

Abstract
The human genome encodes ~750 G‐protein‐coupled receptors (GPCRs), including prokineticin receptor 2 (PROKR2) involved in the regulation of sexual maturation. Previously reported pathogenic gain‐of‐function mutations of GPCR genes invariably encoded aberrant receptors with excessive signal transduction activity. Although in vitro assays demonstrated that an artificially created inactive mutant of PROKR2 exerted paradoxical gain‐of‐function effects when co‐transfected with wild‐type proteins, such a phenomenon has not been observed in vivo. Here, we report a heterozygous frameshift mutation of PROKR2 identified in a 3.5‐year‐old girl with central precocious puberty. The mutant mRNA escaped nonsense‐mediated decay and generated a GPCR lacking two transmembrane domains and the carboxyl‐terminal tail. The mutant protein had no in vitro signal transduction activity; however, cells co‐expressing the mutant and wild‐type PROKR2 exhibited markedly exaggerated ligand‐induced Ca2+ responses. The results indicate that certain inactive PROKR2 mutants can cause early puberty by enhancing the functional property of coexisting wild‐type proteins. Considering the structural similarity among GPCRs, this paradoxical gain‐of‐function mechanism may underlie various human disorders.
Keywords: GPCR, mutation, neuroendocrine
Introduction
The human genome encodes ~750 GPCRs that possibly participate in various biological processes by transmitting extracellular signals to the interior of cells 1. To date, several loss‐of‐function mutations and a few gain‐of‐function mutations in GPCR genes have been identified in patients with congenital disorders, particularly endocrine and neuroendocrine diseases 1. Known pathogenic gain‐of‐function mutations in GPCR genes have invariably encoded hyperactive receptors with constitutive activity, low ligand specificity, excessive expression or defective desensitization 1, 2.
The hypothalamus–pituitary–gonadal axis comprises multiple GPCRs, including PROKR2, gonadotropin‐releasing hormone (GnRH) receptor and KISS1 receptor (KISS1R) 2. Loss‐of‐function mutations in genes for these GPCRs result in hypogonadotropic hypogonadism. In contrast, gain‐of‐function mutations in these genes have not been identified in human beings, except for the p.R386P mutation in KISS1R that was identified in a single patient with precocious puberty 3.
PROKR2 is a 384‐amino acid GPCR that dimerizes and regulates GnRH secretion in the hypothalamus 4, 5, 6, 7. Upon binding of the ligand prokineticin 2 (PROK2), PROKR2 couples with Gαq/11‐type G‐proteins, thereby mobilizing intracellular Ca2+. PROKR2 can also bind to Gαs‐ and Gαi/o‐type G‐proteins, although ligand‐induced signal responses of PROKR2 are more prominent in the Gαq/11‐mediated pathway than in the Gαs‐ and Gαi/o‐mediated pathways 8. PROKR2 mutations account for a certain percentage of the aetiology of hypogonadotropic hypogonadism and Kallmann syndrome 4, 6, 9, 10. In vitro assays have confirmed functional impairment of several PROKR2 mutants 6. In addition, all known nonsense and frameshift mutations in PROKR2 satisfied the conditions for nonsense‐mediated mRNA decay (NMD) 11. Recently, Sposini et al. 12 examined the functional property of artificially created PROKR2 variants. The authors found that a variant (TM1‐5) lacking the last two transmembrane domains and the carboxyl‐terminal tail exerts a unique effect on signal transduction; although the mutant retained no signal transduction activity, co‐transfection of this mutant markedly increased ligand‐induced signal responses of cells expressing wild‐type PROKR2. To date, however, such a paradoxical gain‐of‐function phenomenon of a GPCR has not been observed in vivo. In the present study, we identified a frameshift mutation in PROKR2 in a patient with precocious puberty. This mutation encoded a GPCR structurally equivalent to the TM1‐5 variant.
Materials and methods
Patient
A 3.5‐year‐old girl was referred to our clinic because of early breast budding and accelerated statural growth. She was the sole child of non‐consanguineous Japanese parents. Physical examination revealed bilateral breast development of Tanner stage 2. Blood analyses demonstrated markedly elevated oestradiol and hyper‐responses of gonadotropins to GnRH (Table S1). Bone age was advanced (4.7 years of age). Brain magnetic resonance imaging revealed no abnormalities. The girl was diagnosed with idiopathic central precocious puberty and underwent GnRH analogue treatment from 3.7 to 11.4 years of age. During this treatment, her sexual development was suppressed. She had menarche at 13.0 years of age and regular menses thereafter. Her parents were clinically normal. The mother had menarche at 12 years of age (mean menarcheal age in Japanese women is 12.25 years). Allegedly, no maternal relatives of the patient had a history of precocious puberty.
Molecular analyses
This study was approved by the Institutional Review Board Committee at the National Center for Child Health and Development and was performed after obtaining written informed consent. This study was carried out according to the World Medical Association Declaration of Helsinki. Detailed methods are shown in the Supporting Information of Data S1. We performed molecular analysis of the patient's genomic DNA. Twenty‐nine genes involved in the regulation of GnRH secretion were sequenced. Copy‐number abnormalities in the genome were examined by array‐based comparative genomic hybridization. We also analysed parental DNA and the patient's mRNA.
