

Abstract
Elevated activity of the mechanistic target of rapamycin (mTOR) is associated with poor prognosis and higher incidence of relapse in B-cell acute lymphoblastic leukemia (B-ALL). Thus, ongoing clinical trials are testing mTOR inhibitors in combination with chemotherapy in B-ALL. However, the combination of mTOR inhibitors with standard of care chemotherapy drugs has not been studied extensively in high risk B-ALL subtypes. Therefore, we tested whether mTOR inhibition can augment the efficacy of current chemotherapy agents in Ph+ and Ph-like B-ALL models. Surprisingly, inhibiting mTOR complex 1 (mTORC1) protected B-ALL cells from killing by methotrexate and 6-mercaptopurine, two anti-metabolite drugs used in maintenance chemotherapy. The cytoprotective effects correlated with decreased cell cycle progression and were recapitulated using cell cycle inhibitors, palbociclib or aphidicolin. Dasatinib, a tyrosine kinase inhibitor currently used in Ph+ patients, inhibits ABL kinase upstream of mTOR. Dasatinib resistance is mainly caused by ABL kinase mutations, but is also observed in a subset of ABL unmutated cases. We identified dasatinib-resistant Ph+ cell lines and patient samples where dasatinib can effectively reduce ABL kinase activity and mTORC1 signaling without causing cell death. In these cases, dasatinib protected leukemia cells from killing by 6-mercaptopurine. Using xenograft models, we observed that mTOR inhibition or dasatinib increased the numbers of leukemia cells that emerge after cessation of chemotherapy treatment. These results demonstrate that inhibitors targeting mTOR or upstream signaling nodes should be used with caution when combined with chemotherapeutic agents that rely on cell cycle progression to kill B-ALL cells.
Keywords: Leukemias and lymphomas, Cell signaling, Cellular responses to anti-cancer drugs, Kinase and phosphatase inhibitors, mTOR, B-ALL, dasatinib
Introduction
B-ALL is the most common pediatric cancer and is a significant cause of death in children and adults. While current chemotherapeutic regimens produce complete remissions in the majority of pediatric patients, they are less effective in adults. This is likely due to the higher incidence of Ph+ B-ALL in adults (30–40%) than in children (1–5%) (1,2). Additionally, 10–27% of B-ALL cases have gene-expression profiles that are similar to Ph+ leukemias (termed Ph-like) (3,4), which also confers poor prognosis. Both Ph+ and Ph-like leukemias exhibit elevated oncogenic signaling through tyrosine kinases including ABL, PDGFR and JAK (3). While the use of the ABL tyrosine kinase inhibitor (TKI) imatinib with chemotherapy has improved the initial complete remission rate in Ph+ patients, only 38% of imatinib treated patients survive after 4 years (5). Poor long-term survival is mainly driven by the emergence of TKI resistance due to ABL kinase mutations. The second generation inhibitor dasatinib is active against all imatinib-resistant kinase domain mutations, except T315I. However, a subset of patients has been observed to relapse after dasatinib treatment without ABL mutations (6–8). These dasatinib resistant cells may survive by activating parallel pathways to maintain growth and survival signals driving patient relapse (9,10).
A strategy for treating Ph+ and Ph-like patients is to inhibit survival pathways activated by tyrosine kinases. A promising candidate is mTOR, which integrates growth factors and nutrient signals to promote cell survival and proliferation. mTOR is a kinase that exists in two complexes, mTORC1 and mTORC2, with distinct mechanisms of activation and downstream outputs. mTORC2 is activated by signals from phosphoinositide 3-kinase (PI3K) and promotes activation of the pro-survival protein AKT (11,12). mTORC1 phosphorylates various substrates including ribosomal S6 kinases (S6Ks) and eIF4E-binding proteins (4E-BPs) to promote metabolic pathways that support survival, growth and proliferation (13). Indeed, high 4E-BP1 phosphorylation correlates with increased relapse in B-ALL patients (14). While first generation allosteric mTOR inhibitors (rapamycin and its derivatives, rapalogs) only partially inhibit mTORC1, newer ATP-competitive mTOR kinase inhibitors (TOR-KIs) fully suppress mTOR kinase activity in both complexes (15).
A growing body of evidence indicates that mTOR inhibitors are most effective against cancer cells when used in combination with other agents. For example, TOR-KIs can sensitize B-ALL cells to targeted agents like dasatinib and HDAC inhibitors (16–18). In addition, rapamycin can sensitize B-ALL cells to various chemotherapeutic agents (19–21). Clinical trials of mTOR inhibitors in combination with chemotherapy in B-ALL patients are underway (22). In this study, we specifically addressed whether mTOR inhibition sensitizes Ph+ and Ph-like B-ALL to chemotherapy. Upon combining standard chemotherapeutic agents with mTOR inhibitors, we noted that mTOR inhibition protected cells from methotrexate (MTX) and 6-mercaptopurine (6-MP), antimetabolites mainly used in maintenance chemotherapy. Protection was seen in vitro and in vivo, resulting in faster relapse following cessation of chemotherapy. The mechanism of protection was largely due to slowing cell cycling. In support, both the cell cycle inhibitor palbociclib and mTOR inhibition markedly reduced the amount of DNA damage caused by MTX and 6-MP. Thus, our data suggest that concurrent treatment with mTORC1-targeted agents and certain maintenance drugs may be counterproductive in Ph+ and Ph-like B-ALL patients. Moreover, inhibitors that target kinases upstream of mTORC1 also protect from maintenance drugs depending on how strongly they inhibit downstream mTORC1 activity. Indeed, dasatinib can also be chemoprotective in B-ALL cells where it suppresses mTORC1 activity. These findings highlight that cell cycle inhibition by targeted oncology drugs can cause unintended protective effects when used in certain chemotherapeutic combinations.
Materials and Methods
Chemicals
Dasatinib, GDC-0941, AZD8055, rapamycin and MLN0128 were obtained from LC Laboratories (Woburn, MA); AKT inhibitor VIII from Chemdea (Ridgewood, NJ). InSolution Q-VD-OPh was from EMD Millipore (Billerica, MA); vincristine, doxycycline, etoposide, daunorubicin, MTX, 6-MP, dexamethasone and araC are from Sigma-Aldrich (St. Louis, MO). Palbociclib was purchased from Selleck Chemicals (Houston, TX).
