

Abstract
Background
This study aimed to identify more potential genes and miRNAs associated with the pathogenesis of intracranial aneurysms (IAs).
Material/Methods
The dataset of GSE36791 (accession number) was downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were screened for in the blood samples from patients with ruptured IAs and controls, followed by functional and pathway enrichment analyses. In addition, gene co-expression network was constructed and significant modules were extracted from the network by WGCNA R package. Screening for miRNAs that could regulate DEGs in the modules was performed and an analysis of regulatory relationships was conducted.
Results
A total of 304 DEGs (167 up-regulated and 137 down-regulated genes) were screened for in blood samples from patients with ruptured IAs compared with those from controls. Functional enrichment analysis showed that the up-regulated genes were mainly associated with immune response and the down-regulated DEGs were mainly concerned with the structure of ribosome and translation. Besides, six functional modules were significantly identified, including four modules enriched by up-regulated genes and two modules enriched by down-regulated genes. Thereinto, the blue, yellow, and turquoise modules of up-regulated genes were all linked with immune response. Additionally, 16 miRNAs were predicted to regulate DEGs in the three modules associated with immune response, such as hsa-miR-1304, hsa-miR-33b, hsa-miR-125b, and hsa-miR-125a-5p.
Conclusions
Several genes and miRNAs (such as miR-1304, miR-33b, IRS2 and KCNJ2) may take part in the pathogenesis of IAs.
MeSH Keywords: Gene Regulatory Networks, Intracranial Aneurysm, MicroRNAs, Transcriptional Activation
Background
Intracranial aneurysms (IAs) are a cerebrovascular disease characterized by localized weakened pouches of the arterial wall around bifurcations of the circle of Willis, with disruption of the media and loss of the internal elastic lamina [1,2]. Rupture of IAs can result in subarachnoid hemorrhage (SAH), of which the consequences are dire, carrying an overall mortality rate of about 40% to 50% and a morbidity rate among survivors of approximately 50% [3]. Treatment with both endovascular therapy and microsurgery is effective but can also cause significant morbidity and mortality [4]. Currently, there are no medical alternatives to stabilize aneurysmal progression or prevent rupture. Thus, there is a critical need for a better understanding of the molecular mechanisms underlying the pathogenesis of IAs.
Risk factors predisposing formation of IAs include hypertension, cigarette smoking, heavy alcohol consumption, familial history, cocaine usage, ethnicity, sex, and age [5]. Though the pathophysiology leading to aneurysm formation, growth, and eventual rupture have not been completely elucidated, inflammation is known to play a significant role [6]. Ample evidence has indicated a critical role of TNF-α in the IAs formation and progression to rupture [7,8]. A study demonstrated that hepatocyte growth factor (HGF) concentrations were elevated in blood samples drawn from the lumen of patients with IAs [9]. In addition, Roder et al. performed a meta-analysis of microarray gene expression studies on IAs and revealed that seven genes, including B-cell CLL/lymphoma 2 (BCL2), collagen type I alpha 2 (COL1A2), collagen type III alpha 1 (COL3A1), collagen type V alpha 2 (COL5A2), C-X-C motif chemokine ligand 12 (CXCL12), TIMP metallopeptidase inhibitor 4 (TIMP4), and tenascin C (TNC) were very likely to be involved in the pathogenesis of IAs [10]. On the other hand, the discovery and modulation of microRNAs (miRNAs) provide a novel direction for IA research [11,12]. MiRNAs are small, noncoding RNA molecules of 18 to 25 nucleotides that have fundamental roles in post-transcriptional regulation of gene expression by binding to mRNAs and targeting the mRNA for degradation or translational inhibition [13]. Jiang et al. demonstrated differential expression of 18 miRNAs in IA tissue compared with controls, such as hsa-mir-133b and hsa-mir-143-3p [11]. In addition, the study of Lee et al. showed that miRNAs may take part in cerebral aneurysm formation by affecting multiple target genes and signaling pathways [14]. However, the genes and miRNAs associated with the pathogenesis of IAs were largely unknown.
