and modulate the activity of peptide toxins with multiple cystine bridges, with varying impact on receptor antagonism depending on the geometry of the replacement and which of the disulfide linkages has been replaced. Carba-,14,15 cystathionine-,16 and triazole-bridged17 analogues of other disulfide-bridged biologically active peptides have also been synthesized, again with varying effects on the biological properties of these peptide analogues. Surprisingly, lanthionine-bridged analogues of peptide toxins have not previously been studied. Replacement of cystine by lanthionine results in a thioether side-chain bridge that cannot be cleaved in vivo. This linkage is one bond shorter than a cystine-bridged peptide and will have different conformational preferences. However, replacement of cystine with lanthionine has been carried out with other bioactive peptides such as enkephalin18 and sandostatin19 and has led to potent analogues of these peptides, as has replacement of cystine with selenolanthionine in oxytocin analogues.20
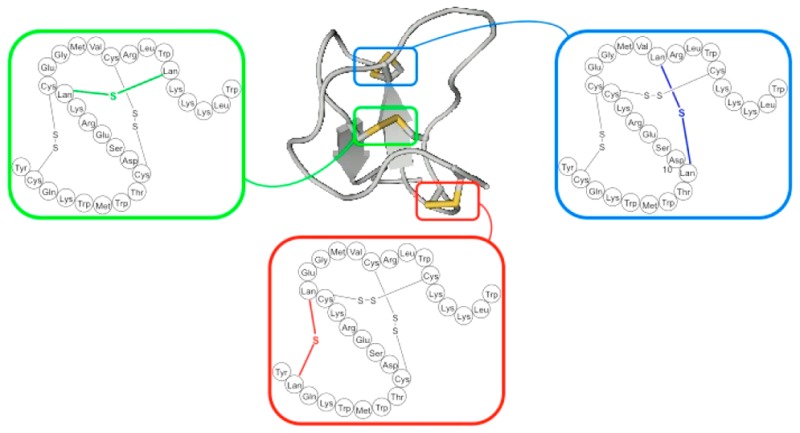
In previous work, we have developed solid-phase methods for synthesizing lanthionine-containing peptides, using orthogonally protected lanthionine,21 which have enabled the synthesis of a range of lantibiotics and analogues,22,23 including Lactocin S24 and Lacticin 3147.25 Looking to extend our methodology for incorporating lanthionine bridges into peptides, we elected to attempt to synthesize lanthionine-containing analogues of ProTx-II, a peptide isolated from the venom of the Peruvian green velvet tarantula (Thrixopelma puriens).26 ProTx-II belongs to a family of spider peptide toxins that contain six cysteine residues. These are oxidized to form three interlinked disulfide bonds, connected together in a 1-4, 2-5, and 3-6 pattern referred to as the inhibitory cystine knot (ICK) scaffold. Other examples of toxins sharing the same pattern include HwTx-IV,27 GpTx-I,28 and CcoTx-I29 (Figure 1). The ICK scaffold has at its core an antiparallel beta-sheet comprising two or three strands, with a third disulfide bridge inserted between the loop formed by the first two disulfide bridges. The number and sequence of amino acids between the cysteine residues are highly variable in these ICK peptides, leading to a wide variety of secondary structural motifs and peptide sequences being displayed between each disulfide bridge.30,31 Effective synthetic routes to ICK peptides with disulfide bond replacements are therefore of considerable interest in the development of peptide toxins with in vivo stability and have the potential to fine-tune receptor selectivity.
Figure 1.

Amino acid sequences and disulfide bond connectivities of the ICK scaffold peptides ProTx-II, HwTx-IV, PaTx-1, GpTx-I, CcoTx-I, and Prialt.
ProTx-II has been the subject of particular interest from researchers as a possible lead for the treatment of chronic nociceptive pain.4 It has been reported to have both the highest potency against the voltage-gated ion channel Nav1.7 (IC50 0.3 nM) and exceptional receptor subtype selectivity (100-fold more selective for the Nav1.7 ion channel compared with other sodium ion channel subtypes).32 Nav1.7 has been implicated as a target for the treatment of chronic pain after loss-of-function mutations in the corresponding SCN9A gene left their carriers unable to feel pain, with seemingly no other detrimental effects on their health.33 By contrast, gain-of-function mutations on the same gene cause sufferers to feel a constant burning sensation, usually in their hands and feet.34 Recently, the NMR structure of ProTx-II and analogues has been solved (PDB ID: 2N9T).35 Park and co-workers have also elucidated the solution structure of ProTx-II by NMR,36 and both structures show that the peptide adopts a classical ICK knot structure with a conformationally labile region at the C-terminus.
Despite these structural studies, much remains unknown about the mechanism of action of ProTx-II on Nav1.7. Structural and mutagenesis investigations of the interaction of ProTx-II with the Nav1.5 ion channel established that ProTx-II does not act by blocking the channel pore.37 Instead, studies into the interaction of ProTx-II and HwTx-IV with hNav1.7 proposed that both peptides interact with the domain II voltage-sensing domain (VSD), with ProTx-II also trapping the domain IV VSD in the resting configuration.38 Mutagenesis experiments on ProTx-II by Park and co-workers,36 and investigations of chimeric peptides incorporating sequences from the related toxin PaTx-I, have revealed that the C-terminal domain is especially important for the specificity and potency of its interaction with Nav1.7, and that certain C-terminal modifications can improve the potency relative to the wild-type (wt) peptide. Henriques and co-workers35 noted that ProTx-II potency is correlated with its membrane-binding ability, and they established the importance of certain residues in the main body of the peptide in binding to membranes. Based on these results, they proposed a model for ProTx-II inhibition of Nav1.7 where binding to the membrane orientates the peptide, which allows for the C-terminal domain to interact with the VSD of Nav1.7. Recently, Flinspach and co-workers39 identified JNJ63955918, a potent and selective inhibitor of Nav1.7, from a library of over 1500 peptides generated using ProTx-II as a scaffold, and Deuis and co-workers have characterized the mode of action of another highly selective hNav1.7 ICK spider venom peptide, Pn3a.40 While these studies have considerably advanced our understanding of ProTx-II inhibition of Nav1.7, we do not yet have a comprehensive understanding of the binding mode of ProTx-II, or how inhibition of Nav1.7 relates to analgesia: injections of ProTx-II failed to elicit analgesia in a mouse model.4 Further studies relating the structure and function of ProTx-II are therefore vital in elucidating its mechanism of action, which will be key to designing future effective treatments for chronic pain.
Our goal in this research was to use and extend our solid-phase synthesis methodology to prepare lanthionine-bridged analogues of ProTx-II and of individual rings of ProTx-II. We sought to explore the effects of subtle changes in peptide conformation arising from replacing each of the naturally occurring disulfide bridged rings in turn with a thioether linked ring on the binding of these peptides to hNav1.7 ion channels in vitro using patch-clamp assays. As part of this study, we have also investigated the effects of using different oxidative folding methods on the disulfide connectivity of the wt peptide sequence and their consequences for the biological activity of ProTx-II. Finally, we report the high-resolution X-ray crystal structure of wt ProTx-II.
Results and Discussion
Synthesis of (2R,6R)-(Allyl, Aloc/Fmoc)-Lanthionine (1)
In the naturally occurring lantibiotics, the lanthionine residues are present as the meso-(2R,6S) diastereoisomer. However, in ProTx-II all the constituent amino acids, including the Cys residues, are the naturally occurring l-enantiomers. In order to synthesize analogues of ProTx-II with the Cys-Cys bridges replaced by lanthionine bridges with the same stereochemistry, we adapted our previously published procedure21 to give the key orthogonally protected (R,R)-lanthionine building block 1. Minor modifications to the original synthetic methodology were made in order to scale up the procedure to give multigram quantities of 1 (Supporting Information).41
Synthesis of Single Ring Truncated Analogues of ProTx-II Containing Thioether or Disulfide Bridges
Before embarking on the synthesis of complex cystine knot structures containing both disulfide and thioether linkages, we elected to synthesize analogues of individual rings of ProTx-II, incorporating either thioether or disulfide bridges (Figure 2). Previous work in our group21 and by others24,25 has enabled cyclic lanthionine-bridged peptides with up to nine residues in the ring to be synthesized using this solid-phase approach. We envisaged that some development of the methodology would be needed to access lanthionine-bridged analogues of ProTx-II with up to 13 residues in the ring. Moreover, producing smaller fragments of ProTx-II would enable us to determine whether any biological activity would remain in the individual rings once removed from the highly ordered framework of the ICK structure.
Figure 2.

Single ring thioether and disulfide analogues of ProTx-II. The positions of the Cys groups, and their Met replacements, are marked according to the numbering from the full-length wt ProTx-II sequence.
Indeed, a trial synthesis of the C-terminal ring 2a using standard peptide coupling conditions at room temperature revealed problems. Linear peptide 6 was prepared on a low-loading NovaSyn TGT resin, and lanthionine derivative 1 was then coupled using PyAOP, HOAt, and DIPEA to give 7. This was then elongated to give the linear peptide 8 (Scheme 1).
Scheme 1. Synthesis of Lanthionine-Bridged Peptide 2a.