Cell Culture
BV-173 cells were purchased from DSMZ (August 2012) and SUP-B15 from ATCC (August 2016) and validated by STR profiling (University of Arizona core facility) on March 2017. B-ALL lines are cultured in RP10 media (RPMI1640 supplemented with 10% FBS, 2 mM L-glutamine, 100 U/mL Pen/strep, and 10 mM HEPES) and used within 3 months of resuscitating viably frozen aliquots.
The Ph-like B-ALL specimen PAUXZX was obtained from the Children Oncology Group through biology protocol AALL16B3-Q. PAUXZX, SFO2, ICN1, and MXP5 B-ALL patient derived xenograft (PDX) lines were cultured as described previously (23). Briefly, cells were cultured on irradiated OP9 stroma in primary media [MEM Alpha (Gibco Cat. No. 12561-056) supplemented with 20% FBS, 1 mM sodium pyruvate, 1X GlutaMAX (Invitrogen), and 100 U/mL Pen/strep]. OP9 cells were irradiated at 2500 Rad and plated onto gelatin-coated T25 flasks one day prior to plating the B-ALL patient cells. Flasks were pre-coated with 1% gelatin (175 g Bloom from Sigma). Primary cells were replated at 1×106 cells/mL on irradiated OP9 cells every 5 days.
Cell Death Assay
Cell lines (human B-ALL and mouse p190 cells) were plated with chemical agents at 4×104 cells in 200 μL RP10 media per well on 96-well U-bottom plate. For short-term patient lines, irradiated OP9 cells were pre-plated on gelatin coated 96-well flat bottom plates. The following day, the media was aspirated and the patient lines were plated with chemical agents at 4×104 cells in 200 μL primary media. See figure legends for drug concentrations and duration. For all treatments, chemotherapy drugs and targeted small molecule inhibitors were added concurrently. For harvesting, both cell lines and patient samples were pelleted, resuspended in 150 μL of Annexin Binding Buffer (10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2-2H2O, pH 7.4) containing Annexin-V Alexa Fluor 647 (ThermoFisher) and 0.4 μg/mL propidium iodide (ThermoFisher). Since OP9 stromal cells adhered strongly, patient cells were dislodged by vigorous pipetting. All FACS data was measured using the FACSCalibur (Becton Dickinson) and processed with FlowJo Software v10.0.8 (FlowJo, LLC).
Western Blot
Cells were plated in 12 well plates at 1×106 cells/mL in 2 mL of drugs/inhibitors. For signaling assays, cells were pelleted 6 hours later. For DNA damaging assays, cells were also treated with 20 μM QVD-OPh (pan-caspase inhibitor) and harvested 48 hours later. For DNA damaging assays, a small aliquot was stained with Annexin-V AlexaFluor647 and PI to confirm cells were fully viable. Cell pellets were washed once with PBS. Pellets were lysed in RIPA lysis buffer (50 mM Tris-HCl, 1% v/v NP-40, 0.5% w/v Na-deoxycholate, 1 mM EDTA, 150 mM NACl, 1 mM EGTA) supplemented with protease inhibitor cocktail (Calbiochem) and phosphatase inhibitor cocktails 2 and 3 (Sigma). Cells were lysed for 30 min on ice and spun down at 10,000 × g for 10 minutes. The supernatants were normalized using Bradford protein assay (Bio-Rad), ran at 20 μg per lane on 10% SDS-PAGE gel and transferred onto nitrocellulose using a Bio-Rad system. The following primary antibodies from Cell Signaling Technology were used for immunoblotting: p-STAT5 (clone C71E5), p-AKT (Ser473) (clone D9E), total AKT (clone 11E7), p-PRAS40 (clone D4D2), GAPDH (clone 14C10), p-S6 (clone D68F8), p-4EBP (clone 236B4), total 4EBP (clone 53H11), p-ABL (cat. No #2861), p-PRAS40 (clone C77D7), total PRAS40 (clone D23C7), p-CHK1 (clone 133D3), and p-histone H2A.X (clone 20E3). HRP conjugated anti-rabbit IgG (Promega), Pierce ECL Western Blotting Substrate, SuperSignal West Femto Maximum Sensitivity Substrate (Life Technologies) and Nikon D700 SLR camera system (24) were used to develop the blots. Images were processed using Adobe Photoshop and densitometry was quantified using ImageJ software.
Intracellular Flow Cytometry
To detect mTORC1 activity, 5×105 cells were plated in 1 mL on a 24-well plate for 6 hours. Cells were fixed in 100 μL of BD Cytofix/Cytoperm (Cat. No. 554722) for 20 min at 4°C, washed with PBS and resuspended in 100 μL 0.5% Tween 20 (in PBS with 10 mM EDTA) containing pS6-APC (Cell Signaling clone D68F8) antibody. Cells were incubated for 20–30 minutes in the dark at 4°C, washed with 1 mL of PBS and resuspended in 150 μL PBS for FACS analysis.
For DNA damage, 1×106 cells were treated with drugs/inhibitors and 20 μM QVD-OPh for 48 hours in 2 mL on 12-well plate, spun down and incubated in 100 μL 4% paraformaldehyde for 10 minutes at 37°C. Cells were washed with PBS, resuspended in 500 μL of 90% ice-cold methanol and stored at −20°C overnight. The following day, cells were washed with PBS and stained with p-histone H2A.X antibody (Cell Signaling clone 20E3) for 30 minutes on ice. Cells were incubated in goat anti-rabbit antibody AlexaFluor488 (Life Technology) for 20 minutes. Cells were resuspended in 150 μL PBS for FACS analysis.
Cell Cycle Assay
Cells were plated at 2×105 cells in 1 mL of RP10 media with various inhibitors (LY2584702, MLN0128, rapamycin, doxycycline, palbociclib or aphidicolin). After 24 hours, cells were pelleted and resuspended in 250 μL of cell cycle buffer (PBS with 5 mM EDTA). With vortexing, another 250 μL of ice-cold ethanol was added. Cells were stored at −20°C overnight. Ethanol fixed cells were pelleted, resuspended in 150 μL of cell cycle buffer with 10 μg/mL RNase and incubated at room temperature for 2 hours. Then 125 μL of cells was combined in a FACS tube with 125 μL of 100 μg/mL propidium iodide in cell cycle buffer for FACS analysis.