Recently, Pera et al. performed an analysis of global gene expression profiles in peripheral blood cells from patients with SAH from ruptured IAs and found that rupture of IAs strongly influenced the transcriptional profiles of peripheral blood cells, involving a depression in lymphocyte response with enhancements in monocyte and neutrophil activities [15]. In this study, we downloaded and reanalyzed the dataset of Pera et al. [15] from a publicly accessible database. Blood samples from patients with ruptured IAs and controls were screened for differentially expressed genes (DEGs), followed by functional and pathway enrichment analyses. In addition, gene co-expression network was constructed and significant modules were extracted from the network. MiRNAs that could regulate DEGs in the modules were screened out and the analysis of regulatory relationships was conducted. This study aimed to identify more potential genes and miRNAs associated with the pathogenesis of IAs, which may provide a valuable resource for future biochemical and genetic studies of IAs pathogenesis and rupture.
Material and Methods
Microarray data
The Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) repository is the largest, fully public gene expression resource for high-throughput microarray and other forms of high-throughput data submitted by the scientific community [16]. In this study, we downloaded the microarray data that deposited by Pera et al. [15] from the GEO database [16] with the accession number of GSE36791. The platform is GPL10558, Illumina HumanHT-12 V4.0 expression beadchip. According to the description in the original study of Pera et al. [15], the dataset included peripheral blood samples from 43 patients with SAH from ruptured IAs and 18 controls who were suffering from headaches. SAH was diagnosed using cranial computed tomography and/or lumbar puncture. The presence and location of IAs were evaluated by digital subtractive angiography and/or angio-computed tomography. Venous whole blood was collected before neurosurgical interventions and total RNA was purified from the blood samples. In this study, 43 blood samples from patients with ruptured IAs and 18 blood samples from controls were applied for the following analysis.
Data preprocessing and screening of DEGs
The expression data which had already been normalized using Robust Multi-Array Average (RMA) [17] were downloaded. Then, probe sets were mapped to gene symbols according to the probe annotation files of the GPL10558 platform. When multiple probes mapped to a same gene symbol, the average value of all probes that mapped to the gene was calculated to represent the gene. Afterwards, the gene expression values were log2 transformed.
Linear Models for Microarray Analysis (limma) is an R-based open-source software package that provides an integrated solution for differential expression analyses for data from experiments involving microarrays and other platforms [18]. In this study, the limma package was used to identify DEGs between blood samples from patients with ruptured IAs and controls. False discovery rate (FDR) was estimated using Benjamini-Hochberg method [19]. The DEGs were as defined by FDR <0.05 and fold change (FC) greater than 2 (|log2FC| >0.58).
Gene ontology (GO) and pathway enrichment analysis for DEGs
Database for Annotation, Visualization, and Integrated Discovery (DAVID) bioinformatics resources, is a web-accessible program that enable investigators to discover biological themes associated with large gene lists [20]. In this study, to further characterize the identified DEGs, we performed GO terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis using the web tool DAVID. Those enriched GO terms and pathways with FDR <0.05 were considered significant.
Construction of gene co-expression network and module extraction
Weighted gene co-expression network analysis (WGCNA) R package provides a comprehensive systems biology method for performing weighted correlation network analysis that describes the correlation patterns among genes and identifying clusters (modules) of highly correlated genes across samples [21]. In this study, in order to identify ruptured IAs-associated co-expression modules and their key constituents, we input expression profiles of the identified DEGs to construct weighted gene co-expression modules using the WGCNA R package [21]. Briefly, the framework for WGCNA involves four steps 1) an unsigned similarity matrix of gene co-expression is initially determined based on pair-wise Pearson correlations for all genes across the samples; 2) the co-expression similarities are transformed into a weighted adjacency matrix of connection strengths using a power adjacency function; 3) a topological overlap matrix that measures the relative interconnectedness (proximity) of the genes is conducted from the adjacency matrix; and 4) average linkage hierarchical clustering of the topological overlap dissimilarity matrix is performed to identify clusters (modules) of co-expressed genes [22,23].