Reagents and conditions: (i) (allyl, Aloc/Fmoc)-lanthionine 1, PyAOP, HOAt, DIPEA, DMF, μwave, 5 min, 60 °C; (ii) incorporation of standard protected amino acids with HBTU, DIPEA, DMF, followed by deprotection with piperidine; (iii) Pd(PPh3)4, 1,3-dimethylbarbituric acid, DMF, CH2Cl2, then 40% piperidine/DMF; (iv) PyAOP, HOAt, DIPEA, DMF, μwave, 5 min, 60 °C; (v) Fmoc-Lys(Boc)-OH, HBTU, DIPEA, DMF, then 20% piperidine/DMF; (vi) TFA, ethanedithiol, iPr3SiH, H2O.
We then envisaged chemoselective removal of the allyl and Aloc groups, followed by Fmoc deprotection, cyclization on-resin, installation of the final amino acid, and cleavage from resin. However, this synthetic sequence yielded only small amounts of the desired 2a, mixed with linear peptide byproducts in which the lanthionine residue had not been incorporated. We therefore investigated conditions for the incorporation of 1 using microwave irradiation coupling conditions.41 Coupling with PyAOP/HOAt/DIPEA using microwave irradiation at 300 W and 60 °C for 5 min proved to be optimal for incorporating 1 to give resin-bound intermediate 7. Standard SPPS was then used to elongate this to give resin-bound peptide 8. The allyl and Aloc groups were then selectively removed with Pd(PPh3)4 using 1,3-dimethylbarbituric acid as an allyl group scavenger;42,43 subsequent removal of the Fmoc group gave 9. This was then selectively cyclized on-resin with PyAOP/HOAt/DIPEA,44 again using microwave irradiation at 300 W and 60 °C for 5 min. Chain extension of 10 to give 11 was followed by resin cleavage to give analogue 2a. Using this approach, we also synthesized a second analogue, 3a, as it has recently been shown36 that the flexible C-terminal fragment of ProTx-II is important for both potency and sodium channel selectivity. Pleasingly, we were also able to prepare the lanthionine analogue of the larger (13 residues) central ring, 4a. However, unfortunately the largest (15 residues) ring, 5a, could not be synthesized. For comparison purposes, the Cys-linked disulfide analogues 2b, 3b, 4b, and 5b were also synthesized. In each case the linear peptides were prepared using standard techniques (Supporting Information) and then cyclized by stirring for 10 days at 4 °C in water, giving the desired peptides in good yield and purity.
Synthesis of Full-Length ProTx-II Analogues with One Disulfide Bridge Replaced by a Thioether Bridge
Having successfully prepared the single ring analogues, we turned our attention to the full length analogues 12, 13, and 14 (Figure 3). Our strategy for these peptides had to take into account two possible challenges. The first is that the thioether bridges in lantibiotics are very susceptible to aerial oxidation,45 and therefore prolonged reaction times when forming the Cys-Cys bridges should be avoided. The second problem that we anticipated, in particular after the formation of the lanthionine bridge, was the folding or aggregation of the resin-bound peptide during synthesis, resulting in the N-terminal amino acid becoming inaccessible to subsequent activated amino acids. In the event this proved to be a more serious problem. Attempted solid-phase synthesis of the linear peptides failed in each case shortly after incorporation of the first lanthionine residue, even when microwave coupling conditions were used, resulting in the isolation of short peptides of between 6 and 12 residues. In order to circumvent this problem, we inserted Hmb-protected amino acids at positions 4, 14, 18, 23, and 28, in order to disrupt hydrogen bonding and prevent on-resin aggregation.46 For analogue 12, we also elected to use the same protecting groups for the Cys residues at positions 2, 9, 16, and 21. We anticipated that the presence of the thioether bridge between positions 15 and 25 would enable the peptide to fold correctly and the Cys-Cys bridges to be formed with the correct connectivity, without the need for an orthogonal Cys side-chain protecting strategy. Thus, (Scheme 2) protected lanthionine 1 was coupled to the resin-bound peptide 15 using the microwave conditions optimized for the single ring analogues. Chain extension of 16 using standard coupling conditions, and Hmb-protected amino acids as indicated, afforded 17. The lanthionine bridge was then formed by sequential removal of the Fmoc and allyl/Aloc protecting groups, followed by microwave-assisted cyclization on-resin, as before.
Figure 3.

Lanthionine-bridged analogues of ProTx-II.
Scheme 2. Synthesis of Lanthionine-Bridged ProTx-II Analogue 12.

Standard protecting groups were used for the amino acids, with additional Hmb protection as indicated; see Experimental Section. Reagents and conditions: (i) 1, PyAOP, HOAt, DIPEA, DMF, μwave, 5 min, 60 °C; (ii) incorporation of standard protected amino acids with HBTU, DIPEA, DMF, followed by deprotection with piperidine; (iii) 40% piperidine/DMF, then Pd(PPh3)4, 1,3-dimethylbarbituric acid, DMF, CH2Cl2; (iv) PyAOP, HOAt, DIPEA, DMF, μwave, 5 min, 60 °C; (v) incorporation of standard protected amino acids with HBTU, DIPEA, DMF, followed by deprotection with piperidine; (vi) TFA, iPr3SiH, H2O.
Chain extension of 18 gave the fully protected, resin-bound peptide intermediate 19. Subsequent cleavage of 19 from the resin, using a deprotection cocktail lacking EDT, gave the desired fully deprotected and cyclized peptide 12 (overall yield 0.4% after purification). This suggests that the partly folded, lanthionine-bridged structure does indeed aid the cyclization of the disulfide bonds during the cleavage and deprotection procedure. The same strategy was used to synthesize 14 in a yield of 1.1% after purification.
For analogue 13, as the third and fourth cysteine residues are next to each other in this sequence, we decided to employ orthogonal protecting group chemistry in order to guarantee the 1-4, 3-6 connectivity. We elected to use S(Tmp)-protected cysteine47 as this protecting group is stable to piperidine but very labile under mild thiolysis conditions, for example in the presence of DTT. Thus, (Scheme 3) 1 was coupled to linear resin-bound peptide 20 using microwave coupling conditions to give 21, and chain extension as before afforded 22. Deprotection of Fmoc and allyl/Aloc protecting groups, followed by microwave-assisted cyclization on-resin, gave 23, which was again chain-extended to give the resin-bound peptide 24. On-resin cleavage of the Tmp protecting groups with DTT was followed by oxidation with NCS to give the bicyclic resin-bound peptide 25. Cleavage and deprotection using the same deprotection cocktail, lacking EDT, followed by stirring in water at 4 °C, gave the desired tricyclic peptide 13 in 7% yield after purification.
Scheme 3. Synthesis of Lanthionine-Bridged ProTx-II Analogue 13.

Standard protecting groups were used for the amino acids, with additional Hmb protection as indicated; see Experimental Section. Reagents and conditions: (i) 1, PyAOP, HOAt, DIPEA, DMF, μwave, 5 min, 60 °C; (ii) incorporation of standard protected amino acids with HBTU, DIPEA, DMF, followed by deprotection with piperidine; (iii) 40% piperidine/DMF, then Pd(PPh3)4, 1,3-dimethylbarbituric acid, DMF, CH2Cl2; (iv) PyAOP, HOAt, DIPEA, DMF, μwave, 5 min, 60 °C; (v) incorporation of standard protected amino acids with HBTU, DIPEA, DMF, followed by deprotection with piperidine; (vi) 5% DTT in 0.1 M NMM, DMF; (vii) NCS (2 equiv), DMF; (viii) TFA, iPr3SiH, H2O.
As a control for the biological testing, we also synthesized the parent ProTx-II structure. The linear sequence was prepared using standard SPPS protocols (Supporting Information) and purified. Despite reports of racemization of cysteine residues under these conditions,48 substitution of DIPEA as the base in the coupling step with 2,4,6-collidine did not noticeably improve the purity of the crude linear ProTx-II produced. Although formation of the correct connectivity of the three disulfide bonds in these toxins is clearly crucial for folding of the peptide into the biologically active structure, achieving regioselective disulfide bond formation is difficult to achieve using orthogonal protecting groups for three pairs of Cys residues.49 Conotoxins with three disulfide bridges are typically synthesized as a linear precursor with all six cysteines unprotected, and then different oxidation methods are attempted until the correct topology is formed. The wide variation in peptide sequence has precluded the development of a single, general method for such cyclizations.50 Intriguingly, in some cases oxidative folding of linear conotoxin precursors has resulted in two disulfide isoforms, both of which have potent Nav channel blocking activity.51 However, for this family of spider toxins there are no reports comparing the effectiveness and selectivity of different oxidative cyclization methods. Initial attempts to cyclize the linear, unprotected ProTx-II peptide sequence using the conditions reported by Middleton26 were unsuccessful in our hands. We next turned to aerobic oxidation,49 and after some investigation we determined that the purified, unprotected linear ProTx-II peptide could be cyclized in H2O at 10 °C and a concentration of 0.1 mg/mL over 7 days to give the cyclized peptide (ProTx-II/7d) cleanly and in high yield. For comparison, ProTx-II was prepared using the conditions recently described by Park and co-workers.36 In this procedure, the unpurified, unprotected linear ProTx-II peptide was stirred at pH 8 in a mixture of glutathione (GSH), glutathione disulfide (GSSG), urea, and Tris-HCl for 24 h, followed by adjustment to pH 3 and purification of the peptide by HPLC to yield the cyclic peptide (ProTx-II/24h). As additional controls, we also purchased commercial samples of ProTx-II from two companies, Sigma-Aldrich and Smartox.
