

Abstract
Defects in the RNA-binding protein, TDP-43, are known to cause a variety of neurodegenerative diseases, including amyotrophic lateral sclerosis and frontotemporal lobar dementia. A variety of experimental systems have shown that neurons are sensitive to TDP-43 expression levels, yet the specific functional defects resulting from TDP-43 dysregulation have not been well described. Using the Drosophila TDP-43 ortholog TBPH, we previously showed that TBPH-null animals display locomotion defects as third instar larvae. Furthermore, loss of TBPH caused a reduction in cacophony, a Type II voltage-gated calcium channel, expression and that genetically restoring cacophony in motor neurons in TBPH mutant animals was sufficient to rescue the locomotion defects. In the present study, we examined the relative contributions of neuromuscular junction physiology and the motor program to the locomotion defects and identified subsets of neurons that require cacophony expression to rescue the defects. At the neuromuscular junction, we showed mEPP amplitudes and frequency require TBPH. Cacophony expression in motor neurons rescued mEPP frequency but not mEPP amplitude. We also showed that TBPH mutants displayed reduced motor neuron bursting and coordination during crawling and restoring cacophony selectively in two pairs of cells located in the brain, the AVM001b/2b neurons, also rescued the locomotion and motor defects, but not the defects in neuromuscular junction physiology. These results suggest that the behavioral defects associated with loss of TBPH throughout the nervous system can be associated with defects in a small number of genes in a limited number of central neurons, rather than peripheral defects.
SIGNIFICANCE STATEMENT TDP-43 dysfunction is a common feature in neurodegenerative diseases, including amyotrophic lateral sclerosis, frontotemporal lobar dementia, and Alzheimer's disease. Loss- and gain-of-function models have shown that neurons are sensitive to TDP-43 expression levels, but the specific defects caused by TDP-43 loss of function have not been described in detail. A Drosophila loss-of-function model displays pronounced locomotion defects that can be reversed by restoring the expression levels of a voltage-gated calcium channel, cacophony. We show these defects can be rescued by expression of cacophony in motor neurons and by expression in two pairs of neurons in the brain. These data suggest that loss of TDP-43 can disrupt the central circuitry of the CNS, opening up identification of alternative therapeutic targets for TDP-43 proteinopathies.
Keywords: amyotrophic lateral sclerosis, calcium channel, frontotemporal lobar dementia, motor program, TDP-43
Introduction
Amyotrophic lateral sclerosis is the most common adult-onset motor neuron disease, for which there is no cure or treatment that significantly extends life. First described as a unique disease by Charcot in 1864, amyotrophic lateral sclerosis is characterized by the progressive, patterned die-back of motor axons and motor neuron death, causing loss of motor function and ultimately death (Guillain and Bailey, 1959). Although patient pathophysiology varies, ∼90% of patient samples show the presence of insoluble protein aggregates in the somas of motor neurons (Arai et al., 2006; Ayala et al., 2008). In 2006, the RNA binding protein TDP-43 was identified as a major protein component in these aggregates (Arai et al., 2006; Neumann et al., 2006).
TDP-43 contains two RNA binding motifs and a nuclear localization motif (Ayala et al., 2005). In healthy cells, it is primarily localized to the nucleus where it functions to regulate gene expression and RNA metabolism (Ayala et al., 2008; Ihara et al., 2013). TDP-43 is also found in the cytoplasm, where it shuttles between the nucleus and the cytoplasm and is present in RNA granules (Dewey et al., 2012). Postmortem tissue samples taken from amyotrophic lateral sclerosis patients show a depletion of TDP-43 from the nucleus, concurrent with the formation of TDP-43 cytoplasmic inclusions (Kwong et al., 2007; Neumann, 2009; Nishimura et al., 2010). The etiology of TDP-43 proteinopathies is poorly characterized, and the relative contributions of loss of nuclear TDP-43 function or a gain of toxicity associated with the formation of inclusions in disease pathogenesis is unknown (Arai et al., 2006; Hirsch, 2007; Kwong et al., 2007; Neumann, 2009; Diaper et al., 2013b). Furthermore, although it is assumed that the dying-back of motor axons is concomitant with a loss of synaptic function at the neuromuscular junction (Bezprozvanny and Hiesinger, 2013; Liu et al., 2013), the specific function of TDP-43 in maintaining synaptic functions has not been elucidated (Hirsch, 2007).
A number of different model systems have been used to study the effect of TDP-43 loss of function, including the fruit fly, Drosophila melanogaster (Casci and Pandey, 2015). The Drosophila ortholog of TDP-43 is named TBPH. Several studies have shown that TBPH is an essential gene, and knock-out mutants die just before or soon after adult eclosion and have severe locomotion defects in third instar larvae (Ayala et al., 2005; Feiguin et al., 2009; Hazelett et al., 2012; Diaper et al., 2013a). To try and understand the molecular basis for these defects, we performed a gene profiling experiment and showed that loss of TBPH led to changes in expression of almost 1000 genes in the CNS of third instar larvae (Hazelett et al., 2012). Subsequent experiments identified the Type II voltage gated calcium channel, cacophony, as a particularly important TBPH-regulated gene. In TBPH mutants, there was a 50% reduction in the protein levels of cacophony, and reversing this reduction in these mutants was sufficient to rescue the larval locomotion defects (Chang et al., 2014).
Cacophony is the major source of presynaptic calcium influx in the presynaptic motor terminals at the neuromuscular junctions (NMJ) (Kawasaki et al., 2004; Peng and Wu, 2007; Lee et al., 2014). It is also a major component of the somatodendritic calcium current in motor neurons (Ryglewski et al., 2012), suggesting that loss of cacophony from either or both of these locations in TBPH mutants could cause defective larval locomotion. In our current study, we extend these observations and find that, in addition to motor neurons, expression of cacophony specifically in the AVM001b and AVM002b neurons is sufficient to rescue TBPH-dependent larval locomotion.
Materials and Methods
Animals.
All Drosophila stocks were reared at 25°C using standard procedures (Greenspan, 1997). For all experiments, animals of both sexes were used. The D42-GAL4 motor neuron driver (w*; P{GawB}D42), UAS-CACOPHONY (w*; P{UAS-cac1-EGFP}422A), UAS-TRPA1 (w*; P{UAS-TrpA1(B).K}attP2), UAS-rpr.c (5824. w1118; P{UAS-rpr.C}14), and UAS-cacRNAi (y1v1; P{TRiP.JF02572}attP2) fly strains were obtained from the Bloomington stock center (http://flystocks.bio.indiana.edu/). The following Janelia GAL4 drivers were also obtain from the Bloomington stock center: R83E12-GAL4 (w*,P{GMR82E12-GAL4}attP2, R12A09-GAL4 (w*;P{GMR12A09-GAL4}attP2, R15A04-GAL4 (w*; P{GMR15A04-GAL4}attP2, R75C05-GAL4 (w*; P{GMR75C05-GAL4}attP2.
The TBPH-null mutant, UAS-TBPH, and TBPH-GAL4 driver lines were described previously (Hazelett et al., 2012). In most experiments, the background control line A1 was used. The A1 line was generated at the same time as the TBPH-null mutant line by precise excision of a transposon located just upstream of the TBPH gene (Hazelett et al., 2012). The GluRIIAsp line was obtained from the Davis Laboratory. In experiments with the GluRIIAsp fly line, the background control line w1118 was used (Petersen et al., 1997). The UAS-mCD8::GFP; UAS-LacZ::NLS fly line was a gift from Dr. Sean Speese.
Crawling assays.
Third instar larvae were rinsed in PBS and placed on 2% agarose plates at either room temperature or 30°C. The crawling paths of the larvae were recorded for 5 min using a Moticam 1000 connected to a PC and using the MIPlus07 software (Motic Images). The surface temperature of the agarose plates was monitored with a temperature sensor connected to a LabQuest Mini (Vernier) connected to a PC. The distance traveled in each video was traced and quantified using ImageJ software (http://imagej.nih.gov/ij/). The number of full posteroanterior peristaltic waveforms and the number of head turns were counted from these videos for each genotype. The distance of displacement for each forward crawl was also measured from these videos, as well as the duration of each wave, using the ImageJ software.
