

Abstract
Passive immunization, the transfer of antibodies to a nonimmune individual to provide immunological protection, has been used for over 100 years to prevent and treat human infectious diseases. The introduction of techniques to produce human monoclonal antibodies (mAbs) has revolutionized the field, and a large number of human mAbs have been licensed for the treatment of cancer, autoimmune and inflammatory diseases. With the recent discovery and production of highly potent broadly neutralizing and other multifunctional antibodies to HIV, mAbs are now being considered for HIV therapy and prophylaxis. In this review, we briefly review recent advances in the anti-HIV mAb field and outline strategies for the selection, engineering and production of human mAbs, including the modification of their structure for optimized stability and function. We also describe results from nonhuman primate studies and Phase 1 clinical trials that have tested the safety, tolerability, PK and efficacy of mAb-based HIV prevention strategies, and discuss the future of parenteral and topical mAb administration for the prevention of HIV transmission.
Keywords: HIV-1, VRC01, monoclonal antibody, passive immunization, sexual transmission, microbicide, vagina, rectal
A BRIEF HISTORY OF PASSIVE IMMUNIZATION
In 1890, Emil von Behring and Shibasaburo Kitasato, working at the Institute of Hygiene in Berlin, reported that serum from rabbits that had been immunized with bacterial toxins protected nonimmune rabbits from infection [1]. Their discovery led to the use of immune serum from horses and other animals to treat tetanus and diphtheria infections in humans and marked the start of the age of “serum therapy”. This treatment was hailed as the most important medical breakthrough of the 19th century, and the inventors received the first Nobel Prize in Physiology or Medicine in 1901 [2]. For approximately 40 years, serum therapy was used as front line treatment for a number of major human bacterial and viral infections including tetanus, diphtheria, pneumococcus, meningococcus, influenza, measles and polio. Following the introduction of potent antibacterial drugs, antibody therapy was restricted to a smaller number of selected treatments for snake venoms, bacterial toxins and some viral infections [2]. However, in recent years, passive immunization has experienced a renaissance with the use of monoclonal antibodies to treat a number of cancers, autoimmune and infectious diseases.
The early days of passive immunization with animal immune sera had been hampered by limited availability of quality antibodies, high cost, and frequent adverse reactions to serum components. In the 1940’s, Cohn significantly advanced the field through the introduction of a procedure to purify immunoglobulins from blood, which, with further improvements, led to the use of potent polyclonal “immune globulin (Ig)” formulations for the prophylaxis and treatment of several viral diseases including measles, polio and infectious hepatitis [3], and for the protection of high risk newborns unable to receive colostrum [4]. These products produced fewer side effects, but supplies were limited and expensive. In 1975, the field of passive immunization was revolutionized with the discovery of a technique to make monoclonal antibodies (mAbs) by Kohler and Milstein [5], and in 2003, by transformative technology which introduced the capability of cloning heavy and light chain immunoglobulin genes amplified from single human B cells and their expression in bacteria, and later in other expression systems as described below [6, 7]. This capability accelerated the discovery of new human antibodies, especially when coupled with rapid manufacturing platforms, and made possible their production on a large scale for clinical applications [8]. By the end of 2014, 47 mAb products had been approved for clinical use, and it is projected that 70 mAb products will be on the market by 2020 with combined sales of $125 billion [9].
USE OF ANTI-HIV MABS TO PREVENT THE SEXUAL TRANSMISSION OF HIV
Most HIV transmission events occur across genital or rectal mucosal surfaces following sexual intercourse with an HIV-infected partner [10, 11]. With the introduction of new intervention strategies, such as male circumcision and treatment-as-prevention (TAP), the global HIV incidence has dropped from its peak in 1997 of 3.5 million new infections per year, to 2.1 million/year [12], but this rate is still unacceptably high. A vaccine may be the ultimate goal for HIV prevention, but this approach has remained elusive. MAbs are currently being explored for HIV therapy and prevention. Approximately one third of HIV-infected individuals make HIV neutralizing antibodies [13], and B cells from these individuals were used to isolate first generation HIV-neutralizing mAbs. These identified conserved epitopes shared between HIV subtypes and isolates; however they had limited breadth and/or potency against global isolates and were only partially effective in SHIV-challenge models. Subsequently, large cohorts of HIV infected individuals were screened for highly effective neutralizing antibodies, and high throughput single cell B-cell receptor amplification techniques and novel soluble trimeric Envs were employed to produce a new generation of extremely potent broadly neutralizing anti-HIV antibodies (bNAbs) [14], and are active across multiple HIV clades. These second generation mAbs are 10–100 fold more potent than the first generation antibodies, and bind to various epitopes on the viral surface (Table 1) which enables the administration of combinations of distinct mAbs to reduce HIV immune escape. Unfortunately, potent HIV neutralization activity is associated with a high degree of somatic hypermutation, making the production of bNAbs through vaccination a challenge [15]. Non-neutralizing HIV mAbs have also been described that may block HIV infection through other antiviral effector functions such as Fc-mediated functional assays (described below). Interest in non-neutralizing anti-HIV antibodies arouse when data from the recent RV144 Thai vaccine trial showed that non-neutralizing antibodies were associated with protection in vaccinated subjects [16]. However, to date non-neutralizing mAbs have been less effective than bNAbs in protecting against SHIV infection in vaginal and rectal transmission models [17–19].
Table 1.
Broadly neutralizing HIV monoclonal antibodies [107]
| Monoclonal Antibody | Epitope |
|---|---|
| 1° Generation | |
| 2F5 | gp41 membrane-proximal external region (MPER) |
| 2G12 | gp120 glycan |
| b12 | Gp120 CD4 binding site (bs) |
| 2° Generation | |
| PGT121 | V3 glycan |
| 10-1074 | V3 glycan |
| 2G12 | gp120 glycan |
| VRC34 | fusion peptide |
| PGT151 | fusion peptide |
| VRC01 | gp120 CD4 bs |
| 3BNC117 | gp120 CD4 bs |
| 4E10 | MPER |
| 10E8 | MPER |
| C5022 | gp120/41 extended glycan |
| PGT151 | gp120/41 extended glycan |
| 3° Generation | |
| VRC07-523 | CD4 bs |
| 3BNC117 | CD4 bs |
| 10-1074 | V3 glycan |
Parenteral MAb Administration
Preclinical studies in animal models
Some of the first studies to demonstrate protective effects of HIV mAbs were conducted in the humanized mouse HIV infection model [20–22]. Following the introduction of the chimeric HIV/SIV (SHIV) virus challenge model, nonhuman primates (NHP) have been extensively used to test the ability of anti-HIV bNAbs to prevent SHIV infection via the vaginal or rectal routes (Table 2 [17, 18, 23–37]). The earliest NHP studies used intravenous (IV) infusion of first-generation bNAbs (e.g., 2G12, b12, 2F5); these mAbs required a high dose (≥25mg/kg) to achieve protection against a single high-dose SHIV vaginal challenge, even with Tier 1 (neutralization sensitive) strains [24, 26, 34]. Passive immunization of various combinations of these bNAbs also protected neonatal macaques against high dose IV or oral challenge with SHIVs, providing further proof-of-concept that mAbs can prevent HIV infection [38]. The introduction of more potent cross-clade bNAbs and Tier 2 (neutralization resistant) SHIV strains have provided a more realistic picture of the potential of passive immunization with HIV antibodies. Second-generation bNAbs (particularly the PG, PGT and VRC series) require lower doses (≤1 mg/kg) for protection in passive immunization studies than first-generation bNAbs; furthermore, they have demonstrated efficacy against both vaginal and rectal SHIV challenges with Tier 2 SHIV [32, 35]. Engineering of bNAbs to enhance neutralization efficacy, Fc function and serum half-life has led to increased protection in some studies [29, 36]. This research has paved the way for clinical trials of parenteral bNAb administration in humans to prevent HIV transmission.
Table 2.