Biological Activity of ProTx-II and Analogues
All compounds were tested against stably transfected cell lines expressing the hNav1.7 receptor, using the hNav1.7-HEK cell IonWorks Population Patch Clamp Assay (Essen) and the QPatch Patch Clamp Assay (B’SYS GmbH). Perhaps unsurprisingly, neither the single ring lanthionine analogues 2a–4a, nor their Cys-Cys counterparts 2b–5b, showed any activity in these assays. Disappointingly, the full-length lanthionine bridged analogues 12, 13, and 14 did not show any activity either. Finally, to our surprise, the wt ProTx-II/7d that we had synthesized was completely inactive, as was the sample purchased from Sigma-Aldrich, whereas the ProTx-II/24h sample, and the sample from Smartox, showed the expected activity (Supporting Information).
Disulfide Connectivity and Conformation of wt ProTx-II Prepared by Different Oxidative Folding Methods
Liquid chromatography–mass spectrometry (LC-MS) showed that all four samples of wt ProTx-II had the correct amino acid composition and initially appeared to indicate that all three disulfide bonds had formed in each sample. The peptide samples were then compared by HPLC, and a clear difference was seen between the retention times of the ProTx-II/7d and the ProTx-II/24h peptides; this was verified by co-injection of the two samples (Figure 4).
Figure 4.
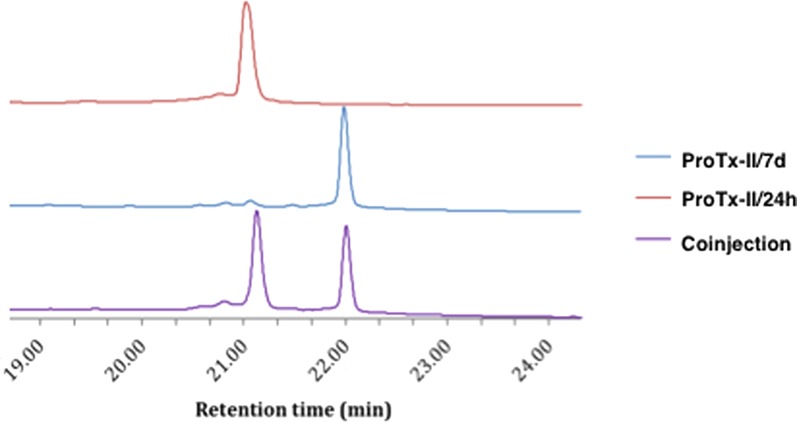
HPLC of ProTx-II/24h and ProTx-II/7d peptides. All experiments were run using analytical HPLC Method B.
This was further confirmed by comparing the HPLC trace of the ProTx-II/24h peptide with the Smartox sample (Supporting Information Figure S1b) and the ProTx-II/7d peptide with the Sigma sample (Supporting Information Figure S1c). This strongly suggested that the ProTx-II/24h peptide had either a different conformation or different disulfide bond connectivity to the ProTx-II/7d peptide.
Investigation of Disulfide Bond Connectivities of the ProTx-II Peptides by Mass Spectrometry
We reasoned that the differences in biological activity and HPLC retention time might arise from differences in disulfide bond connectivity between the two groups of peptides. In particular, because the Cys residues at positions 15 and 16 are adjacent, it was conceivable that one group of peptides might have the 1-4, 2-5, and 3-6 connectivity pattern observed for ICK knot peptides, and the other group might have a different connectivity, such as the 1-3, 2-5, and 4-6 pattern observed in spider toxins such as huwentoxin-II (HwTx-II).52 To investigate this possibility, proteolytic digestion of the peptide samples, followed by sequencing of the resulting products by mass spectrometry, was attempted following the method of Middleton et al.26 In our hands, however, the sequencing experiments could not be reproduced, and enzymatic digestion did not conclusively determine the disulfide bond connectivity.
We therefore decided to examine in more detail the intact forms of the peptide by means of nano-electrospray ionization (nanoESI) and ion mobility mass spectrometry (IM-MS). IM-MS can separate ions based on their mass, charge, and overall shape and has been successfully used to separate peptides of the same sequence but differing modification states.53,54 We hypothesized that any differences observed in the IM-MS data between these two peptides were likely to be due to differences in shape, which could in turn be caused by differences in disulfide bond connectivity. The nanoESI spectra of ProTx-II/24h and ProTx-II/7d samples are shown in Figure 5. The charge state distribution differs between the two samples, with the ProTx-II/24h exhibiting an overall lower charge state which is indicative of a more folded structure.55
Figure 5.

NanoESI spectra of (A) ProTx-II/24h and (B) ProTx-II/7d peptides. The predominant charge states observed are +4 and +5 for the ProTx-II/24h and ProTx-II/7d, respectively. Peaks annotated with * correspond to contaminant species.
A closer inspection of the +4 charge state from both peptides revealed differences in the isotopic distribution (Figure 6). We simulated the theoretical isotopic distribution for a ProTx-II peptide which would be either fully oxidized or fully reduced.
Figure 6.

Zoom-in of the +4 charge states of (A) ProTx-II/24h and (B) ProTx-II/7d peptides and theoretical isotope distribution corresponding to a ProTx-II with (C) all cysteines oxidized and (D) all cysteines reduced.
Comparison of the experimental and theoretical isotope distributions revealed that, while the ProTx-II/24h sample corresponds to a peptide with all cysteines oxidized, the ProTx-II/7d appears to be a mixture of oxidized and fully reduced species. To probe this even further we carried out IM-MS analyses of the +4 charge state. Figure 7 shows the arrival time distributions (ATDs) for the two peptides. While the ATD of ProTx-II/24h comprises one peak, the same is not true for the ProTx-II/7d peptide. The ATD for the ProTx-II/7d peptide is broad and consists of more than one peak, signifying the existence of multiple different species being present. Reconstructing the mass spectra that correspond to each region of the ATD for the ProTx-II/7d sample, further reveals that none of these co-existing forms of this peptide are in the fully oxidized state as indicated by the isotopic ratio.
Figure 7.
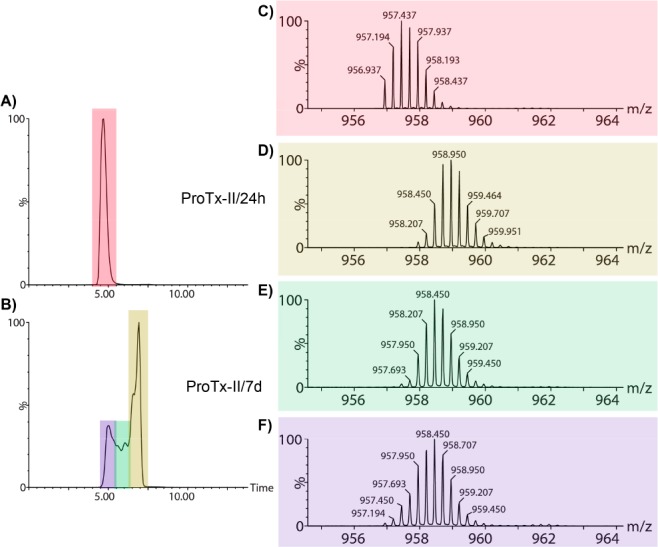
Arrival time distribution (ATD) from the IM-MS analysis of the +4 ion for (A) ProTx-II/24h and (B) ProTx-II/7d peptides. (C) The corresponding mass spectrum for ProTx-II/24h and (D–F) mass spectra corresponding to the different colored ATD regions for ProTx-II/7d.
Our native and IM-MS studies, therefore, show that the two peptides differ in the extent of disulfide bond formation, and this may be responsible for the difference in activity against the target sodium channel.
Crystal Structure of ProTx-II
To confirm the disulfide bond connectivity of the biologically active ProTx-II, we investigated its structure by X-ray crystallography. The structures of spider venom peptides have generally been studied to date by NMR; this is because their relatively small size makes solving the structure by this method feasible, but is also due to the reported difficulty in growing suitable crystals for X-ray crystallography.29 In fact, although the structures of several ICK peptides have been determined using NMR, to date only one ICK spider-venom toxin, ceratotoxin (CcoTx-I) has been successfully characterized by X-ray crystallography (PDB ID: 5EPM);29 moreover, in this structure, the target peptide was bound to a specifically generated high-affinity antibody Fab fragment to aid crystallization. This work represents the first reported crystal structure of ProTx-II and, to our knowledge, is the first crystal structure of any unbound ICK peptide toxin from spider venom.
A sample of ProTx-II known to be active against Nav1.7 was crystallized, and the crystal diffracted well under synchrotron conditions. The atomic resolution structure (0.99 Å) corroborates many features of the previously published structure solved by NMR (PDB ID: 2N9T).35 The disulfide bonds can be clearly resolved in the electron density, with the peptide showing the expected ICK fold and disulfide bond arrangement (1-4, 2-5, 3-6), together with a largely rigid and hydrophobic central core and more conformationally labile C- and N-termini. The RMSD for the full-length peptide all-atom/Cα = 3.19 Å/2.07 Å, whereas with the N- and C-termini removed the RMSD are all-atom/Cα = 2.26 Å /1.36 Å, highlighting the fact that most of the variability in the structure lies in the termini. In contrast to 2N9T, but consistent with other ICK peptides whose structures have been determined, our structure shows evidence of hydrogen bonding consistent with a beta hairpin from residues 19 to 27. The overall fold is similar to that reported in 2N9T, but with some deviations around M6 and L23; these could be a true reflection of the structure or represent the effects of crystal packing.