Electrophysiological methods.
Intracellular recordings were made from the larval body wall muscle 6 in abdominal segment 3 using glass microelectrodes as previously described (Engel, 2008). Recordings were performed at room temperature in extracellular HL3 saline, which contained the following (in mm): 70 NaCl, 5 KCl, 20 MgCl2, 10 NaHCO3 115 sucrose, 5 trehalose, 5 HEPES, and 0.5, 1.0, or 2.0 CaCl2. Membrane potentials were recorded using an Axoclamp-2A amplifier (Molecular Devices), digitized at 10 kHz and stored with a Digidata 1440A digitizer (Molecular Devices) connected to a PC (Dell). Excitatory junctional potentials (EJPs) were generated by injecting current into severed axons, at 0.5 Hz, via a suction electrode and an A310 Accupulser (World Precision Instruments) through an isolation transformer. The average single EJP amplitude of each recording was taken from 30 to 35 EJPs, whose amplitudes were measured using Clampfit 10.2 software (Molecular Devices, Axon Instruments). Spontaneous miniature endplate potentials (mEPPs) were recorded over 3 min and analyzed using Mini Analysis 6.0.0.7 (Synaptosoft). Quantal content was calculated as the ratio of mean EJP amplitude divided by the mean mEPP amplitude, and then averaging recordings across all NMJs for a given genotype. For acute cacophony block, larvae were incubated in plectreurys toxin (PLTX-II, from Alomone Labs) for 5 min.
To record patterns of motor activity from the ventral nerve cord, third instar larvae were cut open along their dorsal midline, pinned open, and extracellular recordings were made from peripheral nerves projecting from the second and seventh neuromeres of the intact CNS. Nerves were suctioned en passant with a glass suction electrode and recorded using an A-M Systems Differential AC Amplifier, digitized at 10 kHz, and stored with the Digidata 1440A digitizer as above. Recordings were made in HL-3.1, which contained the following (in mm): 70 NaCl, 5 KCl, 4 MgCl2, 10 NaHCO3 115 sucrose, 5 trehalose, 5 HEPES, and 1.8 mm CaCl2. To activate the motor program and stimulate fictive crawling, preparations were acutely incubated with 30 μm pilocarpine (Johnston and Levine, 1996). Recordings were made over 10 min and bandpass filtered (100 Hz to 10 kHz) using Clampex software (Molecular Devices).
Sampled data from the nerves in the second and seventh abdominal segments and a time stamp from the Digidata 1400A were passed to a custom program as comma-delimited text files. The program rectified and averaged the data into 1000–1200 50 ms bins. For each epoch of binned data, plots were prepared and auto- and cross-correlations were computed after subtracting the mean value for the epoch, which greatly increased the readability. The autocorrelation can be visualized as sliding a copy of the first half of the epoch across the full epoch and computing a correlation coefficient for each 50 ms increment in delay. Delays at which peaks align in the two data epochs produce high positive correlations, whereas delays at which peaks align with valleys produce high negative correlations. Generally, given measurements, Y1, Y2, …., YN at time X1, X2, …., XN, the lag k autocorrelation function r is defined as follows:
 |
Time of the first peak after t = 0 yields the period of the bursts in each trace. Cross-correlations are computed according to a similar method, except that the two half-data epochs are derived from data from both traces (second and seventh abdominal segments) in the same animal. The cross-correlation r at delay d of two series x(i) and y(i) where I = 0, 1, 2, …, N − 1, is defined as follows:
 |
Where mx and my are the means of the corresponding series.
Again, times at which peaks in two traces align produce high correlations. The first peak of the cross-correlation gave the time delay between the abdominal segment 7 (A7) and abdominal segment 2 (A2) bursts. Averaged, auto-, and cross-correlation data were written as comma-delimited text files that were readable by Microsoft Excel for additional manipulation (sometimes trace amplitudes were normalized) and plotting.
Immunohistochemistry.
To visualize the expression pattern of the GAL4 lines used, each driver line was crossed with the w*; UAS-mCD8::GFP; UAS-LacZ fly line. Third instar larva from these crosses were filleted, while keeping their CNSs intact, in ice-cold HL3 saline and fixed in 4% PFA in PBS with 1% Triton X for 10 min at room temperature, followed by 15 min at room temperature with agitation. Preparations were then washed with PBS with 1% Triton X three times for 5 min with agitation at room temperature. Samples were blocked in SEA Block (Calbiochem) for 15 min at room temperature. The preparations were then incubated 3 times overnight at 4°C in anti-GFP in 50% glycerol (1:200; ThermoFisher).
Following incubation in primary antibodies, the preparations were washed 4 times for 30 min in PBS with 1% Triton X with agitation. The preparations were then incubated 3 times overnight at 4°C in goat anti-rabbit IgG AlexaFluor-488 (ThermoFisher). Following incubation, samples were washed four times for 30 min in PBS with 1% Triton X and incubated for 30 min in Vectashield (Vectorlabs). The samples were then mounted in Vectashield and imaged using an Olympus FV1000 laser scanning confocal.
Experimental design and statistical analysis.
GraphPad Prism 6 software was used to generate the graphs in this work. Datasets were analyzed for significance using either an ordinary one-way ANOVA with a Sidak's multiple-comparison test (see Figs. 1, 3, 6, 7) or multiple t tests with Holm–Sidak multiple-comparison correction (see Fig. 5). A 95% CI was used to determine significance (p < 0.05). See figure legends for sample sizes, p values, and associated F and t values.
Figure 1.
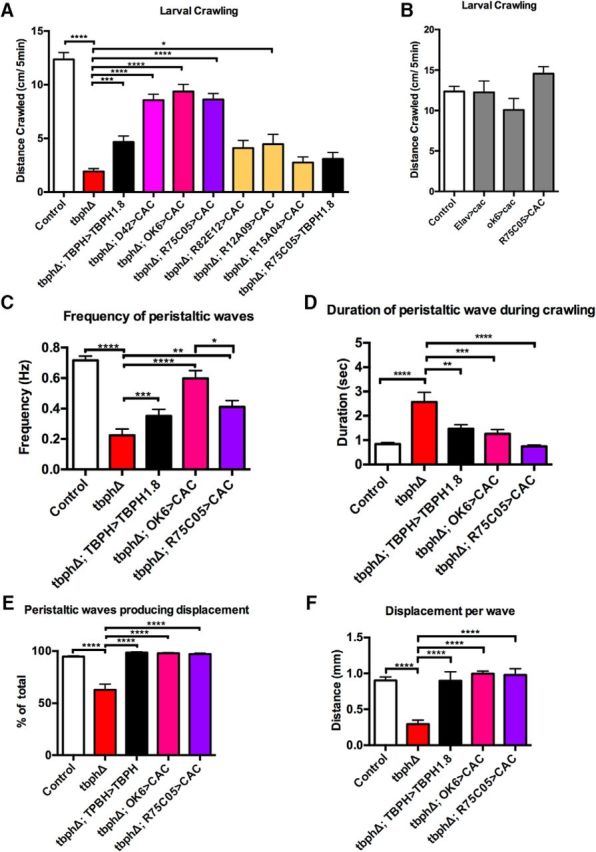
TBPH mutant larvae show defective motor behavior, which can be rescued by cacophony expression in selected neurons. A, TBPH-null mutants (tbphD) crawl shorter distances than control larvae, which is partially rescued with the expression of a TBPH cDNA using a TBPH-GAL4 driver in the TBPH mutant background. Expression of cacophony in motor neurons using either the D42-GAL4 or the more specific OK6-GAL4 driver also rescues this effect. A variety of addition drivers were also used to express cacophony, the most effective of which was the R75C05-GAL4 driver. Expression of tbph with the R75C05-GAL4 driver did not elicit a significant rescue of this effect. Data were analyzed using ordinary one-way ANOVA with a Sidak's multiple-comparisons test (F(7,154) = 48.58, p < 0.0001). ****p < 0.0001. ***p = 0.001. *p = 0.0238. B, Expression of cacophony, with a variety of driver lines in the wild-type background, has no effect on total distance crawled (F(3,53) = 2.492, p = 0.0701). C–F, A more detailed examination of the crawling behavior shows cacophony expression in either motor neurons or using the R75C05-GAL4 driver rescues all measured components of crawling behavior. C, Frequency of peristaltic waves. Loss of TBPH led to an 80% reduction in the number of complete waves that progressed along the body, which was partially rescued with tbph or cacophony expression (F(4,69) = 27.46, p < 0.0001). ****p < 0.0001. ***p = 0.05. **p = 0.0131. *p = 0.0172. D, Duration of peristaltic wave. The time taken for a complete peristaltic wave to traverse the larva was significantly increased in tbph mutants, which was rescued with either tbph or cacophony expression (F(4,44) = 11.40, p < 0.0001). ****p < 0.001. ***p = 0.004. **p = 0.0031. E, Number of waves producing displacement. The peristaltic waves in TBPH mutants were less likely to cause the larvae to move forward compared with controls, which was also rescued by tbph and cacophony expression (F(4,41) = 26.22, p < 0.0001). ****p < 0.001. F, Distance moved per peristaltic wave. For those waves that did produce displacement, the distance traveled was reduced in TBPH mutants, which was again rescued by TBPH and cacophony expression (R(4,41) = 16.8). ****p < 0.001. Data are mean ± SEM of at least 10 animals.