An adjacency matrix (network), A=[aij], was constructed using the following formula:
xi, and xj represent vectors whose components report the gene expression values of gene i and j. cor indicates the Pearson correlation coefficient between xi, and xj. The co-expression similarity matrix was continuously measured with a power (β) as a weight to obtain the weighted adjacency aij. The soft-thresholding parameter b of the power function is selected when the resulting co-expression network (adjacency matrix) best approximates a scale-free topology. In this study, a soft-threshold power of β=6 was used.
Next, the topological overlap-based dissimilarity matrix was computed from the weighted adjacency matrix using the following formula:
where ki indicates the total connectivity of gene i with all other genes in the network.
Average linkage hierarchical clustering was applied using a dissimilarity measure (1-topological overlap matrix) [24,25] and modules which were defined as branches of the resulting hierarchical clustering tree were subsequently identified using the blockwise Modules R function in the WGCNA R package [26]. Each module was subsequently designated by a color. The gene expression profiles of each module were summarized. Moreover, functional enrichment analysis for the DEGs in each enriched module were carried out for identifying overrepresented GO terms and pathways using the DAVID Bioinformatics Resources [20]. A p value <0.05 was set as the criterion.
Screening of disease-related miRNAs and regulatory relationships analysis
The combinatorial Gene Regulatory Networks Builder (cGRNB; http://www.scbit.org/cgrnb/) is a freely available web server to help biologists to model and analyze conditional combinatorial gene regulatory networks through integrated engineering of gene expression datasets and seed-matching sequence information, providing combinatorial regulations involving miRNAs, TFs, and genes [27]. In this study, we downloaded predicted miRNA-gene interactions from cGRNB website [27] with 197,906 regulatory pairs, including 699 mature miRNAs and 8,646 genes. The target genes for a miRNA were defined as Mi. The DEGs enriched in the above modules using WGCNA were defined as N. Fisher’s exact test [28] was applied to measure the significance of the overlap between Mi and N. The algorithm was as follows:
where a is the number of genes that both belong to DEGs in the modules and the target genes of miRNAs; b is the number of genes that belong to the target genes of miRNAs, but not DEGs in the modules; c is the number of genes that belong to DEGs in the modules, but not belong to the target genes of miRNAs; d is the number of genes that neither belong to DEGs in the modules, nor the target genes of miRNAs. In addition, p<0.05 was set as the threshold to identify relevant miRNAs for DEGs in the selected modules. Moreover, the regulatory relationships between the miRNAs and the DEGs in the modules were visualized using Cytoscape software [29].
Results
Data preprocessing and DEGs screening
After data preprocessing, 19,080 genes were obtained. The normalized data for the remaining genes represented similar distribution from sample to sample as shown in the box plot (Figure 1), indicating the reliability of data. Totally, 304 genes were differentially expressed between blood samples from ruptured IAs and samples from controls. There into, 167 DEGs were up-regulated and 137 were down-regulated in blood samples from ruptured IAs.
Figure 1.

A box plot of normalized data for differentially expressed genes (DEGs). (A) Data before normalization. (B) Data after normalization.
Functional and pathway enrichment analysis
After enrichment analysis, we found that the up-regulated genes were only enriched in six over-represented GO biological process (BP) terms that were mainly associated with immune response, including inflammatory response, defense response, response to wounding, innate immune response, immune response, and detection of biotic stimulus. By contrast, the down-regulated DEGs were enriched in different GO terms and pathways. Table 1 represented the top 10 GO terms and pathways (ranged by FDR value) that were enriched by the down-regulated genes. The results showed that the down-regulated DEGs were mainly concerned with structure of ribosome and translation.
Table 1.