Two conformations of the N-terminal Y1 were observed in the electron density map. The crystal structure also shows the same hydrogen-bond interactions between the side chains of D10 and R13 seen in the NMR models (Figure 8), and a hydrogen bond between the side chain of E3 and the backbone of T8, which are not found in 2N9T. Further hydrogen bonds between the peptide backbone atoms, notably E3-K14, C9-L23 and M6-C25 give greater stability to the overall fold. Ultimately, the crystal structure provides conclusive proof that the biologically active form of ProTx-II adopts the ICK fold.
Figure 8.

(A) 2Fo-Fc Electron density map calculated using experimental phases, showing traceable peptide backbone and well-resolved side chains. (B) Fragment of ProTx-II crystal structure showing hydrogen bonding between R13 and D10. (C) Comparison of the X-ray crystal structure (left) with previously published NMR ensemble (PDB ID: 2N9T) showing similar overall folding and structure.
Conclusions
In this paper, we report the synthesis of three synthetic analogues of the ICK scaffold spider toxin ProTx-II, where each of the three disulfide bridges is regioselectively replaced by a thioether linkage. Incorporation of orthogonally protected (2R,6R)-lanthionine via solid-phase synthesis, followed by selective deprotection and on-resin cyclization, has enabled us to achieve for the first time the synthesis of highly complex triply bridged peptides with two cystine bridges and one lanthionine bridge. Moreover, in this work we have also demonstrated that very large lanthionine-bridged rings (up to 15 residues for peptide 14) can be synthesized by this approach.
These lanthionine-bridged peptides have larger ring sizes than those found in the naturally occurring lantibiotics, demonstrating the power of this approach for designing and synthesizing conformationally constrained peptides. It therefore also enables another approach for the modification of naturally occurring spider venom toxins as leads for potent and selective sodium channel blockers with good pharmacokinetic properties, complementary to the mutagenesis and chimeric peptide approaches previously published.28,29,35,36,56
Although lanthionine- and selenolanthionine-bridged analogues of other biologically active cyclic peptides have previously been reported to maintain or increase potency and stability relative to cystine-bridged peptides,18−20 this approach was not successful in delivering potent or selective Nav1.7 channel blockers based on ProTx-II. This could result from a number of factors: the shorter bridge between the α-carbons; the differences between the geometrical constraints imposed by a thioether versus a disulfide bridge; or differences in the correct folding of the resulting peptide. Indeed, a recent NMR study57 comparing thioether analogues of disulfide-bridged cyclic peptides targeting Death Receptor 5 with cystine-bridged peptides concluded that the thioether peptide was slightly less folded than the disulfide peptide.
In this regard, we have also demonstrated the importance of choosing the correct conditions for the oxidative folding of complex peptide toxins. This step is currently a bottleneck in the successful high-throughput production, and scale-up, of peptides containing multiple disulfide bridges.58 For ProTx-II, the presence of a redox buffer containing an excess of GSSG and GSH appears to be mandatory for the formation of the corrected folded toxin with all of the disulfide bridges formed (ProTx-II/24h). Conversely, it is clear from our IM-MS studies that aerial oxidation in water at neutral pH gives a number of products (ProTx-II/7d), none of which is in the fully oxidized state. Whether these partially cyclized peptides result from the kinetic formation of one or more incorrect Cys-Cys linkages, or whether they arise from an incorrectly folded intermediate with one or more correct Cys-Cys linkages, is impossible to determine in this case, as the observed ATD distribution (Figure 7) could arise from differing collision cross sections of both incorrectly folded peptides and incomplete disulfide oxidation.
This also raises the wider issue of whether the use of aerial oxidation, rather than the GSSG/GSH buffer, for folding the lanthionine hybrids 12, 13, and 14 may have been a contributory factor to their lack of activity. An additional complication is that the methodology that we have developed for synthesizing these hybrids necessarily introduces a nonreducible linkage during the synthesis. For multiply disulfide bridged peptides and small proteins, it is widely accepted that there are several different pathways of oxidative folding. These vary from one extreme in which an ensemble of heterogeneous intermediates with many non-native disulfide bonds are initially generated, with these rearranging over time via disulfide shuffling to give the correctly folded and connected peptides, to the other extreme in which the amino acid sequence dictates a folding pathway which leads predominantly or exclusively to native disulfide bonds without subsequent rearrangement being necessary, with intermediate situations in which the formation of a non-native bridge creates a correctly folded peptide in which the required cysteine residues are brought into proximity, allowing reshuffling to give the correct connectivity.59 Moreover, for some peptide sequences such as tick anticoagulant peptide (TAP) it has also been shown that incorrectly folded/connected intermediates can act as kinetic “traps”, giving highly stable structures that are difficult to refold/reconnect into the correct structure.60 Thus, once a nonreducible lanthionine bridge has been introduced during the synthesis of the linear peptide, then the peptide may well have been trapped into a non-natural fold from which it cannot easily refold/rearrange to the native fold/connectivity.
Importantly, in this work we have also demonstrated the power of IM-MS studies for rapidly determining whether the oxidative folding method employed for synthetic ICK fold toxins gives a single, correctly folded and oxidized peptide or a mixture of incompletely oxidized species. We envisage that this will be particularly useful when screening libraries of peptides29,39 based on lead peptides from spider venoms. Such libraries, whether produced by either solid-phase synthesis or recombinant expression, usually rely on aerial oxidation to form the disulfide bridges, and our present work suggests that for some sequences in these libraries, a lack of biological activity might result from misfolding or incomplete oxidation of the particular peptide.
We have also reported here the first crystal structure of an ICK peptide toxin from spider venom not bound to a substrate. Whereas the X-ray crystal structure of a much larger (278 residues) phospholipase D enzyme spider venom peptide has recently been reported,61 smaller (30–35 residue) peptides such as ProTx-II are surprisingly difficult to crystallize. Hitherto all structural studies with these ICK fold spider toxins have been carried out using NMR techniques. The high resolution of this X-ray crystal structure structure of ProTx-II has enabled hydrogen bonds which could not be seen in the NMR structures to be observed. In turn, this will shed additional light on the stabilization of the ICK fold, and may help with further analysis of the structure–activity relationships between these toxins and the sodium channel receptors.
Finally, having demonstrated the potential of this solid-phase peptide synthesis approach to the preparation of ICK fold peptide toxin analogues with Cys-Cys linkages replaced by unnatural bridges, this paves the way for the preparation of other peptide structures with different ring sizes and bridge geometry. This opens up the potential for the design of more potent and selective Nav1.7 channel blockers with better pharmacokinetic properties and metabolic stability. In addition, peptides with one side-chain bridge that is regioselectively installed and cannot be reduced may also act as tools for investigating the folding pathways of these multiply bridged peptides, in combination with the IM-MS methodology reported here. Although the presence of a nonreducible side-chain bridge could be problematic for the correct folding of some peptide sequences, conversely in some cases it also has the potential to streamline the folding pathways by diminishing the number of nonproductive conformations available10,58 and thus improving the synthetic routes to these peptides.
Experimental Section
General Experimental for Peptide Synthesis
Protected amino acids and coupling reagents were purchased from Novabiochem; coupling reagents, bases, and solvents were purchased from Sigma-Aldrich. All water used was either distilled using an Elga Purelab Option R 7 water purifier or used directly from a bottle of HPLC-grade water. The peptides were either synthesized using an automated peptide synthesizer or manually, using the same timings and solution volumes. Amino acid and reagent concentrations were calculated based on the quantity and loading of the resin. The total volume of all reagents in each step was 1.5 mL. All reagents were dissolved in HPLC grade DMF.
Microwave couplings were carried out using a Personal Chemistry Smith Creator microwave loaded with 5 mL reaction vials. In all cases, the vial was irradiated for 5 min at 60 °C and 300 W before transfer of the resin back to the reaction syringe. DMF was used as the primary solvent throughout the peptide synthesis.
Peptides were centrifuged using an Eppendorf Centrifuge, model 5810R, and were lyophilized using a Thermo Scientific Heto PowerDry LL1500 freeze-dryer.
Automated Peptide Synthesis
Peptides were synthesized on a MultiSynTech Syro Peptide Synthesizer (Model MP-60).
Manual Peptide Synthesis
All manual coupling and deprotection steps, and complete manual peptide syntheses, were carried out using an IKA KS130 basic platform shaker to agitate the solutions. Reactions were carried out in 5 mL syringes with frits, which were evacuated by hand at the end of each reaction.
Fmoc Deprotection
For the automated peptide syntheses, all Fmoc deprotections were carried out using a solution of 40% piperidine in DMF, which was added to the syringe containing the resin. The mixture was agitated for 20 s every minute for a total of 3 min before removal of reagent by filtration under vacuum. The resin was washed with DMF (4 × 1.5 mL) before addition of a second portion of piperidine in DMF solution (40% v/v, 0.75 mL), followed by DMF (0.75 mL) to make an overall 20% v/v solution of piperidine in DMF. This mixture was agitated for 20 s every minute for a total of 10 min. The reagents were removed by filtration under vacuum, and the resin was washed with DMF (6 × 1.5 mL). For the manual peptide syntheses, all Fmoc deprotections were carried out using a solution of 40% piperidine in DMF, which was added to the syringe containing the resin. The mixture was agitated for 3 min before removal of reagent by filtration. A second portion of piperidine in DMF solution (40% v/v, 0.75 mL) was added, followed by DMF (0.75 mL) to make an overall 20% v/v solution of piperidine in DMF. This mixture was agitated for a total of 10 min. The reagents were removed by filtration, and the resin was washed with DMF (6 × 1.5 mL).