Protection against SHIV mucosal challenge following intravenous parenteral administration of HIV-MAbs
| Reference | MAbs | MAb Dosage | SHIV Challenge | Protection | Notes |
|---|---|---|---|---|---|
| Mascola [30] | 2G12 2G12 + 2F5 |
15 mg/kg 15 mg/kg @ |
Vaginal, sHDa SHIV89.bPD ″ |
2/4 2/5 |
First demonstration of modest protection against mucosal SHIV challenge w/mAbs. |
| Parren [34] | b12 ″ ″ |
1 mg/kg 5 mg/kg 25 mg/kg |
Vaginal, sHD SHIV162.P4 ″ ″ |
0/4 2/4 4/4 |
MAb protection was dose-dependent. |
| Hessell [24] | b12 b12KA b12LALA |
25mg/kg ″ ″ |
Vaginal, sHD SHIVSF162P3 | 8/9 8/9 5/9 |
Decreased protection without Fc function. |
| Hessell [25] | b12 b12LALA |
1 mg/kg (weekly) ″ |
Vaginal, rLD SHIV162.P4 ″ |
20X RRb 10X RR |
First repeated low dose SHIV challenge study: less mAb required for protection. Fc important. |
| Hessell [26] | 2G12 | 40 mg/kg | Vaginal, sHD, SHIVSF16273 | 3/5 | Half-life of 2G12 in serum: 7.7-12.2 days. Half-life in vaginal secretions: <24 hrs. |
| Hessell [27] | 2F5 4E10 |
50 mg/kg 50 mg/kg |
Rectal, sHD SHIVBa-L ″ |
5/6 5/6 |
MPER mAbs are also protective. |
| Burton [18] | b12 b6 |
25 mg/kg 25 mg/kg |
Vaginal, sHD SHIVSF162P2 ″ |
3/4 0/4 |
Strong neutralizing mAb (b12) more protective than weakly neutralizing mAb (b6). |
| Moldt [32] | PGT121 ″ ″ |
0.2 mg/kg 1 mg/kg 5 mg/kg |
Vaginal, sHD, SHIVSF162P3 ″ ″ |
3/5 5/5 5/5 |
Potently neutralizing mAb (PGT121) achieved protection at low dose |
| Moldt [33] | b12 NFb12 |
1 mg/kg (weekly) | Vaginal, rLDc, SHIVSF162 ″ |
4.5X RR 3.0X RR |
Nonfucosylated b12 (higher affinity for Fcγ RIIIa and ↑ADCC) did not show enhanced protection. |
| Klein [28] | 2F5 ″ ″ 2F5 (Fab) |
5 mg/kg 25 mg/kg 50 mg/kg 25 mg/kg |
Vaginal, sHD SHIVBaL ″ ″ ″ |
3/5 5/5 5/5 0/4 |
Serum and vaginal PK and PD: serum mAb levels were higher and more predictive of protection than vaginal mAbs. Fab mAb not protective. |
| Ko [29] | VRC01 VRC01-LS |
0.3 mg/kg ″ |
Rectal, sHD SHIVBaLP45 ″ |
2/12 7/12 |
VRC01-LS, engineered for enhanced FcRn binding, had 3X longer serum half-life and was more protective than VRC01. |
| Pegu [35] | VRC01 ″ ″ 10E8 ″ ″ PG9 ″ ″ |
0.3 mg/kg 5 mg/kg 20 mg/kg 0.3 mg/kg 5 mg/kg 20 mg/kg 0.3 mg/kg 5 mg/kg 20 mg/kg |
Rectal, sHD SHIVBaLP4 ″ ″ Rectal, sHD SHIVBaLP4 ″ ″ Rectal, sHD SHIVBaLP4 ″ ″ |
4/10 6/6 6/6 3/6 6/6 6/6 0/6 3/6 4/6 |
Various bNAbs directed against different epitopes on HIV Env were protective. |
| Rudicell [36] | VRC01 ″ VRC07-523 |
0.05 mg/kg 0.2 mg/kg 0.3 mg/kg |
Rectal, sHD SHIVBaLP4 ″ ″ |
0/4 3/4 5/12 |
VRC07-523, a clonal relative of VRC01, engineered to have increased neutralization potency, was more protective than VRC01. EC50: 0.47 μg/ml vs. 2.5 μg/ml. |
| Shingai [37] | VRC01 ″ ″ PGT 121 ″ ″ 10-1074 ″ ″ |
20 mg/kg 30 mg/kg 50 mg/kg 1 mg/kg 5 mg/kg 20 mg/kg 1 mg/kg 5 mg/kg 20 mg/kg |
Rectal, sHD SHIVDH12-V3AD8 or SHIV AD8E0 ″ ″ |
0/2 1/2 1/2 3/4 1/2 6/6 1/4 4/4 4/4 |
Plasma protective neutralization titer (50% animals) ~ 1:100. [3BNC117 and 45-46m2 mAbs showed little to no protection in same concentration range.] |
| Gautam [23] | VRC01 VRC01-LS 3BNC 117 10-1074 |
20 mg/kg ″ ″ ″ |
Rectal, rLD SHIVAD8-EO ″ ″ ″ |
RR=2.6 RR=4.8 RR=4.3 RR=4.2 |
A single mAb injection provided protection against several weeks of low dose SHIV rectal challenge (n=6/group). |
| Moldt [31] | PGT126 ″ ″ PGT126 ″ ″ |
0.4 mg/kg 2 mg/kg 10 mg/kg 0.4 mg/kg 2 mg/kg 10 mg/kg |
Vaginal, sHD SHIVSF163P3 ″ ″ Rectal, sHD SHIVSF163P3 ″ ″ |
1/5 2/5 5/5 0/4 2/4 3/4 |
bNAbs protect against both vaginal and rectal challenge routes. |
| Astronomo [17] | CH54 IgG CH38 mIgA2 |
50 mg/kg ″ |
Rectal, sHD SHIVBaLP4 | 0/8 1/6 |
These non-neutralizing mAbs were not protective against rectal SHIV challenge. |
sHD: Single high dose SHIV challenge;
RR: Relative risk, Cox Proportional Hazard Model;
rLD: Repeated low dose SHIV challenge
Clinical trials
Three Phase 1 clinical trials have been conducted to determine the safety, tolerability, and serum antibody PK in adults receiving IV infusions of the bNAb VRC01 with and without supplemental subcutaneous (SC) injections of antibody. VRC601 and 602 were dose escalation and PK studies of IV vs. SC administration of VRC01 in HIV-infected and uninfected subjects respectively [39, 40]. HVTN104 was a larger Phase 1 trial evaluating the safety of multiple doses of VRC01 administered over 6 months in regimens hypothesized to result in drug levels corresponding to protection against HIV. The study recruited a total of 88 healthy HIV-uninfected volunteers (44 men, 43 women and 1 transgender person). The bNAb infusions and injections were well tolerated and no severe adverse events were reported. PK results showed that: 1) after a 40 mg/kg IV loading dose, VRC01 levels in blood were maintained at >10 μg/ml for 6 months through biweekly 5mg/kg SC injections; 2) 20 mg/kg VRC01 given IV monthly after a 40 mg/kg loading dose maintained blood levels >40 μg/ml; 3) bimonthly VRC01 administered IV at 10, 30 or 40 mg/kg resulted in peak concentrations between 80 and >400 μg/ml and nadirs between 12 and >20 μg/ml. All three of these protocols maintained antibody titers well above the minimum effective HIV neutralization concentration of VRC01 (IC50 = 1 μg/ml). Potentially effective antibody titers were sustained from 53 to 81 days after mAb dosing was terminated [41]. Furthermore, preliminary data indicate that the infused antibody enters vaginal and rectal mucosal tissues at levels conferring protection in ex vivo HIV challenge studies [42].
Two large Phase 2B clinical trials, referred to as the Antibody Mediated Prevention (AMP) study (www.ampstudy.org), have been initiated to evaluate the efficacy of the VRC01 bNAbs in reducing acquisition of HIV-1 infection in high risk populations [43]. HVTN704/HPTN085 will test parenteral administration of VRC01 in 2,700 men-that-have-sex-with-men (MSM) and transgender people in the Americas, and HVTN703/HPTN081 will use the same approach in 1,500 heterosexual women at risk for HIV acquisition in sub-Saharan Africa. The mAb will be infused IV at doses of either 10 or 30 mg/kg bimonthly for a total of 8 infusions, and participants will be tested for HIV infection through 80 weeks after initiation of mAb treatment. It is hoped that data from this study will provide additional information on safety, tolerability and efficacy of VRC01 IV infusion, as well as antibody concentrations needed for protection against HIV. Furthermore, if this approach demonstrates protection against HIV acquisition, it may be used to protect vulnerable populations going forward. Other potent bNAbs are under study which could result in combination prophylactic regimens using several bNAbs at the same time to inhibit HIV binding via complementary mechanisms of action [44]. One particularly potent bNAb, 3BNC117, has been shown to suppress HIV rebound after treatment interruption in chronically-infected patients [45]. Other refinements include the development of bispecific antibodies that could enhance the breadth and potency of protection against diverse HIV strains [46].