Growth Rate Measurements
B-ALL cell lines were plated at 2×105 cells in 1 mL of RP10 media with various inhibitors in the presence or absence of 20 μM QVD-OPh. After 72 hours culture, cells were spun down and resuspended in 150 μL of Annexin Binding Buffer (10 mM HEPES, 140 mM NaCl and 2.5 mM CaCl2-2H2O, pH 7.4) containing Annexin-V Alexa Fluor 647 (ThermoFisher) and 0.4 μg/mL propidium iodide (ThermoFisher) for FACS analysis. Each sample was run for 10 seconds to equalize the flow rate and then events were read for 30 seconds on high flow rate. The number of viable cells was calculated from events collected at this constant time for each condition.
Animals
Mice were studied in compliance with protocols approved by the Institutional Animal Care and Use Committees of the University of California, Irvine.
4E-BP1 5A inducible p190 cells
Using Rosa26-rtTA control mice and Rosa26-rtTA/TRE-4E-BP1 5A mice described in So et al (25), we made pre-B leukemia cells (p190 cells) driven by the p190 isoform of BCR-ABL as described in Kharas et al (26).
In vitro regrowth assay
Cell lines were plated at 2.5 ×105 cells/mL in 3 mL RP10 media on 6-well plate with 30 nM MTX and inhibitors for 3 days. Cells were then washed and resuspended in 1 mL RP10 media. Cells were grown for 7 additional days with regular passaging. The volume during each passaging was recorded for later back-calculation of growth rate. After regrowth, equal volume of cells were resuspended in 150 μL of Annexin Binding Buffer with Annexin-V AlexaFluor647 and PI. Cells were ran on BD FACSCalibur to count number of viable cells at equal flow rates and time collected, similar to growth rate method described above. The number of viable cells was calculated using the viable events counted and passaging records.
In vivo xenograft
NSG mice were obtained from JAX (NOD-SCID-IL2Rγ-null, stock 005557). Animal studies were approved by the Institutional Animal Care and Use Committee at UC Irvine. NSG mice at 1–3 months of age were retro-orbital injected with 2.5 million SFO2 or PAUXZX patient cells. Facial vein bleeds were done to monitor engraftment. Upon detection of greater than 1% leukemia was observed in all mice, mice were dosed once daily for 5 days with 30 mg/kg 6-MP, 20 mg/kg AZD8055, 10 mg/kg dasatinib, or combinations. 6-MP was dissolved in 0.5% carboxymethylcellulose (medium viscosity, Sigma) in water and injected with a 26 gauge-needle by intraperitoneal injection. AZD8055 was given orally in 0.5% hydroxypropylmethylcellulose with 0.1% Tween-80 in water. Dasatinib was given orally dissolved in a 50:50 mix of polypropylene glycol and water. Dosing schedules are outline in Figure 6A for PAUXZX and 6D for SFO2.
Figure 6. mTOR inhibitor protects B-ALL in vivo causing greater relapse after chemo.
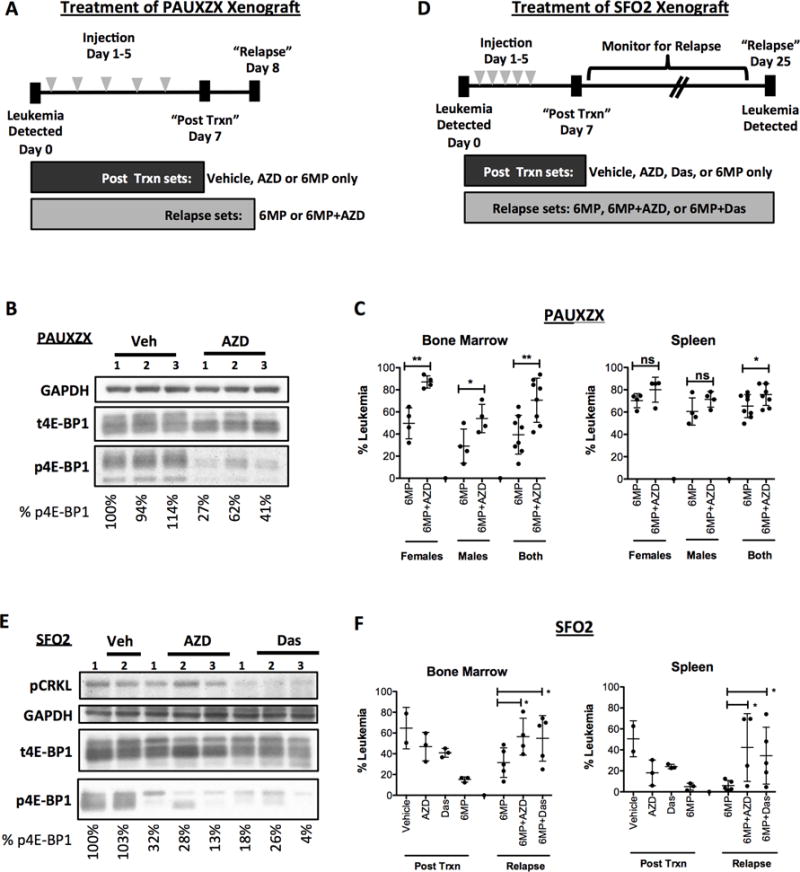
(A) For in vivo relapse assays, NSG mice were treated for 5 days (dosed once a day) upon detection of at least 1% PAUXZX leukemia in their peripheral blood. Control mice were treated with vehicle, AZD8055 or 6-MP only. Experimental mice were all dosed with 6-MP in addition to vehicle or AZD8055. The post treatment control mice were sacrificed 7 days after treatment was started. The experimental mice were sacrificed on day 8. (B) Control post-treatment mice were dosed with vehicle or AZD8055 2 hours before sacrifice on day 7. Spleen cells were harvested and Western blot analysis was done to determine effect of AZD8055 on mTORC1 activity as measured by p4E-BP1. (C) The leukemia burden in the bone marrow and spleen of the “relapse” PAUXZX sets were detected using human CD19 versus mouse CD45. (D) NSG mice were treated for 5 days (dosed once a day) upon detection of at least 1% SFO2 leukemia in their peripheral blood. Control mice were treated with vehicle, AZD8055, dasatinib or 6-MP as single agents. Experimental mice were all dosed with 6-MP in addition to vehicle, AZD8055 or dasatinib. The post treatment control mice were sacrificed 7 days after treatment was started. The experimental mice were monitored until relapse was detected in their peripheral blood, upon which time they were sacrificed (day 25). Note that during this period, one mouse in the 6MP+AZD set died, likely due to toxicity of the 6MP+AZD combination. (E) Control Ph+ SFO2 injected mice were treated with vehicle, AZD8055 alone or dasatinib alone. On day 7, mice were dosed with one final dose of inhibitor and sacrificed 2 hours after dosing. Western blot was performed on their bone marrow cells to detect inhibition of mTOR pathway by inhibitors. (F) The bone marrow and spleen of the control “post-treatment” and experimental “relapse” mice were obtained. The percentage of leukemia in bone or spleen were detected by staining human CD19 versus mouse CD45. Unpaired t-test was done for each comparison, mean±SD, *p<0.05, **p<0.005, ***p<0.0005.
