

Abstract
Abscission is the last step of cytokinesis that physically separates the cytoplasm of sister cells. As the final stage of cell division, abscission is poorly characterized during animal development. Here, we show that Aurora B and Survivin regulate the number of germ cells in each Drosophila egg chamber by inhibiting abscission during differentiation. This inhibition is mediated by an Aurora B-dependent phosphorylation of Cyclin B, as a phosphomimic form of Cyclin B rescues premature abscission caused by a loss-of-function of Aurora B. We show that Cyclin B localizes at the cytokinesis bridge, where it promotes abscission. We propose that mutual inhibitions between Aurora-B and Cyclin-B regulate the duration of abscission and thereby the number of sister cells in each cyst. Finally, we show that inhibitions of Aurora B and Cdk-1 activity in vertebrate cells also have opposite effects on the timing of abscission, suggesting a possible conservation of these mechanisms.
Keywords: cytokinesis, abscission, oogenesis, gametogenesis, cell cycle, development, stem cell, Drosophila, HeLa, Cdk-1, CPC
Graphical abstract
INTRODUCTION
During animal development, the canonical cell cycle is modulated and adapted in different cell types. How specific developmental programs alter different steps of the cell cycle remains to be understood in most cases. In this respect, the last stages of cell division, when daughter cells become separated, are probably the most diverse, but also the least explored. In sea urchin embryos, the timing of cytokinesis is shifted, and the completion of cell division only occurs during the S phase of the next cycle (Sanger et al., 1985). Cytokinesis altogether is absent during megakaryocyte differentiation, and can also be arrested at a late stage in spermatocytes of most species (Pepling et al., 1999; Vitrat et al., 1998). Cytokinesis starts by the specification of a cleavage plane and is followed by the ingression of an actomyosin contractile ring. During this transition, the mitotic spindle rearranges at the midzone to form an electron-dense structure known as the midbody, at the center of the intercellular bridge. Daughter cells then become physically separated, by a process called abscission. It remains unclear what regulates the timing of abscission during animal development, which can vary from minutes to hours, or can even stay incomplete, as in germ cells (Pepling et al., 1999).
The timing and order of the cell cycle is driven by oscillations in the activities of conserved cyclin-dependent kinases (Cdk). A high Cdk-1 activity, mostly driven by CyclinB/Cdk-1 in higher eukaryotes, allows the cell to enter and perform mitosis (Malumbres and Barbacid, 2009). Rising levels of Cyclin B and its translocation into the nucleus prepare cells to enter M phase (Gavet and Pines, 2010a, b). CycB/Cdk-1 then becomes fully active when inhibitory phosphorylations by Wee1 and Myt1 are removed by Cdc25 phosphatase on Cdk-1 activating sites (Lindqvist et al., 2009). Exit from mitosis is then initiated by the degradation of Cyclin B in metaphase, when the spindle assembly checkpoint is satisfied (Clute and Pines, 1999; Sigrist et al., 1995; Sullivan and Morgan, 2007). Consequently, CycB/Cdk-1 activity remains low until the next G1 phase. However, it is unknown whether these low levels of CycB/Cdk-1 have any function after anaphase (Wurzenberger and Gerlich, 2011). The sharp changes in CycB/Cdk-1 activity are due to positive and negative feedback loops between Cdk-1 and the kinases/phosphatases mentioned above. Additional families of kinases, such as Polo (Plk) and Aurora (Aur), impinge on these loops to further regulate CycB/Cdk-1 activity and to order multiple events downstream of the core regulators (Lindqvist et al., 2009). Among those, Aurora B (AurB) is known to regulate chromosome orientation and attachment to the mitotic spindle at metaphase. AurB is the active kinase of a biochemical complex, named the Chromosomal Passenger Complex (CPC). This complex is highly conserved in many species and also contains the Survivin, Incenp and Borealin subunits, which regulate AurB localization and kinase activity (Ruchaud et al., 2007). In contrast to Cdk-1, Polo and Aur B are known to play later functions during cell division, as they translocate from centromeres to the spindle midzone to participate in the early steps of cytokinesis, such as the ingression of the furrow (Adams et al., 2001; Burkard et al., 2007; Gruneberg et al., 2004; Neef et al., 2003; Petronczki et al., 2007; Terada et al., 1998). The translocation of Aurora B to the midzone is, however, inhibited while CycB/Cdk-1 activity is still high at the metaphase-anaphase transition (Parry et al., 2003). Indeed, recent results showed that direct phosphorylation of the CPC by Cdk-1 targets Aurora B to the inner centromeres and prevents its localization to the spindle midzone through Mklp2 (Hummer and Mayer, 2009; Tsukahara et al., 2010). At the midbody, Aurora B was shown recently to have an additional function near the very end of cytokinesis, during abscission. Studies in yeast and human cells demonstrated that Aurora B delays completion of cytokinesis until chromosomes are well separated after anaphase (Norden et al., 2006; Steigemann et al., 2009). This checkpoint, named the NoCut pathway, prevents lagging DNA to be cut by the cleavage furrow in order to avoid chromosome breakage. Aurora B thus has two different functions during cytokinesis, as it allows furrow ingression during the early steps, but can also delay the completion of cytokinesis. It remains, however, unknown whether Aurora B also regulates abscission in vivo during normal animal development, and if Aurora B acts at this stage with other mitotic kinases, such as Plks and Cdks.
Cytokinesis is blocked in germ cells of most species during at least some stage of their normal development (Pepling et al., 1999). A classic example is the Drosophila egg chamber, which is a syncytium of 16 cells produced by four rounds of mitosis of a single precursor, called a cystoblast (CB) (Huynh and St Johnston, 2004). Cystoblasts are produced throughout the life of adult females, by germline stem cells (GSCs) located at the anterior tip of each ovary in the germarium (Fig. 1A). Each stem cell self-renews by dividing asymmetrically to generate one stem cell, which stays in contact with support cells in the niche and receives signals that prevent differentiation (Chen et al., 2011). The second daughter cell is positioned outside of the niche, does not receive these signals, and thus starts to transcribe the bam gene, which is necessary and sufficient to trigger the transcription program of the cystoblast. This differentiation is characterized by four rounds of synchronous divisions, which form a 16-cell cyst, made of 15 nurse cells and one oocyte. In the resulting cyst, each cytokinesis is arrested and all sister cells share the same cytoplasm through ring canals. In contrast, cytokinesis between the GSC and the CB is complete. It is, however, very slow, and GSCs and CBs remain synchronized until abscission is completed during the G2 phase of the next cycle, about 24h later (de Cuevas and Spradling, 1998). How abscission is regulated differently in GSCs and CBs is unknown. The orientation and synchrony of these divisions is controlled by a germline-specific organelle, called the fusome, which is made of ER-derived vesicles (Huynh, 2005). The fusome is partly inherited from the spectrosome of the GSCs (also made of ER-derived vesicles), and partly newly formed at the midbody during each division. Fusion between fusome precursors creates a continuum of vesicles going through each canal and connecting all the cells within a cyst (de Cuevas and Spradling, 1998; Snapp et al., 2004). Interestingly, cell cycle regulators such as Cyclin A, Cyclin E and subunits of the proteasome localize on the fusome, which may help to synchronize their activation and destruction in all cells (Lilly et al., 2000; Lilly and Spradling, 1996; Ohlmeyer and Schupbach, 2003). The pattern of divisions is invariant with 8 cells with one ring canal, 4 cells with two, 2 cells with three and 2 cells with four. This pattern is important, as the oocyte always differentiates from one of the two cells with four ring canals, which are called the pro-oocytes (Spradling, 1993b). Incidentally, the number of ring canals in each cell can be used as a marker for the number of divisions (Fig. 1A).
Figure 1. Identification of survivin and aurora-B loss of function alleles.
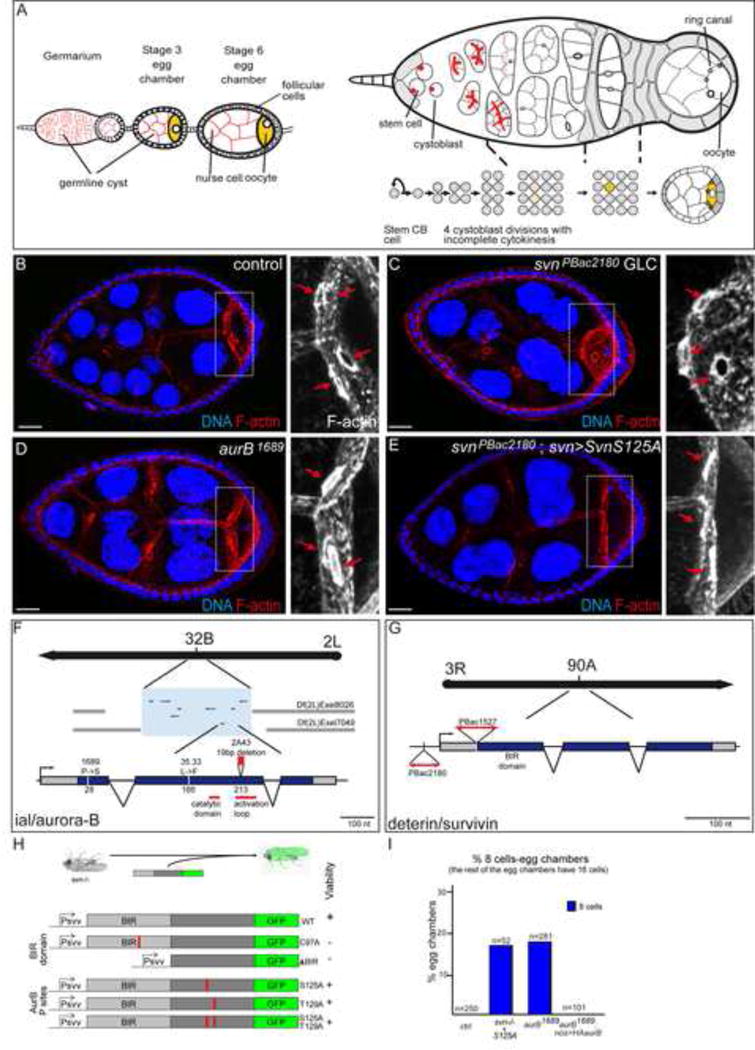
(A) Scheme showing an ovariole with a germarium linked to 2 growing egg chambers (left), and a close up on a germarium (right). The egg chambers are formed of 16 germline cells, 15 nurse cells and one oocyte (in yellow), surrounded by a follicular epithelium. The egg chamber matures from germline stem cell to germline cyst (left to right), and encapsulation of the cyst by follicular cells in the germarium. At the anterior tip of the germarium, the germline stem cell divides asymmetrically and produces a cystoblast (CB). The spectrosome in the GSC and the fusome in its progeny (in red in the right scheme) are germline specific organelles. Anterior is on the left, posterior on the right.