Figure 3.

Synaptic physiology at the larval NMJ is defective in TBPH mutants. A, Representative examples of EJPs from control and TBPH mutant larvae. B, EJP amplitude is unchanged in all genotypes tested. Data were analyzed using ordinary one-way ANOVA with a Sidak's multiple-comparison test (F(4,80) = 0.5015, p = 0.7347). C, Representative example of mEPP control and TBPH mutant larvae showing smaller and less frequency mEPPs in TBPH mutants. D, The mEPP amplitude is reduced in TBPH mutants (red), which is rescued by expression of TBPH (black), but not by expression of cacophony (pink). ***p < 0.001. *p = 0.0127. F(4,80) = 6.074 (p = 0.0003). E, The mEPP frequency is reduced in TBPH mutants and is not rescued by TBPH expression, but is rescued by expression of cacophony in motor neurons, but not with the R75C05-GAL4 driver. ***p = 0.0458. **p < 0.0001. F(4,79) = 7.644 (p < 0.0001). F, Quantal content. The quantal content of the EJPs was calculated as the ratio of EJP amplitude to mEPP amplitude as was increased in both TBPH and glutamate receptor mutants. The double mutant showed no further increase in quantal content. **p = 0.0143. *p = 0.047. R(2,61) = 3.940 (p = 0.0246). All experiments were performed in 1 mm calcium. Data are mean ± SEM of at least 10 animals.
Figure 6.

The motor output from the CNS of TBPH-null mutants is unpatterned and uncoordinated. A, Representative examples of the motor output show that control larvae exhibit regular, patterned motor bursts that progress from A7 and A2. By contrast, recordings from TBPH mutant larvae show infrequent, poorly defined, uncoordinated bursts (recordings from 3 different TBPH mutant larvae are shown). B, Representative examples of recordings from TBPH mutant larvae expressing TBPH and cacophony in motor neurons (OK6>CAC), and Cacophony in AV001b/2b cells (R75C05>CAC) showing that the patterned output is restored. C, D, Quantification of patterned bursting parameters using autocorrelation and cross-correlation of rectified recordings. Data were analyzed using ordinary one-way ANOVA with a Sidak's multiple-comparison test. C, Frequency of motor bursts. An autocorrelation analysis was performed on each recording to test whether the bursting showed nonrandomness. Less than 50% of the recordings from TBPH mutant larvae showed nonrandom bursting. For the TBPH mutant larvae that showed nonrandom bursting, the frequency of bursts was reduced by ∼50% compared with control larvae. This reduction was restored by expression of either tbph or cacophony in both motor neurons and the AVM001b/2b cells (R75C05>CAC; D). *p = 0.0002. **p = 0.004. ***p < 0.0001. R(4,43) = 11.39 (p < 0.0001). D, Bursting delay between A7 and A2. The offset between bursts in A7 and A2 was calculated from cross-correlations between A7 and A2 traces in each animal. The nonrandom pattern recordings from TBPH mutant larvae exhibited an offset between A7 and A2 motor neurons of approximately twice that of the control animals. This increased delay was restored by expression of both TBPH and cacophony. ****p = 0.0001. ***p = 0.0006. **p = 0.0016. *p = 0.0039. F(4,36) = 6.214 (p = 0.0007). All recordings were done in HL3.1 saline solution containing 1.8 mm calcium and 30 μm pilocarpine. Recordings were taken from A2 and A7 motor nerves of at least 10 animals of each genotype. Data are mean ± SEM of at least 10 animals.
Figure 7.

TrpA1 activation of AVM001b and AVM002b cells does not rescue crawling in TBPH mutants. A, Larval crawling was measured at 30°C over 5 min and total distance crawled recorded. TBPH mutants show no increase in larval crawling at 30°C. Driving UAS-TrpA1 with the R75C05-GAL4 driver in the TBPH mutant background showed no increase in larval crawling at 30°C compared with the parental controls. Driving cacophony with the R75C05-GAL4 driver, however, showed a significant increase in distance crawled at 30°C compared with the parental controls and restoring the total distance crawled to the same level as the wild-type controls. Driving UAS-TrpA1 expression with the OK6-GAL4 driver in a wild-type background produced immediate paralysis. Data were analyzed using ordinary one-way ANOVA with a Sidak's multiple-comparison test: ****p < 0.0001. ns1, p = 0.09; ns2, p = 0.0855; R(5,51) = 116.9, p < 0.0001. B, Larval crawling at room temperature over 5 min before being placed at 30°C. TBPH mutants show reduced crawling. Driving UAS-TrpA1 with the R75C05-GAL4 driver in the TBPH mutant background had no effect on larval crawling compared with the parental controls, whereas driving cacophony expression with the R75C05-GAL4 driver restored crawling at room temperature. ****p < 0.0001 (F(5,50) = 20.78, p < 0.0001). C, Elimination of AVM001b/2b neurons has no effect on larval crawling. Embryos in which UAS-rpr was driven with either ApplX-GAL4 or OK6-GAL4 failed to hatch into the first instar (denoted by Ο). By contrast, third instar animals in which UAS-rpr was driven by R75C05-GAL4 showed no defects in crawling behavior. Driving UAS-cacRNAi with the R75C05-GAL4 driver had no effect on crawling distance (F(3,50) = 2.251, p = 0.0938). D, Driving UAS-rpr and UAS-mCD8::GFP with R75C05-GAL4 does not produce fluorescence in the area occupied by the AVM001b/2b cells, indicating that those cells have been killed in development. Data are mean ± SEM of at least 10 animals.
Figure 5.