In the near term, the AMP studies will be pivotal for the assessment of the role of systemic bNAb infusions for HIV immunoprophylaxis, and the findings will inform future efficacy trials of this approach using other bNAbs alone or in combination. If VRC01 infusions are found to protect against HIV acquisition, this will facilitate moving quickly forward to study other bNAbs that have other favorable characteristics, e.g., longer half-lives, and/or the ability to neutralize a broader array of HIV strains [45]. Analyses of viral isolates from individuals in the AMP trials who received either 30 mg/kg or 10 mg/kg of VRC01 who became HIV-infected will help to inform future dosing regimens of VRC01 and other bNAbs, and provide insights into optimal systemic antibody concentrations needed for protection against HIV acquisition. The pharmacokinetics of the dosing regimens used in the AMP studies has been thoroughly delineated in HVTN 104 [45], so it will be feasible to correlate expected drug concentrations with HIV protection once the AMP data are available. The findings from the AMP studies can be correlated with earlier work with non-human primates, and will allow for enhanced appreciation of the extent to which bNAb protection in the non-human primate model correlates with anti-HIV protection in humans. HVTN 104 and the AMP studies have included mucosal sampling substudies, which will be informative for the determination of bNAb levels necessary for mucosal protection [45], which should facilitate the development of mucosal antibody approaches including preventative HIV vaccines, vectored immunoprophylaxis and topical mAb administration.
Topical MAb Administration
Topical administration of mAbs directly to sites of mucosal HIV transmission in the genital tract or rectum is another promising approach for HIV prevention. Topical mAb-based microbicides potentially offer several advantages including: use of synergistic mAb combinations, ease of application, delivery of concentrated product directly to site where needed, reversibility, few if any side effects, and cost effectiveness.
Preclinical studies in animal models
Several studies have demonstrated that topical administration of anti-HIV mAbs can protect macaques from SHIV mucosal challenge (Table 3 [17–19, 47–50]). MAbs have been administered vaginally [18, 19, 48, 50] or rectally [17, 49], followed by high dose or repeated low dose SHIV challenges to the site shortly after mAb delivery. The half-life of mAbs in mucosal secretions after topical delivery of 5–20 mg mAb was about 4 hours; animals were protected to varying degrees against the SHIV challenges, and no side effects were noted. Efficacy of topically administered mAbs has also been demonstrated in the humanized mouse model [51].
Table 3.
Protection against SHIV mucosal challenge following topical administration of HIV-MAbs
| Reference | MAb(s) | MAb Dosage | Route | SHIV Challenge | Protection | Notes |
|---|---|---|---|---|---|---|
| Veazey [48] | b12 | 5 mg | Vaginal | Vaginal, sHDa SHIV162P4 | 9/12 | First topical mAb efficacy study. |
| Burton [18] | b12 b6 F240 b12 b6 F240 |
5 mg ″ ″ 5 mg ″ ″ |
Vaginal ″ ″ Vaginal ″ ″ |
Vaginal, sHD SHIV162P4 ″ ″ Vaginal, sHD SHIV162P3 ″ ″ |
5/5 0/5 2/5 1/4 1/4 1/4 |
Strong neutralizing mAb (b12) more protective than weakly neutralizing mAbs (b6, F240). MAbs less effective against Tier 2 virus (SHIV162P3) |
| Watkins [49] | HGN 194 IgG1 HGN 194 dIgA1 HGN 194 dIgA2 |
1.25 mg ″ ″ |
Rectal ″ ″ |
Rectal, sHD SHIV1157ipEL-p ″ ″ |
2/6 5/6 1/6 |
dIgA1 was more protective than IgG1 or dIgA2 mAbs. |
| Moog [19] | 2G12 + 2F5 + 4E10 246-D + 4B3 |
20 mg @ mAb 30 mg @ mAb |
Vaginal Vaginal |
Vaginal, sHD SHIV162P3 ″ |
10/15 0/15 |
Neutralizing mAbs (2G12, 2F5, 4E10) were more effective than non-neutralizing mAbs (246-D and 4B3). Tier 2 SHIV was used. |
| Sholukh [47] | HGN 194 IgG1 HGN 194 IgG1 + dIgA2 |
1.45 mg/kg 1.45 mg/kg + 1.25 mg |
IV IV + Rectal |
Rectal, sHD SHIV1157ipEL-p ″ |
0/6 6/6 |
Rectal administration of dIgA2 mAb enhanced protection against rectal SHIV challenge. |
| Zhao [50] | 4E10-Nb ″ ″ VRC01-N ″ ″ |
1.25 mg 5 mg 20 mg 1.25 mg 5 mg 20 mg |
Vaginal ″ ″ Vaginal ″ ″ |
Vaginal, rLDc SHIV162P3 ″ ″ Vaginal, rLD SHIV162P3 ″ ″ |
0/5 0/5 0/5 3/5 4/5 5/5 |
First study to use “plantibodies”, repeated low dose challenge with Tier 2 SHIV. |
| Astronomo [17] | CH31 IgG1 CH31 m-IgA2 CH31 d-IgA2 CH31 s-IgA2 |
5 mg ″ ″ ″ |
Rectal ″ ″ ″ |
Rectal, sHD SHIVSF162P3 ″ ″ ″ |
6/6 3/6 5/6 4/6 |
IgG1 was more protective than IgA2 forms. |
sHD: Single high dose SHIV challenge;
-N, mab made in Nicotiana benthamiana;
rLD: Repeated low dose SHIV challenge
Clinical trials
MAb-based vaginal microbicides have been evaluated for safety in at least 3 Phase 1 clinical trials. A vaginal microbicide containing a combination of 4E10, 2F5 and 2G12 (MABGEL), developed by the European Microbicides Programme, was recently tested for safety and pharmacokinetics (PK) [52]. The mAbs were produced in Chinese hamster ovary (CHO) cells as human IgG1, were formulated at 20 mg/ml (high dose) or 10 mg/ml (low dose) in gel, and 2.5 ml of gel was administered vaginally to women daily for 12 days. None of the women reported serious adverse events, and effective concentrations of mAbs could be observed in cervicovaginal secretions up to 8 hours post treatment. Furthermore, there was no evidence of systemic absorption of the mAbs. Another European consortium recently conducted a first-in-human, double-blind, placebo-controlled Phase 1 clinical trial with a plant-derived human HIV bNAb, 2G12, documenting the safety of a single vaginal administration of this mAb [53]. Our team of U.S. scientists is developing mAb-based vaginal microbicides using a transient expression system in Nicotiana (tobacco plants) [54]. A prototype vaginal microbicide, called MB66, containing the HIV bNAb VRC01 and the anti-HSV mAb HSV-8 [55], has been formulated into gels and films and tested for efficacy and stability in vitro and in vivo. MB66 film is currently undergoing evaluation in a Phase 1 clinical trial. Segment A of the study, a single dose of MB66 film in 8 women, was recently completed with no serious adverse effects; Segment B, 7 daily vaginal doses of MB66 or placebo film in 15 women/group, is currently underway. The next generation MB66 microbicide may contain additional mAbs, such as more potent bNAbs and mAbs that block cell-associated HIV transmission, to improve efficacy. In addition, mAbs that agglutinate sperm may be added to provide contraceptive protection [56]. A prototype MB66 vaginal ring has been developed and tested in macaques [57]. Sustained release of mAbs from a vaginal ring could improve user compliance, overcoming the significant disadvantage of poor adherence to study protocols that has plagued other vaginal microbicide trials [58].
PRODUCTION AND ENGINEERING OF ANTI-HIV MABs
Engineering mAbs for enhanced performance in the mucosal environment
Recent research indicates that passive immunization with anti-HIV mAbs may be a viable approach for HIV prevention and therapy. Non-traditional antibodies such as s-IgA, and mAbs engineered with Fab or Fc modifications to improve function may in the future be significantly more efficacious and cost effective, further enhancing the feasibility of this approach.
Engineering mAb isotype
Most monoclonal antibodies are manufactured as IgG1, which is the most abundant Ig subclass in blood [59]. However, polymeric secretory IgA (s-IgA) is the predominant immunoglobulin type in most mucosal secretions [60], and has structural characteristics that enhance its presence, stability and function at mucosal sites [61].
It is possible to manufacture IgA, dimeric (d) IgA and s-IgA mAbs, but this is currently an industrialization challenge [62]. However, several studies provide evidence that IgA mAbs may provide superior protection in the mucosal environment, indicating that it may be worthwhile to produce IgA mAbs for passive mucosal protection.
Dimeric and secretory forms of IgA have double the number of available antigen-binding sites as IgG, and multiple antigen-binding sites are crucial for the formation of viral aggregates [63]. Furthermore, the flexibility of diametrically opposed F(ab′) portions of dIgA with a higher extension span overcomes steric hindrance when accessing vulnerable epitopes on the viral envelope [64].
IgA1 subclass antibodies have a more flexible hinge region and a broader reach than IgG and IgA2 subclass antibodies (16.3 nm vs 10.2 nm). In a recent study the dIgA1 form of HCGN194 captured approximately twice as many virions as dIgA2 in a virion capture assay, and inhibited the transcytosis of cell-free SHIV across an epithelial monolayer in vitro whereas IgA2 and IgG mAbs did not [49].
s-IgA is particularly suited to function in the mucosal environment due to resistance to protease digestion and its ability to anchor onto the apical side of epithelial tissues by interacting with mucins [49, 65].