Amino Acid Coupling
Fmoc-protected amino acid (4 equiv) was added to the reaction syringe with HBTU (4 equiv) and DIPEA (8 equiv). The mixture was agitated for 20 s every 3 min for a total of 40 min. The reagents were removed by filtration under vacuum, and the resin was washed with DMF (4 × 1.5 mL).
Lanthionine Coupling
(S)-Allyl 3-((S)-2-((9-fluorenyl-methoxycarbonyl)amino-3-tert-butoxy-3-oxopropylthio)-2-(allyloxycarbonylamino)propanoic acid (1) (3 equiv), PyAOP (5 equiv), and HOAt (5 equiv) were dissolved in DMF (4 mL). To this, N,N-diisopropylethylamine (10 equiv) was added and left to react for 1 min before addition to a microwave vial containing the resin. The vial was irradiated for 5 min at 60 °C before transfer of the resin back to the reaction syringe. The resin was then washed with DMF (4 × 1.5 mL).
Allyl/Alloc Deprotection
Simultaneous deprotection of both the allyl and Alloc ester groups was performed using tetrakistriphenylphosphine palladium(0) (1 equiv) and 1,3-dimethylbarbituric acid (10 equiv) in a 1:1 solution of CHCl3: DMF (2 mL). This was left to react for 2 h in the dark under argon before removal of the solution by filtration. The resin was then washed with CH2Cl2 (10 × 1.5 mL), sodium diethyldithiocarbamate (5% w/v, 15 × 1.5 mL), and DMF (10 × 1.5 mL).
Lanthionine Cyclization
N,N-Diisopropylethylamine (10 equiv) was added to a solution of PyAOP (5 equiv) and HOAt (5 equiv) in DMF (4 mL). The solution was left to react for 1 min before addition to a microwave vial containing the resin. The vial was irradiated for 5 min at 60 °C before transfer of the resin back to the reaction syringe. The resin was then washed with DMF (4 × 1.5 mL).
Cleavage from the Resin
Peptides were first washed with DMF (4 × 1.5 mL), CH2Cl2 (4 × 1.5 mL), methanol (4 × 1.5 mL), and diethyl ether (4 × 1.5 mL) before drying in a desiccator for 45 min. A solution of 94% TFA, 2.5% water, 2.5% EDT, and 1% TIPS (2.5 mL) was then added to the resin and left to agitate for 30 min on the platform shaker. After this time, the entire solution was transferred to a Falcon tube, and 12 mL of diethyl ether was added to precipitate the peptide. The cleavage procedure was then repeated with fresh cleavage solution (2.5 mL containing 94% TFA, 2.5% water, 2.5% EDT, and 1% TIPS) and left to agitate for a further 40 min. The entire solution was again transferred to a Falcon tube before addition of 12 mL of diethyl ether to precipitate the peptide.
The Falcon tubes were then centrifuged for 10 min at 4000 rpm and 4 °C before decanting off the diethyl ether solution. This process was performed three times in total before re-dissolving the peptide in water and lyophilizing.
HPLC Purification
The peptides were analyzed and purified via reverse phase HPLC using either a Varian ProStar system with a model 210 solvent delivery module and a model 320 UV detector or a Dionex system with a PDA-100 photodiode array detector and a model ASI-100 automated sample injector. The preparative purification was performed using an ACE C8-300 semipreparative column (150 × 10 mm, flow rate of 8.0 mL/min), with UV detection at 215 and 254 nm, loaded with 200–1850 μL aliquots of a 10–20 mg/mL solution of peptide dissolved in water. Gradient conditions are reported for each peptide. The fractions containing the correct peak were pooled, the solvent was removed under reduced pressure to approximately 2 mL, and the solution was freeze-dried.
HPLC Analysis
All peptides were analyzed using UV detection at 215 and 254 nm, using the conditions shown below. Retention times are reported for each peptide.
HPLC Method A
ACE C8-300 analytical column (150 × 10 mm, flow rate of 1.0 mL/min). Gradient: 2–98% B over 20 min (A = water, B = acetonitrile, 0.1% TFA).
HPLC Method B
Dr Maisch C8 column (Reprosil Gold 200 C8, 5 μm, 250 × 4.6 mm, flow rate of 1.0 mL min–1). Gradient: 0–2 min on 95% A; then 5–95% B over 48 min; then 95% B over 2 min; then 95–5% B over 3 min; then 5% B over 5 min (A = water, B = acetonitrile, 0.1% TFA).
ESI-MS Analysis
This was performed on a Waters Acquity Ultra Performance LC/MS machine using a linear gradient of 5–95% B over 5 min (A = water, B = acetonitrile, 0.1% TFA). The analysis of the chromatograms was conducted using MassLynx software version 4.0.
Single Ring Thioether Analogue 2a
The synthesis was carried out on a 100 mg scale using preloaded Fmoc-Lys(Boc)-NovaSyn resin (loading 0.24 mmol/g). The following protected amino acids were used: Fmoc-Glu(OtBu)-OH; Fmoc-Gly-OH; Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Met-OH: Fmoc-Arg(Pbf)-OH; Fmoc-Val-OH; Fmoc-Trp(Boc)-OH. The peptide was assembled following the strategy outlined in Scheme 1. Amino acid coupling steps, Fmoc deprotection, coupling of lanthionine 1, removal of the allyl/Aloc groups, and cyclization were all carried out according to the general procedures above. The peptide was cleaved under standard conditions, washed with ether, and lyophilized. The peptide was purified by semipreparative HPLC using a gradient of 5–50% B over 15 min (A = water, B = acetonitrile) to yield 3 mg of product (8%). m/z (ES+) 855.66 ([M + 2H]2+), 570.68 ([M + 3H]3+), 428.23 ([M + 4H]4+). HPLC Method A:tR = 6.9 min.
Single Ring Thioether Analogue 3a
The synthesis was carried out on a 100 mg scale using preloaded Fmoc-Trp(Boc)-NovaSyn resin (loading 0.20 mmol/g). The following protected amino acids were used: Fmoc-Glu(OtBu)-OH; Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Met-OH: Fmoc-Arg(Pbf)-OH; Fmoc-Val-OH; Fmoc-Trp(Boc)-OH. The following Hmb-protected amino acids were also incorporated: Fmoc-(FmocHmb)-Gly-OH (position 5); Fmoc-(FmocHmb)-Leu-OH (position 10); Fmoc-(FmocHmb)-Lys(Boc)-OH (position 15). The peptide was assembled following the strategy outlined in Scheme 1. Amino acid coupling steps, Fmoc deprotection, coupling of lanthionine 1, removal of the allyl/Aloc groups, and cyclization were all carried out according to the general procedures above. The peptide was cleaved under standard conditions, washed with ether, and lyophilized. The peptide was purified by semipreparative HPLC using a gradient of 10–55% B over 8 min (A = water, B = acetonitrile) to yield 4 mg of product (9%). m/z (ES+) 538.27 ([M + 4H]4+). HPLC Method A: tR = 2.4 min.
Single Ring Thioether Analogue 4a
The synthesis was carried out on a 100 mg scale using preloaded Fmoc-Trp(Boc)-NovaSyn resin (loading 0.20 mmol/g). The following protected amino acids were used: Fmoc-Asp(OtBu)-OH; Fmoc-Glu(OtBu)-OH; Fmoc-Gly-OH; Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Met-OH: Fmoc-Arg(Pbf)-OH; Fmoc-Ser(tBu)-OH; Fmoc-Thr(tBu)-OH; Fmoc-Val-OH; Fmoc-Trp(Boc)–OH. The peptide was assembled following the strategy outlined in Scheme 1. Amino acid coupling steps, Fmoc deprotection, coupling of lanthionine 1, removal of the allyl/Aloc groups, and cyclization were all carried out according to the general procedures above. The peptide was cleaved under standard conditions, washed with ether, and lyophilized. The peptide was purified by semipreparative HPLC using a gradient of 5–50% B over 15 min (A = water, B = acetonitrile) to yield 0.5 mg of product (1%). m/z (ES+) 1021.71 ([M + 2H]2+). HPLC Method A: tR = 2.6 min.