Top 10 gene ontology (GO) terms and pathways enriched by the down-regulated genes.
| Category | Term | FDR |
|---|---|---|
| GOTERM_BP_FAT | GO: 0006414~translational elongation | 1.14E-20 |
| KEGG_PATHWAY | hsa03010: Ribosome | 1.11E-15 |
| GOTERM_MF_FAT | GO: 0003735~structural constituent of ribosome | 6.80E-14 |
| GOTERM_CC_FAT | GO: 0022626~cytosolic ribosome | 2.66E-13 |
| GOTERM_CC_FAT | GO: 0033279~ribosomal subunit | 5.33E-13 |
| GOTERM_CC_FAT | GO: 0005840~ribosome | 2.24E-11 |
| GOTERM_BP_FAT | GO: 0006412~translation | 8.18E-11 |
| GOTERM_CC_FAT | GO: 0044445~cytosolic part | 3.14E-09 |
| GOTERM_MF_FAT | GO: 0005198~structural molecule activity | 1.64E-07 |
| GOTERM_CC_FAT | GO: 0030529~ribonucleoprotein complex | 3.71E-07 |
The top 10 GO terms and pathways were ranged by FDR value. BP – biological process; CC – cellular component; MF – molecular function; KEGG – Kyoto Encyclopedia of Genes and Genomes; FDR – false discovery rate.
Identification of gene co-expression modules
WGCNA analysis of the DEGs and the following module selection produced six modules significantly altered between ruptured IAs and controls. As shown in Table 2, four out of six modules were enriched by up-regulated genes, whereas genes distributed in two modules were down-regulated. To understand the biological meaning of the modules of up-regulated genes and the modules of down-regulated genes, we carried out functional enrichment analyses. From the results of enrichment analysis, we found that the blue module of up-regulated genes was mainly linked with immune response and anti-apoptosis process. The brown module of up-regulated genes was mainly involved in chromatin assembly and the yellow module of up-regulated genes was correlated with interleukin-1 receptor activity. While the turquoise module of up-regulated genes was concerned with inflammatory response. On the other hand, the blue module of down-regulated genes was mainly enriched in cytolysis and cellular defense response. The turquoise module of down-regulated genes was mainly involved in the translation process. Table 2 only shows the top two GO terms or pathways that were ranked by p values. Collectively, the functions of the blue, yellow, and turquoise modules of up-regulated genes were similar, which were connected with immune response, and had differences with the functions of the brown module of up-regulated genes. The two modules of down-regulated genes were enriched in different functional categories.
Table 2.
The top 2 GO terms or pathways enriched by DEGs in the modules.
| Module label | Number of genes | Function | P value | |
|---|---|---|---|---|
| Up_modules | Blue | 28 | GO: 0000267~cell fraction | 0.00278 |
| GO: 0019864~IgG binding | 0.011041 | |||
| Brown | 21 | GO: 0006334~nucleosome assembly | 0.0037 | |
| GO: 0031497~chromatin assembly | 0.004 | |||
| Turquoise | 95 | GO: 0045087~innate immune response | 1.42E-05 | |
| GO: 0006954~inflammatory response | 1.00E-04 | |||
| Yellow | 20 | GO: 0004908~interleukin-1 receptor activity | 0.0085 | |
| GO: 0019966~interleukin-1 binding | 0.012 | |||
| Down_modules | Blue | 16 | GO: 0019835~cytolysis | 1.70E-04 |
| GO: 0006968~cellular defense response | 0.00159 | |||
| Turquoise | 119 | GO: 0006414~translational elongation | 2.30E-25 | |
| hsa03010: Ribosome | 5.60E-20 |
Up_modules represent the modules enriched by up-regulated genes. Down_modules represent the modules enriched by the down-regulated genes.
Screening of ruptured IAs-related miRNAs
Totally, we identified miRNAs for only three modules (blue, yellow, and turquoise) with up-regulated genes of the identified 6 functional modules (Table 3, Figure 2). We found that miR-1304 was significantly enriched with less p value. Among the significant miRNAs, miR-33b could regulate up-regulated genes of two modules (yellow module and turquoise module), such as insulin receptor substrate 2 (IRS2), potassium voltage-gated channel subfamily J member 2 (KCNJ2), and monoamine oxidase A (MAOA). The up-regulated target genes in these two modules for miR-33b ate represented in Figure 3.