Association of the polymeric Ig receptor (pIgR) with HIV-bound IgA mAbs mediates HIV virion excretion from polarized epithelial cells suggesting a possible mechanism of viral efflux and subsequent reduction in viral load from mucosal epithelia [66].
Evidence for superior protection by IgA anti-HIV antibodies has been recently reported: a neutralizing monoclonal dIgA1 provided better protection than IgG in a SHIV rectal challenge study [49], in vitro studies showed intracellular abrogation of HIV replication/transcytosis by HIV-specific IgA mAbs [67], and vaginal transmission of HIV-1 in humanized mice was inhibited by polymeric recombinant forms of IgA but not monomeric IgG mAbs [68].
Despite considerable evidence favoring IgA for mucosal protection, contrary evidence also exists. A recent study that compared the protective effects of a number of neutralizing and nonneutralizing mAbs of IgG vs. IgA2 isotypes in both in vitro and in vivo NHP studies did not find an advantage to the use of IgA mAbs for protection against HIV and SHIV infection [17]. Furthermore, the RV144 HIV vaccine trial showed an in inverse relationship between the presence of IgA HIV antibodies in serum and protection in vaccine recipients [69]; in vitro experiments using serum from vaccinated subjects indicated that HIV Env-specific IgA antibodies competed for Env binding sites with vaccine-elicited IgG antibodies and diminished their ADCC antiviral effector function [70]. In light of this evidence, Ruprecht and colleages administered both IgG and IgA isotype HIV-specific mAbs to NHP to determine if the dIgA2 mAbs compete with IgG mAbs to affect protection in a rectal SHIV challenge experiment; they found that instead of competing, IgG and dIgA2 mAbs worked synergistically to completely protect macaques from rectal SHIV challenge [47]. Clearly, this is an important research area that calls for further study.
Engineering Fab
A variety of approaches for enhancing the function of anti-HIV mAbs have been devised through engineering of the Fab region. Passive immunization has demonstrated a strong correlation between anti-HIV mAb neutralizing potency, a property of the Fab region, and protection against SHIV mucosal challenge in vivo [18]. VRC07-523, a clonal relative of VRC01 engineered to have increased neutralization potency, was 5X more effective in a SHIV mucosal challenge study [36]. Antibody valence is another important feature. Additional F(ab′) fragments provide increased possible orientations for maximal ligation and crosslinking of HIV-1 envelope spikes [63]. In addition, bispecific antibodies have been engineered that demonstrate extremely potent and broadly neutralizing activity due to their ability to bind to more than one HIV Env epitope [71], and chimeric molecules such as one consisting of a PG16 antibody heavy chain and CCR5-like peptide have also been shown to be highly effective at neutralizing HIV [72].
Engineering Fc
Engineering of the immunoglobulin Fc region provides another approach to potentially enhance mAb protective functions. There are at least four Fc-mediated antibody activities that are important in regulating HIV infection (Figure 1). The Fc region can activate complement, leading to the enzymatic destruction of virions or infected cells [73]. Antibody-dependent cellular cytotoxicity (ADCC) is another principal Fc-mediated protective immune effector mechanism. Classic experiments illustrated killing of HIV antigen-coated or infected cells by immune cells that had captured antibodies from immune serum from HIV-infected individuals [74, 75], and this ADCC mechanism has been recapitulated in a number of studies using mAbs instead of immune serum [76–78]. Antibody-dependent cellular phagocytosis (ADCP), another Fc-mediated antibody function, similar to ADCC but involving phagocytosis of infected cells and immune complexes, has also been demonstrated for HIV antigen-coated beads in vitro [79], and is associated with improved clinical outcomes in chronically infected HIV patients [80]. Antibody-dependent cell-mediated viral inhibition (ADCVI) is an Fc receptor-mediated effector function that mediates viral reduction not only through cytotoxicity but also by inhibition with antiviral factors such as cytokines [81].
Figure 1. HIV broadly neutralizing monoclonal antibodies and their binding sites.
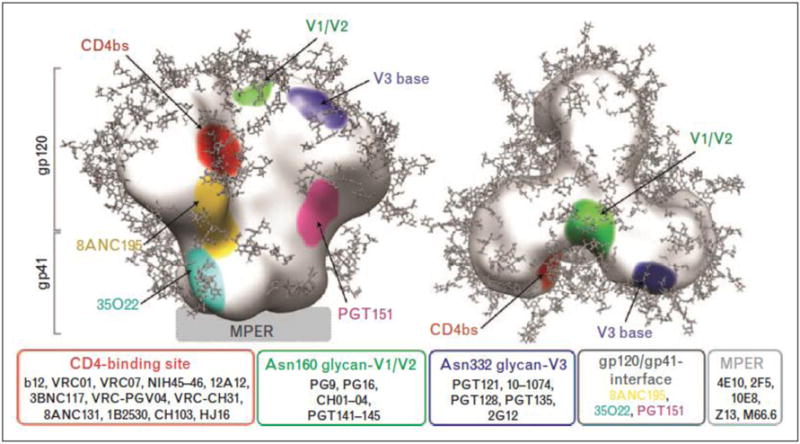
From: Sievers, Schaarf, West, Bjorkman; Curr Opin HIV AIDS 2015.
Passive immunization studies have implicated the Fc region in protection against HIV/SHIV challenge in vivo. Fab fragments of the 2F5 anti-HIV mAb were not protective against vaginal SHIV challenge whereas whole mAbs (with Fc region) were protective [28]. Furthermore, b12 mAbs with diminished Fc function due to the LALA mutation were less protective than unmodified mAbs [24, 25]. On the other hand, HIV mAbs engineered for enhanced Fc function have been more protective in SHIV mucosal challenge studies [29, 33]. Various engineering approaches have been used to modify the Fc region of mAbs for enhanced function:
There is a direct link between Fc glycosylation patterns and mAb effector function [79, 82]. IgG molecules with Fc glycans lacking the core fucose residue display an increased affinity for Fcγ RIIIa and enhanced ADCC [82, 83]. Nonfucosylated glycan forms on antibodies from elite controllers were shown to minimize viral load during chronic HIV-1 infection through ADCVI [84], and nonfucosylated HIV bNAbs have been made that demonstrate higher affinity for Fcγ RIIIa receptors and enhanced ADCC activity [33]. Furthermore, Fc glycosylation also affects antibody interactions with mucins; antibodies with shorter glycan profiles preferentially associated with MUC16 and captured more virions [85]. These data suggest that modification of Fc glycosylation could improve antibody function in the mucosal environment. However, one in vivo SHIV vaginal challenge experiment was conducted comparing nonfucosylated b12 mAb to normally glycosylated b12, and the results were inconclusive [33].
Another approach has been to modify the protein structure of the Fc region. GASDALIE is an Fc domain variant that enhances FcRγ binding and function [86]. GASDALIE-modified anti-HIV bNAbs demonstrated enhanced protection against HIV infection in the humanized mouse model [87].
The neonatal Fc receptor (FcRn) is another receptor of interest in designing mAbs with enhanced circulation time in blood and improved function at the mucosal surface. This receptor is expressed by mucosal epithelial cells, and is especially abundant in the placenta and newborn intestine where it directs the polarized uptake of maternal IgG antibodies for fetal immune protection [88]. MAbs engineered with point mutations that alter the binding of IgG to FcRn for greater stability have been shown to have a prolonged half-life in blood [89, 90]; this activity could extend to the mucosa as Fc-engineered mAbs with enhanced FcRn function showed enhanced protection against SHIV vaginal challenge in animal studies [29].
Overview of mAb production platforms
Mammalian cell platforms and alternative systems are rapidly being developed and optimized for more efficient mAb production, and promise to deliver clinical grade mAbs at reduced cost in the future (Figure 2). Evaluation of various technologies and production options is best accomplished early in product development. Ideally this occurs after product feasibility is established but well before product-related process development is initiated. Cost analysis for production and processing needs to be applicable to the complete platform process from initiation to harvest and downstream processing. The employed analytical methods are applicable to a variety of users and organizations and contain standardized and relevant cost data [91].
Figure 2.