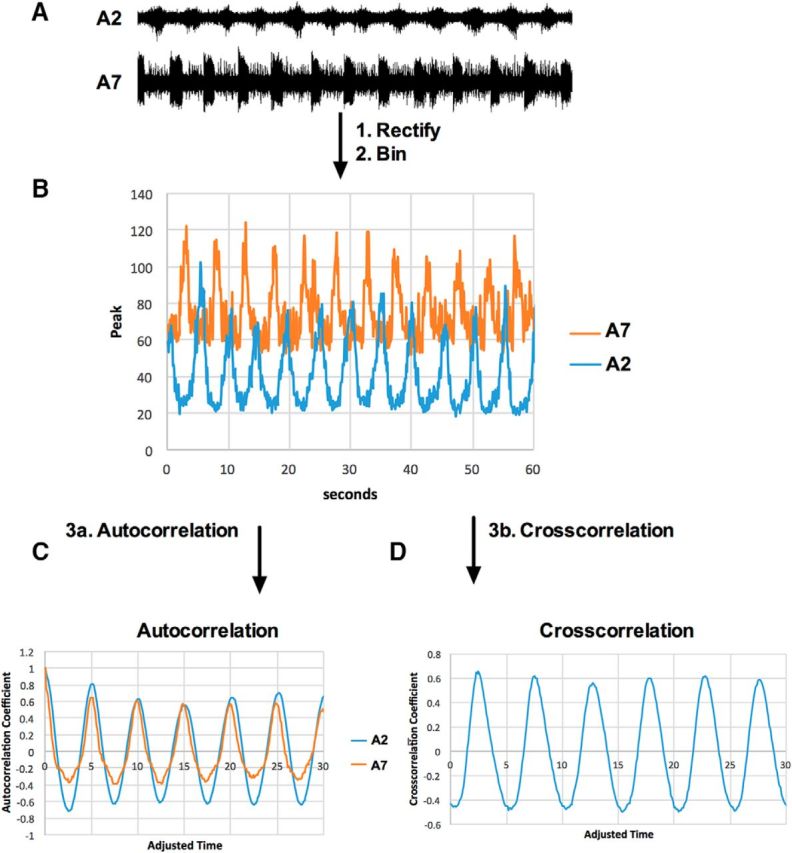
Extracellular recordings of motor bursts from intact nerves were analyzed via autocorrelation and cross-correlation. A, Extracellular recordings were made of intact nerves innervating A2 and A7. The example trace was taken over 60 s of a control animal. B, To smooth and decrease the size of the data file, each trace was rectified and data averaged into 1200 bins of 50 ms. C, To calculate the frequency of motor bursts in each recording, an autocorrelation was performed and the autocorrelation coefficient taken from the peak of the first lag (lag 1). The peak at t = 0 is 1.0 because the undelayed data correlate perfectly with themselves. D, To calculate the time between A7 and A2 bursts, a cross-correlation was performed on each set of recordings. The time between bursts was extrapolated from the first peak of the cross-correlation.
Results
Expressing cacophony in centrally located neurons rescues the TBPH mutant crawling defect
Larval crawling is a highly stereotyped behavior characterized by the synchronous contraction of muscles on the left and right side of the body (Fox et al., 2006). Forward crawling is generated by peristaltic waves of muscle contractions, which travel from posterior to anterior body wall segments (Fox et al., 2006). These peristaltic waves are generated by central pattern generators and are dependent upon firing of motor neurons in the ventral nerve cord (Inada et al., 2011). We have previously shown that loss of TBPH causes a dramatic reduction in the distance that larvae crawl, which can be rescued by driving cacophony expression in motor neurons using the D42-GAL4 driver (Chang et al., 2014). These results suggested that the reduced crawling distances were therefore due to a motor neuron-specific defect in the TBPH mutants. However, closer inspection of the D42-GAL4 driver has revealed that it shows expression not only in motor neurons of the ventral nerve cord, but also cells in the protocerebrum of the brain and peripheral sensory neurons along the body wall (Sanyal, 2009). Therefore, we examined another motor neuron driver described as being more specific for motor neurons, OK6-GAL4 (Sanyal, 2009). As previously described, expressing TBPH with the TBPH-GAL4 driver only partially rescued the larval crawling defects (Fig. 1A). By contrast, expressing cacophony in motor neurons using the OK6-GAL4 driver in the TBPH mutant was, like the D42-GAL4 driver, more effective at restoring the distance crawled than rescue with TBPH (Fig. 1A).
Although OK6-GAL4 showed more restricted expression specific to the motor neurons in the ventral nerve cord, it also showed expression in the brain hemispheres (Sanyal, 2009). To explore in more detail which cells require expression of cacophony to restore locomotion in the TBPH mutants, we tested a variety of additional driver lines that showed restricted expression in discreet sets of cells in the ventral nerve cord and brain and had been shown to affect larval crawling behaviors (Vogelstein et al., 2014). The drivers tested were as follows: R75C05-GAL4, which shows expression in the AVMOO1b/2b cells in the brain; R82E12-GAL4, which shows expression in the A08n cells in the CNS; R12A09-GAL4, which shows expression in the TINa cells in the ventral nerve cord; and R15A09-GAL4, which shows expression in the SeIN161 cells in the CNS (Vogelstein et al., 2014). Unlike using the TBPH driver to restore TBPH levels, which partially rescued adult lethality (Hazelett et al., 2012), none of the drivers used to express cacophony was able to rescue lethality.
These experiments revealed two additional drivers, R75C05-GAL4 and R12A09-GAL4, that were sufficient to rescue locomotion when used to express cacophony in the TBPH mutant background (Fig. 1A). Of the two, only R75C05-GAL4 was sufficient to rescue the crawling defect to the same extent as the motor neuron driver OK6-GAL4. R75C05-GAL4 drives expression in the AVM001b/2b neurons whose cell bodies are located in the protocerebrum of the Drosophila brain. Activation of these cells had previously been shown to produce a 20% increased probability of larval escape behavior during larval crawling (Vogelstein et al., 2014). Restoring TBPH only in the AVM001b/2b neurons using the R75C05-GAL4 driver in the TBPH mutant background increased the distance crawled relative to the TBPH mutant, although this increase was not statistically significant. To test whether increased levels of cacophony in a wild-type background was sufficient to increase crawling distance, we also expressed cacophony with Elav-GAL4, OK6-GAL4, and R75C05-GAL4 in a wild-type background. None of these larvae showed any difference in crawling distance compared with controls (Fig. 1B).
The total distance crawled is a relatively crude measure of the output of the motor program and is insufficient to identify specific defects in the motor program of TBPH mutant animals. To examine the TBPH mutant crawling behavior in more detail, we quantified the frequency of posterior-anterior peristaltic waves, the time in which it took these waves to traverse the body completely, and the total displacement resulting from each wave. We then examined how driving cacophony in all motor neurons and more discreetly in the AV00M1b/2b cells altered these parameters (Fig. 1).
TBPH mutants show an 80% reduction in the number of complete posterior-anterior peristaltic waves in the 5 min observation period compared with background controls (Fig. 1C). This reduction was partially rescued by expressing tbph with the tbph driver, and a more complete rescue was achieved by expressing cacophony in motor neurons using the OK6-GAL4 driver (Fig. 1C). Similarly, driving cacophony with R75C05-GAL4 also partially rescued this parameter, although not as fully as the motor neuron driver (Fig. 1C). The time taken for a complete peristaltic wave to travel the length of the larvae took approximately twice as long in TBPH mutant larvae compared with control animals (Fig. 1D). This parameter was also rescued by tbph and by driving cacophony with both the motor neuron and R75C05-GAL4 drivers. Of the 50 complete posterior-anterior peristaltic waves TBPH mutants completed over 5 min, ∼60% of these failed to produce forward displacement (Fig. 1D), which was measured as averaging 0.25 mm (Fig. 1F). This contrasts with the control and all the rescues in which almost each complete peristaltic wave produced displacement of ∼1.0 mm (Fig. 1E,F).
These results show that the behavioral defects in TBPH mutants can be rescued not only by restoring cacophony broadly in all motor neurons, but also, surprisingly, by selectively restoring it in the discreet set of AVM001b/2b neurons located in the brain. There appear to be relatively little qualitative or quantitative differences in the restoration of the behavior by expressing cacophony in these two different populations of neurons.