Full-Length C-Terminal Thioether Analogue 12
The synthesis was carried out on a 100 mg scale using preloaded Fmoc-Trp(Boc)-NovaSyn resin (loading 0.20 mmol/g). The following protected amino acids were used: Fmoc-Cys(Trt)-OH; Fmoc-Asp(OtBu)-OH; Fmoc-Glu(OtBu)-OH; Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Met-OH: Fmoc-Gln(Trt)-OH; Fmoc-Arg(Pbf)-OH; Fmoc-Ser(tBu)-OH; Fmoc-Thr(tBu)-OH; Fmoc-Val-OH; Fmoc-Trp(Boc)-OH; Fmoc-Tyr(tBu)-OH. The following Hmb-protected amino acids were also incorporated: Fmoc-(FmocHmb)-Lys(Boc)-OH (positions 4, 14, 28); Fmoc-(FmocHmb)-Gly-OH (position 18); Fmoc-(FmocHmb)-Leu-OH (position 23). The peptide was assembled following the strategy outlined in Scheme 2. Amino acid coupling steps, Fmoc deprotection, coupling of lanthionine 1, removal of the allyl/Aloc groups, and cyclization were all carried out according to the general procedures above. The peptide was cleaved from the resin using a solution of 96.5% TFA, 2.5% water, and 1% TIPS (2.5 mL) and left to stir for 1 h. After this time, the entire solution was transferred to a Falcon tube, and 12 mL of diethyl ether was added to precipitate the peptide. The Falcon tube was then centrifuged for 10 min at 4000 rpm and 4 °C before decanting off the diethyl ether solution. This process was performed three times in total before re-dissolving the peptide in water and lyophilizing. The peptide was purified by semipreparative HPLC using a gradient of 10–55% B over 15 min (A = water, B = acetonitrile, 0.1% TFA) to yield 0.3 mg of product (0.4%). m/z (ES+) 758.50 ([M + 5H]5+). HPLC Method B: tR = 19.16 min.
Full-Length Middle Ring Thioether Analogue 13
The synthesis was carried out on a 100 mg scale using preloaded Fmoc-Trp(Boc)-NovaSyn resin (loading 0.20 mmol/g). The following protected amino acids were used: Fmoc-Cys(Trt)-OH (positions 15, 25); Fmoc-Cys(S(Tmp))-OH (positions 2, 16); Fmoc-Asp(OtBu)-OH; Fmoc-Glu(OtBu)-OH; Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Met-OH: Fmoc-Gln(Trt)-OH; Fmoc-Arg(Pbf)-OH; Fmoc-Ser(tBu)-OH; Fmoc-Thr(tBu)-OH; Fmoc-Val-OH; Fmoc-Trp(Boc)-OH; Fmoc-Tyr(tBu)–OH. The following Hmb-protected amino acids were also incorporated: Fmoc-(FmocHmb)-Lys(Boc)-OH (positions 4, 14, 28); Fmoc-(FmocHmb)-Gly-OH (position 18); Fmoc-(FmocHmb)-Leu-OH (position 23). The peptide was assembled following the strategy outlined in Scheme 3. Amino acid coupling steps, Fmoc deprotection, coupling of lanthionine 1, removal of the allyl/Aloc groups, and cyclization were all carried out according to the general procedures above. Selective deprotection of the S(Tmp) groups from the orthogonally protected cysteine residues was carried out using 5% DTT in 0.1 M NMM in DMF (1.5 mL) (3 × 30 min) before washing with DMF (4 × 1.5 mL). The deprotected cysteines were then cyclized using N-chlorosuccinimide (53.4 mg, 2 equiv) in DMF (2 mL) for 1.5 h before again washing with DMF (4 × 1.5 mL). The peptide was cleaved from the resin using a solution of 96.5% TFA, 2.5% water, and 1% TIPS (2.5 mL) and left to stir for 1 h. After this time, the entire solution was transferred to a Falcon tube, and 12 mL of diethyl ether was added to precipitate the peptide. The Falcon tube was then centrifuged for 10 min at 4000 rpm and 4 °C before decanting off the diethyl ether solution. This process was performed three times in total before re-dissolving the peptide in water and lyophilizing. The peptide was purified by semipreparative HPLC using a gradient of 10–55% B over 15 min (A = water, B = acetonitrile, 0.1% TFA) to yield 5 mg of product (7%). This was then dissolved in water (pH 7.7) at 1 mg/10 mL concentration, stirred for a further 11 days at 4 °C to ensure complete disulfide bond formation, then lyophilized. m/z (ES+) 544.64 ([M + 7H]7+). HPLC Method B: tR = 19.15 min.
Full-Length N-Terminal Thioether Analogue 14
The synthesis was carried out on a 100 mg scale using preloaded Fmoc-Trp(Boc)-NovaSyn resin (loading 0.20 mmol/g). The following protected amino acids were used: Fmoc-Cys(Trt)-OH; Fmoc-Asp(OtBu)-OH; Fmoc-Glu(OtBu)-OH; Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Met-OH: Fmoc-Gln(Trt)-OH; Fmoc-Arg(Pbf)-OH; Fmoc-Ser(tBu)-OH; Fmoc-Thr(tBu)-OH; Fmoc-Val-OH; Fmoc-Trp(Boc)-OH; Fmoc-Tyr(tBu)-OH. The following Hmb-protected amino acids were also incorporated: Fmoc-(FmocHmb)-Lys(Boc)-OH (positions 4, 14, 28); Fmoc-(FmocHmb)-Gly-OH (position 18); Fmoc-(FmocHmb)-Leu-OH (position 23). The peptide was assembled following the general strategy outlined in Scheme 2, but with the protected lanthionine 1 incorporated at position 16 and cyclized at position 2. Amino acid coupling steps, Fmoc deprotection, coupling of lanthionine 1, removal of the allyl/Aloc groups, and cyclization were all carried out according to the general procedures above. The peptide was cleaved from the resin using a solution of 96.5% TFA, 2.5% water, and 1% TIPS (2.5 mL) and left to stir for 1 h. After this time, the entire solution was transferred to a Falcon tube, and 12 mL of diethyl ether was added to precipitate the peptide. The Falcon tube was then centrifuged for 10 min at 4000 rpm and 4 °C before decanting off the diethyl ether solution. This process was performed three times in total before re-dissolving the peptide in water and lyophilizing. The peptide was purified by semipreparative HPLC using a gradient of 10–55% B over 15 min (A = water, B = acetonitrile, 0.1% TFA) to yield 0.8 mg of product (1%). m/z (ES+) 544.64 ([M + 7H]7+). HPLC Method B: tR = 19.11 min.
Synthesis of ProTx-II
The synthesis was carried out on a 100 mg scale using preloaded Fmoc-Trp(Boc)-NovaSyn resin (loading 0.20 mmol/g). The following protected amino acids were used: Fmoc-Cys(Trt)-OH, Fmoc-Asp(OtBu)-OH; Fmoc-Glu(OtBu)-OH; Fmoc-Gly-OH; Fmoc-Lys(Boc)-OH; Fmoc-Leu-OH; Fmoc-Met-OH: Fmoc-Gln(Trt)-OH; Fmoc-Arg(Pbf)-OH; Fmoc-Ser(tBu)-OH; Fmoc-Thr(tBu)-OH; Fmoc-Val-OH; Fmoc-Trp(Boc)-OH; Fmoc-Tyr(tBu)-OH. Amino acid coupling steps and Fmoc deprotection were all carried out according to the general procedures above. The peptide was cleaved under standard conditions, washed with ether, and lyophilized.
ProTx-II/7d
The crude peptide was purified using a gradient of 35–60% B over 15 min (A = water, B = acetonitrile, 0.1% TFA) to give the linear peptide: m/z (ES+) 765.93 ([M + 5H]5+), 639.57 ([M + 6H]6+. Analytical HPLC: tR = 2.4 min. The peptide was then allowed to cyclize for 7 days in pure water at 4 °C and a concentration of 0.1 mg/mL. The solution was then concentrated to approximately 2 mL, flash frozen, and lyophilized to yield the cyclized peptide as a white solid (10 mg, 13%) m/z (ES+) 1276.51 ([M + 3H]3+), 957.43 ([M + 4H]4+), 765.93 ([M + 5H]5+), 638.72 ([M + 6H]6+). HPLC Method B: tR = 21.80 min.
ProTx-II/24h
Glutathione (0.15 mM) and glutathione disulfide (0.3 mM) were added to a mixture of urea (2 M) and Tris-HCl (0.1 M) in water. The pH was adjusted to 8.0 using saturated aqueous sodium bicarbonate. A solution of crude linear ProTx-II was added at a concentration of 0.1 mg/mL and left to stir for 24 h at room temperature. After this time, the pH of the solution was adjusted to pH 3 using HCl (2 M). The peptide was purified by semipreparative HPLC: gradient: 0–1.5 min on 95% A; then 5–70% B over 38 min; then 70% B over 2.0 min; then 70–5% B over 1.1 min; then 5% B over 2.4 min (A = water, B = acetonitrile, 0.1% TFA). The peptide was further purified using analytical HPLC Method B: tR = 21.03 min.
Automated and Manual Patch Clamp Assays
Peptides were diluted to a concentration of 100 nM and tested against stably transfected cell lines expressing the hNav1.7 receptor. Peptides 2a, 2b, 4a, 4b, 5b, 12, 13, 14, ProTx-II/7d, Sigma, and Smartox were tested against stably transfected cell lines expressing the hNav1.7 receptor, using the hNav1.7-HEK cell IonWorks Population Patch Clamp Assay, as designed by Essen Bioscience. These peptides were tested at 11 different points to make an IC50 curve using 3-fold dilution from 1 μM to 0.000001 μM in triplicate, using tetracaine as the control molecule. The samples were allowed to incubate for 5–7 min before testing occurred and were tested in the presence of 0.1% bovine serum albumin to prevent the samples adhering to the side of the microtiter plate. The percentage inhibition between the first and 20th pulses was then analyzed and plotted on a graph. Peptides 3a, 3b, 14, ProTx-II/24h, ProTx-II/7d, Sigma, Smartox, and the linear sequence of ProTx-II were tested using the QPatch Patch Clamp Assay as designed by B’SYS GmbH. hNav1.7 inward peak currents were recorded in CHO cells stably transfected with cDNA encoding this sodium channel in the resting, fast and slow inactivated state. Lidocaine was used as the reference. Full details of these experiments are in the Supporting Information.