(B–E) Stage 7 egg chambers from WT, svnPBac2180 GLC, ial1689 or svnPBac2180; svn>SvnS125A females, stained with DAPI (DNA) and phalloidin (F-actin). On the right, close up on oocytes. Red arrows indicate the 4 ring canals in the control oocyte, and the 3 ring canals in the mutant backgrounds.
(F–G) Mapping of the ial/aur-B and deterin/svn alleles.
(H) Structure of the svn transgenes generated, and their ability to rescue the viability of the svnPBac2180 mutants.
(I) Fraction of egg chambers exhibiting less than 16 cells (8 cells here) on the Y axis. Genotypes are on the X axis.
RESULTS
1) Loss-of-function mutations in aurora B and survivin reduce the number of germ cells per egg chamber
We identified two complementation groups of several alleles affecting the early steps of germ cell development in Drosophila ovaries (see Supplementary Experimental procedures for details). In hypomorphic conditions, mutant egg chambers were made of 8 germ cells instead of 16, and the oocyte was linked to only three nurse cells by three ring canals instead of four (Fig. 1B–D). These results suggested that these mutant egg chambers had gone through three divisions instead of four. In strong loss-of-function conditions, induced using the FLP/FRT system, we observed the formation of giant GSCs filling the entire germarium (Fig. S2A–C). These mutant cells had highly polyploid nuclei, enlarged spectrosomes and did not come out of the germarium.
The first complementation group was made of three EMS-induced alleles called 2A43, 35.33 and 1689. We genetically mapped these mutations to the ial (Ipl1-aurora-like kinase) locus, which encodes the Drosophila homolog of Aurora B (the locus is referred as aurB hereafter) (Fig. 1F). 2A43 and 35.33 are the strongest alleles and both are homozygous lethal. They correspond to alterations in the most conserved part of the protein, with a Leucine to Phenylalanine substitution at position 166 (35.33) and a frame shift followed by a truncation in the kinase activation loop (2A43) (Fig. 1F). In contrast, 1689 is a hypomorphic and viable allele, corresponding to a Proline to Serine substitution in the non-conserved N-terminal part of the protein (Fig. 1F). The second complementation group is made of two PiggyBac insertions, that we generated (PBac2180, (Mathieu et al., 2007)) or found in public stock collection (PBac{RB}e01527). Both transposons are inserted in the 5′ regulatory region of the deterin locus (also called dSurvivin), which encodes the Drosophila homolog of Survivin, referred as svn hereafter (Fig. 1G)(Jones et al., 2000). Both alleles produced very little svn RNA (data not shown) and were homozygous lethal at the pupal stage. We confirmed that all phenotypes were only due to the lack of aurB or svn, as wild type genomic transgenes encoding aurora B or svn could rescue viability and mitotic phenotypes (Fig. S1D,E). Aurora B and Survivin are known to be part of the highly conserved Chromosomal Passenger Complex (CPC), and as expected from the well-described roles of the CPC in mitosis, highly polyploid nuclei were not specific to mutant germ cells, but were found in any dividing cell types, such larval neural stem cells (Fig. S2D, E and Fig S2F–M).
We took advantage of the absence of endogenous Svn in our strong loss-of-function mutants to perform a structure/function analysis. We generated wild type and mutant GFP-tagged transgenes based on a genomic rescue construct containing the endogenous promoter and regulatory sequences; and we tested their ability to rescue viability, ploidy and the number of germ cells per cyst (Fig. 1H). We found that a wild type form of Svn could rescue perfectly viability and diploidy. It also localized as expected at centromeres during prophase and prometaphase, and at the midzone and intercellular bridge during later mitosis (Fig. S1A–C). In contrast, variants of Svn with a defective BIR domain were not able to rescue viability (Fig. 1H), consistent with previous studies in cell culture (Lens et al., 2006; Yue et al., 2008). It has been proposed that Survivin could be phosphorylated by Aurora B in vitro on T117 in human cells (Wheatley et al., 2004). We found two residues (S125 and T129) fitting the consensus of phosphorylation by Aurora B in Drosophila Survivin. However, they both appeared dispensable for fly viability. Indeed, non-phosphorylatable transgenes (SvnS125A and SvnT129A either mutated together or separately) rescued localization (not shown), viability and diploidy to wild type level (Fig. 1H and S2N). However, they did not rescue the number of germ cells per egg chambers (Fig. 1E,I). Both svn−/−; svn>SvnS125A and svn−/−; svn>SvnS125A,T129A females produced around 20% (17%; n=52) of egg chambers with only 8 cells, as we observed in hypomorphic aurB1689 mutant flies (18%; n=281) (Fig.1I). The remaining cysts had 16 cells as in wild type condition. Furthermore, svn−/−; svn>SvnWT females were perfectly rescued: 100% of cysts with 16 cells, n=115. svn−/−; svn>SvnS125A flies thus correspond to a weak loss-of-function condition. We concluded that our strong loss-of-function alleles of aurB and svn recapitulated the known functions of the CPC in a multicellular organism, while the weaker alleles revealed a developmental requirement of the CPC in the regulation of the number of germ cells per egg chamber.
2) Gain-of-function mutations in aurora B and survivin increase the number of germ cells per egg chamber
Next we performed the converse experiment by overexpressing wild type forms of Svn or AurB using the nanos-Gal4 driver (nos>), which is expressed specifically in all germ cells of the germarium (Fig. 2E). We found that in these nos>Svn or nos>AurB females, a significant number of egg chambers had 32 cells and an oocyte with 5 ring canals, suggesting that they resulted from one extra division (Fig. 2A,B,G). As a control experiment, overexpression of Aurora-A with the same driver did not induce any extra germ cell (Fig. 2G). To mimic constitutive phosphorylation of Svn at the AurB consensus site, we overexpressed a SvnS125E form. We observed a dramatic increase in both the number of germ cells per cyst and the penetrance of the phenotype (Fig. 2C,G). Almost 50% of nos>SvnS125E egg chambers had 32 cells or even more, as we found oocytes with 6 ring canals (Fig. 2C,G). Furthermore, an insertion of SvnS125E expressed at a low level gave the same percentage of extra germ cells than a strongly expressed wild type Svn, indicating that SvnS125E is more potent than Svn (Fig. 2D,G). Importantly, the activity of SvnS125E still depended on the CPC, as removing one copy of the endogenous svn, aurB or Incenp gene, partially suppressed the extra-germ cells phenotype (Fig. S3A). These results indicated that SvnS125E fulfills the genetic definition of a hypermorphic allele of the CPC in our system. We thus concluded that loss-of-function of the CPC led to less germ cells per cyst, while gain-of-function of the CPC increased the number of germline cells per cyst.
Figure 2. Svn and the CPC regulate the number of germ cell per egg chamber.
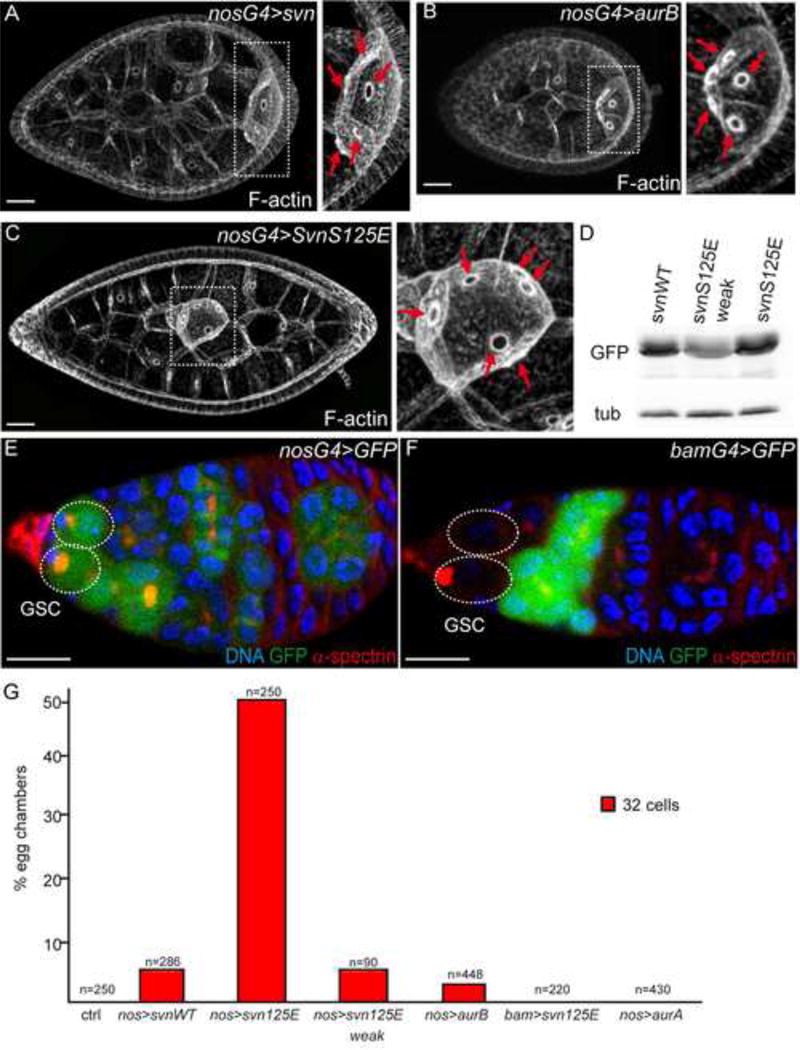
(A–C) Stage 7 egg chambers of females expressing SvnWT-GFP (A), HA-AurB (B) or SvnS125E-GFP (C) under the control of the nanos-Gal4 driver (nosG4) stained with phalloidin. On the right, close up on the oocyte. Red arrows indicate the 5, 5 and 6 oocyte ring canals in SvnWT-GFP (A), HA-AurB (B) or SvnS125E-GFP (C) respectively.
(D) Ovaries from females expressing Svn-GFP variants, WT or S125E, were processed for Western blot analyses. Two insertions of SvnS125E-GFP (2nd and 3rd lanes) are shown.