To verify the expression patterns of the OK6-GAL4 and R75C05-GAL4 driver lines used in our experiments, both driver lines were crossed with flies expressing UAS-mCd8::GFP. Animals expressing OK6>mCD8::GFP show broad expression within lateral motor neurons of the neuropil, as well as expression in cells located in the brain (Fig. 2B). This expression pattern matches those previously reported by Vogelstein et al. (2014). Animals expressing R75C05>mCD8::GFP show localized, bilateral expression in two cells whose cell bodies appear to be housed in the protocerebrum of the brain (Fig. 2C). This expression is identical to the expression pattern reported by Vogelstein et al. (2014), in which those cells were identified as AVM001b and AVM002b. It should be noted that axonal projections from those cell bodies were occasionally observed extending into the ventral nerve cord, but never leaving the CNS. To verify the AVM001b/2b cells were present in tbph mutants, we also expressed R75C05>mCD8::GFP in the TBPH mutant background (Fig. 2D). The AVM001b/2b cells appear to be intact in the tbph mutant background.
Figure 2.

Expression patterns of the GAL4 drivers. A, Expression pattern of D42-GAL4 driving UAS-mCD8::GFP expression. Signal is enhanced with GFP antibody. B, Expression pattern of OK6-GAL4 driving UAS-mCD8::GFP expression; signal is enhanced with GFP antibody. C, Expression of R75C05-GAL4 driving UAS-mCD8::GFP; signal is enhanced with GFP antibody. D, Expression of R75C05-GAL4 driving UAS-mCD8::GFP in the tbph mutant background (TBPHΔ). Ten larvae were visualized for each construct. Scale bars, 50 μm.
Synaptic transmission at the NMJ
Cacophony is required for evoked neurotransmitter release at the larval NMJ (Kawasaki et al., 2004; Kuromi et al., 2004). As we measured a significant reduction of presynaptically localized cacophony at the larval NMJ in tbph mutants (Chang et al., 2014), it seemed likely that evoked synaptic transmission would be defective in tbph mutants. Surprisingly, when we measured the amplitude of the evoked EJPs in TBPH mutant larvae, we found no significant difference compared with controls (Fig. 3A). We made these measurements at a variety of calcium concentrations as the size of the cacophony-dependent EJP amplitude is dependent on the external calcium concentration (Lee et al., 2014), but detected no significant difference in the EJP amplitude comparing tbph mutants with controls in 1 mm extracellular calcium (Fig. 3A). We also failed to see a difference at calcium concentrations between 0.5 and 2 mm (data not shown). In addition to measuring evoked neurotransmitter release, we analyzed spontaneous neurotransmitter release by measuring the amplitude and frequency of mEPPs. At 1 mm calcium, tbph mutants had significantly smaller mEPPs compared with control animals (Fig. 3B,C). This effect was also detected at 2 mm calcium, although not at 0.5 mm (data not shown). These observations were similar to those reported for another tbph mutant allele (Diaper et al., 2013a, b). To determine whether this reduction was due to the loss of TBPH, we expressed tbph in the tbph mutants using the TBPH-GAL4 driver. The mEPP amplitude was significantly increased, suggesting that the reduced size was due to loss of tbph. However, expressing cacophony either in the motor neurons or with the R75C05-GAL4 driver failed to rescue this phenotype (Fig. 3C).
We also quantified the frequency of spontaneous vesicle release in tbph mutants. The frequency of mEPPs in tbph mutants was significantly lower than that of control animals (Fig. 3E). This reduction was not rescued by expressing tbph under the control of the tbph promoter (Fig. 3E). It was, however, restored by expressing cacophony in motor neurons using the OK6 driver (Fig. 3E). Notably, there was no significant increase in the frequency of mEPPs when we expressed cacophony with the R12A09-GAL4 (data not shown) or R75C05-GAL4 drivers (Fig. 3D), suggesting that the alterations in mEPP frequency were not associated with the behavioral defects of tbph mutants.
The finding that the tbph mutants have unchanged EJP amplitude despite having reduced mEPP amplitude suggested that synaptic homeostasis had compensated for the reduced mEPP amplitude. Synaptic homeostasis is the process whereby synaptic strength is maintained within physiological ranges, especially when faced with perturbations of synaptic function, such as the loss of presynaptic voltage-gated calcium channels or postsynaptic glutamate receptors (Turrigiano, 2007). At the Drosophila NMJ, there is accumulating evidence for several parallel signaling pathways acting to generate changes in quantal content to compensate for an altered postsynaptic response (Turrigiano, 2007). Based on these findings, we calculated quantal content of the EJP in TBPH mutants show that there is a significant increase in quantal content (Fig. 3E), confirming that the NMJ in tbph mutants is capable of synaptic homeostasis. Previous studies have shown that the signaling pathways that generate homeostasis converge on cacophony to enhance presynaptic calcium influx and thus neurotransmitter release (Frank et al., 2006; Müller and Davis, 2012; Frank, 2014).
As cacophony levels are reduced in tbph mutants and they nevertheless exhibit synaptic homeostasis, it is possible that no further homeostasis was possible. To test this, we imposed a postsynaptic challenge by genetically expressing a loss-of-function allele of the α-2 subunit of the glutamate receptor (GluRIIAsp16) in the tbph mutant (Petersen et al., 1997). As expected, the EJP amplitude in the GluRIIASP16 mutants was unchanged while the mEPP amplitude was reduced, thereby indicating the expected higher quantal content compared with controls (Fig. 3E) as previously described (Petersen et al., 1997). When this mutation was combined with the tbph mutant, there was no additional increase in quantal content (Fig. 3E).
An unexpected result from the analysis of synaptic transmission was the apparent cacophony dependence of the frequency of mEPPs (Fig. 3E) as this parameter is not normally thought to be directly regulated by voltage-dependent calcium channels (Lee et al., 2014). To confirm that the frequency of mEPPs was cacophony dependent, we used the small peptide PLTX-II to block cacophony function. PLTX-II is a specific, irreversible inhibitor of Type II voltage-gated calcium channels and blocks cacophony function specifically (King, 2007). We measured both EJP amplitudes and mEPP frequencies in the presence of a high concentration (2 nm) of PLTX-II (Fig. 4). In the presence of PLTX-II, EJP amplitude was, as expected, reduced by >95% in both the TBPH mutant and control animals, confirming the cacophony dependence of the EJPs (Fig. 4A). The frequencies of mEPPs were similarly reduced by >60% in both the TBPH mutant and control animals, suggesting that acute cacophony function is also required to regulate the spontaneous release of vesicles (Fig. 4B). The amplitude of the mEPPs was, as expected, unaffected by PLTX-II (Fig. 4C).
Figure 4.

Effect of the Cacophony irreversible antagonist PLTX-II on NMJ synaptic transmission. A, The EJP amplitude is blocked by >90% in both control and TBPH mutant larvae. Data were analyzed using multiple t tests with Holm–Sidak multiple-comparison correction. ****p < 0.00001 (t(18) = 11.8485, t(20) = 7.13139). B, PLTX-II significantly reduces mEPP frequency in both control and TBPH larvae. ****p = 0.000268908 (t(18) = 4.45932). ***p = 0.000231752 (t(20) = 4.7188). C, PLTX-II has no effect on mEPP amplitudes in either control or TBPH larvae (p = 0.0947586, t(18) = 1.75391; p = 0.151073, t(20) ratio = 1.54309). Data are mean ± SEM of at least 10 animals.
These results demonstrate some defects in synaptic transmission due to the loss of TBPH, some of which can be reversed by expression of cacophony in motor neurons. However, the lack of concordance between the cacophony dependence of synaptic physiology and behavior suggests that these defects are not sufficient to explain the defects in larval locomotion.
CNS motor output
Forward larval crawling is generated by peristaltic waves traversing the length of the larval body wall in an posterior to anterior fashion (Fox et al., 2006). The rhythmicity of these waves is likely to be generated by a central pattern generator (CPG) (Marder and Bucher, 2001; Kohsaka et al., 2012). The CPG generates rhythmic bursts of activity within a segment and also coordinates patterns of bursts between segments to generate a delay between bursts in adjacent segments (Inada et al., 2011; Kohsaka et al., 2012; Fushiki et al., 2016). Experiments blocking the activity of motor neurons have shown that motor neurons in the ventral nerve cord are not merely downstream effectors of the CPG, but function to regulate the propagation of the patterned activity between adjacent segments (Inada et al., 2011). Our behavioral analysis has shown that tbph mutants show far fewer anteroposterior peristaltic waves (Fig. 1B), a higher occurrence of incomplete waves (Fig. 1D), and longer times to completions of their peristaltic waves (Fig. 1C) than background controls. These components of the behavior can be restored by expressing cacophony in both motor neurons and neurons in the brain. To determine whether restoring expression of cacophony in these populations of neurons had the same effect on the output from the CPG, we monitored the motor output from the CNS in semi-intact larvae.