Table 3.
The predicted miRNAs that regulated the 3 modules of up-regulated genes.
| miRNA | Module | P value | Up.DEGs_targets |
|---|---|---|---|
| hsa-miR-1304 | Blue | 0.003169 | CNIH4, CYP1B1, SH3GLB1, YOD1 |
| hsa-miR-373* | Blue | 0.004342 | PFKFB3, SH3GLB1 |
| hsa-miR-514 | Blue | 0.012935 | ADM, SH3GLB1, YOD1 |
| hsa-miR-33b | Yellow | 0.014884 | IRS2, KLHL8, MAOA |
| hsa-miR-568 | Turquoise | 0.022219 | ACSL1, B4GALT5, CREB5, DUSP1, ETS2, GAS7, KLHL2, MEGF9, PHF21A, SIPA1L2 |
| hsa-miR-139-3p | Turquoise | 0.023429 | BASP1, GAS7 |
| hsa-miR-621 | Blue | 0.023761 | PFKFB3, YOD1 |
| hsa-miR-125b | Turquoise | 0.024502 | CREB5, FAM126B, KIF1B, MEGF9, MTF1, RNF144B, ZNF281 |
| hsa-miR-575 | Yellow | 0.033528 | IRS2, MANSC1 |
| hsa-miR-125a-5p | Turquoise | 0.033995 | CREB5, FAM126B, KIF1B, MEGF9, MTF1, RNF144B, ZNF281 |
| hsa-miR-33b | Turquoise | 0.03954 | FAM126B, KCNJ2, KIF1B, PCNX, RNF144B, ZNF281 |
| hsa-miR-1197 | Yellow | 0.039541 | DAAM2, MAOA |
| hsa-miR-432* | Turquoise | 0.041785 | ETS2, RNF144B, ST3GAL4 |
| hsa-miR-1234 | Turquoise | 0.046918 | CEBPB, ZNF281 |
| hsa-miR-545 | Blue | 0.04737 | ADM, CNIH4, SH3GLB1, YOD1 |
| hsa-miR-1208 | Yellow | 0.048709 | DAAM2, IRS2 |
Figure 2.
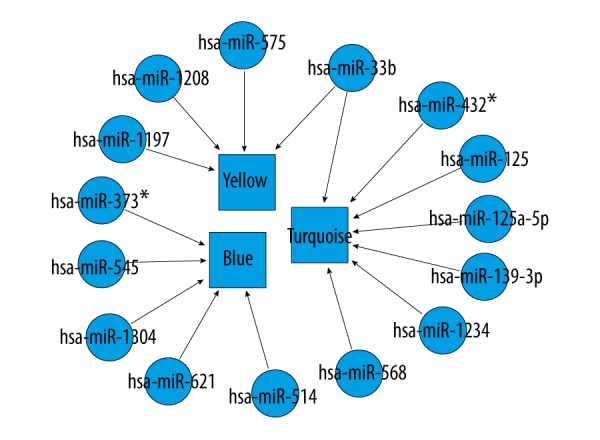
The miRNAs that regulated the three modules of up-regulated genes. Circular nodes indicate miRNAs. Rectangular nodes denote functional modules.
Figure 3.
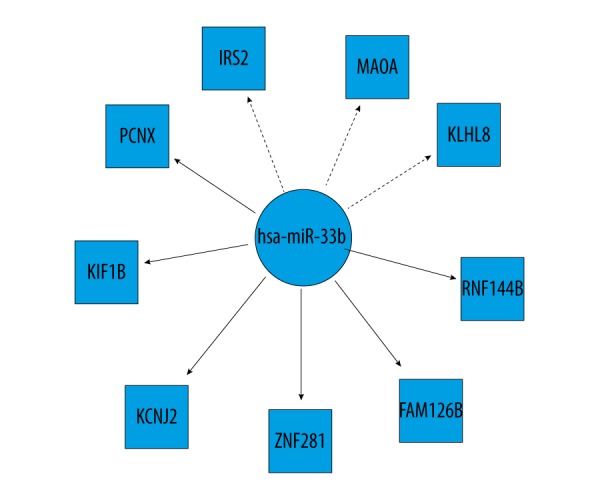
The up-regulated genes in yellow module and turquoise module targeted by miR-33b. The dotted line denotes the target genes in the yellow module. The solid line denotes the target genes in the turquoise module.