(E–F) Germaria of females expressing GFP under the control of the nanos-Gal4 (E, nosG4) and bam-Gal4 (F, bamG4) drivers, stained with DAPI and α-spectrin. The Germline Stem Cells (GSC, surrounded by dotted lines) are GFP positive with nosG4, not with bamG4.
(G) Fraction of egg chambers exhibiting more than 16 cells on the Y axis. Genotypes are on the X axis.
We reasoned that if extra germ cells were caused by additional mitoses in the cyst, one would expect the phenotype to be autonomous to the cyst. We thus expressed SvnS125E only in dividing germline cysts using the bam-Gal4 driver (bam>), which is expressed from cystoblasts to 8-cell cysts, but not in GSCs (Fig. 2F). To our surprise, we did not detect any extra germ cells (Fig. 2G). Our results strongly suggested that it was the activity of SvnS125E in cells expressing nanos but not bam, i.e. mainly GSCs and some pre-cystoblasts, which induced extra germ cells in the cyst.
3) Gain-of-function of the CPC leads to the formation of stem-cysts
How might stem cells regulate the number of divisions of their daughter cells? To address this question, we performed live-imaging of GSCs expressing nos>SvnS125E-GFP. Surprisingly, these GSCs were dividing synchronously with several neighboring germ cells (Fig. 3A, Movie 3, n=9/10), suggesting that they may be physically connected, which is never seen in a wild type situation (n>50 in WT, Fig.S7, Movie7). We then analyzed the spectrosome in these GSCs and found that spectrosomes were branched as fusomes, and linked GSCs with up to 8 other germ cells (44,7%, n=85) (Fig. 3B). These results pointed to an arrest of cytokinesis and showed that these cells were connected by a fusome, which could explain their synchrony in mitosis. To characterize whether these groups of cells were stem cells or cyst cells, we used several markers of identity. These clusters did not express Bam, a differentiation marker of the cyst (Fig. 3C,D), but weakly expressed Nanos like in stem cells (Fig. 3E,F) (Gilboa and Lehmann, 2004; McKearin and Ohlstein, 1995). Furthermore, only the cell in direct contact with niche cells was positive for p-Mad, a reporter of the Dpp pathway activation (Fig. 3G,H) (Song et al., 2004). These synchronous cells thus had characteristics of both stem cells and cyst cells; we thus named them “stem-cysts”. Stem-cysts may also represent an intermediate state of differentiation as postulated previously (Gilboa et al., 2003).
Figure 3. SvnS125E generates stem cysts.
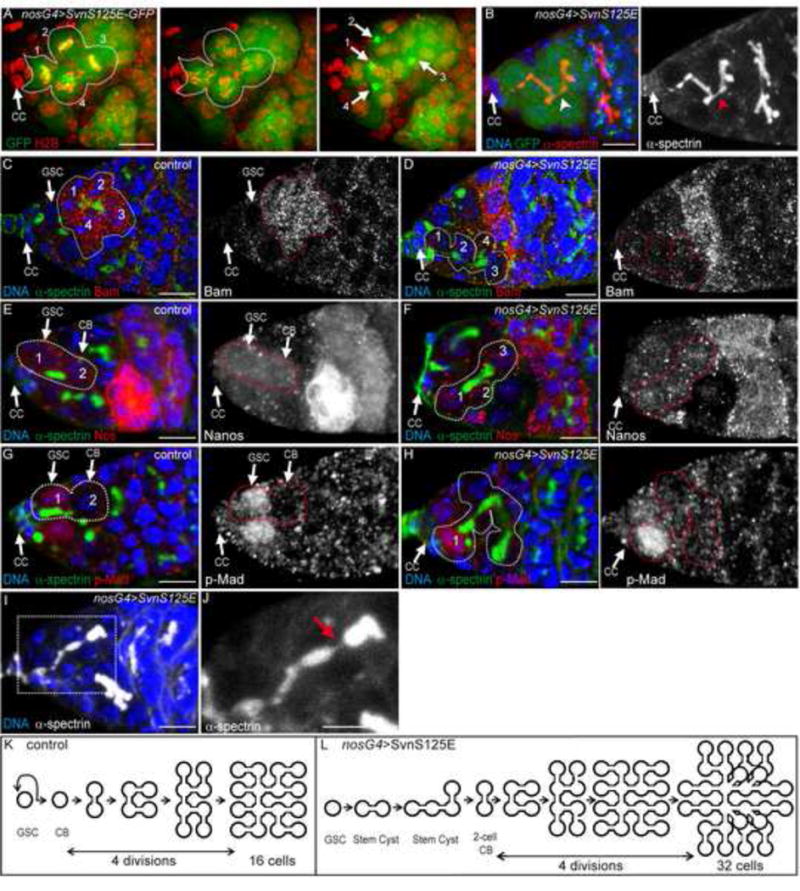
(A) H2B-RFP in red and SvnS125E-GFP in green. One GSC (attached to the Cap Cells) and 3 neighboring cells undergo mitosis synchronously.
(B) Expression of SvnS125E-GFP with nanos-Gal4 induces the formation of branched fusome (α-spectrin, red) in GSC attached to CC.
(C–H) Germaria of a WT females (C, E, G) or females expressing SvnS125E under the control of nanos-Gal4 driver (nosG4), stained for α-spectrin (green), and either Bag of marble (Bam C,D, red), Nanos (E–F, red), or p-Mad (G, H, red).
(I–J) Germarium expressing nos>SvnS125E. A branched fusome (white) is breaking at the posterior part (close up in J). The red arrow indicates a thin thread of α-spectrin, probably the breaking point.
(K–L) Schemes representing the mitotic events in WT (K) and in SvnS125E (L) expressing germaria. (K)
These stem-cysts did not grow indefinitely, however, and we observed that their fusome eventually broke down (Fig. 3I, J, arrow), indicating that SvnS125E delayed abscission, but did not block it completely We thus hypothesized that complete abscission of the oldest links in stem cysts will generate “cystoblast-like” precursors made of two or more cells. In this model, a two-cell cystoblast will go through the regular four mitoses and give rise to a 32-cell cyst indistinguishable from a 32-cell cyst generated by 5 rounds of division of a single-cell precursor (Fig. 3K,L). We thus proposed that 32-cell cysts did not come from extra mitoses in the cyst, but that they could be generated by an abscission delay in GSCs and the formation of two-cell cystoblasts.
4) The CPC regulates abscission in the germline stem cell lineage
The CPC is known to be required for furrow ingression during cytokinesis, but an excess of its activity later on at the midbody was recently proposed to delay abscission (Norden et al., 2006; Steigemann et al., 2009). In the germline, we noticed that Svn localized on the fusome, which forms at the midbody in both GSCs and differentiated cysts (Fig. 5A). We further observed that SvnS125E localized like Svn on the fusome (Fig. 4A). We reasoned that this concentration of SvnS125E at the fusome/midbody may cause the abscission delay in GSCs. To test this idea, we expressed nos>SvnS125E in hts mutant ovaries, which totally lack a fusome (Yue and Spradling, 1992), and counted the number of synchronous cells dividing, one being attached to the niche. On average, we found that 3 cells were dividing simultaneously in nos>SvnS125E stem-cysts. In the absence of fusome, we mostly found single GSC dividing like in the wild type control (Fig. 4B–E and Fig.S7, Movie7). We thus concluded that the fusome played a key role in the formation of stem-cysts and the inhibition of abscission by SvnS125E.
Figure 5. Aurora-B is required for phosphorylation of Cyclin B during mitosis.
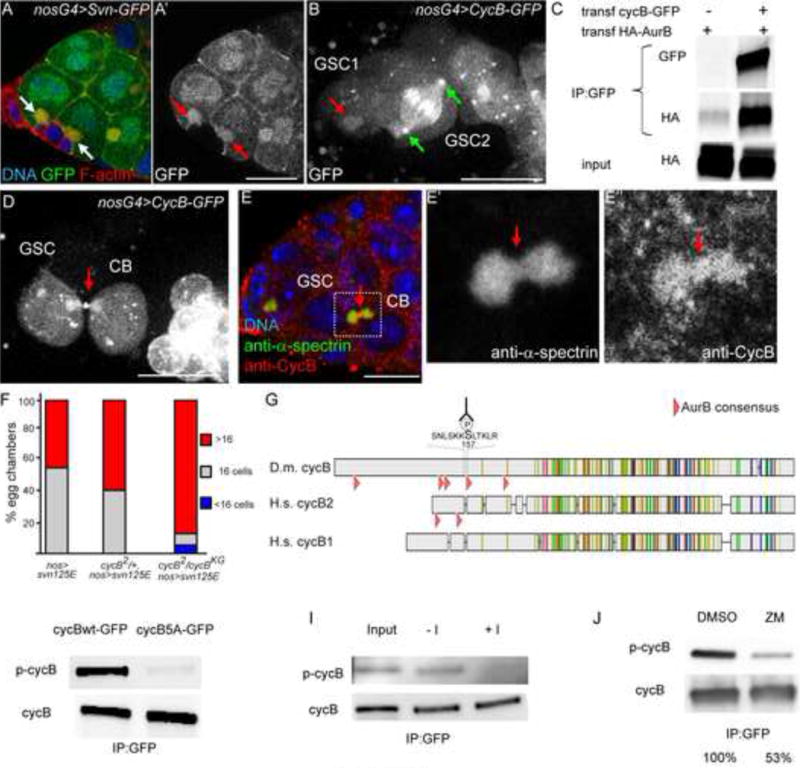
(A) Germarium expressing nos>SvnWT-GFP stained with phalloidin (red) and DAPI (blue). GFP is in green. The spectrosome (arrows white in A, red in A′) is enriched in F-actin and SvnWT-GFP.
(B) Germarium expressing nos>CycB-WT-GFP, which localizes on the spectrosome (red arrow in GSC1), and centrosomes in metaphase (green arrows in GSC2).
(C) HEK293T cells were transfected with HA-AurB alone or with cycB-WT-GFP expressing vectors, and were processed for immuno-precipitation (IP) with anti-GFP antibody, and western blot analyses, with the GFP and HA antibodies. HA-AurB and CycB-GFP co-immunoprecipitate.
(D) Germarium expressing nos>CycB-WT-GFP. Note the presence of GFP at the midbody linking the GSC and its CB (red arrow).
(E) Germarium of a WT female stained with anti-α-spectrin (green in overlay) and anti-CyclinB (red in overlay) antibodies. Endogenous CycB localizes on the fusome and at the midbody (red arrows in E′ and E″).