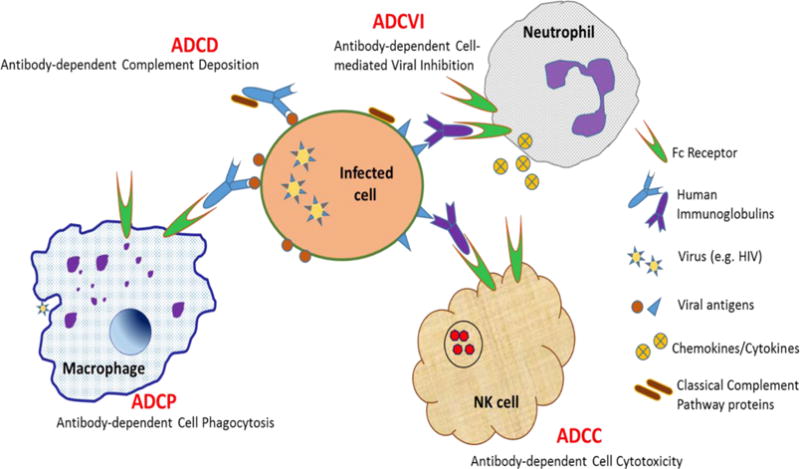
Fc Receptor-Mediated Antibody Dependent Functions that Boost the Immune Response to HIV and HIV-infected Cells
Understanding the cost implications of new technologies that may include continuous processes and modular facilities can be difficult. This evaluation at different production scales adds to the complexity. One example is mAb expression in mammalian cells. The production levels have increased dramatically in the last two decades and now titers of 3–5 g/L are not uncommon [92]. During the past 10 years, an industry-wide emphasis on upstream process optimization has resulted in up to 100- fold increases in productivity. Based on current rates of optimization, it would not be surprising to witness further increases in the range of 10–15 g/L. Due to the success of upstream process optimization, attention has shifted toward new or improved technologies that will augment the productivity of the downstream purification process. In particular, single use systems that have high throughput and minimize expensive chromatography resin turnover are being developed. Examples include continuous, multicolumn fed streams using disposable columns (such as simulated moving-bed chromatography (SMB) [93] as well as novel membrane chromatography technologies [94]. Single use technologies offer the advantages of low capital costs. This can ultimately reduce the initial investment as well as distribute cost expenditures across the entire lifetime of the product [95]. In this scenario, the increased cost burden of consumables results in an enhanced response rate when market demand changes. This flexibility is now increasingly important as drug pipelines transition from traditional mAbs with average demands of 250 kg/yr to more heterogeneous product capabilities such as fusion proteins, nanobodies, bispecifics and other emerging antibody structures, with production rates ranging up to 500 kg/yr.
Mammalian Cell MAb Production Systems
The prevalent stably transformed mammalian cell lines for recombinant mAb production are Chinese Hamster Ovary (CHO), NS0, Sp2/0, HEK293, and PER.C6 [96]. The majority (~70%) of mAbs approved for human therapy are produced in CHO, although NS0 and Sp2/0 have also been used for clinical mAb production [97]. The primary importance of mammalian cells compared to microbial or plant production systems lies in the mammalian post-translational modifications such as glycosylation [98]. Despite an excellent safety and efficacy record, the use of mammalian cells for recombinant mAb expression does have some drawbacks including a relatively slow growth rate, potential risk of viral contamination, and complex manufacturing process. Furthermore, the current standard production process is expensive, cumbersome and time-consuming [99].
Alternative MAb Production Systems
In the interest of cost-savings and potential increased scale of production, numerous transgenic technologies have been explored during the last two decades. These transgenic systems have involved yeast, bacteria, insects, animals, and plants [100]. Challenges for these transgenic platforms include immunogenic glycosylation, a susceptibility to viral pathogen co-propagation, developmental instability, complex genetics, environmental containment concerns, and most significantly, prolonged development times. One or more of these deficiencies is associated with every transgenic system, including current mammalian cell-based techniques. In addition, major GMP challenges for large-scale production can also contribute to significantly longer development times and unfavorable economics. For global health indications, the existing mammalian manufacturing platform for antibodies is viable, but emerging platforms like yeast/fungus, plants, and transient mammalian transfection could potentially play a complementary role when cost, scale and speed are critical factors.
GAPS, CHALLENGES AND CONCLUSIONS
Recent mAb HIV protection studies have measured antibody levels in female genital tract and rectal tissue to correlate antibody titers with protection. The male genital tract has not been studied but also requires protection from female-to-male HIV transmission. It is unknown whether systemically-administered mAbs reach transmission sites in the male genital tract (foreskin and urethra), and whether mAbs can effectively be administered topically to the male genital tract.
Infectious virus was detected in draining lymph nodes and gastrointestinal tissues from macaques that had been infused with protective levels of bNAbs and then challenged vaginally with SHIV [101]. This indicates that protective mAb titers must be sustained in tissues distal from the site of viral entry. MAb titers have not been monitored at these sites, and implications of this finding for topical mAb-based microbicides are unknown.
More studies are needed to identify synergistic combinations of bNAbs to maximize efficacy against a wide range of HIV strains and to minimize viral immune escape.
There is a growing interest in the use of mAbs to block cell-associated HIV transmission [102]. HIV-infected cells are present in genital secretions of HIV-infected men and women, and could play a role in the sexual transmission of HIV [103]. Cell-cell HIV transmission is 10 to >1,000-fold more efficient than cell-free transmission in vitro, and since cell-cell viral transmission entails transfer of HIV though specialized intercellular synapses, the virions are partially protected from environmental agents including antibodies [104, 105]. A number of studies have ranked existing bNAbs for their ability to block cell-cell HIV transmission [102]; the outcome of these studies has varied depending on the timing and concentration of antibodies and the model systems used. More research is needed with improved mucosal transmission model systems to identify anti-HIV mAbs that effectively block cell-associated HIV transmission for addition to HIV-prevention mAb cocktails. Such antibodies could prevent early events in HIV acquisition such as spreading of virus from HIV-infected cells in genital secretions, and between cells in the genital mucosal epithelium [103, 106].
MAb-based prevention methods may never enter the main stream due to cost considerations.
Existing mammalian cell manufacturing platforms are very costly for clinical grade mAb production. Emerging platforms such as yeast/fungus, plants, and transient mammalian transfection may prove advantageous when cost, scale and speed are critical factors. Use of potent mAbs engineered for enhanced function and half-life could also reduce costs.
Figure 3. Manufacturing Estimates for mAb Drug Substance.
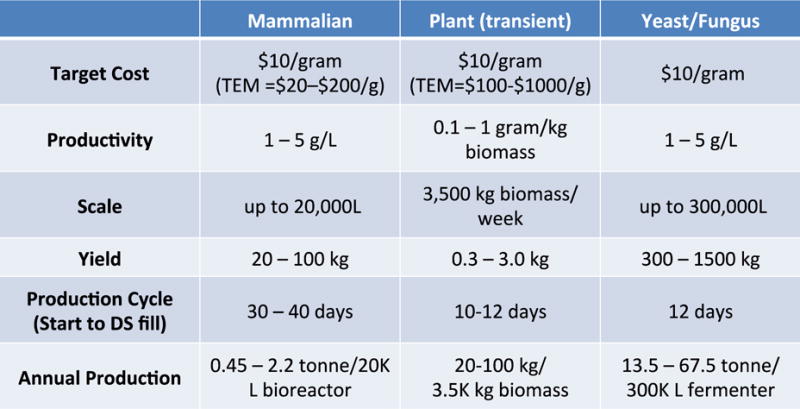
TEM = Techno-economic models
Acknowledgments
This review is based on information presented at the workshop “Systemic and Topical Delivery of Anti-HIV Monoclonal Antibodies to Prevent HIV Transmission” held on October 17, 2016 in conjunction with the HIV Research For Prevention (R4P) Meeting in Chicago, IL. All authors contributed substantively to the writing of this article. The workshop on which this article is based was supported by NIH grant U19 AI096398 (DA).
Footnotes
The authors have no conflicts of interest.
All authors contributed substantially to the preparation and revision of this manuscript.