After final dosing on day 5, control mice were given one day of rest to clear dying cells that would make accurate detection of leukemia burden difficult. Post-treatment mice given only single agents controls or vehicle were sacrificed on day 7. Two hours before sacrifice, single agent (vehicle, AZD8055 and dasatinib) control mice were dosed one more time to detect inhibitor effects on mTORC1 signaling.
Relapse mouse sets were monitored by facial bleeds for signs of leukemia relapse. Upon detection of more than 1% leukemia in peripheral blood of any group, all relapse sets were sacrificed at once.
Bone marrow and spleen were obtained. Red blood cells were lysed in ACK buffer (150 mM NH4Cl, 10 mM KHCO3 and 0.1 mM Na2EDTA in water) for 5 minutes at room temperature and washed with cold PBS + 1% FBS. One million cells were stained with human CD19-PE (Biolegend clone HIB19) and mouse CD45-APC (eBioscience clone 30-F11) for 20 minutes on ice in PBS + 1% FBS. Cells were spun down at 500 × g for 5 minutes and resuspended in cold PBS for FACS analysis. Remaining samples were analyzed according to Western blot section above.
Results
Inhibition of mTORC1 protects from MTX and 6-MP
Since high mTORC1 activity is predictive of increased relapse in B-ALL patients, we tested whether inhibiting mTOR would sensitize B-ALL cell lines to chemotherapeutic agents. Using MLN0128 (a TOR-KI compound), we found that mTOR inhibition protected Ph+ B-ALL cell lines BV173 and SUP-B15 from the cytotoxic effects of both MTX and 6-MP (Fig. 1A and B). A titration of rapamycin, MLN1028 (27) and AZD8055 (28) (a chemically distinct TOR-KI) revealed that all mTORC1 inhibitors protect from MTX in a dose-dependent manner (Supplementary Fig. S1A). Moreover, mTOR inhibition shifted the dose response curve for MTX and 6MP towards greater survival even at higher doses (Supplementary Fig. S1B). Treatment with MLN0128 alone did not affect viability of BV173 or SUP-B15 (Supplementary Fig. S1A and D). The dual PI3K/mTOR inhibitor NVP-BEZ235 also exhibited protection (Supplementary Fig. S1C).
Figure 1. mTOR inhibition protects from methotrexate and 6-mercaptopurine.
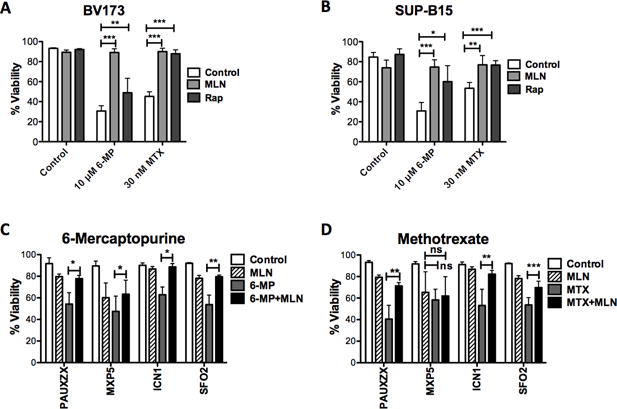
(A) BV173 and (B) SUP-B15 Ph+ B-ALL cells are treated with MTX and 6MP for 48 hours in presence of 100 nM MLN0128. Viability is measured by Annexin-V and PI staining. Unpaired t-tests of were performed, n=3, mean±SD, *p<0.05, **p<0.005, ***p<0.0005. Patient lines were treated with (C) MTX or (D) 6-MP for 5 days in presence or absence of 100 nM MLN0128. Viability is measured by Annexin-V and PI staining. Unpaired t-tests of were performed, n=3, mean±SD, *p<0.05, **p<0.005, ***p<0.0005.
To confirm that mTOR inhibition preserves more viable cells, we assessed the outgrowth of leukemia cells after treatment cessation. Therefore, we treated BV173 and SUP-B15 cells with MTX in presence or absence of mTOR inhibition for 3 days. Cells were allowed to regrow for 7 days in the absence of drug. Indeed, mTOR inhibition combined with MTX increased the number of recovered cells compared to MTX alone (Supplementary Fig. S1E and F, note the log scale).
We further tested these combinations on B-ALL lines (Supplementary Table S1) established as patient-derived xenografts (PDX) and then grown on OP9 stromal cells. mTOR inhibition protected from killing by MTX and 6-MP in the Ph-like sample PAUXZX as well as two Ph+ samples ICN1 and SFO2 (Fig. 1C and D). In a third Ph+ sample, MXP5, MLN0128 slightly rescued from killing by 6MP but not MTX.
mTOR inhibition had variable affects on the cytotoxicity of other chemotherapeutic agents in BV173 and SUP-B15 cell lines (Supplementary Fig. S2A and B), with slight but statistically significant protection observed for some agents, for example araC. However, we did not observe similar protection from araC in any of the PDX lines (Supplementary Fig. S2C). Together these results show that in both cell lines and stromal-dependent B-ALL cells, mTOR inhibition consistently protects from killing by MTX and 6-MP but not other chemotherapeutic agents.
Inhibition of kinases upstream of mTOR protects B-ALL cells
Inhibitors of various kinases in the PI3K/AKT/mTOR network are actively being explored as therapeutic agents (Fig. 2A) (15,29–31). We tested if these inhibitors would similarly have the adverse effect of chemoprotection. We titrated each inhibitor to find the minimum concentration needed to fully and selectively inhibit its intended target (Supplementary Fig. S3), then treated the Ph+ cell line BV173 with each inhibitor in the presence or absence of MTX (Fig. 2B and C). No inhibitor caused cell death as a single agent. However, when combined with MTX, most inhibitors protected from the chemotherapy agent. Notably, direct mTORC1 inhibitors (MLN0128, rapamycin) provided the greatest protection. The TKI dasatinib, which blocked p-STAT5 and reduced mTOR signaling in BV173 cells (Fig. 2B), also protected from MTX. Quantifying mTOR inhibition by intracellular staining of S6 phosphorylation (Fig. 2D) revealed that the degree of mTORC1 inhibition by each compound correlates with how well it protects from MTX (Fig. 2E). These effects were not restricted to BV173 cells since upstream inhibitors also protected from MTX in Ph+ SUP-B15 cells (Fig. 2F). Notably, dasatinib minimally affected p-S6 in SUP-B15 cells despite fully suppressing STAT5 phosphorylation. Correspondingly, dasatinib did not protect SUP-B15 from MTX but rather enhanced the effect of the chemotherapy (Fig. 2F, lower graph). Thus, inhibition of mTORC1 either directly or indirectly can be protective.