(F) Fraction of egg chambers exhibiting less than 16 cells (blue), 16 cells (grey) and more than 16 cells (red) on the Y axis. Genotypes are on the X axis. The penetrance of the SvnS125E phenotype (more than 16 cells) is enhanced by a reduction in CycB level.
(G) Alignment of Drosophila melanogaster Cyclin B with human Cyclin B1 and Cyclin B2. The pink arrowheads indicate Aurora-B consensus site of the N-terminal regions of the different Cyclins. The sequence of the phospho-peptide used to generate the phospho-specific Cyclin B antibody is shown (the p-Serine is indicated by P)
(H) Embryos expressing CycB-WT-GFP or CycB-5E-GFP were processed for immuno-precipitation (IP) and western blot analyses, with the p-CycB antibody or total CycB. CycB-5A is poorly recognized by the anti p-CycB antibody.
(I) Embryos expressing CycB-WT-GFP were processed for immuno-precipitation (IP) and λ-phosphatase treatment, followed by western blot analyses, with the p-CycB antibody or total CycB. Upon λ-phosphatase treatment, the p-CycB positive band is not detected anymore.
(I) HEK293T cells transfected with CycB-WT-GFP expression vector were treated with ZM447439 or DMSO as a control, processed for immuno-precipitation (IP), and analyzed by western blot, with anti- p-CycB antibody or anti-CycB. p-CycB level is diminished upon ZM447439 treatment. The percentages represent the amount of p-cycB detected relative to the one in DMSO treated cells.
Scale bar: 10 μm. See also Figure S5
Figure 4. The CPC regulates abscission in the germline.
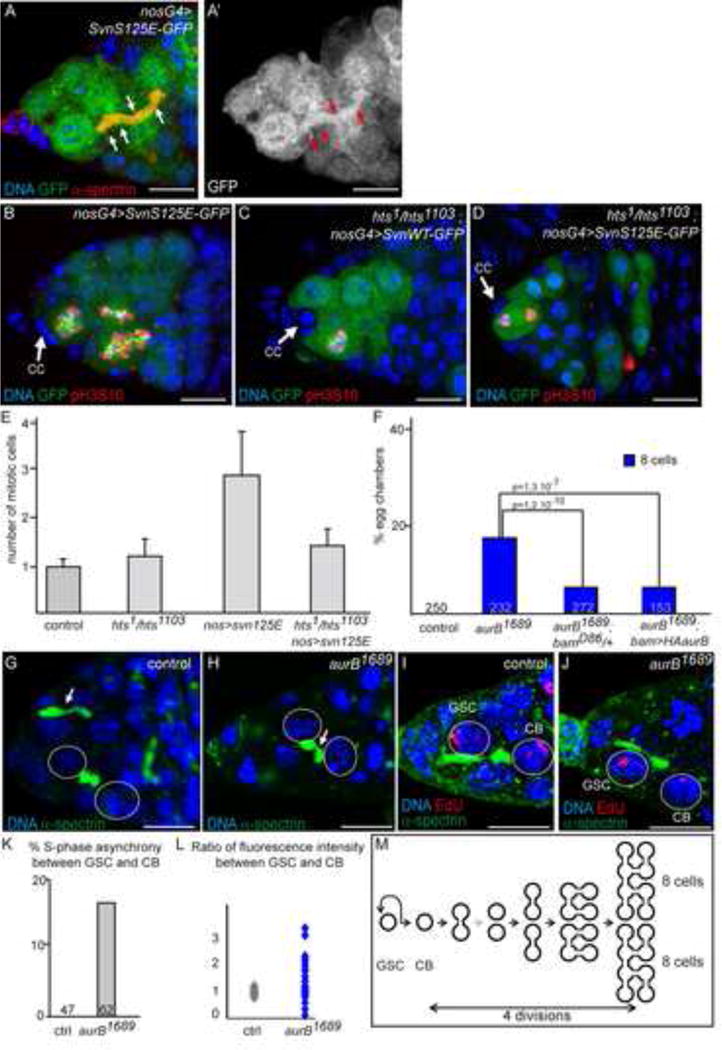
(A) Germarium expressing nos>SvnS125E. SvnS125E-GFP (green and white in A′) colocalizes with α-spectrin (red) on the fusome (arrows).
(B–D) Germaria expressing either nos>SvnS125E-GFP (C, E) or nos>SvnWT-GFP (D) in a WT (C) or hts mutant background (D, E), stained with pH3S10 (red) to highlight mitotic cells.
(E) Number of synchronously dividing cells, counted only when the most anterior is a GSC.
(F) Fraction of egg chambers exhibiting less than 16 cells on the Y axis. Genotypes are on the X axis. The penetrance of aurB1689 phenotype is rescued by a reduction in Bam level, or by addition of AurB in the cyst only (bam>AurB).
(G–H) Germaria of WT (H) and aurB1689 (I) females, stained for α-spectrin (green) and DNA (blue). (G) A snowman-shape fusome observed in a 2-cell cyst. Abscission is occuring between a GSC and its daughter CB (arrow). (H) Breaking fusome in a aurB1689 mutant 2-cell cyst. Arrows point to a thin thread of fusome.
(I–J) Germaria of a WT (J) and an aurB1689 (K) females, stained for Edu (red) and α-spectrin (green), showing S-phase synchrony between the GSC and its daughter CB in WT (J), but not in aurB1689 (K) females.
(K) Fraction of GSC/CB pair showing S-phase asynchrony on the Y axis, in %. Genotypes are on the X axis.
(L) GSC/CB ratios of EdU fluorescence in WT (grey) and in aurB1689 mutant (blue). S-phase synchrony is revealed by a ratio close to 1.
(M) Scheme representing the mitotic events and premature abscission in aurB1689 germaria.
Scale bar: 10 μm. Error bars are SD. See also Figure S7, Movie 7.
Overall, our results showed that the extra germ cells induced by a gain-of-function of the CPC are caused by an abscission delay in GSCs. We thus hypothesized that the reduction of germ cell number upon loss-of-function of the CPC may be caused by premature abscission in the cyst. In this model, complete abscission of a 2-cell cyst will give rise to two single cells carrying on to make the three remaining divisions, thus generating two cysts of 8 cells (Fig. 4M). In support of this model, we observed in hypomorphic aurB1689 mutants, 2-cell cysts linked by a thin thread of fusome, which indicates that abscission is about to be completed (Fig. 4G,H; arrow). These threads of fusome are never seen in wild type cysts, but have been described during de-differentiation experiments when cysts complete abscission and breaks into single cells (Kai and Spradling, 2004). We further noticed that 8-cell cysts were found in pairs at a frequency 3 times higher than expected if their distribution was random (0.048 observed vs 0.016 expected p < 0.005; see Supplementary Experimental procedures), suggesting that each pair of 8-cell cysts was derived from a single precursor. In addition, we found that removing one copy of bam strongly rescued aurB1689 phenotype (from 17% to 4% of 8-cell cysts), indicating that the phenotype is caused by the reduction of AurB activity in the cyst and not in the GSC (Fig. 4F). To validate this conclusion, we expressed a wild type AurB transgene driven by the bam promoter in an aurB1689 homozygous background and found a strong rescue of the 8-cell cyst phenotype (4%; n=153; Fig. 4F). Altogether, our results demonstrate that the CPC is required in the wild type cyst to inhibit abscission during the first mitosis of the cystoblast (i.e. at the 2-cell stage), and that abscission becomes precocious in aurB1689 mutant cysts. We further tested this conclusion in GSCs, which are known to remain connected and synchronous with their daughter CB until at least the G2 phase of the next cycle (de Cuevas and Spradling, 1998). Indeed, a 30 min pulse of EdU equally labels GSC and CB indicating that they replicated their DNA at the same time, and thus that they were still connected during S phase of the following cycle. As reported previously, we found that all pairs of wild type GSC and CB showed equal staining of EdU (100%, n=47; Fig. 4K,L) (de Cuevas and Spradling, 1998). In contrast, we found that GSC and CB were asymmetrically stained in 17% of aurB1689 mutant pairs (Fig. 4K,L; n=62), revealing asynchronous behaviors already before or during S phase and suggesting complete abscission. We concluded that a reduction in Aurora B activity induced a precocious abscission in both GSCs and germline cysts.
5) Aurora B is required for Cyclin B phosphorylation
Next we asked what the targets of Aurora B that regulate the timing of abscission might be. To find putative substrates, we performed immuno-precipitations of Svn-GFP, AurB-RFP and HA-AurB from embryos, and sequenced the interactors by mass-spectrometry. We repeatedly found peptides matching CycB sequence, which caught our attention as CycB was previously shown to regulate cell number of germline cysts (Lilly et al., 2000). We confirmed this interaction by co-immunoprecipitation of HA-AuroraB with Cyclin B-GFP (Fig. 5C). We generated a full length CycB-GFP transgene, which could rescue cycB null mutant flies, and found that it localized like Svn on the GSC spectrosome and cyst fusome during interphase (red arrow on GSC1; Fig. 5B). We analyzed its dynamic localization during mitosis and observed that it marked the centrosomes and kinetochores as expected (green arrows for the centrosomes; Fig. 5B). Interestingly, we noticed that a pool of CycB-GFP accumulated at the intercellular bridge during late cytokinesis (red arrow; Fig. 5D). A similar localization was found for endogenous CycB (Fig. 5E). Reducing the level of CycB greatly enhanced nos>SvnS125E gain-of-function phenotype, demonstrating a negative genetic interaction between the CPC and CycB (Fig. 5F). We also noticed 5 sites fitting the phosphorylation consensus of Aurora B in the N-terminal part of CycB. Two consensus sites are also present in the N-terminal region of human CycB2 (Fig. 5G). In order to examine whether Drosophila Cyclin B was phosphorylated in vivo, we generated an antibody against a phospho-peptide corresponding to one of these sites (pS157) (Fig. 5G). This antibody recognized CycB from wild type extracts, but not a non-phosphorylatable CycB-5A form, where all 5 sites were changed into Alanines (Fig. 5H). The Alanine substitutions, however, may not be recognized by our antibody. To further test the specificity of our antibody, we treated wild type ovarian extracts with lambda phosphastase. We found that after this treatment the wild type form of Cyclin B was not recognized anymore by our anti-pCycB (Fig. 5I), indicating that this antibody marked the phosphorylated form of CycB but not the unphosphorylated CycB (at least at S157). This result further demonstrated that the phosphorylated form of CycB at S157 existed in vivo in ovarian extracts. We next tested whether this phosphorylation event was dependent on Aurora B activity. We inhibited Aurora B with the widely used ZM447439 drug. We found a strong reduction of almost 50% in the pS157-CycB signal for equal amounts of total CycB (Fig. 5J). We concluded that this phosphorylation depended on Aurora B activity. Taken together our results demonstrate that AurB and CycB interact in vivo, that they both localize at the fusome and the midbody in the germline, and that phosphorylation of CycB at S157 depends on AurB activity.