Discussion
In this study, a total of 304 DEGs (167 up-regulated and 137 down-regulated genes) were screened for in blood samples from patients with ruptured IAs compared with those from controls. Functional enrichment analysis showed that the up-regulated genes were mainly associated with immune response and the down-regulated DEGs were mainly concerned with structure of ribosome and translation. Besides, six functional modules were significantly identified, including four modules enriched by up-regulated genes and two modules enriched by down-regulated genes. Therein, the blue, yellow, and turquoise modules of up-regulated genes were all linked with immune response. Additionally, 16 miRNAs were predicted to regulate DEGs in the three modules associated with immune response, such as hsa-miR-1304, hsa-miR-33b, hsa-miR-125b, and hsa-miR-125a-5p.
MiR-1304 is a primate-specific miRNA which has low expression in human brain cortex, peripheral blood, and embryonic stem cells [30,31]. Very few reports are available on the exact roles of miR-1304 to date. Recently, the study of Li et al. demonstrated that miR-1304 suppressed cell growth and survival through inducing cell apoptosis and cell cycle arrest in non-small cell lung cancer [32]. Evidence reports that structural degeneration of aortic tissue with apoptosis of vascular smooth muscle cells (VSMCs) contributes to aneurysmal formation. In the present study, we found that miR-1304 could regulate the up-regulated genes in the blue module with most significance. A single miRNA may regulate several genes as its targets, while one gene may be targeted by many miRNAs. Although the exact role of miR-1304 is unclear, it is reasonable to conclude that miR-1304 may be involved in the pathogenesis of IAs possibly by affecting VSMCs apoptosis.
In this study, miR-33b could regulate the up-regulated genes of two modules (yellow module and turquoise module), such as IRS2, and KCNJ2. Martino et al. indicated that circulating miR-33b were up-regulated in familial hypercholesterolemia in pediatric age cases [33]. In addition, hypercholesterolemia was identified to be risk factor for aneurysms [34]. Thus, we suggested that miR-33b may play a critical role in the development and progression of IAs. In addition, IRS2 encodes the insulin receptor substrate 2, a cytoplasmic signaling molecule which mediates effects of insulin-like growth factor 1, insulin, and other cytokines [35]. A study demonstrated that insulin treatment for glycemic control was safe in patients with aneurysmal SAH [36]. The protein encoded by KCNJ2 is an integral membrane protein and inward-rectifier type potassium channel [37]. Additionally, activation of calcium-activated potassium channels and ATP-sensitive potassium channels may be a major mechanism which regulates vasodilatation of cerebral arteries and arterioles in response to some stimuli [38]. Taken together, these data strongly suggested that miR-33b may be essential in the development and progression of IAs by regulating several genes, such as IRS2 and KCNJ2. However, further studies are warranted to verify these findings.
Our study still had some limitations. First, the sample size was relatively small and further investigations with a larger size of samples are needed. Second, data cross-check and experimental verifications were not included in this study. We will further strengthen our study by further experimental verifications in the future.
Conclusions
This study suggested that several genes and miRNAs (such as miR-1304, miR-33b, IRS2 and KCNJ2) may take part in the pathogenesis of IAs. MiR-1304 may be involved in the pathogenesis of IAs possibly by affecting VSMCs apoptosis. In addition, miR-33b may be essential in the development and progression of IAs by regulating several genes such as IRS2 and KCNJ2. This preliminary work may be a valuable resource, after further characterization in a wider sample set, for designing future studies of both the pathogenesis and pathology of IAs.
Footnotes
Conflict of interest
None.