Figure 2. Upstream inhibitors protect relative to their degree of mTOR inhibition.
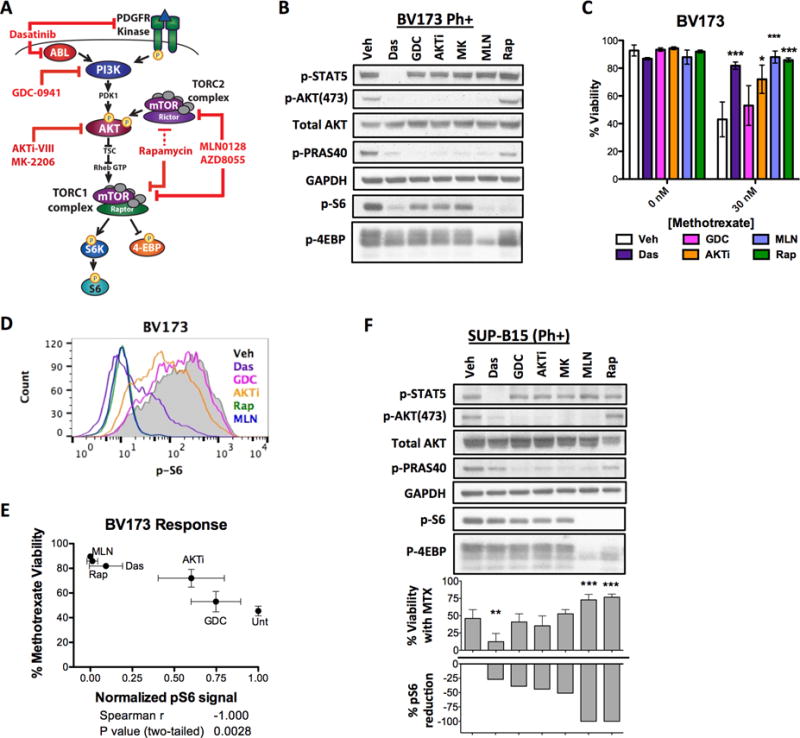
(A) The various kinase inhibitors of the PI3K/AKT/mTOR network tested in this project. Note: AKT is not the only input to mTORC1, and inhibition of PI3K or AKT only partially reduces mTORC1 output. (B) BV173 cells were treated for 6 hours with different kinase inhibitors followed by Western blot detection of BCR-ABL (p-STAT5), mTORC1 (p-S6, p-4E-BP) and mTORC2 (p-AKT, p-PRAS40) activity. (C) BV173 viability after treatment with MTX in combination with various kinase inhibitors. BV173 cells were not killed by dasatinib as a single agent. Unpaired t-tests of “chemo” versus “chemo+ inhibitor” were performed, n=3, mean±SD, *p<0.05, ***p<0.0005. (D) mTOR inhibition by each kinase inhibitor was measured by intracellular staining of p-S6 after 6 hours. (E) The mean fluorescent intensity (MFI) of p-S6 correlated (two-tailed Spearman correlation) with the degree of protection from killing by MTX. n=3, mean±SD. (F) Western blot of BCR-ABL, mTORC1, and mTORC2 substrates after 6 hours of treatment in Ph+ SUP-B15 cell lines. Below the blots, the reduction of p-S6 with various kinase inhibitors is inversely graphed against the viability of cells when these inhibitors were combined with MTX. n=3, mean±SD. For all experiments the following inhibitor concentrations were used: 3 nM dasatinib, 200 nM GD-0941, 1 μM AKTi-VIII, 300 nM MK-2206, 100 nM MLN0128, 10 nM rapamycin. For viability assays, BV173 and SUP-B15 were treated with 30 nM MTX for 48 hours.
Dasatinib protects only when it inhibits mTORC1
Currently, TKIs improve the clinical outcome of most Ph+ B-ALL patients when added to the standard chemotherapy regimen (5). However, our data suggest that dasatinib may be chemo-protective in some contexts (Fig. 2C). Therefore, we evaluated the context where TKIs like dasatinib would be counterproductive. At a dose of 3 nM dasatinib, which was sufficient to inhibit phosphorylation of ABL and STAT5 (Supplementary Fig. S4A and B), dasatinib did not reduce mTORC1 activity and correspondingly sensitized SUP-B15 cells to all chemotherapies tested (Fig. 3A and B). Rapamycin was used as a positive control to suppress pS6 downstream of mTORC1 (Fig. 3B). However, in BV173 cells, where dasatinib suppressed mTORC1 activity, dasatinib protected from 6-MP and MTX (Fig. 3C and D). Using a cell counting assay, we confirmed that dasatinib and MLN0128 protected from 6-MP and MTX in BV173 cells whereas only MLN0128 protected in SUP-B15 cells (Supplementary Figure S4C – F). Since 3 nM dasatinib did not significantly kill Ph+ SUP-B15 or BV173 cell lines as a single agent, we tested whether higher doses would more fully block ABL kinase and override the protective effect. Interestingly, higher doses maintained similar biochemical and viability effects in both cell lines tested (Supplementary Fig. S4G and H). Thus both lines belong to the category of Ph+ B-ALL cells that are resistant to killing by dasatinib despite inhibition of ABL kinase activity.
Figure 3. Dasatinib protects if it inhibits mTOR.