6) Cyclin B phosphorylation regulates abscission in the germline stem cell lineage
In order to test in vivo the potential role of the AurB-dependent phosphorylation of Cyclin B, we generated transgenic flies expressing a phosphomimic form of CycB at all five sites (CycB-5E). We found that nos>CycB-5E phenocopied nos>SvnS125E flies with a high percentage of 32-cell cysts containing an oocyte with 5 ring canals (Fig. 6A, B). These 32-cell cysts were also generated by the formation of stem-cysts rather than a fifth division, as we found branched fusomes originating from GSC in 55% of germaria (n=47; Fig. 6C) and synchronous divisions in stem cells expressing CycB-5E (Fig. 6D, Movie 4), while these cells remained Bam negative (Fig. 6E). In addition, we co-expressed a photoactivable-GFP fused to α-tubulin, which could freely diffuse in the cytoplasm. We found that photoactivation with a 2-photon laser of any cell of a stem-cyst in G1/S phase (CycB-5E-GFP is absent in G1/S) led to a rapid diffusion of the fluorescence throughout the stem-cyst including the most anterior stem cell, which had not been activated (Fig. 6F, Movies 5 and 6). We concluded that these cells were sharing the same cytoplasm and that abscission was incomplete in nos>CycB-5E stem-cysts. Furthermore, expression of CycB-5E only in the cyst (bam>CycB-5E) had a wild type phenotype, as observed when SvnS125E is also driven by bam (Fig. 6B). In contrast, wild type CycB or non-phosphorylatable CycB-5A induced a significant number of 32-cell cysts when expressed in the cyst (driven by the bam promoter) but not driven by the nanos promoter, indicating instead a fifth division in the cyst (Fig. 6B).
Figure 6. Cyclin B phosphorylation regulates abscission in the germline.
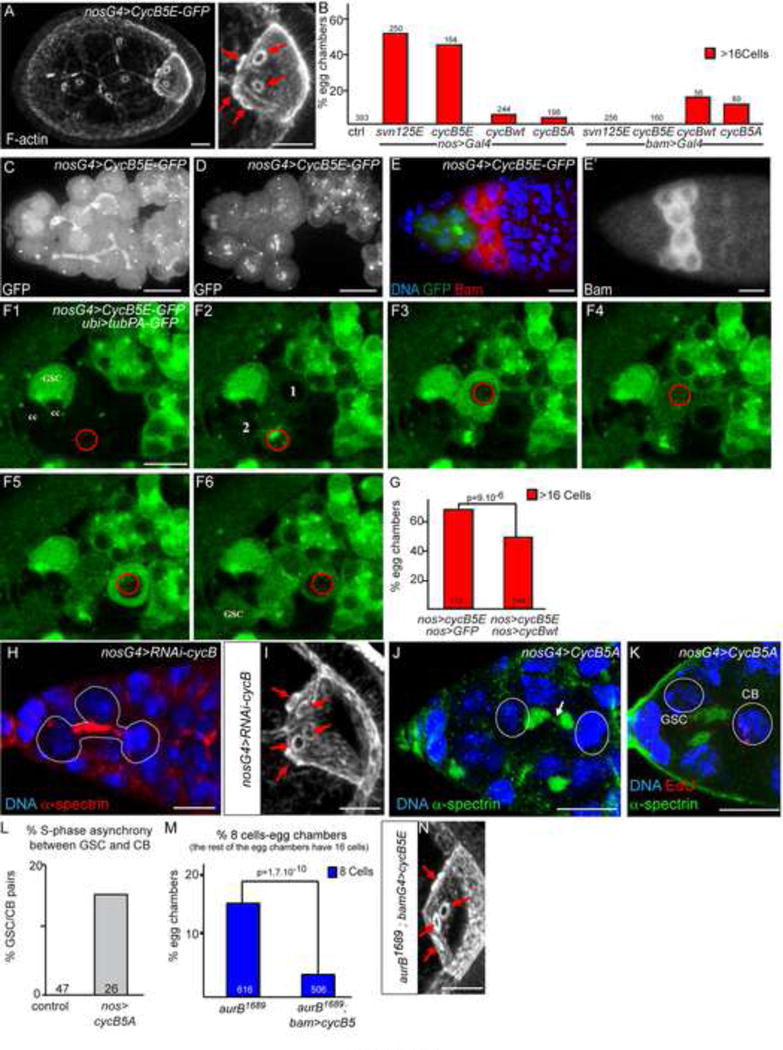
(A) Stage 7 egg chambers expressing nos>CycB5E-GFP stained with phalloidin (F-actin). Red arrows indicate the 5 ring canals of the oocyte.
(B) Fraction of egg chambers exhibiting more than 16 cells on the Y axis. Genotypes are on the X axis. CycB5E phenocopies SvnS125E.
(C–D) nos>CycB-5E-GFP localizes on the fusome in interphase. Stem cysts with branched fusomes form (C), and mitosis are synchronous within the stem cyst (D). In D, 4 metaphase cells are visible, with GFP accumulation at the centrosomes, centromeres, and weakly on the fusome.
(E) Germarium expressing nos>CycB5E-GFP stained with DAPI (blue) and Bam (red, and white in E′). The stem cyst does not express Bam.
(F) Selected time points of a germarium expressing nos>CycB5E-GFP and nos>Tubulin-PA-GFP. In F1, no CycB5E-GFP is visible in the selected cyst because cells are in G1 or S phase. In F2, Tub-PA-GFP is photo-activated in a region defined by a red circle, and the fluorescence diffuses to cell 1 and cell 2 very rapidly. In F3, PA-GFP is activated in cell 1, and in cell 3 in F5. In F6, all cells have the same level of fluorescence, including the GSC, which has not been directly activated. It demonstrates that Tub-PA-GFP could diffuse in all cells and that abscission remained incomplete in cyst expressing CycB5E.
(G) Fraction of egg chambers exhibiting more than 16 cells on the Y axis. Genotypes are on the X axis. Expression of CycB-WT can partially suppress the CycB5E overexpression phenotype.
(H) Germarium expressing RNAi directed against cycB in germ cells (nanos driver), stained with DAPI (blue) and fusome (red). A stem cyst, formed of 3 cells, is shown (dotted line).
(I) nos>CycB-RNAi induces the formation of oocytes with 5 ring canals (arrows). F-actin stained with phalloidin.
(J) Germarium expressing nos>CycB-5E shows a fusome (green) breaking (arrow) in a 2-cell cyst (dotted line). α-spectrin (green).
(K) Germarium expressing nos>CycB-5E stained for Edu (red) and α-spectrin (green), showing that S-phase synchrony between the GSC (Edu negative) and its CB (EdU positive) is lost in these females.
(L) Fraction of GSC/CB pair showing S-phase asynchrony on the Y axis, in %. Genotypes are on the X axis.
(M) Fraction of egg chambers exhibiting less than 16 cells on the Y axis. Genotypes are on the X axis. The loss of AurB can be rescued by expression of CycB5E only in the cyst with the bam-Gal4 driver.
(N) Example of a stage 7 “rescued” oocyte, mutant for aurB1689 and expressing bam>CycB-5E. Red arrows indicate the 4 ring canals in the oocyte.
Scale bar: 10 μm. See also Figure S4, Movie 4, Movie 5, Movie 6.
CycB-5E delays abscission as a gain-of-function of the CPC, which we showed to negatively interact with CycB (Fig. 5F). CycB-5E thus behaved as a dominant-negative form for Cdk-1 activity, at least regarding abscission. In support, co-expression of a wild type UAS-CycB, but not of a neutral UAS-GFP, partially rescued the number of 32-cell cysts induced by CycB-5E (Fig. 6G). If CycB-5E behaved as a dominant negative form, then one should expect the same phenotype in CycB loss-of-function. In the complete absence of CycB, mutant flies are viable, but sterile with almost no germ cells as there is a specific requirement for CycB in germline stem cells self-renewal (Wang and Lin, 2005). To circumvent this problem, we expressed a newly developed RNAi TRiP line against CycB in GSCs and we occasionally found stem-cysts with branched fusomes (6.25%; n=127), and resulting egg chambers made of 32 cells (13%; n=44), as well as the expected 8-cell cysts (Fig. 6H–I) (Ni et al., 2011). These results are in agreement with CycB-5E being a dominant negative form. Nevertheless, CycB-5E could rescue cycB mutants sterility (data not shown), and cycled like wild-type CycB (Movie 4; Fig. S4).
Overall, our results showed that CycB promotes abscission and that its phosphorylation by Aurora B inhibits this activity. If Cyclin B was the main target of Aurora B during this process, one could make at least two predictions: 1) expression of the non-phosphorylatable CycB-5A should mimic the absence of AurB/Svn; and 2) expression of phospho-mimic CycB-5E should compensate for the absence of AurB/Svn. We found that the expression of CycB-5A induced the appearance of breaking fusome in 2-cell cysts as seen in aurB1689mutant cysts (Fig. 6J). We also detected asynchronous pairs of GSC/CB (16%; n=26) similar to aurB1689mutant pairs (17%; n=62; Fig 6K,L). We concluded that, like in aurB mutants, abscission is precocious in GSC and 2-cell cysts expressing CycB-5A. To test the second prediction, we expressed CycB-5E in aurB1689 mutant cyts, in which we had found a premature abscission at the 2-cell cyst stage (leading to the formation of 8-cell cysts). We found a strong reduction in the number of 8-cell cysts when aurB1689 flies also expressed bam>CycB-5E (from 14,5% to 3,5% of 8-cells cyst, Fig. 6M,N). Importantly, this rescue was not the consequence of a combination of two phenotypes (32-cell cyst induced by CycB-5E, broken into two 16-cell cysts by aurB1689 mutation), as we expressed CycB-5E with the bam promoter, i.e. only in the cyst and not in GSCs, and bam>CycB-5E had no phenotype on its own (Fig. 6B). In contrast, bam>CycB and bam>CycB-5A induced a significant number of cysts with 32-cells indicating that these forms were able to force an extra-division in cysts (Fig. 6B), and could not be used in an aurB1689 mutant background. We thus concluded that loss of phosphorylation of CycB accounted for most phenotypes observed in aurB1689 mutants and that CycB is a major target of the CPC in the regulation of abscission. Overall, our results showed that Aurora B delays abscission by antagonizing Cyclin B activity in Drosophila germ cells.