We made focal extracellular recordings en passant from intact peripheral nerves projecting on muscle 6/7 in A2 and A7 (Figs. 5, Fig. 6). When the preparation was bathed only in saline solution, we observed spontaneous bursting in control animals that became more reliable with the addition of pilocarpine. Subsequent experiments used pilocarpine (30 μm) to activate the motor program and has been shown previously to stimulate fictive crawling (Johnston and Levine, 1996; Baudoux et al., 1998). No consistent bursting was observed in mutant animals, even with the addition of pilocarpine, although nonpatterned activity was clearly visible (Fig. 6A).
To quantify the motor patterns from the nerves innervating A7 and A2, we first rectified each trace and grouped each data point into bins of 50 ms (Fig. 5A,B). Each rectified trace was then run through an autocorrelation function (Fig. 5C). Autocorrelation is a mathematical tool for finding nonrandom patterns within a time-dependent dataset and can be used to measure the similarity between observations as a function of the time lag between them (Ryan, 2006). From the lag time of the first peak, we calculated the frequency of bursts for the nerves innervating A7 and A2 (Fig. 5C). To calculate the lag time between corresponding A7 and A2 bursts, the A7 and A2 recordings were cross-correlated. Cross-correlation is a mathematical tool in which the similarity of two series is measured as a function of the displacement of one relative to the other (Ryan, 2006). The lag time between A7 and A2 bursts was calculated from the cross-correlation coefficient (Fig. 5D).
Of the recordings made on the tbph-null mutant, only 30% of recordings elicited an autocorrelation peak. These animals showed an average frequency of 5 bursts per minute, a 50% reduction from the 10 burst per minute of the control (Fig. 6C). Restoring TBPH with the TBPH-GAL4 driver reversed this effect. Restoring cacophony with either the OK6 and R75C05 driver also reversed this effect. It should be noted that, because these recordings are made en passant, there is likely intact sensory neuron feedback acting on the motor program.
Of the 30% of tbph-null mutant recordings eliciting a cross-correlation peak, the average delay between segments 7 and 2 was 8 s (Fig. 6D). This delay is twice as long as the 4 s delay measured in controls. Restoring TBPH with the TBPH-GAL4 driver reversed this effect. Restoring cacophony with the OK6 and R75C05 driver also reversed this effect. Indeed, restoring cacophony with the R75C05 driver produced the most robust reversal of this effect.
The AVM001b/2b cells are not an integral component of the motor circuit
Activation of AV001b/2b cells has been reported to drive escape behavior in crawling larvae (Vogelstein et al., 2014), and we have shown genetically driving cacophony in these cells, in the TBPH mutant restores larval crawling behavior and rhythmic motor bursts. We therefore asked whether this rescue could be reproduced by acute activation of these cells in the TBPH mutant background. To answer this question, we drove the temperature-activated channel, TrpA1, under UAS/GAL4 control with the R75C05-GAL4 driver in the TBPH mutant background. TrpA1 (transient receptor potential A1) is one of three thermosensation channels in Drosophila that detect subtle changes in ambient temperature and can be activated by warm temperatures (Neely et al., 2011; Luo et al., 2017).
Larval crawling was measured at 25°C, when TrpA1 is inactive, and 30°C, a temperature that activates TrpA1, over 5 min (Fig. 7). To confirm that the UAS-TrpA1 was being activated on our apparatus, we expressed it using the OK6-GAL4 driver and observed that, at 30°C, the animals were, as expected, completely paralyzed (Fig. 7A). At room temperature, they crawled the same distance as controls (Fig. 7B). Driving UAS-TrpA1 with the R75C05-GAL4 driver in the TBPH mutant background had no effect on crawling distance at 30°C (Fig. 7A), crawling no further than either parental control. Because we could not rule out the possibility of a sensory defect in the TBPH mutants that could be causing them to simply not sense the elevated temperature, we repeated the experiment with animals in which we drove UAS-cacophony with the R75C05-GAL4 driver. These animals show an increase in distance crawled over time at 30°C, a similar increase to that observed in the control animals (Fig. 7A). These results suggest that the cacophony rescue is not solely due to the activation of the AVM001b/2b cells, but rather is likely dependent upon the chronic restoration of cacophony in the TBPH mutants throughout development. To test whether reducing cacophony in the AVM001b/2b neurons had the opposite effect, we expressed UAS-cacRNAi in these cells and found that this had no effect on larval crawling (Fig. 7C). Similarly, driving this UAS line with either the D42-GAL4 or Elav-GAL4 drivers also had no effect on crawling distance (data not shown).
As the AVM001b/2b cells act on the motor circuit, we tested whether eliminating these cells would inhibit larval crawling behavior (Fig. 7C). To kill these cells, we made use of the gene reaper (rpr), the protein that activates programmed cell death in Drosophila (Chen et al., 1996; Mohseni et al., 2009). We drove UAS-rpr in a wild-type background under the pan neuronal driver Appl-GAL4, in addition to the OK6-GAL4 and R75C05-GAL4 drivers. To verify that expression of UAS-rpr with R75C05-GAL4 was indeed killing the cells, we simultaneously drove UAS-mCD8::GFP with rpr and tested for fluorescence in the brain (Fig. 7D). We saw no fluorescence in the brain, verifying that expression of UAS-rpr was sufficient to kill the cells. Embryos in which UAS-rpr was driven with ApplX-GAL4 or OK6-GAL4 failed to hatch into the first instar. Driving UAS-rpr with R75C05-GAL4, however, produced third instars that crawled normally (Fig. 7C). It therefore appears that killing these cells is not sufficient to inhibit larval crawling.
Discussion
We have shown that genetically restoring the Type II voltage gated calcium channel cacophony in the AVM001b and AVM002b cells in our TBPH mutant is sufficient to restore larval crawling and rhythmic motor bursts. TBPH mutants show significant defects in larval crawling (Fig. 1), subtle changes in spontaneous release at the neuromuscular junction (Fig. 3), and significant loss of rhythmic motor bursts (Fig. 6). Surprisingly, driving cacophony in the AVM001b/2b cells, in the TBPH mutant background, was sufficient to rescue both larval crawling and rhythmic motor pattern. It was not, however, sufficient to rescue the changes in synaptic physiology at the neuromuscular junction, suggesting that changes in physiology at this location are not sufficient to drive whole-animal motor defects.
TBPH has previously been reported to function in regulating both mEPP amplitude and frequency of spontaneous release (Diaper et al., 2013a). Here, we failed to elicit a rescue in the frequency of spontaneous release, but we did observe a TBPH-dependent rescue in mEPP amplitude, confirming a function for TBPH in regulating the amplitude of mEPPs. Spontaneous neurotransmitter release is a core element of synaptic communication in mature neurons and is thought to function in modulating activity-dependent transmission and synaptic homeostasis (Fatt and Katz, 1952; Frank et al., 2006; Lee et al., 2010). Our inability to rescue changes in mEPP amplitude while rescuing rhythmic motor bursts suggests that this parameter does not directly regulate larger, systems-dependent behavior, such as crawling. This conclusion is further supported by the fact that, although driving cacophony with the R75C05-GAL4 driver rescued TBPH mutant crawling, these animals showed no rescue in frequency of spontaneous release at the neuromuscular junction. Therefore, it appears that TBPH-dependent changes in physiology at the neuromuscular junction are not driving the observed crawling defects.