Source of support: Departmental sources
References
- 1.Sadasivan C, Fiorella DJ, Woo HH, Lieber BB. Physical factors effecting cerebral aneurysm pathophysiology. Ann Biom Eng. 2013;41(7):1347–65. doi: 10.1007/s10439-013-0800-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chalouhi N, Hoh BL, Hasan D. Review of cerebral aneurysm formation, growth, and rupture. Stroke. 2013;44(12):3613–22. doi: 10.1161/STROKEAHA.113.002390. [DOI] [PubMed] [Google Scholar]
- 3.Etminan N, Rinkel GJE. Cerebral aneurysms: Cerebral aneurysm guidelines – more guidance needed. Nat Rev Neurol. 2015;11(9):490–91. doi: 10.1038/nrneurol.2015.146. [DOI] [PubMed] [Google Scholar]
- 4.Starke RM, Chalouhi N, Ding D, et al. Vascular smooth muscle cells in cerebral aneurysm pathogenesis. Transl Stroke Res. 2014;5(3):338–46. doi: 10.1007/s12975-013-0290-1. [DOI] [PubMed] [Google Scholar]
- 5.Penn DL, Witte SR, Komotar RJ, Connolly ES. The role of vascular remodeling and inflammation in the pathogenesis of intracranial aneurysms. J Clin Neurosci. 2014;21(1):28–32. doi: 10.1016/j.jocn.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Chalouhi N, Ali MS, Jabbour PM, et al. Biology of intracranial aneurysms: Role of inflammation. J Cereb Blood Flow Metab. 2012;32(9):1659–76. doi: 10.1038/jcbfm.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starke RM, Chalouhi N, Jabbour PM, et al. Critical role of TNF-α in cerebral aneurysm formation and progression to rupture. J Neuroinflammation. 2014;11(4):1–10. doi: 10.1186/1742-2094-11-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Starke RM, Raper DMS, Ding D, et al. Tumor necrosis factor-α modulates cerebral aneurysm formation and rupture. Transl Stroke Res. 2014;5(2):269–77. doi: 10.1007/s12975-013-0287-9. [DOI] [PubMed] [Google Scholar]
- 9.Peña-Silva RA, Chalouhi N, Wegman-Points L, et al. Novel role for endogenous hepatocyte growth factor in the pathogenesis of intracranial aneurysms. Hypertension. 2014;65(3):587–93. doi: 10.1161/HYPERTENSIONAHA.114.04681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roder C, Kasuya H, Harati A, et al. Meta-analysis of microarray gene expression studies on intracranial aneurysms. Neuroscience. 2012;201(1):105–13. doi: 10.1016/j.neuroscience.2011.10.033. [DOI] [PubMed] [Google Scholar]
- 11.Jiang Y, Zhang M, He H, et al. MicroRNA/mRNA profiling and regulatory network of intracranial aneurysm. BMC Med Genom. 2013;6(1):280–86. doi: 10.1186/1755-8794-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeyaseelan K, Kai YL, Armugam A. MicroRNA expression in the blood and brain of rats subjected to transient focal ischemia by middle cerebral artery occlusion. Stroke. 2008;39(3):959–66. doi: 10.1161/STROKEAHA.107.500736. [DOI] [PubMed] [Google Scholar]
- 13.Vidigal JA, Ventura A. The biological functions of miRNAs: Lessons from in vivo studies. Trends Cell Biol. 2015;25(3):137–47. doi: 10.1016/j.tcb.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee HJ, Yi JS, Lee HJ, et al. Dysregulated expression profiles of MicroRNAs of experimentally induced cerebral aneurysms in rats. J Korean Neurosurg Soc. 2013;53(2):72–76. doi: 10.3340/jkns.2013.53.2.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pera J, Korostynski M, Golda S, et al. Gene expression profiling of blood in ruptured intracranial aneurysms: In search of biomarkers. J Cereb Blood Flow Metab. 2013;33(7):1025–31. doi: 10.1038/jcbfm.2013.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barrett T, Wilhite SE, Ledoux P, et al. NCBI GEO: Archive for functional genomics data sets – update. Nucleic Acids Res. 2013;41(Database issue):D991–95. doi: 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4(2):249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 18.Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acid Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferreira JA, Zwinderman AH. On the Benjamini-Hochberg method. Annals of Statistics. 2006;34(4):1827–49. [Google Scholar]