7) Cyclin B2 localizes at the intercellular bridge, and Aurora B and Cdk-1 have opposite effects on abscission timing in human cell culture
As mentioned previously Cyclin B2 in vertebrates, but not Cyclin B1, exhibits two consensus sites for Aurora B in its N-terminal domain (Fig. 5G). We found that in mouse embryonic fibroblasts (MEF) endogenous CycB2 localized at the intercellular bridge where MT are less dense (Fig. 7A). This signal was specific as MEF derived from CycB2 Knock-Out mice did not show this staining (Fig 7B). Human CycB2 tagged with GFP also localized at the intercellular bridge in HeLa cells (Fig. 7C,D,E). Next, we tested directly whether CycB2/Cdk-1 was required for abscission by inhibiting Cdk-1 activity only after the ingression of the cytokinetic furrow of Hela cells. Drugs used to inhibit Cdk-1(Cdk inhibitor III and RO-3306 (Gavet and Pines, 2010b)) were added well after metaphase (around 60 min), i.e. well after any known function of Cdk-1 (Fig. 7F). We found that both drugs induced a strong delay in the completion of abscission (Fig. 7G and not shown for RO-3306). Abscission duration was increased by 50% on average (216 min in control vs 317 min in treated cells p=0,0056; Fig. 7H). We concluded that Cdk-1 activity promotes abscission during late cytokinesis. Conversely, inhibition of Aurora B with the ZM447439 drug had the opposite effect and induced precocious abscission (171 min; Fig. 7G), as previously reported (Steigemann et al., 2009). Furthermore, we found that the simultaneous inhibition of Aurora B and Cdk-1 restored normal timing (216 min in control vs 235 min in treated cells p=0,135 (not significant); Fig. 7G,H). We concluded that Aurora B and Cdk-1 have opposite activities on abscission timing in mammalian cells.
Figure 7. CycB2 localizes at the intercellular brigde; and Cdk activity regulates abscission timing in mammalian cells.
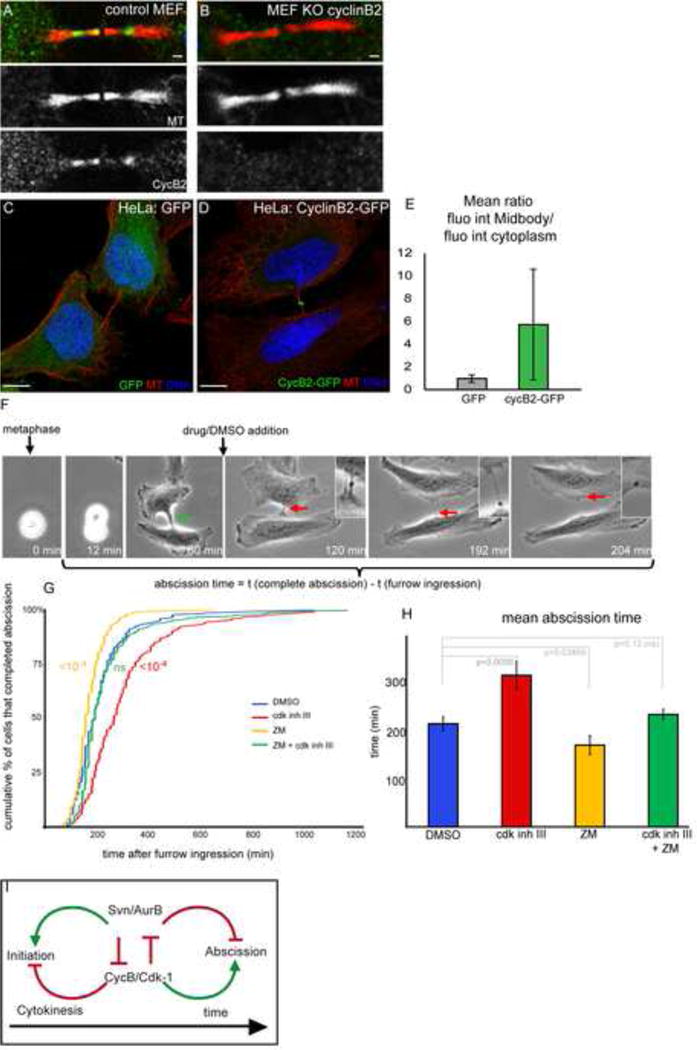
(A–B) Confocal sections of control (A) or cyclin B2 knock out (B) MEFs stained for Tubulin (red) and Cyclin B2 (green).
(C–D) Confocal sections of HeLa cells transfected with GFP (E) or Cyclin B2-GFP (F) expressing vectors, stained for Tubulin (red) and DNA (DAPI, blue).
(E) Mean ratio of the fluorescence at the midbody/fluorescence in the cytoplasm of HeLa cells transfected with GFP (left, grey bar) or Cyclin B2-GFP (right, green bar).
(F) Cells were imaged from metaphase (tp 0min) till abscission (tp 204min). Drugs or DMSO were added only once furrow had ingressed into a thin bridge (green arrow, tp 60min). Abscission duration was determined as the time between complete abscission and start of furrow ingression (tp 12min). The midbody is visible as a dense structure in the bridge (see insets). At tp 204min, the bridge is cut.
(G) Cumulative plot showing the fraction of HeLa cells (%) that have completed abscission in function of the duration of abscission (min) upon DMSO (n=223), cdk1/2 inhibitor III (n=215), ZM447439 (n=223), or both drugs (n=247). p values (Kolmogorov-Smirnov Test) are indicated.
(H) Mean abscission duration of HeLa cells treated with cdk1/2 inhibitor III, ZM447439, or both drugs. p values (student test) are indicated.
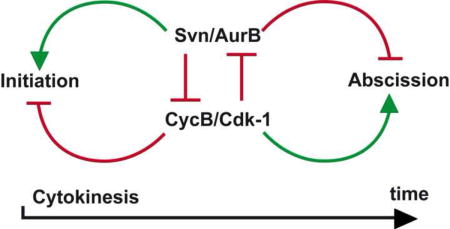
(I) Schematic model of the interactions between the CPC and CycB/Cdk1 during early and late cytokinesis. See text for more details.
Scale bar: 10 μm. Error bars are SD.
DISCUSSION
1) Aurora B and Cyclin B regulate abscission
Collectively, our data point to a simple model for the regulation of abscission in the germline. We propose that the accumulation of Aurora B on the fusome at the site of abscission creates a local activity of the CPC, which phosphorylates Cyclin B and delays abscission. In support of this model, we found that increasing the level and activity of the CPC delayed abscission in GSCs, while a decrease led to premature abscission in 2-cell cysts. Conversely, a decrease in Cyclin B slowed down abscission in GSCs and counteracted premature abscission in mutant cysts with reduced Aurora B level (Fig. 7I). Furthermore, inhibition of Cdk-1 after furrow ingression in HeLa cells delayed abscission, while inhibition of Aurora B led to faster abscission (Fig. 7G,H). Our work also demonstrates that phoshorylation of CycB depends on Aurora B and inhibits CycB activity during late cytokinesis. Interestingly, this antagonistic relationship is reverse during early cytokinesis (Fig. 7I), as high CycB/Cdk-1 blocks the initiation of cytokinesis (Echard and O’Farrell, 2003; Sigrist et al., 1995), while Aurora B is known to promote it (Ruchaud et al., 2007). Furthermore, it was recently shown that CycB-Cdk-1 can directly phosphorylate the CPC to trigger its localization to inner centromeres and inhibit its localization to the spindle midzone through Mklp2 (Gruneberg et al., 2004; Hummer and Mayer, 2009; Tsukahara et al., 2010). On the one hand, CycB/Cdk-1 negatively regulates the localization of the CPC to the midzone and Aurora B function in the early steps of cytokinesis, and on the other hand, we found that the CPC negatively regulates CycB to inhibit abscission. We propose that a balance between the CPC and CycB/Cdk-1 mutual inhibition by phosphorylation plays a central role in the regulation of cytokinesis and abscission (Fig. 7I).
How could this balance be shifted from complete cytokinesis in GSCs to arrest of abscission in differentiating cysts? One hypothesis is based on the difference of duration of the cell cycle between GSCs and germline cysts. GSCs enter mitosis and finish abscission every 24 hr. In contrast, differentiating cysts cycle much faster and undergo four cell cycles also in 24h (Morris and Spradling, 2011; Spradling, 1993a). Aurora B could delay abscission equally in GSCs and cystoblasts, however, GSCs would have the time to complete abscission, while cystoblasts would have already completed their four mitoses and differentiation program. Consistent with this hypothesis, when cyst differentiation is blocked at the 2-cell stage in bam mutant ovaries, pairs of cells can be seen connected only by a thin-thread of breaking fusome (McKearin and Ohlstein, 1995), similar to those we observed in aurB1689 mutant cysts. In the absence of Bam, mutant ovaries accumulate single cells and pair of cells, which are unable to differentiate and are delayed in that state (McKearin and Ohlstein, 1995). We thus propose that Aurora B delays abscission in wild type GSCs and cystoblasts, and we speculate that cytokinesis can be completed even in cystoblasts, if abscission becomes faster such as in aurB1689 mutants, or if differentiation is slower or arrested such as in bam mutants.
2) Cyclin B localization and function at the midbody
One surprising finding of our study is the presence and function of CycB at the end of cell division, which contrasts with the widely accepted view that all CycB is degraded during anaphase (Pines, 2006). Our live-imaging data showed that most of the wild type and phosphomimic forms of CycB were indeed degraded at anaphase onset. However, a small pool of both forms accumulated later on at midbodies and at ring canals in differentiating cysts (Fig 5D,E and S4B). This localization of CycB is conserved in several species as we report that CycB2 also accumulates at the midbody in mouse and human cells (Fig. 7A–D). Interestingly, Clb2, a CycB homologue in yeast, was shown to localize at the bud neck (Eluere et al., 2007). Finally, Cdk-1 was also isolated from biochemically purified midbodies in a proteomic screen for functional components of cytokinesis (Skop et al., 2004). CycB and Cdk-1 thus localize at the midbody in several model systems.