References
- 1.von Behring E, Kitasato S. Uber das zustandekommen der diptherie-immunitat und der tetanus-immunitat bei thieren. Deutsche Medizinische Wochenschrift. 1890;16:1113–1114. [PubMed] [Google Scholar]
- 2.Hey A. History and Practice: Antibodies in Infectious Diseases. Microbiol Spectr. 2015;3 doi: 10.1128/microbiolspec.AID-0026-2014. AID-0026-2014. [DOI] [PubMed] [Google Scholar]
- 3.Cohn EJ. The history of plasma fractionation. In: Andrus EC, editor. Advances in Military Medicine. Boston: Little, Brown and Co; 1948. [Google Scholar]
- 4.Keller MA, Stiehm ER. Passive immunity in prevention and treatment of infectious diseases. Clin Microbiol Rev. 2000;13:602–614. doi: 10.1128/cmr.13.4.602-614.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495–497. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 6.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–554. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 7.Steinitz M, Klein G, Koskimies S, Makel O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature. 1977;269:420–422. doi: 10.1038/269420a0. [DOI] [PubMed] [Google Scholar]
- 8.Tiller T, Meffre E, Yurasov S, Tsuiji M, Nussenzweig MC, Wardemann H. Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods. 2008;329:112–124. doi: 10.1016/j.jim.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs. 2015;7:9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.UNAIDS. The GAP Report. 2014 [Google Scholar]
- 11.UNAIDS. Local Epidemics Issues Brief. 2014 [Google Scholar]
- 12.UNAIDS. UNAIDS Fact Sheet November 2016. UNAIDS; 2016. [Google Scholar]
- 13.Mascola JR, Haynes BF. HIV-1 neutralizing antibodies: understanding nature’s pathways. Immunol Rev. 2013;254:225–244. doi: 10.1111/imr.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stephenson KE, Barouch DH. Broadly Neutralizing Antibodies for HIV Eradication. Curr HIV/AIDS Rep. 2016;13:31–37. doi: 10.1007/s11904-016-0299-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haynes BF, Mascola JR. The quest for an antibody-based HIV vaccine. Immunol Rev. 2017;275:5–10. doi: 10.1111/imr.12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 17.Astronomo RD, Santra S, Ballweber-Fleming L, Westerberg KG, Mach L, Hensley-McBain T, et al. Neutralization Takes Precedence Over IgG or IgA Isotype-related Functions in Mucosal HIV-1 Antibody-mediated Protection. EBioMedicine. 2016;14:97–111. doi: 10.1016/j.ebiom.2016.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burton DR, Hessell AJ, Keele BF, Klasse PJ, Ketas TA, Moldt B, et al. Limited or no protection by weakly or nonneutralizing antibodies against vaginal SHIV challenge of macaques compared with a strongly neutralizing antibody. Proceedings of the National Academy of Sciences. 2011;108:11181–11186. doi: 10.1073/pnas.1103012108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moog C, Dereuddre-Bosquet N, Teillaud JL, Biedma ME, Holl V, Van Ham G, et al. Protective effect of vaginal application of neutralizing and nonneutralizing inhibitory antibodies against vaginal SHIV challenge in macaques. Mucosal Immunol. 2014;7:46–56. doi: 10.1038/mi.2013.23. [DOI] [PubMed] [Google Scholar]
- 20.Gauduin MC, Parren PW, Weir R, Barbas CF, Burton DR, Koup RA. Passive immunization with a human monoclonal antibody protects hu-PBL-SCID mice against challenge by primary isolates of HIV-1. Nat Med. 1997;3:1389–1393. doi: 10.1038/nm1297-1389. [DOI] [PubMed] [Google Scholar]
- 21.Gauduin MC, Safrit JT, Weir R, Fung MS, Koup RA. Pre- and postexposure protection against human immunodeficiency virus type 1 infection mediated by a monoclonal antibody. J Infect Dis. 1995;171:1203–1209. doi: 10.1093/infdis/171.5.1203. [DOI] [PubMed] [Google Scholar]
- 22.Parren PW, Ditzel HJ, Gulizia RJ, Binley JM, Barbas CF, 3rd, Burton DR, et al. Protection against HIV-1 infection in hu-PBL-SCID mice by passive immunization with a neutralizing human monoclonal antibody against the gp120 CD4-binding site. AIDS. 1995;9:F1–6. doi: 10.1097/00002030-199506000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Gautam R, Nishimura Y, Pegu A, Nason MC, Klein F, Gazumyan A, et al. A single injection of anti-HIV-1 antibodies protects against repeated SHIV challenges. Nature. 2016;533:105–109. doi: 10.1038/nature17677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hessell AJ, Hangartner L, Hunter M, Havenith CEG, Beurskens FJ, Bakker JM, et al. Fc receptor but not complement binding is important in antibody protection against HIV. Nature. 2007;449:101–104. doi: 10.1038/nature06106. [DOI] [PubMed] [Google Scholar]
- 25.Hessell AJ, Poignard P, Hunter M, Hangartner L, Tehrani DM, Bleeker WK, et al. Effective, low-titer antibody protection against low-dose repeated mucosal SHIV challenge in macaques. Nat Med. 2009;15:951–954. doi: 10.1038/nm.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hessell AJ, Rakasz EG, Poignard P, Hangartner L, Landucci G, Forthal DN, et al. Broadly neutralizing human anti-HIV antibody 2G12 is effective in protection against mucosal SHIV challenge even at low serum neutralizing titers. PLoS Pathog. 2009;5:e1000433. doi: 10.1371/journal.ppat.1000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hessell AJ, Rakasz EG, Tehrani DM, Huber M, Weisgrau KL, Landucci G, et al. Broadly neutralizing monoclonal antibodies 2F5 and 4E10 directed against the human immunodeficiency virus type 1 gp41 membrane-proximal external region protect against mucosal challenge by simian-human immunodeficiency virus SHIVBa-L. J Virol. 2010;84:1302–1313. doi: 10.1128/JVI.01272-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein K, Veazey RS, Warrier R, Hraber P, Doyle-Meyers LA, Buffa V, et al. Neutralizing IgG at the portal of infection mediates protection against vaginal simian/human immunodeficiency virus challenge. J Virol. 2013;87:11604–11616. doi: 10.1128/JVI.01361-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ko S-Y, Pegu A, Rudicell RS, Yang Z-Y, Joyce MG, Chen X, et al. Enhanced neonatal Fc receptor function improves protection against primate SHIV infection. Nature. 2014;514:642–645. doi: 10.1038/nature13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mascola JR, Stiegler G, VanCott TC, Katinger H, Carpenter CB, Hanson CE, et al. Protection of macaques against vaginal transmission of a pathogenic HIV-1/SIV chimeric virus by passive infusion of neutralizing antibodies. Nature Medicine. 2000;6:207–210. doi: 10.1038/72318. [DOI] [PubMed] [Google Scholar]
- 31.Moldt B, Le KM, Carnathan DG, Whitney JB, Schultz N, Lewis MG, et al. Neutralizing antibody affords comparable protection against vaginal and rectal simian/human immunodeficiency virus challenge in macaques. AIDS. 2016;30:1543–1551. doi: 10.1097/QAD.0000000000001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moldt B, Rakasz EG, Schultz N, Chan-Hui PY, Swiderek K, Weisgrau KL, et al. Highly potent HIV-specific antibody neutralization in vitro translates into effective protection against mucosal SHIV challenge in vivo. Proc Natl Acad Sci U S A. 2012;109:18921–18925. doi: 10.1073/pnas.1214785109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moldt B, Shibata-Koyama M, Rakasz EG, Schultz N, Kanda Y, Dunlop DC, et al. A nonfucosylated variant of the anti-HIV-1 monoclonal antibody b12 has enhanced FcgammaRIIIa-mediated antiviral activity in vitro but does not improve protection against mucosal SHIV challenge in macaques. J Virol. 2012;86:6189–6196. doi: 10.1128/JVI.00491-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parren PW, Marx PA, Hessell AJ, Luckay A, Harouse J, Cheng-Mayer C, et al. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J Virol. 2001;75:8340–8347. doi: 10.1128/JVI.75.17.8340-8347.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pegu A, Yang ZY, Boyington JC, Wu L, Ko SY, Schmidt SD, et al. Neutralizing antibodies to HIV-1 envelope protect more effectively in vivo than those to the CD4 receptor. Sci Transl Med. 2014;6:243ra288. doi: 10.1126/scitranslmed.3008992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudicell RS, Kwon YD, Ko SY, Pegu A, Louder MK, Georgiev IS, et al. Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J Virol. 2014;88:12669–12682. doi: 10.1128/JVI.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shingai M, Donau OK, Plishka RJ, Buckler-White A, Mascola JR, Nabel GJ, et al. Passive transfer of modest titers of potent and broadly neutralizing anti-HIV monoclonal antibodies block SHIV infection in macaques. J Exp Med. 2014;211:2061–2074. doi: 10.1084/jem.20132494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braibant M, Barin F. The role of neutralizing antibodies in prevention of HIV-1 infection: what can we learn from the mother-to-child transmission context? Retrovirology. 2013;10:103. doi: 10.1186/1742-4690-10-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ledgerwood JE, Coates EE, Yamshchikov G, Saunders JG, Holman L, Enama ME, et al. Safety, pharmacokinetics and neutralization of the broadly neutralizing HIV-1 human monoclonal antibody VRC01 in healthy adults. Clin Exp Immunol. 2015;182:289–301. doi: 10.1111/cei.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lynch RM, Boritz E, Coates EE, DeZure A, Madden P, Costner P, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med. 2015;7:319ra206. doi: 10.1126/scitranslmed.aad5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mayer KH, Seaton K, Huang Y, Grunenberg N, Hural J, Ledgerwood J, et al. CROI. Boston: 2016. Clinical safety and pharmacokinetics of IV and SC VRC01, a broadly neutralizing mAb. [Google Scholar]