To clarify the functional consequences of a PROKR2 mutation identified in the patient, we performed Ca2+ mobilization analyses. Chinese hamster ovary‐K1 cells were transiently transfected with the expression vectors for the wild‐type and/or mutant PROKR2. Ca2+ responses were measured before and after ligand stimulation.
Results
A heterozygous 4‐bp deletion in PROKR2 (c.724_727delTGCT, p.C242fsX305) was identified in the girl and her mother (Fig. 1A). The girl had no additional mutations or copy‐number alterations. The PROKR2 mutation was hitherto unreported and was absent from 121,412 alleles in the exome database. RT‐PCR products from the girl contained almost equal amounts of wild‐type and mutant PROKR2 alleles, indicating that the mutant mRNA escaped NMD (Fig. 1A).
Figure 1.
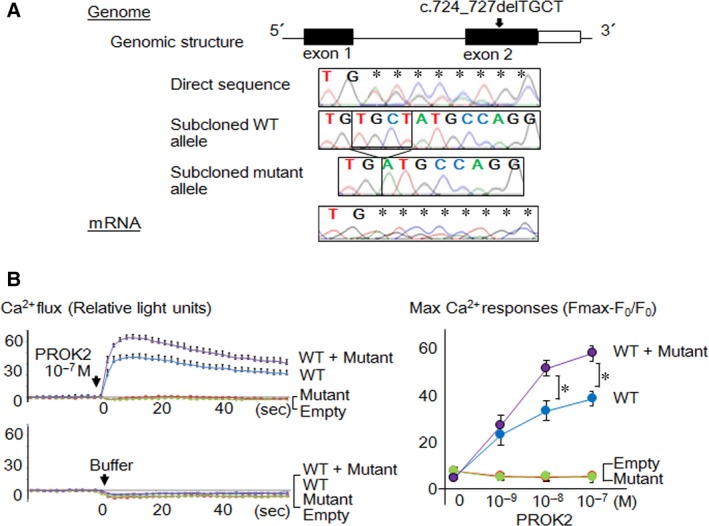
Identification and characterization of the PROKR2 mutation. (A) Results of the molecular analyses. Black and white boxes in the genomic structure depict coding and non‐coding exons, respectively. The girl carried a heterozygous c.724_727delTGCT mutation in PROKR2 exon 2. RT‐PCR revealed the presence of both wild‐type (WT) and mutant mRNA. (B) Representative results of Ca2+ mobilization assays. The left panel shows Ca2+ flux curves of cells transfected with WT alone (blue lines), mutant alone (green lines), both wild‐type and mutant (WT + Mutant, purple lines), and an empty vector (red lines) (the mean ± 1.0 standard deviation). The cells were treated with 10−7 M PROK2 or buffer (Hank's balanced salt solution plus HEPES buffer). The right panel shows max Ca2+ responses to different doses of PROK2 (0, 10−9, 10−8 and 10−7 M) calculated from the ratio between the change in fluorescence signal intensity (Fmax‐F0) and the baseline intensity (F0). Asterisks indicate significant differences between WT alone and WT + Mutant (P ≤ 0.05).
Ca2+ mobilization assays revealed paradoxical gain‐of‐function effects of the mutant PROKR2 (Fig. 1B). Basal signal activity was similar among cells transfected with empty and PROKR2 expression vectors. Cells transfected with wild‐type PROKR2 showed ligand‐induced Ca2+ flux, while cells transfected with the mutant did not respond to ligand stimulation. When treated with >10−8 M PROK2, cells co‐transfected with both proteins showed significantly greater Ca2+ responses than cells transfected with the wild‐type alone. The Ca2+ flux curves of the wild‐type‐expressing cells and those of the co‐expressing cells showed parallel changes after ligand administration.
Discussion
We identified a heterozygous p.C242fsX305 mutation in PROKR2 in a girl with precocious puberty. Mutant mRNA escaped NMD and appeared to encode a truncated PROKR2 lacking two transmembrane domains and the carboxyl‐terminal tail. Although the mutant protein retained no in vitro signal transduction activity, cells co‐expressing the wild‐type and the mutant showed greater ligand‐induced Ca2+ flux than cells expressing the wild‐type alone. Thus, p.C242fsX305 likely enhances the activity of the coexisting wild‐type receptor. Additionally, p.C242fsX305 may suppress desensitization of the receptor dimers, because the mutant lacked the carboxyl‐terminal tail that mediates receptor internalization 13. However, as Ca2+ flux curves of the co‐transfected cells paralleled that of the wild‐type‐transfected cells, enhanced signal transduction rather than delayed desensitization appears to be the major effect of p.C242fsX305. These results are consistent with those of a previous study by Sposini et al. 12. Our p.C242fsX305 mutant may modify the spatial arrangement of PROKR2 protomers to form hyperactive heterodimers, as in the case of the TM1‐5 variant 12.