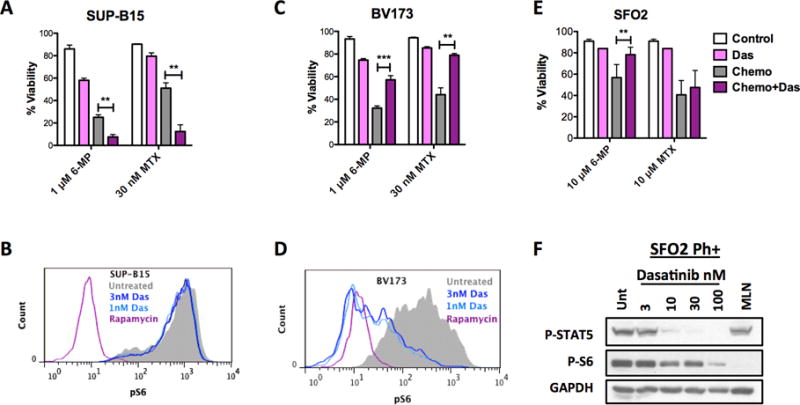
Viability of (A) SUP-B15 or (C) BV173 treated with 6-MP for 72 hours and MTX for 48 hours in combination with 3 nM dasatinib. Effect of dasatinib on mTORC1 activity in SUP-B15 (B) or BV173 (D) cells as detected by intracellular staining of p-S6. Rapamycin 10 nM is used as positive control. (E) Viability of Ph+ SFO2 following dasatinib (100 nM) treatment combined with 6-MP or MPX both for 5 days. (F) Western blot analysis of dasatinib effect on ABL (pSTAT5) or mTORC1 kinase activity (p-S6) in SFO2 cells after 6 hours of treatment. For death assays, unpaired t-tests were performed for each drug. Results are from at least 3 replicates for each drug, mean±SD, *p<0.05, **p<0.005, ***p<0.0005.
To determine whether protection by dasatinib was unique to BV173 cells, we also assessed the combination of dasatinib with chemotherapy in stromal dependent PDX lines. While dasatinib reduced mTORC1 activity in all cells tested (Supplementary Fig. S5A and C), its protective effect was only observed in patient cells that were innately resistant to killing by dasatinib. In Ph+ SFO2 cells, which were obtained from a patient who relapsed while on dasatinib without an ABL mutation (Supplementary Table 1), dasatinib was not cytotoxic as a single agent and protected from 6-MP (Fig. 3E and F). Similarly, in Ph+ ICN1 cells, dasatinib had a minor killing effect (Supplementary Fig. S5A and B) and protected from 6-MP. In both SFO2 and ICN1, dasatinib did not protect from MTX. However, in Ph+ MXP5 and Ph-like PAUXZX cells, where dasatinib killed as a single agent, there was no protection from 6-MP or MTX (Supplementary Fig. S5C and D). Overall, these results suggest that protection from 6-MP by dasatinib is only observed in B-ALL cells where dasatinib is not cytotoxic and mTOR is inhibited.
Protection by mTOR inhibition correlates with cell cycle inhibition
Since MTX and 6-MP both inhibit production of nucleotides necessary for cell proliferation, we tested whether protection by mTORC1 inhibition correlated with slowing of cell proliferation. By counting the accumulation of cells after 72 hours, we found that the degree of growth inhibition by different mTORC1 inhibitors significantly correlated with the degree of protection from both MTX and 6-MP (Fig. 4A and B). Notably, dasatinib reduced cell growth to a similar extent as MLN0128 in BV173 (Fig. 4C), where it protected from MTX. However, dasatinib did not greatly reduce growth of SUP-B15 cells, where it is not protective.
Figure 4. mTOR inhibition protects by inhibiting cell cycling.
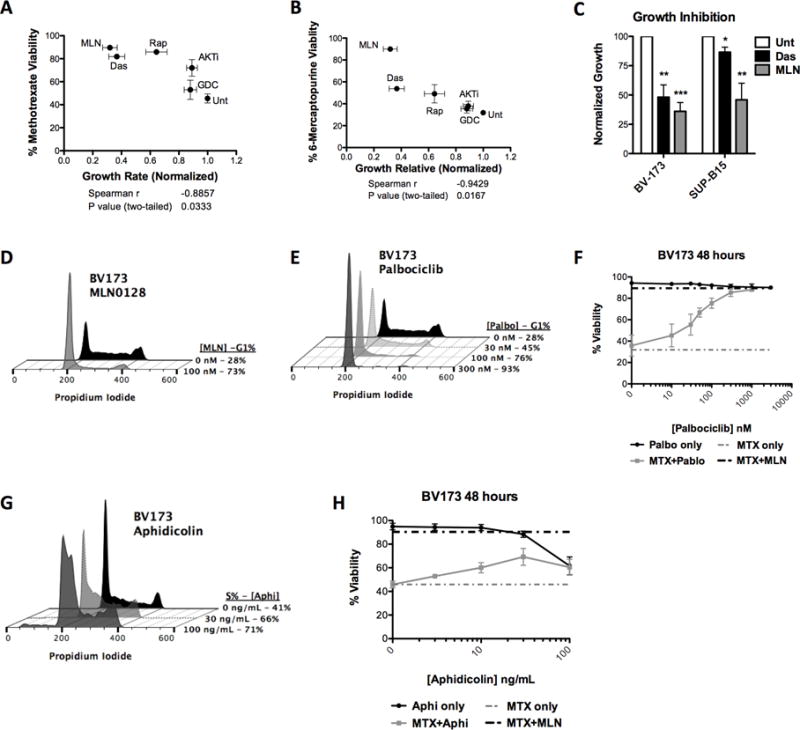
(A) Protection from 30 nM MTX (48 hr) or (B) 10 μM 6-MP (72 hr) by each kinase inhibitor was correlated (two-tailed Spearman correlation) with the inhibitor’s effect on growth inhibition as measured by number of viable cells after 72 hours of culture. Drug concentrations were: 3 nM dasatinib, 200 nM GDC-0941, 1 μM AKTi-VIII, 300 nM MK-2206, 100 nM MLN0128, and 10 nM rapamycin. Caspase inhibitor (QVD-Oph 20 μM) was added to rule out the effect of death by each inhibitor on growth measurements. n=4, mean±SD. (C) Cell growth effect of 100 nM MLN and 3 nM dasatinib at 72 hours. One sample t-test was done for each inhibitor against untreated (Unt) control, n=4, mean±SD. (D) Effect of MLN0128 on cell cycle after 24 hours treatment. (E) Effect of palbociclib, CDK 4/6 inhibitor, on cell cycle after 24 hours of treatment. (F) Viability of BV173 cells treated with MTX and increasing dose of palbociclib was measured after 48 hours of treatment. n=3, mean±SD. (G) Effect of aphidicolin on cell cycle after 24 hours of treatment. (H) Viability of BV173 cells treated with MTX and increasing dose of aphidicolin was measured after 48 hours of treatment. n=3, mean±SD.