- 42.Astronomo RD, Lemos MP, Narpala S, Czartoski J, Westerberg K, Fleming L, et al. HIV Research for Prevention. Chicago: 2016. Rectal and vaginal biopsies from men and women infused intravenously with VRC01 show protection in ex vivo HIV-1 challenge. [Google Scholar]
- 43.HVTN and HPTN Announce Initiation of Antibody Mediated Prevention (AMP) Study. 2016 [Google Scholar]
- 44.Mascola JR. Defining the protective antibody response for HIV-1. Curr Mol Med. 2003;3:209–216. doi: 10.2174/1566524033479799. [DOI] [PubMed] [Google Scholar]
- 45.Scheid JF, Horwitz JA, Bar-On Y, Kreider EF, Lu CL, Lorenzi JC, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535:556–560. doi: 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bournazos S, Gazumyan A, Seaman MS, Nussenzweig MC, Ravetch JV. Bispecific Anti-HIV-1 Antibodies with Enhanced Breadth and Potency. Cell. 2016;165:1609–1620. doi: 10.1016/j.cell.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sholukh AM, Watkins JD, Vyas HK, Gupta S, Lakhashe SK, Thorat S, et al. Defense-in-depth by mucosally administered anti-HIV dimeric IgA2 and systemic IgG1 mAbs: complete protection of rhesus monkeys from mucosal SHIV challenge. Vaccine. 2015;33:2086–2095. doi: 10.1016/j.vaccine.2015.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Veazey RS, Shattock RJ, Pope M, Kirijan JC, Jones J, Hu Q, et al. Prevention of virus transmission to macaque monkeys by a vaginally applied monoclonal antibody to HIV-1 gp120. Nature Medicine. 2003;9:343–346. doi: 10.1038/nm833. [DOI] [PubMed] [Google Scholar]
- 49.Watkins JD, Sholukh AM, Mukhtar MM, Siddappa NB, Lakhashe SK, Kim M, et al. Anti-HIV IgA isotypes: differential virion capture and inhibition of transcytosis are linked to prevention of mucosal R5 SHIV transmission. AIDS (London, England) 2013;27:F13–20. doi: 10.1097/QAD.0b013e328360eac6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao C, Connor-Stroud F, Oviendo-Moreno P, Zeitlin L, Whaley KJ, Bohorov O, et al. Prevention of vaginal SHIV acquisition by a microbicide containing Nicotiana-produced broadly neutralizing anti-HIV monoclonal antibodies. 2015 [Google Scholar]
- 51.Veselinovic M, Neff CP, Mulder LR, Akkina R. Topical gel formulation of broadly neutralizing anti-HIV-1 monoclonal antibody VRC01 confers protection against HIV-1 vaginal challenge in a humanized mouse model. Virology. 2012;432:505–510. doi: 10.1016/j.virol.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morris GC, Wiggins RC, Woodhall SC, Bland JM, Taylor CR, Jespers V, et al. MABGEL 1: first phase 1 trial of the anti-HIV-1 monoclonal antibodies 2F5, 4E10 and 2G12 as a vaginal microbicide. PLoS One. 2014;9:e116153. doi: 10.1371/journal.pone.0116153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ma JK, Drossard J, Lewis D, Altmann F, Boyle J, Christou P, et al. Regulatory approval and a first-in-human phase I clinical trial of a monoclonal antibody produced in transgenic tobacco plants. Plant Biotechnol J. 2015;13:1106–1120. doi: 10.1111/pbi.12416. [DOI] [PubMed] [Google Scholar]
- 54.Whaley KJ, Zeitlin L. Preventing transmission: plant-derived microbicides and mucosal vaccines for reproductive health. Vaccine. 2005;23:1819–1822. doi: 10.1016/j.vaccine.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 55.De Logu A, Williamson RA, Rozenshteyn R, Ramiro-Ibanez F, Simpson CD, Burton DR, et al. Characterization of a type-common human recombinant monoclonal antibody to herpes simplex virus with high therapeutic potential. J Clin Microbiol. 1998;36:3198–3204. doi: 10.1128/jcm.36.11.3198-3204.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diekman AB, Norton EJ, Westbrook VA, Klotz KL, Naaby-Hansen S, Herr JC. Anti-sperm antibodies from infertile patients and their cognate sperm antigens: a review. Identity between SAGA-1, the H6–3C4 antigen, and CD52. Am J Reprod Immunol. 2000;43:134–143. doi: 10.1111/j.8755-8920.2000.430302.x. [DOI] [PubMed] [Google Scholar]
- 57.Zhao C, Gunawardana M, Villinger F, Baum M, Remedios-Chan R, Moench T et al. Pharmacokinetics and safety of pod-intravaginal rings delivering VRC01-N for HIV prophylaxis in a macaque model. Antimicrob Agents Chemother. 2017 doi: 10.1128/AAC.02465-16. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gengiah TN, Moosa A, Naidoo A, Mansoor LE. Adherence challenges with drugs for pre-exposure prophylaxis to prevent HIV infection. Int J Clin Pharm. 2014;36:70–85. doi: 10.1007/s11096-013-9861-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25:1369–1372. doi: 10.1038/nbt1207-1369. [DOI] [PubMed] [Google Scholar]
- 60.Delacroix DL, Hodgson HJ, McPherson A, Dive C, Vaerman JP. Selective transport of polymeric immunoglobulin A in bile. Quantitative relationships of monomeric and polymeric immunoglobulin A, immunoglobulin M, and other proteins in serum, bile, and saliva. The Journal of Clinical Investigation. 1982;70:230–241. doi: 10.1172/JCI110610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brandtzaeg P. Secretory IgA: Designed for Anti-Microbial Defense. Frontiers in Immunology. 2013;4:222. doi: 10.3389/fimmu.2013.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Virdi V, Juarez P, Boudolf V, Depicker A. Recombinant IgA production for mucosal passive immunization, advancing beyond the hurdles. Cell Mol Life Sci. 2016;73:535–545. doi: 10.1007/s00018-015-2074-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stieh DJ, King DF, Klein K, Liu P, Shen X, Hwang KK, et al. Aggregate complexes of HIV-1 induced by multimeric antibodies. Retrovirology. 2014;11 doi: 10.1186/s12977-014-0078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bonner A, Furtado PB, Almogren A, Kerr MA, Perkins SJ. Implications of the near-planar solution structure of human myeloma dimeric IgA1 for mucosal immunity and IgA nephropathy. Journal of Immunology (Baltimore, Md: 1950) 2008;180:1008–1018. doi: 10.4049/jimmunol.180.2.1008. [DOI] [PubMed] [Google Scholar]
- 65.Phalipon A, Corthésy B. Novel functions of the polymeric Ig receptor: well beyond transport of immunoglobulins. Trends in Immunology. 2003;24:55–58. doi: 10.1016/s1471-4906(02)00031-5. [DOI] [PubMed] [Google Scholar]
- 66.Wright A, Lamm ME, Huang YT. Excretion of human immunodeficiency virus type 1 through polarized epithelium by immunoglobulin A. Journal of Virology. 2008;82:11526–11535. doi: 10.1128/JVI.01111-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang YT, Wright A, Gao X, Kulick L, Yan H, Lamm ME. Intraepithelial cell neutralization of HIV-1 replication by IgA. Journal of Immunology (Baltimore, Md: 1950) 2005;174:4828–4835. doi: 10.4049/jimmunol.174.8.4828. [DOI] [PubMed] [Google Scholar]
- 68.Hur EM, Patel SN, Shimizu S, Rao DS, Gnanapragasam PNP, An DS, et al. Inhibitory effect of HIV-specific neutralizing IgA on mucosal transmission of HIV in humanized mice. Blood. 2012;120:4571–4582. doi: 10.1182/blood-2012-04-422303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, et al. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N Engl J Med. 2012;366:1275–1286. doi: 10.1056/NEJMoa1113425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomaras GD, Ferrari G, Shen X, Alam SM, Liao H-X, Pollara J, et al. Vaccine-induced plasma IgA specific for the C1 region of the HIV-1 envelope blocks binding and effector function of IgG. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:9019–9024. doi: 10.1073/pnas.1301456110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang Y, Yu J, Lanzi A, Yao X, Andrews CD, Tsai L, et al. Engineered Bispecific Antibodies with Exquisite HIV-1-Neutralizing Activity. Cell. 2016;165:1621–1631. doi: 10.1016/j.cell.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chiang JJ, Gardner MR, Quinlan BD, Dorfman T, Choe H, Farzan M. Enhanced recognition and neutralization of HIV-1 by antibody-derived CCR5-mimetic peptide variants. J Virol. 2012;86:12417–12421. doi: 10.1128/JVI.00967-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu Q, Yu R, Qin X. The good and evil of complement activation in HIV-1 infection. Cell Mol Immunol. 2010;7:334–340. doi: 10.1038/cmi.2010.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ahmad A, Menezes J. Antibody-dependent cellular cytotoxicity in HIV infections. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1996;10:258–266. doi: 10.1096/fasebj.10.2.8641559. [DOI] [PubMed] [Google Scholar]