HPLC Analysis of ProTx-II Samples
ProTx-II samples were mixed as indicated and co-injected into a Dr Maisch C8 column, eluting with HPLC Method B.
NanoESI and IM-MS Analyses
All measurements were carried out using a G2Si mass spectrometer (Waters, UK). The TOF analyzer was operated in V-optic mode and tuned for an operating resolution of 20 000 fwhm. The instrument was mass calibrated using a 2 mg/mL solution of CsI. ProTx-II samples were dissolved in 50% MeOH with 0.1% HCOOH and 2.5 μL aliquots, of 10 μM concentration, were delivered to the mass spectrometer by means of nanoESI using gold-coated capillaries, prepared in house. Instrumental parameters were as follows unless otherwise specified: capillary voltage 0.9 kV, cone voltage 40 V, trap energy 4 V, transfer energy 2 V, bias voltage 45 V, IMS pressure 3 mbar, IMS wave velocity 650 m/s, IMS wave height 35 V. Data acquisition, processing, and calculation of theoretical isotopic distributions were carried out using MassLynx (v4.1) software (Waters Corp., Milford, MA). IM-MS data were processed in Driftscope v2.8 and exported for further analysis to MassLynx.
Acknowledgments
We thank Eisai for a Ph.D. studentship (to Z.V.F.W.), the Wellcome Trust for a studentship (to S.M., 109073/Z/15/Z), the EPSRC for a studentship (to R.D.), and BBSRC/Waters for a CASE studentship (to A.C.). The G2Si ion mobility mass spectrometer was purchased with a grant from the Wellcome Trust (104913/Z/14/ZBM). Diffraction data were collected at the Berkeley Center for Structural Biology (Berkeley, CA). The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. The Gonen laboratory is supported by the Howard Hughes Medical Institute. We also thank Dr. Carolyn Hyde (Bio-Analysis Centre, London Bioscience Innovation Centre), Dr. Ian Kilford (Eisai), Prof. Rob Liskamp and Dr. Helmus Van De Langemheen (Department of Chemistry, University of Glasgow), Dr. Abil Aliev, Dr. Jamie Baker and Prof. Erik Årstad (Department of Chemistry, UCL), Dr. Srinivasan Kanumilli, Dr. Tim Dale, and Dr. Derek Trezise (Essen Biosciences), and Dr. Daniel Konrad and Dr. Alexandra Illy (B’SYS GmbH) for numerous helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b06506. The X-ray crystal structure of ProTx-II has been deposited in the Protein Data Bank with accession code 5O0U.
X-ray crystallographic data for ProTx-II (CIF)
Experimental procedures for the multigram-scale synthesis of protected lanthionine 1 and for the synthesis of peptides 2b, 3b, 4b, and 5b; 1H NMR spectra for key synthetic intermediates and for 1; mass spectral and HPLC data for all peptides after purification; crystallization and diffraction methods for ProTx-II; complete materials and methods, and full data from the patch clamp assays (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fosgerau K.; Hoffmann T. Drug Discovery Today 2015, 20, 122. 10.1016/j.drudis.2014.10.003. [DOI] [PubMed] [Google Scholar]
- King G. F.Venoms to drugs: venom as a source for the development of human therapeutics; Royal Society of Chemistry: Cambridge, 2015. [Google Scholar]
- Pope J. E.; Deer T. R. Expert Opin. Pharmacother. 2013, 14, 957. 10.1517/14656566.2013.784269. [DOI] [PubMed] [Google Scholar]
- King G. F.; Vetter I. ACS Chem. Neurosci. 2014, 5, 749. 10.1021/cn500171p. [DOI] [PubMed] [Google Scholar]
- Harvey A. L. Toxicon 2014, 92, 193. 10.1016/j.toxicon.2014.10.020. [DOI] [PubMed] [Google Scholar]
- Armishaw C. J.; Daly N. L.; Nevin S. T.; Adams D. J.; Craik D. J.; Alewood P. F. J. Biol. Chem. 2006, 281, 14136. 10.1074/jbc.M512419200. [DOI] [PubMed] [Google Scholar]
- Muttenthaler M.; Nevin S. T.; Grishin A. A.; Ngo S. T.; Choy P. T.; Daly N. L.; Hu S.-H.; Armishaw C. J.; Wang C.-I. A.; Lewis R. J.; Martin J. L.; Noakes P. G.; Craik D. J.; Adams D. J.; Alewood P. F. J. Am. Chem. Soc. 2010, 132, 3514. 10.1021/ja910602h. [DOI] [PubMed] [Google Scholar]
- MacRaild C. A.; Illesinghe J.; van Lierop B. J.; Townsend A. L.; Chebib M.; Livett B. G.; Robinson A. J.; Norton R. S. J. Med. Chem. 2009, 52, 755. 10.1021/jm8011504. [DOI] [PubMed] [Google Scholar]
- Van Lierop B. J.; Robinson S. D.; Kompella S. N.; Belgi A.; McArthur J. R.; Hung A.; MacRaild C. A.; Adams D. J.; Norton R. S.; Robinson A. J. ACS Chem. Biol. 2014, 8, 1815. 10.1021/cb4002393. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Sun D.-M.; Wang F.-L.; He Y.; Liu L.; Tian C.-L. Angew. Chem., Int. Ed. 2015, 54, 14276. 10.1002/anie.201500699. [DOI] [PubMed] [Google Scholar]
- Gleeson E. C.; Jackson W. R.; Robinson A. J. Tetrahedron Lett. 2016, 57, 4325. 10.1016/j.tetlet.2016.08.032. [DOI] [Google Scholar]
- Hargittai B.; Solé N. A.; Groebe D. R.; Abramson S. N.; Barany G. J. Med. Chem. 2000, 43, 4787–4792. 10.1021/jm990635c. [DOI] [PubMed] [Google Scholar]
- Dekan Z.; Vetter I.; Daly D. L.; Craik D. J.; Lewis R. J.; Alewood P. F. J. Am. Chem. Soc. 2011, 133, 15866. 10.1021/ja206408q. [DOI] [PubMed] [Google Scholar]
- Stymiest J. L.; Mitchell B. F.; Wong S.; Vederas J. C. J. Org. Chem. 2005, 70, 7799. 10.1021/jo050539l. [DOI] [PubMed] [Google Scholar]
- Derksen D. J.; Stymiest J. L.; Vederas J. C. J. Am. Chem. Soc. 2006, 128, 14252. 10.1021/ja066203q. [DOI] [PubMed] [Google Scholar]
- Knerr P. J.; Tzekou A.; Ricklin D.; Qu H. C.; Chen H.; van der Donk W. A.; Lambris J. D. ACS Chem. Biol. 2011, 6, 753. 10.1021/cb2000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Liu C.; Song H.; Wang F.-L.; Zou Y.; Wu Q.-Y.; Hu H.-G. RSC Adv. 2017, 7, 2110. 10.1039/C6RA26617G. [DOI] [Google Scholar]
- Polinsky A.; Cooney M. G.; Toy-Palmer A.; Ösapay G.; Goodman M. J. Med. Chem. 1992, 35, 4185. 10.1021/jm00100a026. [DOI] [PubMed] [Google Scholar]
- Ösapay G.; Prokai L.; Kim H.-S.; Medzihradszky K. F.; Coy D. H.; Liapakis G.; Reisine T.; Melacini G.; Zhu Q.; Wang S. H.-H.; Mattern R.-H.; Goodman M. J. Med. Chem. 1997, 40, 2241. 10.1021/jm960850i. [DOI] [PubMed] [Google Scholar]
- Dantas de Araujo A.; Mobli M.; Castro J.; Harrington A. M.; Vetter I.; Dekan Z.; Muttenthaler M.; Wan J.-J.; Lewis R. J.; King G. F.; Brierley S. M.; Alewood P. F. Nat. Commun. 2014, 5, 3165. 10.1038/ncomms4165. [DOI] [PubMed] [Google Scholar]
- Bregant S.; Tabor A. B. J. Org. Chem. 2005, 70, 2430. 10.1021/jo048222t. [DOI] [PubMed] [Google Scholar]
- Tabor A. B. Org. Biomol. Chem. 2011, 9, 7606. 10.1039/c1ob05946g. [DOI] [PubMed] [Google Scholar]
- Tabor A. B. Bioorg. Chem. 2014, 55, 39. 10.1016/j.bioorg.2014.04.004. [DOI] [PubMed] [Google Scholar]
- Ross A. C.; Liu H.; Pattabiraman V. R.; Vederas J. C. J. Am. Chem. Soc. 2010, 132, 462. 10.1021/ja9095945. [DOI] [PubMed] [Google Scholar]
- Liu W.; Chan A. S. H.; Liu H.; Cochrane S. A.; Vederas J. C. J. Am. Chem. Soc. 2011, 133, 14216. 10.1021/ja206017p. [DOI] [PubMed] [Google Scholar]