To determine whether cell cycle inhibition was sufficient to protect B-ALL cells from maintenance drugs, we used the CDK4/6 inhibitor, palbociclib (32), to selectively inhibit cell cycling without suppressing mTORC1. Similar to mTOR inhibition (Fig. 4D), palbociclib caused dose-dependent G1 arrest (Fig. 4E) and protection from MTX (Fig. 4F) without affecting viability as a single agent (Supplementary Fig. S6). Aphidicolin (33), which causes early S phase arrest, similarly protected against MTX in a dose dependent manner (Fig. 4G and H). Thus, cytotoxicity of maintenance drugs is highly affected by cell cycle inhibition.
The 4E-BP-eIF4F axis downstream of mTORC1 regulates protein translation and cell cycle progression (34,35). Active mTORC1 phosphorylates the 4E-BPs, inhibiting their ability to suppress the formation of the eIF4F translation initiation complex. Therefore, we tested whether inhibition of eIF4F function through 4E-BP activity was sufficient to cause G1 arrest and protect from maintenance drugs. We utilized mice expressing the reverse tetracycline transactivator (rtTA) under control of the Rosa26 promoter. Additionally, the mice have a mutant form of 4E-BP1, which has all five of its mTORC1 phosphorylation sites mutated to alanines (termed “4E-BP1 5A”) (36), controlled by a tetracycline responsive element (TRE) (25,37). This 4E-BP1 5A cannot be phosphorylated by mTOR, allowing it to constitutively suppress eIF4F formation. From these mice, we generated pre-B leukemia cells driven by the p190 isoform BCR-ABL (termed p190 cells) (26,38), where expression of 4E-BP1 5A can be induced by the addition of doxycycline (Dox, Fig. 5A).
Figure 5. Active 4E-BP cause cell cycle inhibition and protection.
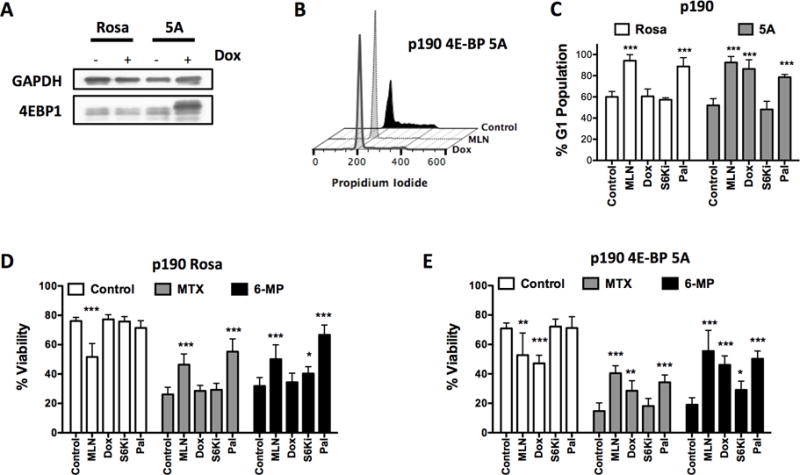
(A) p190 BCR-ABL expressing mouse leukemia cells were made to express constitutively-active 4E-BP1 5A mutant upon doxycycline induction. Western blot of 4E-BP1 5A mutant expression induction after 4 hours doxycycline treatment. (B-C) Effect on G1 arrest of p190 cells treated with doxycycline (1 μg/mL) for 24 hours. (D) Control or (E) inducible 4E-BP1 5A p190 cells were treated with MTX or 6-MP for 48 hours in presence or absence of doxycycline. For p190 assays, controls were mTOR inhibitor (100 nM MLN0128) or cell cycle inhibitor (300 nM palbociclib). Unpaired t-tests of “chemo” versus “chemo+inhibitor” were performed for each drug in cell cycle and viability assays, n=3, mean±SD, *p<0.05, **p<0.005, ***p<0.0005.
Dox induction of 4E-BP1 5A expression caused cell cycle arrest in p190 cells similar to MLN0128 and palbociclib treatment (Fig. 5B and C). Dox had no effect in RosaA26-rtTA control cells (Fig. 5D). In the 4E-BP1 5A inducible p190 cells MLN0128, Dox, and palbociclib protected cells from MTX and 6-MP (Fig. 5E), suggesting that the 4E-BP axis can account for most of the cell cycle and protective effects of mTOR inhibition.
Cell cycle inhibition reduces DNA damage by maintenance drugs
MTX and 6-MP cause DNA breaks, which activates ATR leading to the phosphorylation of Chk1 (39,40). Therefore, we asked whether cell cycle inhibition reduces DNA damage by these drugs. We measured DNA damage caused by MTX using phosphorylation of histone H2A.X as a DNA damage marker. We added the pan-caspase inhibitor QVD-OPh to eliminate apoptosis-mediated DNA damage. After 48 hours, MTX increased p-H2A.X in both BV173 and SUP-B15 cells, which was reduced by addition of MLN0128 (Supplementary Fig. S7A and B). However, dasatinib only reduced the amount of DNA damage in BV173 but not in SUP-B15 cells, suggesting that this effect is dependent on suppression of mTORC1 activity and cell proliferation.
To determine whether cell cycle inhibition without mTORC1 inhibition is sufficient to reduce DNA damage by maintenance drugs, we treated BV173 and SUP-B15 cells with 6-MP or MTX with increasing concentrations of palbociclib. Both 6-MP and MTX caused single stranded DNA breaks indicated by phosphorylation of Chk1. Increasing concentrations of palbociclib decreased p-Chk1 and p-Histone H2A.X (Supplementary Fig. S7C and D). Similarly, p-Chk1 and p-histone H2A.X were not observed in in the presence of MLN1028. Thus, cell cycle inhibition is sufficient to reduce DNA damage by 6-MP and MTX. In agreement, others have found that palbociclib protected cells from irradiation-induced DNA damage (41).
mTOR inhibition increases relapse after 6-MP treatment in vivo
To determine whether mTOR inhibition protects B-ALL cells from 6-MP in an in vivo model, we injected Ph-like PAUXZX cells into immunodeficient NOD-SCID-IL2Rγ−/− (NSG) mice. After engraftment was detected in the NSG mice, we treated mice with 6-MP, the mTOR kinase inhibitor AZD8055, or the combination for 5 days. After treatment, we sacrificed control vehicle and AZD8055 treated mice and verified that mTOR was indeed inhibited in the spleen cells of AZD8055 treated mice (Fig. 6A and B). Three days after the last dose, we sacrificed the 6MP and 6MP+AZD groups to determine leukemic burden. Both male and female mice were used in this experiment to determine if gender has an effect on the outcome. In both male and female mice, AZD8055 significantly increased the percentage of leukemia cells in the bone marrow (Fig. 6C). Thus in a Ph-like B-ALL xenograft, mTOR inhibition protects leukemia from 6-MP leading to faster outgrowth after cessation of treatment.