Our genetic analysis showed that CycB plays an important function in counteracting Aurora B inhibition of abscission. This novel function may help to solve a lasting paradox of the regulation of the cell cycle in germ cells. Indeed, overexpression of string/cdc25, an activator of Cdk-1, which should force extra mitoses, instead induces mostly 8-cell cysts (Mata et al., 2000). Even more puzzling, a few 32-cell cysts are also produced in the same experiment. In contrast, overexpression of Tribbles (and Wee1 and Myt1 ((Jin et al., 2005) and S. Campbell, pers. comm.), which are known inhibitors of Cdc25 and Cdk-1, produce 32-cell cysts rather than the expected 8-cell cysts (Mata et al., 2000). In agreement with these counter-intuitive results, we found that reducing CycB levels could also produce some 32-cell cysts (Fig. 6I). However, in light of our model, activation of Cdk-1 could lead to premature abscission in germline cysts and explain the 8-cell cysts, whereas inhibition of Cdk-1 could delay abscission in GSCs and produce 32-cell cysts. Furthermore, activation or inhibition of CycB/cdk-1 in both GSC and cyst at the same time (as in homozygous mutants or overexpression using the nanos promoter) would lead to various combinations of opposite phenotypes (8-, 16- and 32- cells), because loss- or gain-of-function of cycB/cdk-1 have opposite effects in the two cell types.
3) Inhibition of abscission in germline and somatic cells
One intriguing observation in our results is that a decrease of the CPC activity leads to the formation of cysts made of precisely 8 cells, but not of 4 or 2 cells. It shows that the first division of the cystoblast is particularly sensitive to a reduction of Aurora B activity, but not the three remaining divisions. In agreement, we only found breaking fusomes in 2-cell cysts, but not in cysts with more branched fusomes. As described above, this phenotype is reminiscent of defects found in bam mutant ovaries (McKearin and Ohlstein, 1995). We thus propose that inhibition of abscission in the first division of the cystoblast is different from the three following mitosis. We suggest that initially this first division is very similar to a GSC division and both abscissions are delayed by Aurora B, however, when Bam starts to be expressed in cystoblasts, it triggers a developmental program, which completely blocks abscission of the first and following divisions, as previously proposed by McKearin and Ohlstein.
We believe that these findings have important implications for our understanding of abscission in germ cells, but also in somatic cells in general. Cytokinesis can also be arrested in follicle cells and other somatic tissues where there is no fusome or transcription of bam (Airoldi et al., 2011; de Cuevas and Spradling, 1998; Haglund et al., 2010). We further demonstrated that CycB2 localizes at the intercellular bridge, and that Aurora B and Cdk-1 have opposite effects on abscission in vertebrate somatic cells. Interestingly, although mice KO for Cyclin B2 are viable, males are less fertile and have small litter size (Brandeis et al., 1998). We speculate that germ cells may be more sensitive to abscission defects than somatic cells, but that the underlying mechanisms are very similar.
SUMMARY of EXPERIMENTAL PROCEDURES
Fly Strains
The PBac2180 insertion was identified in a previous screen (Mathieu et al., 2007), and PBac1527 was obtained at the Bloomington Stock Center. Mutations in aurB were generated during two independent EMS mutagenesis and screens. The following alleles and transgenes were used: bamΔ86 (Bopp, 1993); hts1; hts01103 (Yue, 1992), cycB2 and cycB 3(Jacobs, 1998); cycBKG08886 (Bloomington Stock Center); UAS-PA-GFP (Murray, 2007); UAS-Trip cycB (Ni, 2011); Ubq-RFP-α-Tubulin (Basto, 2008); cid-RFP (Schuh, 2007), H2B-RFP (Schuh, 2007) and jupiter-GFP (Karpova, 2006).
Cell culture, transfection and drug treatments
Transfection of ATCC cells were performed using FuGENE-6 (Roche). The plasmid encoding myc-CycB2-GFP was obtained from J. Pines via J.Sobczack.
HEK 293T (2.106) cells were transfected by the plasmids pCMV-HA-AurB and/or pCMV-cycB-GFP (4 μg total) using PEI (Polyethylenimine).
Cells have been treated with 2 μM ZM447439 (TOCRIS Bioscience) and/or with 300 nM cdk1/2 inhibitor III (Merk).
Constructs and Antibodies
To generate the genomic rescue construct of survivin, a PCR fragment corresponding to the survivin locus was amplified from genomic DNA. The making of svn>svnΔBIR, svn>svnC97A, svn>svnS125A, svn>svnT129A, svn>svnS125A,T129A, UASp>svnWT-GFP, UASp>svn125E-GFP, tub>RFP-aurB, pUASp>RFP-aurA, pUASp>CycB-WT-GFP, pUASp>CycB-5A-GFP, and pUASp>CycB-5E-GFP are detailed in the Supplementary Files.
A phosphorylated Cyclin B peptide, SNLSKKS157(PO3H2)LTKLR, corresponding to the 4th potential Aurora-B phosphorylation site, was synthesized and used for immunization of two rabbits (Eurogentec).
Quantification and Statistics
We counted the number of nuclei with the DAPI staining. In addition, the number of ring canals stained by Phalloidin was counted for the oocyte to discard encapsulation defects. Chi-square tests were used to compare the proportions of egg chambers having 8, 16 or 32 cells.
Microscopy
Acquisition of Z-stacks on fixed sample was carried out on Zeiss LSM710 or LSM780 confocal microscopes. For live imaging of germarium, ovaries were dissected and mounted in oil and were imaged with an inverted Confocal Spinning Disk Roper/Nikon.
HeLa cells were plated on 35mm glass dishes, and time-lapse sequences were recorded every 10 or 12 min for 24 h on a Nikon Eclipse Ti microscope with a ×20 0.45 NA Plan Fluor ELWD objective lens controlled by Metamorph 6.1 software (Universal Imaging).
Photo-activation was done with a 2-photon laser at 820nm. Imaging was done with a Zeiss LSM 710.
Supplementary Material
Highlights.
Aurora B delays abscission in Drosophila germline cysts
Cyclin B localizes on the fusome and promotes abscission
Aurora B opposes CycB-Cdk1 activity by phosphorylating Cyclin B
Aurora B and CycB2-Cdk1 have opposite effect on abscission timing in HeLa cells
Acknowledgments
We are grateful to M. Bettencourt-Dias, S. Campbell, M. Carmena, M. Carrington, O. Gavet, D. Glover, B. Earnshaw, I. Ferreira, M. Gho, J. Raff, S. Ruchaud, J. Sobszack, P. ten Dijke for helpful discussions, advices and materials. We thank members of the Huynh laboratory, A. Gonzalez-Reyes and E. Heard for critical reading of the manuscript. DSHB (Iowa University) for antibodies and Bloomington Drosophila Stock center for fly stocks. We thank the imaging facility (PICT@BDD) and the proteomic platform for excellent technical help. This work was supported by the CNRS, ANR (ANR-06-JCJC- 0092), ARC post-doctoral fellowship (J.M.), FSER (Schlumberger), Ville de Paris and Fondation BNP-Paribas to JRH. A.E. (CNRS, Institut Pasteur, equipe FRM2012 et FSER (Schlumberger).
References
- Adams RR, Maiato H, Earnshaw WC, Carmena M. Essential roles of Drosophila inner centromere protein (INCENP) and aurora B in histone H3 phosphorylation, metaphase chromosome alignment, kinetochore disjunction, and chromosome segregation. J Cell Biol. 2001;153:865–880. doi: 10.1083/jcb.153.4.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airoldi S, McLean P, Shimada Y, Cooley L. Intercellular protein movement in syncytial Drosophila follicle cells. J Cell Sci. 2011;124:4077–4086. doi: 10.1242/jcs.090456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandeis M, Rosewell I, Carrington M, Crompton T, Jacobs MA, Kirk J, Gannon J, Hunt T. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc Natl Acad Sci U S A. 1998;95:4344–4349. doi: 10.1073/pnas.95.8.4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkard ME, Randall CL, Larochelle S, Zhang C, Shokat KM, Fisher RP, Jallepalli PV. Chemical genetics reveals the requirement for Polo-like kinase 1 activity in positioning RhoA and triggering cytokinesis in human cells. Proc Natl Acad Sci U S A. 2007;104:4383–4388. doi: 10.1073/pnas.0701140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang S, Xie T. Restricting self-renewal signals within the stem cell niche: multiple levels of control. Curr Opin Genet Dev. 2011;21:684–689. doi: 10.1016/j.gde.2011.07.008. [DOI] [PubMed] [Google Scholar]
- Clute P, Pines J. Temporal and spatial control of cyclin B1 destruction in metaphase. Nat Cell Biol. 1999;1:82–87. doi: 10.1038/10049. [DOI] [PubMed] [Google Scholar]
- de Cuevas M, Spradling AC. Morphogenesis of the Drosophila fusome and its implications for oocyte specification. Development. 1998;125:2781–2789. doi: 10.1242/dev.125.15.2781. [DOI] [PubMed] [Google Scholar]
- Echard A, O’Farrell PH. The degradation of two mitotic cyclins contributes to the timing of cytokinesis. Curr Biol. 2003;13:373–383. doi: 10.1016/s0960-9822(03)00127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eluere R, Offner N, Varlet I, Motteux O, Signon L, Picard A, Bailly E, Simon MN. Compartmentalization of the functions and regulation of the mitotic cyclin Clb2 in S. cerevisiae. J Cell Sci. 2007;120:702–711. doi: 10.1242/jcs.03380. [DOI] [PubMed] [Google Scholar]
- Gavet O, Pines J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol. 2010a;189:247–259. doi: 10.1083/jcb.200909144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell. 2010b;18:533–543. doi: 10.1016/j.devcel.2010.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilboa L, Forbes A, Tazuke SI, Fuller MT, Lehmann R. Germ line stem cell differentiation in Drosophila requires gap junctions and proceeds via an intermediate state. Development. 2003;130:6625–6634. doi: 10.1242/dev.00853. [DOI] [PubMed] [Google Scholar]