The phenotype of our patient is compatible with the excessive PROKR2 function in the hypothalamus 6. As this patient exhibited regular menstrual cycles after 13.0 years of age, gain‐of‐function of PROKR2 unlikely affects feedback regulation of the hypothalamus–pituitary–gonadal axis. Notably, none of the patient's family members, including the mother with the same mutation as the patient, manifested apparent precocious puberty. These results can be explained by assuming that the clinical consequences of this mutation are determined by the balance between hetero and homodimers in the cells; a relatively small proportion of PROKR2 proteins may have formed heterodimers in the hypothalamic cells of the mother. Alternatively, the phenotypic variation in this family may reflect the incomplete penetrance or variable expressivity of PROKR2 abnormalities, because previously reported PROKR2 mutations were associated with a wide phenotypic spectrum 4, 6, 9, 10.
This study has two limitations. First, we did not examine whether p.C242fsX305 actually dimerizes to the wild‐type PROKR2 in vivo. Thus, although Sposini et al. 12 have shown that the TM1‐5 variant dimerizes with the wild‐type protein, the physical interaction between p.C242fsX305 and wild‐type PROKR2 needs to be confirmed by future studies. Second, subcellular localization of the wild‐type and mutant PROKR2 was not examined this study. Thus, we cannot exclude the possibility that p.C242fsX305 exerts a high signal transduction activity by affecting the internalization of the heterodimerized wild‐type protein.
Collectively, our results indicate that some inactive PROKR2 mutants can cause precocious puberty by enhancing the functional property of coexisting wild‐type proteins. This notion challenges the current understanding that all pathogenic gain‐of‐function mutations in GPCR genes encode hyperactive receptors 1. Considering the structural similarity among the ~750 known GPCRs 1, this paradoxical gain‐of‐function mechanism may underlie various human disorders.
Conflict of interest
The authors confirm that there are no conflicts of interest.
Author contributions
MF, JY and TO designed the study; MF, ES, YI, TT, SN, MI, MM, MK, KN, KH, AU, YM and JY performed molecular analyses; MF, ES and SN analysed the data; YF and TO collected clinical data; and MF wrote the manuscript.
Supporting information
Table S1 Hormonal findings of the girl
Data S1 Supporting Methods
Acknowledgements
This work was supported by the Japan Society for the Promotion of Science; the National Center for Child Health and Development; the Takeda Foundation; the Ministry of Health, Labour and Welfare; and the Japan Agency for Medical Research and Development. This study was also supported by the National Center Biobank Network. We thank Ms. Emma L. Barber and Dr. Julian Tang (National Center for Child Health and Development) for English editing.
MF, ES and YI contributed equally to this work.
References
- 1. Vassart G, Costagliola S. G protein‐coupled receptors: mutations and endocrine diseases. Nat Rev Endocrinol. 2011; 7: 362–72. [DOI] [PubMed] [Google Scholar]
- 2. Noel SD, Kaiser UB. G protein‐coupled receptors involved in GnRH regulation: molecular insights from human disease. Mol Cell Endocrinol. 2011; 346: 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Teles MG, Bianco SD, Brito VN, et al A GPR54‐activating mutation in a patient with central precocious puberty. N Engl J Med. 2008; 358: 709–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cole LW, Sidis Y, Zhang C, et al Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin‐releasing hormone deficiency: molecular genetics and clinical spectrum. J Clin Endocrinol Metab. 2008; 93: 3551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marsango S, Bonaccorsi di Patti MC, Barra D, et al Evidence that prokineticin receptor 2 exists as a dimer in vivo . Cell Mol Life Sci. 2011; 68: 2919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martin C, Balasubramanian R, Dwyer AA, et al The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr Rev. 2011; 32: 225–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Soga T, Matsumoto SI, Oda T, et al Molecular cloning and characterization of prokineticin receptors. Biochim Biophys Acta. 2002; 1579: 173–9. [DOI] [PubMed] [Google Scholar]
- 8. Sbai O, Monnier C, Dodé C, et al Biased signaling through G‐protein‐coupled PROKR2 receptors harboring missense mutations. FASEB J. 2014; 28: 3734–44. [DOI] [PubMed] [Google Scholar]
- 9. McCabe MJ, Gaston‐Massuet C, Gregory LC, et al Variations in PROKR2, but not PROK2, are associated with hypopituitarism and septo‐optic dysplasia. J Clin Endocrinol Metab. 2013; 98: E547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miraoui H, Dwyer AA, Sykiotis GP, et al Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet. 2013; 92: 725–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kuzmiak HA, Maquat LE. Applying nonsense‐mediated mRNA decay research to the clinic: progress and challenges. Trends Mol Med. 2006; 12: 306–16. [DOI] [PubMed] [Google Scholar]
- 12. Sposini S, Caltabiano G, Hanyaloglu AC, et al Identification of transmembrane domains that regulate spatial arrangements and activity of prokineticin receptor 2 dimers. Mol Cell Endocrinol. 2015; 399: 362–72. [DOI] [PubMed] [Google Scholar]
- 13. Ma L, Pei G. Beta‐arrestin signaling and regulation of transcription. J Cell Sci. 2007; 120: 213–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Hormonal findings of the girl
Data S1 Supporting Methods