- 75.Fanger MW, Shen L, Graziano RF, Guyre PM. Cytotoxicity mediated by human Fc receptors for IgG. Immunology Today. 1989;10:92–99. doi: 10.1016/0167-5699(89)90234-X. [DOI] [PubMed] [Google Scholar]
- 76.Klein JS, Webster A, Gnanapragasam PNP, Galimidi RP, Bjorkman PJ. A dimeric form of the HIV-1 antibody 2G12 elicits potent antibody-dependent cellular cytotoxicity. AIDS (London, England) 2010;24:1633–1640. doi: 10.1097/qad.0b013e32833ad8c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mabondzo A, Boussin F, Raoul H, Le Naour R, Gras G, Vaslin B, et al. Antibody-dependent cellular cytotoxicity and neutralization of human immunodeficiency virus type 1 by high affinity cross-linking of gp41 to human macrophage Fc IgG receptor using bispecific antibody. The Journal of General Virology. 1994;75(Pt 6):1451–1456. doi: 10.1099/0022-1317-75-6-1451. [DOI] [PubMed] [Google Scholar]
- 78.Perez LG, Costa MR, Todd CA, Haynes BF, Montefiori DC. Utilization of immunoglobulin G Fc receptors by human immunodeficiency virus type 1: a specific role for antibodies against the membrane-proximal external region of gp41. Journal of Virology. 2009;83:7397–7410. doi: 10.1128/JVI.00656-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ackerman ME, Dugast A-S, McAndrew EG, Tsoukas S, Licht AF, Irvine DJ, et al. Enhanced phagocytic activity of HIV-specific antibodies correlates with natural production of immunoglobulins with skewed affinity for FcγR2a and FcγR2b. Journal of Virology. 2013;87:5468–5476. doi: 10.1128/JVI.03403-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dugast A-S, Tonelli A, Berger CT, Ackerman ME, Sciaranghella G, Liu Q, et al. Decreased Fc receptor expression on innate immune cells is associated with impaired antibody-mediated cellular phagocytic activity in chronically HIV-1 infected individuals. Virology. 2011;415:160–167. doi: 10.1016/j.virol.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asmal M, Sun Y, Lane S, Yeh W, Schmidt SD, Mascola JR, et al. Antibody-dependent cell-mediated viral inhibition emerges after simian immunodeficiency virus SIVmac251 infection of rhesus monkeys coincident with gp140-binding antibodies and is effective against neutralization-resistant viruses. J Virol. 2011;85:5465–5475. doi: 10.1128/JVI.00313-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. The Journal of Biological Chemistry. 2002;277:26733–26740. doi: 10.1074/jbc.M202069200. [DOI] [PubMed] [Google Scholar]
- 83.Ferrara C, Grau S, Jager C, Sondermann P, Brunker P, Waldhauer I, et al. Unique carbohydrate-carbohydrate interactions are required for high affinity binding between FcgammaRIII and antibodies lacking core fucose. Proc Natl Acad Sci U S A. 2011;108:12669–12674. doi: 10.1073/pnas.1108455108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ackerman ME, Crispin M, Yu X, Baruah K, Boesch AW, Harvey DJ, et al. Natural variation in Fc glycosylation of HIV-specific antibodies impacts antiviral activity. The Journal of Clinical Investigation. 2013;123:2183–2192. doi: 10.1172/JCI65708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gunn BM, Schneider JR, Shansab M, Bastian AR, Fahrbach KM, Smith ADt, et al. Enhanced binding of antibodies generated during chronic HIV infection to mucus component MUC16. Mucosal Immunol. 2016 doi: 10.1038/mi.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ahmed AA, Keremane SR, Vielmetter J, Bjorkman PJ. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcgammaRIIIa. J Struct Biol. 2016;194:78–89. doi: 10.1016/j.jsb.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. 2014;158:1243–1253. doi: 10.1016/j.cell.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nature Reviews Immunology. 2007;7:715–725. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 89.Euler Z, Alter G. Exploring the Potential of Monoclonal Antibody Therapeutics for HIV-1 Eradication. AIDS Research and Human Retroviruses. 2015;31:13–24. doi: 10.1089/aid.2014.0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gong R, Wang Y, Ying T, Dimitrov DS. Bispecific engineered antibody domains (nanoantibodies) that interact noncompetitively with an HIV-1 neutralizing epitope and FcRn. PloS One. 2012;7:e42288. doi: 10.1371/journal.pone.0042288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.David Pollard MB, Yuki Abe, Lopes Adriana G, Sinclair Andrew. Standardized Economic Cost Modeling for Next-Generation MAb Production. BioProcess International. 2016;14:14–23. [Google Scholar]
- 92.Jagschies Gea. Technical and Economical Evaluation of Downstream Processing Options for Monoclonal Antibody (MAb) Production. BioPharm Int. 2006 [Google Scholar]
- 93.M. B. BioSMB™ Technology MB— Continuous Countercurrent Chromatography Enabling a Fully Disposable Process. Biopharmaceutical Production Technology. 2012 [Google Scholar]
- 94.Hou Y, Brower M, Pollard D, Kanani D, Jacquemart R, Kachuik B, et al. Advective hydrogel membrane chromatography for monoclonal antibody purification in bioprocessing. Biotechnol Prog. 2015;31:974–982. doi: 10.1002/btpr.2113. [DOI] [PubMed] [Google Scholar]
- 95.Sinclair AMM. Biomanufacturing for the 21st Century: Designing a Concept Facility Based on Single-Use Systems. BioProcess Int. 2004;2:S26–S31. [Google Scholar]
- 96.Kunert R, Reinhart D. Advances in recombinant antibody manufacturing. Appl Microbiol Biotechnol. 2016;100:3451–3461. doi: 10.1007/s00253-016-7388-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dumont J, Euwart D, Mei B, Estes S, Kshirsagar R. Human cell lines for biopharmaceutical manufacturing: history, status, and future perspectives. Crit Rev Biotechnol. 2015:1–13. doi: 10.3109/07388551.2015.1084266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hossler P, Khattak SF, Li ZJ. Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology. 2009;19:936–949. doi: 10.1093/glycob/cwp079. [DOI] [PubMed] [Google Scholar]
- 99.Spadiut O, Capone S, Krainer F, Glieder A, Herwig C. Microbials for the production of monoclonal antibodies and antibody fragments. Trends Biotechnol. 2014;32:54–60. doi: 10.1016/j.tibtech.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Baker M. Upping the ante on antibodies. Nat Biotechnol. 2005;23:1065–1072. doi: 10.1038/nbt0905-1065. [DOI] [PubMed] [Google Scholar]
- 101.Liu J, Ghneim K, Sok D, Bosche WJ, Li Y, Chipriano E, et al. Antibody-mediated protection against SHIV challenge includes systemic clearance of distal virus. Science. 2016;353:1045–1049. doi: 10.1126/science.aag0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Anderson DJ. Modeling mucosal cell-associated HIV type 1 transmission in vitro. J Infect Dis. 2014;210(Suppl 3):S648–653. doi: 10.1093/infdis/jiu537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anderson DJ, Politch JA, Nadolski AM, Blaskewicz CD, Pudney J, Mayer KH. Targeting Trojan Horse leukocytes for HIV prevention. AIDS. 2010;24:163–187. doi: 10.1097/QAD.0b013e32833424c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sattentau Q. Avoiding the void: cell-to-cell spread of human viruses. Nat Rev Microbiol. 2008;6:815–826. doi: 10.1038/nrmicro1972. [DOI] [PubMed] [Google Scholar]
- 105.Zhong P, Agosto LM, Ilinskaya A, Dorjbal B, Truong R, Derse D, et al. Cell-to-cell transmission can overcome multiple donor and target cell barriers imposed on cell-free HIV. PLoS One. 2013;8:e53138. doi: 10.1371/journal.pone.0053138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Politch JA, Marathe J, Anderson DJ. Characteristics and quantities of HIV host cells in human genital tract secretions. J Infect Dis. 2014;210(Suppl 3):S609–615. doi: 10.1093/infdis/jiu390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Burton DR, Hangartner L. Broadly Neutralizing Antibodies to HIV and Their Role in Vaccine Design. Annu Rev Immunol. 2016;34:635–659. doi: 10.1146/annurev-immunol-041015-055515. [DOI] [PMC free article] [PubMed] [Google Scholar]