- Middleton R. E.; Warren V. A.; Kraus R. L.; Hwang J. C.; Liu C. J.; Dai G.; Brochu R. M.; Kohler M. G.; Gao Y.-D.; Garsky V. M.; Bogusky M. J.; Mehl J. T.; Cohen C. J.; Smith M. M. Biochemistry 2002, 41, 14734. 10.1021/bi026546a. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Bingham J.-P.; Zhu W.; Moczydlowski E.; Liang S.; Cummins T. R. J. Biol. Chem. 2008, 283, 27300. 10.1074/jbc.M708447200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray J. K.; Ligutti J.; Liu D.; Zou A.; Poppe L.; Li H.; Andrews K. L.; Moyer B. D.; McDonough S. I.; Favreau P.; Stöcklin R.; Miranda L. P. J. Med. Chem. 2015, 58, 2299. 10.1021/jm501765v. [DOI] [PubMed] [Google Scholar]
- Shcherbatko A.; Rossi A.; Foletti D.; Zhu G.; Bogin O.; Casas M. G.; Rickert M.; Hasa-Moreno A.; Bartsevich V.; Crameri A.; Steiner A. R.; Henningsen R.; Gill A.; Pons J.; Shelton D. L.; Rajpal A.; Strop P. J. Biol. Chem. 2016, 291, 13974. 10.1074/jbc.M116.725978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akondi K. B.; Muttenthaler M.; Dutertre S.; Kaas Q.; Craik D. J.; Lewis R. J.; Alewood P. F. Chem. Rev. 2014, 114, 5815. 10.1021/cr400401e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrat G.; Darbon H. Toxin Rev. 2005, 24, 361. 10.3109/15569540509162659. [DOI] [Google Scholar]
- Schmalhofer W. A.; Calhoun J.; Burrows R.; Bailey T.; Kohler M. G.; Weinglass A. B.; Kaczorowski G. J.; Garcia M. L.; Koltzenburg M.; Priest B. T. Mol. Pharmacol. 2008, 74, 1476. 10.1124/mol.108.047670. [DOI] [PubMed] [Google Scholar]
- Cox J. J.; Reimann F.; Nicholas A. K.; Thornton G.; Roberts E.; Springell K.; Karbani G.; Jafri H.; Mannan J.; Raashid Y.; Al-Gazali L.; Hamamy H.; Valente E. M.; Gorman S.; Williams R.; McHale D. P.; Wood J. N.; Gribble F. M.; Woods C. G. Nature 2006, 444, 894. 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Wang Y.; Li S.; Xu Z.; Li H.; Ma L.; Fan J.; Bu D.; Liu B.; Fan Z.; Wu G.; Jin J.; Ding B.; Zhu X.; Shen Y. J. Med. Genet. 2004, 41, 171. 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques S. T.; Deplazes E.; Lawrence N.; Cheneval O.; Chaousis S.; Inserra M.; Thongyoo P.; King G.; Mark A. E.; Vetter I.; Craik D. J.; Schroeder C. I. J. Biol. Chem. 2016, 291, 17049. 10.1074/jbc.M116.729095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. H.; Carlin K. P.; Wu G.; Ilyin V. I.; Musza L. L.; Blake P. R.; Kyle D. J. J. Med. Chem. 2014, 57, 6623. 10.1021/jm500687u. [DOI] [PubMed] [Google Scholar]
- Smith J. J.; Cummins T. R.; Alphy S.; Blumenthal K. M. J. Biol. Chem. 2007, 282, 12687. 10.1074/jbc.M610462200. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Blumenthal K.; Jackson J. O.; Liang S.; Cummins T. R. Mol. Pharmacol. 2010, 78, 1124. 10.1124/mol.110.066332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flinspach M.; Xu Q.; Piekarz A. D.; Fellows R.; Hagan R.; Gibbs A.; Liu Y.; Neff R. A.; Freedman J.; Eckert W. A.; Zhou M.; Bonesteel R.; Pennington M. W.; Eddinger K. A.; Yaksh T. L.; Hunter M.; Swanson R. V.; Wickenden A. D. Sci. Rep. 2017, 7, 39662. 10.1038/srep39662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuis J. R.; Dekan Z.; Wingerd J. S.; Smith J. J.; Munasinghe N. R.; Bhola R. F.; Imlach W. L.; Herzig V.; Armstrong D. A.; Rosengren K. J.; Bosmans F.; Waxman S. G.; Dib-Hajj S. D.; Escoubas P.; Minett M. S.; Christie M. J.; King G. F.; Alewood P. F.; Lewis R. J.; Wood J. N.; Vetter I. Sci. Rep. 2017, 7, 40883. 10.1038/srep40883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen S. L.; Tofteng A. P.; Malik L.; Jensen K. J. Chem. Soc. Rev. 2012, 41, 1826. 10.1039/C1CS15214A. [DOI] [PubMed] [Google Scholar]
- Machauer R.; Waldmann H. Chem. - Eur. J. 2001, 7, 2933.. [DOI] [PubMed] [Google Scholar]
- Mothia B.; Appleyard A. N.; Wadman S.; Tabor A. B. Org. Lett. 2011, 13, 4216. 10.1021/ol201548m. [DOI] [PubMed] [Google Scholar]
- Albericio F.; Cases M.; Alsina J.; Triolo S. A.; Carpino L. A.; Kates S. A. Tetrahedron Lett. 1997, 38, 4853. 10.1016/S0040-4039(97)01011-3. [DOI] [Google Scholar]
- Wilson-Stanford S.; Kalli A.; Håkansson K.; Kastrantas J.; Orugunty R. S.; Smith L. Appl. Environ. Microbiol. 2009, 75, 1381. 10.1128/AEM.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson T.; Quibell M.; Sheppard R. C. J. Pept. Sci. 1995, 1, 11. 10.1002/psc.310010104. [DOI] [PubMed] [Google Scholar]
- Postma T. M.; Albericio F. Org. Lett. 2013, 15, 616. 10.1021/ol303428d. [DOI] [PubMed] [Google Scholar]
- Park J. H.; Carlin K. P.; Wu G.; Ilyin V. I.; Kyle D. J. J. Pept. Sci. 2012, 18, 442. 10.1002/psc.2407. [DOI] [PubMed] [Google Scholar]
- Annis I.; Hargittai B.; Barany G. Methods Enzymol. 1997, 289, 198. 10.1016/S0076-6879(97)89049-0. [DOI] [PubMed] [Google Scholar]
- Steiner A. M.; Bulaj G. J. Pept. Sci. 2011, 17, 1. 10.1002/psc.1283. [DOI] [PubMed] [Google Scholar]
- Khoo K. K.; Gupta K.; Green B. R.; Zhang M.-M.; Watkins M.; Olivera B. M.; Balaram P.; Yoshikami D.; Bulaj G.; Norton R. S. Biochemistry 2012, 51, 9826. 10.1021/bi301256s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Q.; Lu S.-H.; Gu X.-C.; Liang S.-P. Protein Sci. 2002, 11, 245. 10.1110/ps.30502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalassinos K.; Grabenauer M.; Slade S. E.; Hilton G. R.; Bowers M. T.; Scrivens J. H. Anal. Chem. 2009, 81, 248. 10.1021/ac801916h. [DOI] [PubMed] [Google Scholar]
- Santos L. F.; Iglesias A. H.; Pilau E. J.; Gomes A. F.; Gozzo F. C. J. Am. Soc. Mass Spectrom. 2010, 21, 2062. 10.1016/j.jasms.2010.08.017. [DOI] [PubMed] [Google Scholar]
- Scarff C. A.; Thalassinos K.; Hilton G. R.; Scrivens J. H. Rapid Commun. Mass Spectrom. 2008, 22, 3297. 10.1002/rcm.3737. [DOI] [PubMed] [Google Scholar]
- Murray J. K.; Ligutti J.; Liu D.; Zou A.; Poppe L.; Li H.; Andrews K. L.; Moyer B. D.; McDonough S. I.; Favreau P.; Stöcklin R.; Miranda L. P. J. Med. Chem. 2015, 58, 2299. 10.1021/jm501765v. [DOI] [PubMed] [Google Scholar]
- Pulka-Ziach K.; Pavet V.; Chekkat N.; Estieu-Gionnet; Rohac R.; Lechner M.-C.; Smulski C.; Zeder-Lutz G.; Altschuh D.; Gronemeyer H.; Fournel S.; Odaert B.; Guichard G. ChemBioChem 2015, 16, 293. 10.1002/cbic.201402485. [DOI] [PubMed] [Google Scholar]
- Upert G.; Mourier G.; Pastor A.; Verdenaud M.; Alili D.; Servent D.; Gilles N. Chem. Commun. 2014, 50, 8408. 10.1039/C4CC02679A. [DOI] [PubMed] [Google Scholar]
- Salamanca S.; Chang J.-Y. Protein J. 2006, 25, 275. 10.1007/s10930-006-9011-x. [DOI] [PubMed] [Google Scholar]
- Chang J.-Y.; Li L. Arch. Biochem. Biophys. 2005, 437, 85. 10.1016/j.abb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Lajoie D. M.; Roberts S. A.; Zobel-Thropp P. A.; Delahaye J. L.; Bandarian V.; Binford G. J.; Cordes M. H. J. J. Biol. Chem. 2015, 290, 10994. 10.1074/jbc.M115.636951. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.