- 20.Huang DW, Sherman BT, Tan Q, et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35(Web Server issue):169–75. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9(1):1–13. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mason MJ, Fan G, Plath K, et al. Signed weighted gene co-expression network analysis of transcriptional regulation in murine embryonic stem cells. BMC Genom. 2009;10:327. doi: 10.1186/1471-2164-10-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mulligan MK, Rhodes JS, Crabbe JC, et al. Molecular profiles of drinking alcohol to intoxication in C57BL/6J mice. Alcohol Clin Exp Res. 2011;35(4):659–70. doi: 10.1111/j.1530-0277.2010.01384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yip AM, Horvath S. Gene network interconnectedness and the generalized topological overlap measure. BMC Bioinformatics. 2007;8:22. doi: 10.1186/1471-2105-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Jong S, Boks MP, Fuller TF, et al. A gene co-expression network in whole blood of schizophrenia patients is independent of antipsychotic-use and enriched for brain-expressed genes. PLoS One. 2012;7(6):e39498. doi: 10.1371/journal.pone.0039498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu H, Yu H, Tu K, et al. cGRNB: A web server for building combinatorial gene regulatory networks through integrated engineering of seed-matching sequence information and gene expression datasets. BMC Systems Biology. 2013;7(Suppl 2):S7. doi: 10.1186/1752-0509-7-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crans GG, Shuster JJ. How conservative is Fisher’s exact test? A quantitative evaluation of the two-sample comparative binomial trial. Stat Med. 2008;27(18):3598–611. doi: 10.1002/sim.3221. [DOI] [PubMed] [Google Scholar]
- 29.Kohl M, Wiese S, Warscheid B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 30.Vaz C, Ahmad HM, Sharma P, et al. Analysis of microRNA transcriptome by deep sequencing of small RNA libraries of peripheral blood. BMC Genomics. 2010;11(1):624–25. doi: 10.1186/1471-2164-11-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martí E, Pantano L, Bañezcoronel M, et al. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010;38(20):7219–35. doi: 10.1093/nar/gkq575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aletaha D, Neogi T, Silman AJ, et al. 2010 Rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2016;62(2):2569–81. doi: 10.1002/art.27584. [DOI] [PubMed] [Google Scholar]
- 33.Martino F, Carlomosti F, Avitabile D, et al. Circulating miR-33a and miR-33b are up-regulated in familial hypercholesterolaemia in paediatric age. Clin Sci. 2015;129(11):963–72. doi: 10.1042/CS20150235. [DOI] [PubMed] [Google Scholar]
- 34.Wardlaw JM, White PM. The detection and management of unruptured intracranial aneurysms. Brain. 2000;123( Pt 2):205–21. doi: 10.1093/brain/123.2.205. [DOI] [PubMed] [Google Scholar]
- 35.Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317(5836):369–72. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- 36.Schlenk F, Graetz D, Nagel A, et al. Insulin-related decrease in cerebral glucose despite normoglycemia in aneurysmal subarachnoid hemorrhage. Critical Care. 2008;12(1):1–7. doi: 10.1186/cc6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munoz C, Almilaji A, Setiawan I, et al. Up-regulation of the inwardly rectifying K + Channel Kir2.1 (KCNJ2) by protein kinase B (PKB/Akt) and PIKfyve. J Membr Biol. 2013;246(3):189–97. doi: 10.1007/s00232-012-9520-9. [DOI] [PubMed] [Google Scholar]
- 38.Kitazono T, Faraci FM, Taguchi H, Heistad DD. Role of potassium channels in cerebral blood vessels. Stroke. 1995;26(9):1713–23. doi: 10.1161/01.str.26.9.1713. [DOI] [PubMed] [Google Scholar]