Cacophony is the primary voltage-gated calcium channel at the Drosophila NMJ and is known to regulate evoked neurotransmission (Lee et al., 2014). It localizes with other presynaptic active zone proteins, bruchpilot and RIM, at active zones; and expression of hypomorphic versions of the channel, such as cacs, causes significant reductions in EJP amplitude (Kawasaki et al., 2004; Fouquet et al., 2009; Graf et al., 2012; Lee et al., 2014). Blocking cacophony channels irreversibly with PLTX-II similarly caused a reduction in EJP amplitude (Fig. 4). Although cacophony is known to regulate evoked neurotransmitter release, it is not known to directly regulate spontaneous release (Lee et al., 2014). Therefore, not only was it initially surprising that our TBPH mutant showed no reduction in evoked release, but equally surprising that driving UAS-cac with the OK6-GAL4 driver was sufficient to rescue the frequency of spontaneous release. To test directly whether cacophony regulates the frequency of spontaneous release, we pharmacologically blocked endogenous cacophony channels with PLTX-II. Blocking cacophony with PLTX-II reduces the frequency of spontaneous release, while not reducing mEPP amplitude (Fig. 4). Therefore, it appears that cacophony does indeed function in regulating the basal frequency of spontaneous release at the NMJ.
Activation of AVM001b and AVM002b cells with channel rhodopsin caused a slightly higher probability of larvae to present escape behavior during crawling (Vogelstein et al., 2014). The function of these cells in third instar larvae or adults has not been characterized beyond that report. Therefore, it was quite surprising that restoring cacophony in these cells, in the TBPH mutant, was sufficient to rescue both the crawling and rhythmic motor bursts. This result suggests that these cells are either part of, or act on, the motor program in such a way as to regulate the rhythmic pattern of motor neuron bursts. How they do this remains unclear. Restoring TBPH only in these cells was not sufficient to rescue the crawling defect. However, this could have been due to the counteracting effect of cytotoxicity of TBPH overexpression (Hanson et al., 2010; Vanden Broeck et al., 2013; Casci and Pandey, 2015).
To determine whether the restoration of locomotion is specific to cacophony expression or whether it could also occur with general activation of these cells, we tested whether activation of the AVM001b/2b cells with TrpA1 in the TBPH mutant could also restore locomotion. We observed no rescue of the crawling defect, suggesting that it was dependent upon the restoration of cacophony specifically, not just an acute activation of these cells (Fig. 7). The possibility of a thermosensory defect in TBPH mutants was also ruled out, as animals expressing cacophony under the R75C05-GAL4 driver completely rescued the increase in distance crawled at 30°C. We also showed that eliminating these cells had no effect on crawling (Fig. 7). Therefore, it seems that the rescue of crawling behavior is specific to the chronic restoration of cacophony in the AVM001b/2b cells through development.
In conclusion, our data show that TBPH-dependent defects at the NMJ are insufficient to explain the TBPH-dependent locomotion observed in Drosophila larvae. Furthermore, restoring the TBPH-dependent loss of cacophony in either motor neurons or a discrete set of central neurons is sufficient to restore the locomotion defects. If these effects are mirrored in human TDP-43 proteinopathies, our observations could open new avenues to investigate alternative therapeutic targets for these neurodegenerative diseases.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant NS071186 and the Amyotrophic Lateral Sclerosis Association. Stocks obtained from the Bloomington Drosophila Stock Center (National Institutes of Health P40OD018537) were used in this study. We thank members of the D.B.M. laboratory, especially Dr. Jer-Cherng Chang, for critical comments on the manuscript and helpful discussions during the course of this work; Dr. Graeme Davis for the gift of the GluRIIAsp fly line; and Dr. Sean Speese for the gift of the UAS-mcd8::GFP; UAS-LacZ::NLS fly line.
The authors declare no competing financial interests.
References
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351:602–611. 10.1016/j.bbrc.2006.10.093 [DOI] [PubMed] [Google Scholar]
- Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE (2005) Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 348:575–588. 10.1016/j.jmb.2005.02.038 [DOI] [PubMed] [Google Scholar]
- Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci 121:3778–3785. 10.1242/jcs.038950 [DOI] [PubMed] [Google Scholar]
- Baudoux S, Duch C, Morris OT (1998) Coupling of efferent neuromodulatory neurons to rhythmical leg motor activity in the locust. J Neurophysiol 79:361–370. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Hiesinger PR (2013) The synaptic maintenance problem: membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol Neurodegener 8:23. 10.1186/1750-1326-8-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casci I, Pandey UB (2015) A fruitful endeavor: modeling ALS in the fruit fly. Brain Res 1607:47–74. 10.1016/j.brainres.2014.09.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Hazelett DJ, Stewart JA, Morton DB (2014) Motor neuron expression of the voltage-gated calcium channel cacophony restores locomotion defects in a Drosophila, TDP-43 loss of function model of ALS. Brain Res 1584:39–51. 10.1016/j.brainres.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Lee P, Otto L, Abrams J (1996) Apoptotic activity of REAPER is distinct from signaling by the tumor necrosis factor receptor 1 death domain. J Biol Chem 271:25735–25737. 10.1074/jbc.271.42.25735 [DOI] [PubMed] [Google Scholar]
- Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G (2012) TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res 1462:16–25. 10.1016/j.brainres.2012.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaper DC, Adachi Y, Lazarou L, Greenstein M, Simoes FA, Di Domenico A, Solomon DA, Lowe S, Alsubaie R, Cheng D, Buckley S, Humphrey DM, Shaw CE, Hirth F (2013a) Drosophila TDP-43 dysfunction in glia and muscle cells cause cytological and behavioural phenotypes that characterize ALS and FTLD. Hum Mol Genet 22:3883–3893. 10.1093/hmg/ddt243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaper DC, Adachi Y, Sutcliffe B, Humphrey DM, Elliott CJ, Stepto A, Ludlow ZN, Vanden Broeck L, Callaerts P, Dermaut B, Al-Chalabi A, Shaw CE, Robinson IM, Hirth F (2013b) Loss and gain of Drosophila TDP-43 impair synaptic efficacy and motor control leading to age-related neurodegeneration by loss-of-function phenotypes. Hum Mol Genet 22:1539–1557. 10.1093/hmg/ddt005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG. (2008) Synaptic electrophysiology of the Drosophila neuromuscular junction. In: Drosophila neurobiology, a laboratory handbook (Bing Z, Freeman MR, Waddell S, eds), pp 103–120. New York: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Fatt P, Katz B (1952) Spontaneous subthreshold activity at motor nerve endings. J Physiol 117:109–128. 10.1113/jphysiol.1952.sp004735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F, Godena VK, Romano G, D'Ambrogio A, Klima R, Baralle FE (2009) Depletion of TDP-43 affects Drosophila motor neurons terminals synapsis and locomotive behavior. FEBS Lett 583:1586–1592. [DOI] [PubMed] [Google Scholar]