- Gilboa L, Lehmann R. Repression of primordial germ cell differentiation parallels germ line stem cell maintenance. Curr Biol. 2004;14:981–986. doi: 10.1016/j.cub.2004.05.049. [DOI] [PubMed] [Google Scholar]
- Gruneberg U, Neef R, Honda R, Nigg EA, Barr FA. Relocation of Aurora B from centromeres to the central spindle at the metaphase to anaphase transition requires MKlp2. J Cell Biol. 2004;166:167–172. doi: 10.1083/jcb.200403084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Nezis I, Lemus D, Grabbe C, Wesche J, Liestøl K, Dikic I, Palmer R, Stenmark H. Cindr interacts with anillin to control cytokinesis in Drosophila melanogaster. Curr Biol. 2010;20:944–950. doi: 10.1016/j.cub.2010.03.068. [DOI] [PubMed] [Google Scholar]
- Hummer S, Mayer TU. Cdk1 negatively regulates midzone localization of the mitotic kinesin Mklp2 and the chromosomal passenger complex. Curr Biol. 2009;19:607–612. doi: 10.1016/j.cub.2009.02.046. [DOI] [PubMed] [Google Scholar]
- Huynh JR. Fusome as a Cell-Cell Communication Channel of Drosophila Ovarian Cyst. In: Baluska F, Volkmann D, Barlow PW, editors. Cell-cell Channels. 2005. Landes Biosciences. [Google Scholar]
- Huynh JR, St Johnston D. The origin of asymmetry: early polarisation of the Drosophila germline cyst and oocyte. Curr Biol. 2004;14:R438–449. doi: 10.1016/j.cub.2004.05.040. [DOI] [PubMed] [Google Scholar]
- Jin Z, Homola EM, Goldbach P, Choi Y, Brill JA, Campbell SD. Drosophila Myt1 is a Cdk1 inhibitory kinase that regulates multiple aspects of cell cycle behavior during gametogenesis. Development. 2005;132:4075–4085. doi: 10.1242/dev.01965. [DOI] [PubMed] [Google Scholar]
- Jones G, Jones D, Zhou L, Steller H, Chu Y. Deterin, a new inhibitor of apoptosis from Drosophila melanogaster. J Biol Chem. 2000;275:22157–22165. doi: 10.1074/jbc.M000369200. [DOI] [PubMed] [Google Scholar]
- Kai T, Spradling A. Differentiating germ cells can revert into functional stem cells in Drosophila melanogaster ovaries. Nature. 2004;428:564–569. doi: 10.1038/nature02436. [DOI] [PubMed] [Google Scholar]
- Lens SM, Rodriguez JA, Vader G, Span SW, Giaccone G, Medema RH. Uncoupling the central spindle-associated function of the chromosomal passenger complex from its role at centromeres. Mol Biol Cell. 2006;17:1897–1909. doi: 10.1091/mbc.E05-08-0727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilly MA, de Cuevas M, Spradling AC. Cyclin A associates with the fusome during germline cyst formation in the Drosophila ovary. Dev Biol. 2000;218:53–63. doi: 10.1006/dbio.1999.9570. [DOI] [PubMed] [Google Scholar]
- Lilly MA, Spradling AC. The Drosophila endocycle is controlled by Cyclin E and lacks a checkpoint ensuring S-phase completion. Genes Dev. 1996;10:2514–2526. doi: 10.1101/gad.10.19.2514. [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Rodriguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol. 2009;185:193–202. doi: 10.1083/jcb.200812045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Mata J, Curado S, Ephrussi A, Rorth P. Tribbles coordinates mitosis and morphogenesis in Drosophila by regulating string/CDC25 proteolysis. Cell. 2000;101:511–522. doi: 10.1016/s0092-8674(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Mathieu J, Sung HH, Pugieux C, Soetaert J, Rorth P. A sensitized PiggyBac-based screen for regulators of border cell migration in Drosophila. Genetics. 2007;176:1579–1590. doi: 10.1534/genetics.107.071282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKearin D, Ohlstein B. A role for the Drosophila bag-of-marbles protein in the differentiation of cystoblasts from germline stem cells. Development. 1995;121:2937–2947. doi: 10.1242/dev.121.9.2937. [DOI] [PubMed] [Google Scholar]
- Morris LX, Spradling AC. Long-term live imaging provides new insight into stem cell regulation and germline-soma coordination in the Drosophila ovary. Development. 2011;138:2207–2215. doi: 10.1242/dev.065508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef R, Preisinger C, Sutcliffe J, Kopajtich R, Nigg EA, Mayer TU, Barr FA. Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J Cell Biol. 2003;162:863–875. doi: 10.1083/jcb.200306009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JQ, Zhou R, Czech B, Liu LP, Holderbaum L, Yang-Zhou D, Shim HS, Tao R, Handler D, Karpowicz P, et al. A genome-scale shRNA resource for transgenic RNAi in Drosophila. Nat Methods. 2011;8:405–407. doi: 10.1038/nmeth.1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norden C, Mendoza M, Dobbelaere J, Kotwaliwale CV, Biggins S, Barral Y. The NoCut pathway links completion of cytokinesis to spindle midzone function to prevent chromosome breakage. Cell. 2006;125:85–98. doi: 10.1016/j.cell.2006.01.045. [DOI] [PubMed] [Google Scholar]
- Ohlmeyer JT, Schupbach T. Encore facilitates SCF-Ubiquitin-proteasome-dependent proteolysis during Drosophila oogenesis. Development. 2003;130:6339–6349. doi: 10.1242/dev.00855. [DOI] [PubMed] [Google Scholar]
- Parry DH, Hickson GR, O’Farrell PH. Cyclin B destruction triggers changes in kinetochore behavior essential for successful anaphase. Curr Biol. 2003;13:647–653. doi: 10.1016/s0960-9822(03)00242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepling ME, de Cuevas M, Spradling AC. Germline cysts: a conserved phase of germ cell development? Trends Cell Biol. 1999;9:257–262. doi: 10.1016/s0962-8924(99)01594-9. [DOI] [PubMed] [Google Scholar]
- Petronczki M, Glotzer M, Kraut N, Peters JM. Polo-like kinase 1 triggers the initiation of cytokinesis in human cells by promoting recruitment of the RhoGEF Ect2 to the central spindle. Dev Cell. 2007;12:713–725. doi: 10.1016/j.devcel.2007.03.013. [DOI] [PubMed] [Google Scholar]
- Pines J. Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol. 2006;16:55–63. doi: 10.1016/j.tcb.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Ruchaud S, Carmena M, Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007;8:798–812. doi: 10.1038/nrm2257. [DOI] [PubMed] [Google Scholar]
- Sanger JM, Pochapin MB, Sanger JW. Midbody sealing after cytokinesis in embryos of the sea urchin Arabacia punctulata. Cell Tissue Res. 1985;240:287–292. doi: 10.1007/BF00222337. [DOI] [PubMed] [Google Scholar]
- Sigrist S, Jacobs H, Stratmann R, Lehner CF. Exit from mitosis is regulated by Drosophila fizzy and the sequential destruction of cyclins A, B and B3. EMBO J. 1995;14:4827–4838. doi: 10.1002/j.1460-2075.1995.tb00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skop AR, Liu H, Yates J, 3rd, Meyer BJ, Heald R. Dissection of the mammalian midbody proteome reveals conserved cytokinesis mechanisms. Science. 2004;305:61–66. doi: 10.1126/science.1097931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapp EL, Iida T, Frescas D, Lippincott-Schwartz J, Lilly MA. The fusome mediates intercellular endoplasmic reticulum connectivity in Drosophila ovarian cysts. Mol Biol Cell. 2004;15:4512–4521. doi: 10.1091/mbc.E04-06-0475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Wong MD, Kawase E, Xi R, Ding BC, McCarthy JJ, Xie T. Bmp signals from niche cells directly repress transcription of a differentiation-promoting gene, bag of marbles, in germline stem cells in the Drosophila ovary. Development. 2004;131:1353–1364. doi: 10.1242/dev.01026. [DOI] [PubMed] [Google Scholar]
- Spradling A. Developmental genetics of oogenesis. In: Bate M, Martinez-Arias A, editors. The development of Drosophila melanogaster. New-York: Cold Spring Harbor Laboratory Press; 1993a. pp. 1–70. [Google Scholar]
- Spradling AC. Germline cysts: communes that work. Cell. 1993b;72:649–651. doi: 10.1016/0092-8674(93)90393-5. [DOI] [PubMed] [Google Scholar]
- Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell. 2009;136:473–484. doi: 10.1016/j.cell.2008.12.020. [DOI] [PubMed] [Google Scholar]
- Sullivan M, Morgan DO. Finishing mitosis, one step at a time. Nat Rev Mol Cell Biol. 2007;8:894–903. doi: 10.1038/nrm2276. [DOI] [PubMed] [Google Scholar]
- Terada Y, Tatsuka M, Suzuki F, Yasuda Y, Fujita S, Otsu M. AIM-1: a mammalian midbody-associated protein required for cytokinesis. EMBO J. 1998;17:667–676. doi: 10.1093/emboj/17.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara T, Tanno Y, Watanabe Y. Phosphorylation of the CPC by Cdk1 promotes chromosome bi-orientation. Nature. 2010;467:719–723. doi: 10.1038/nature09390. [DOI] [PubMed] [Google Scholar]
- Vitrat N, Cohen-Solal K, Pique C, Le Couedic JP, Norol F, Larsen AK, Katz A, Vainchenker W, Debili N. Endomitosis of human megakaryocytes are due to abortive mitosis. Blood. 1998;91:3711–3723. [PubMed] [Google Scholar]
- Wang Z, Lin H. The division of Drosophila germline stem cells and their precursors requires a specific cyclin. Curr Biol. 2005;15:328–333. doi: 10.1016/j.cub.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Wheatley SP, Henzing AJ, Dodson H, Khaled W, Earnshaw WC. Aurora-B phosphorylation in vitro identifies a residue of survivin that is essential for its localization and binding to inner centromere protein (INCENP) in vivo. J Biol Chem. 2004;279:5655–5660. doi: 10.1074/jbc.M311299200. [DOI] [PubMed] [Google Scholar]
- Wurzenberger C, Gerlich DW. Phosphatases: providing safe passage through mitotic exit. Nat Rev Mol Cell Biol. 2011;12:469–482. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- Yue L, Spradling AC. hu-li tai shao, a gene required for ring canal formation during Drosophila oogenesis, encodes a homolog of adducin. Genes Dev. 1992;6:2443–2454. doi: 10.1101/gad.6.12b.2443. [DOI] [PubMed] [Google Scholar]
- Yue Z, Carvalho A, Xu Z, Yuan X, Cardinale S, Ribeiro S, Lai F, Ogawa H, Gudmundsdottir E, Gassmann R, et al. Deconstructing Survivin: comprehensive genetic analysis of Survivin function by conditional knockout in a vertebrate cell line. J Cell Biol. 2008;183:279–296. doi: 10.1083/jcb.200806118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.