- Fouquet W, Owald D, Wichmann C, Mertel S, Depner H, Dyba M, Hallermann S, Kittel RJ, Eimer S, Sigrist SJ (2009) Maturation of active zone assembly by Drosophila Bruchpilot. J Cell Biol 186:129–145. 10.1083/jcb.200812150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox LE, Soll DR, Wu CF (2006) Coordination and modulation of locomotion pattern generators in Drosophila larvae: effects of altered biogenic amine levels by the tyramine beta hydroxlyase mutation. J Neurosci 26:1486–1498. 10.1523/JNEUROSCI.4749-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA. (2014) How voltage-gated calcium channels gate forms of homeostatic synaptic plasticity. Front Cell Neurosci 8:40. 10.3389/fncel.2014.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW (2006) Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron 52:663–677. 10.1016/j.neuron.2006.09.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushiki A, Zwart MF, Kohsaka H, Fetter RD, Cardona A, Nose A (2016) A circuit mechanism for the propagation of waves of muscle contraction in Drosophila. Elife 5:e13253. 10.7554/eLife.13253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Valakh V, Wright CM, Wu C, Liu Z, Zhang YQ, DiAntonio A (2012) RIM promotes calcium channel accumulation at active zones of the Drosophila neuromuscular junction. J Neurosci 32:16586–16596. 10.1523/JNEUROSCI.0965-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenspan RJ. (1997) Fly pushing: the theory and practice of Drosophila genetics. Cold Spring Harb Lab, pp3–23. [Google Scholar]
- Guillain G, Bailey P (1959) J.-M. Charcot, 1825–1893: his life, his work. New York: Hoeber. [Google Scholar]
- Hanson KA, Kim SH, Wassarman DA, Tibbetts RS (2010) Ubiquilin modifies TDP-43 toxicity in a Drosophila model of amyotrophic lateral sclerosis (ALS). J Biol Chem 285:11068–11072. 10.1074/jbc.C109.078527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelett DJ, Chang JC, Lakeland DL, Morton DB (2012) Comparison of parallel high-throughput RNA sequencing between knockout of TDP-43 and its overexpression reveals primarily nonreciprocal and nonoverlapping gene expression changes in the central nervous system of Drosophila. G3 (Bethesda) 2:789–802. 10.1534/g3.112.002998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch NP. (2007) Neuromuscular junction in health and disease. Br J Anaesth 99:132–138. 10.1093/bja/aem144 [DOI] [PubMed] [Google Scholar]
- Ihara R, Matsukawa K, Nagata Y, Kunugi H, Tsuji S, Chihara T, Kuranaga E, Miura M, Wakabayashi T, Hashimoto T, Iwatsubo T (2013) RNA binding mediates neurotoxicity in the transgenic Drosophila model of TDP-43 proteinopathy. Hum Mol Genet 22:4474–4484. 10.1093/hmg/ddt296 [DOI] [PubMed] [Google Scholar]
- Inada K, Kohsaka H, Takasu E, Matsunaga T, Nose A (2011) Optical dissection of neural circuits responsible for Drosophila larval locomotion with Halorhodopsin. PLoS One 6:e29019. 10.1371/journal.pone.0029019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RM, Levine RB (1996) Crawling motor patterns induced by pilocarpine in isolated larval nerve cords of Manduca sexta. J Neurophysiol 76:3178–3195. [DOI] [PubMed] [Google Scholar]
- Kawasaki F, Zou B, Xu X, Ordway RW (2004) Active zone localization of presynaptic calcium channels encoded by the cacophony locus of Drosophila. J Neurosci 24:282–285. 10.1523/JNEUROSCI.3553-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GF. (2007) Modulation of insect Ca(v) channels by peptidic spider toxins. Toxicon 49:513–530. 10.1016/j.toxicon.2006.11.012 [DOI] [PubMed] [Google Scholar]
- Kohsaka H, Okusawa S, Itakura Y, Fushiki A, Nose A (2012) Development of larval motor circuits in Drosophila. Dev Growth Differ 54:408–419. 10.1111/j.1440-169X.2012.01347.x [DOI] [PubMed] [Google Scholar]
- Kuromi H, Honda A, Kidokoro Y (2004) Ca2+ influx through distinct routes controls exocytosis and endocytosis at Drosophila presynaptic terminals. Neuron 41:101–111. 10.1016/S0896-6273(03)00815-8 [DOI] [PubMed] [Google Scholar]
- Kwong LK, Neumann M, Sampathu DM, Lee VM, Trojanowski JQ (2007) TDP-43 proteinopathy: the neuropathology underlying major forms of sporadic and familial frontotemporal lobar degeneration and motor neuron disease. Acta Neuropathol 114:63–70. 10.1007/s00401-007-0226-5 [DOI] [PubMed] [Google Scholar]
- Lee J, Ueda A, Wu CF (2014) Distinct roles of Drosophila cacophony and Dmca1D Ca2+ channels in synaptic homeostasis: genetic interactions with slowpoke Ca2+-activated BK channels in presynaptic excitability and postsynaptic response. Dev Neurobiol 74:1–15. 10.1002/dneu.22120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Yasuda R, Ehlers MD (2010) Metaplasticity at single glutamatergic synapses. Neuron 66:859–870. 10.1016/j.neuron.2010.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Brännström T, Andersen PM, Pedrosa-Domellöf F (2013) Distinct changes in synaptic protein composition at neuromuscular junctions of extraocular muscles versus limb muscles of ALS donors. PLoS One 8:e57473. 10.1371/journal.pone.0057473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Shen WL, Montell C (2017) TRPA1 mediates sensation of the rate of temperature change in Drosophila larvae. Nat Neurosci 20:34–41. 10.1038/nn.4416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, Bucher D (2001) Central pattern generators and the control of rhythmic movements. Curr Biol 11:R986–R996. 10.1016/S0960-9822(01)00581-4 [DOI] [PubMed] [Google Scholar]
- Mohseni N, McMillan SC, Chaudhary R, Mok J, Reed BH (2009) Autophagy promotes caspase-dependent cell death during Drosophila development. Autophagy 5:329–338. 10.4161/auto.5.3.7444 [DOI] [PubMed] [Google Scholar]
- Müller M, Davis GW (2012) Transsynaptic control of presynaptic Ca2+ influx achieves homeostatic potentiation of neurotransmitter release. Curr Biol 22:1–7. 10.1016/j.sbi.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely GG, Keene AC, Duchek P, Chang EC, Wang QP, Aksoy YA, Rosenzweig M, Costigan M, Woolf CJ, Garrity PA, Penninger JM (2011) TrpA1 regulates thermal nociception in Drosophila. PLoS One 6:e24343. 10.1371/journal.pone.0024343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M. (2009) Molecular neuropathology of TDP-43 proteinopathies. Int J Mol Sci 10:232–246. 10.3390/ijms10010232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314:130–133. 10.1126/science.1134108 [DOI] [PubMed] [Google Scholar]
- Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, Gallo JM, Hortobágyi T, Shaw CE, Rogelj B (2010) Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain 133:1763–1771. 10.1093/brain/awq111 [DOI] [PubMed] [Google Scholar]
- Peng IF, Wu CF (2007) Drosophila cacophony channels: a major mediator of neuronal Ca2+ currents and a trigger for K+ channel homeostatic regulation. J Neurosci 27:1072–1081. 10.1523/JNEUROSCI.4746-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19:1237–1248. 10.1016/S0896-6273(00)80415-8 [DOI] [PubMed] [Google Scholar]
- Ryan TP. (2006) Modern engineering statistics. New York: Wiley. [Google Scholar]
- Ryglewski S, Lance K, Levine RB, Duch C (2012) Ca(v)2 channels mediate low and high voltage-activated calcium currents in Drosophila motoneurons. J Physiol 590:809–825. 10.1113/jphysiol.2011.222836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal S. (2009) Genomic mapping and expression patterns of C380, OK6 and D42 enhancer trap lines in the larval nervous system of Drosophila. Gene Expr Patterns 9:371–380. 10.1016/j.gep.2009.01.002 [DOI] [PubMed] [Google Scholar]
- Turrigiano G. (2007) Homeostatic signaling: the positive side of negative feedback. Curr Opin Neurobiol 17:318–324. 10.1016/j.conb.2007.04.004 [DOI] [PubMed] [Google Scholar]
- Vanden Broeck L, Naval-Sánchez M, Adachi Y, Diaper D, Dourlen P, Chapuis J, Kleinberger G, Gistelinck M, Van Broeckhoven C, Lambert JC, Hirth F, Aerts S, Callaerts P, Dermaut B (2013) TDP-43 loss-of-function causes neuronal loss due to defective steroid receptor-mediated gene program switching in Drosophila. Cell Rep 3:160–172. 10.1016/j.celrep.2012.12.014 [DOI] [PubMed] [Google Scholar]
- Vogelstein JT, Park Y, Ohyama T, Kerr RA, Truman JW, Priebe CE, Zlatic M (2014) Discovery of brainwide neural-behavioral maps via multiscale unsupervised structure learning. Science 344:386–392. 10.1126/science.1250298 [DOI] [PubMed] [Google Scholar]