To determine the effect of dasatinib in vivo, we injected Ph+ SFO2 patient cells into NSG mice. Upon detection of leukemia we treated the mice for 5 days with single or combined regimens of 6-MP with AZD8055 or dasatinib (Fig. 6D). Mice were separated into post-treatment (assess efficacy of drugs as single agents) or relapse groups (assess regrowth effect of drug combinations). After treatment (Day 7), mice post-treatment groups were assessed for leukemia burden and mTOR inhibition. Compared to vehicle, AZD8055 and dasatinib reduced mTORC1 activity in bone marrow cells indicated by a reduction in phosphorylation of 4E-BP1 (Fig. 6E). Both AZD8055 and dasatinib as single agents slightly reduced leukemic burden (Fig. 6F, Post RX group), presumably due of reduction in cell cycle since neither drug alone is cytotoxic to SFO2 cells in vitro. 6-MP alone greatly reduced the leukemia burden in the bone marrow and spleen of the post-treatment mice.
Additional groups (6MP only, 6MP+AZD, and 6MP+Das groups) were monitored for relapsing leukemia in the peripheral blood. Mice were sacrificed on day 25 when more than 1% leukemia was detected in peripheral blood. Both AZD8055 and dasatinib significantly increased the leukemic burden in the bone marrow of 6-MP treated mice (Fig. 6F). Thus, inhibiting mTORC1, either direct or indirectly, can protect B-ALL leukemia cells from 6-MP and accelerate relapse in vivo.
Discussion
We found that direct or indirect mTORC1 inhibition in Ph+ and Ph-like B-ALL cells can cause protection from maintenance drugs MTX and 6-MP. ABL, PI3K and AKT inhibitors protect depending on how strongly these inhibitors reduce mTORC1 activity. Since many pathways activate mTOR, these upstream inhibitors vary in how fully they reduce mTOR activity among various cell lines. Importantly, in a subset of Ph+ B-ALL cells where dasatinib reduces ABL signaling but does not reduce leukemia survival, dasatinib can protect from 6-MP in vitro and in vivo.
Protection by mTORC1 inhibition is in large part due to cell cycle inhibition and prevention of DNA damage. MTX and 6-MP both inhibit the synthesis of nucleotides necessary for DNA replication in dividing cells (40,42). Their mechanism of killing therefore is highly dependent on cell cycle progression. With synthesis of new nucleotides blocked, dividing cells deplete their pool of nucleotides and DNA damage occurs. Indeed, established B-ALL cell lines divide faster than cultured stroma-dependent patient cells and require less time (48 hours versus 5 days) and less drug (30 nM versus 10 μM MTX) to be killed. Similarly, Li et al also noted that MTX produces less DNA damage and cell death in growth-arrested cells (43).
Interestingly, in patient cells mTOR inhibition consistently protects from 6-MP, but not as consistently from MTX. Teachey et al reported that the allosteric mTOR inhibitor temsirolimus combines with MTX to reduce leukemia burden in a mouse B-ALL xenograft model (20). They found that mTOR inhibition reduced DHFR, the target of MTX, making cells more sensitive to MTX. In agreement, we found that DHFR is reduced by mTOR inhibition in most PDX cells (Supplementary Fig. S8). Likely, mTOR inhibition causes antagonizing cell cycle related protection and MTX sensitizing effects, with the overall outcome differing from patient to patient. Whether mTORC1 inhibition rescues survival in distinct subclones of a leukemia remains to be determined. Results from the current trial NCT01162551 of sirolimus and MTX in relapsed/refractory lymphoblastic leukemia will determine if mTOR inhibition predominantly sensitizes or protects from MTX in most B-ALL patients.
The effect of mTOR inhibition on the cytotoxicity of induction phase chemotherapy agents is highly variable. mTOR inhibition tends to be protective in the cases of vincristine and etoposide (Supplementary Fig. S2). It tends to sensitize to daunorubicin. The inconsistencies may be a product of two counteracting effects of mTOR inhibition: cell cycle inhibition and mitochondrial priming. Using BH3 profiling, we previously found that mTOR inhibition affects the BCL-2 family of proteins to make B-ALL cells more sensitive to apoptosis initiation signals at the mitochondria (“mitochondrial priming”) (16). However for a cytotoxic drug that is highly cell cycle dependent, mTOR inhibition slows cell cycling to prevent cellular damage and apoptosis initiation signals are not even triggered. Though this model needs further validation, it can explain why mTOR inhibition sensitizes to dasatinib and HDAC inhibitors (16,18), which are not dependent on cell cycling to initiate apoptosis. For induction chemotherapy drugs, the outcome of these competing effects is hard to predict. A Phase I/II study has shown that mTOR inhibitor everolimus combined with intensive HyperCVAD induction chemotherapy provided no significant improvement compared to historical response to HyperCVAD alone in B-ALL patients, while improvement was only seen in T-ALL cases (22). Trials testing other induction regimens with mTOR inhibitors are underway (NCT01614197 and NCT01523977).
Kano and colleagues found that BCR/ABL inhibitor STI571 (imatinib) protects the Ph+ chronic myeloid leukemia cell line KU812 from methotrexate (44). Similarly, we found that dasatinib can protect against 6-MP in Ph+ B-ALL patient cells. This protection is most prominent in B-ALL cells where dasatinib doesn’t kill as a single agent but still inhibits ABL kinase and downstream mTORC1. Since most newly diagnosed Ph+ B-ALL patients are sensitive to dasatinib, protection would most likely occur in patients who have developed resistance to TKIs over time. Indeed, protection by dasatinib was observed in SFO2 cells, obtained from a patient who had multiple relapses while on TKIs without an ABL mutation. Interestingly, many cancer centers use dasatinib with 6-MP concurrently during maintenance for Ph+ B-ALL patient cells, while other centers don’t overlap these drugs during treatment. If protection by mTOR inhibition is transient and not giving rise to resistant subclones, a non-overlapping protocol might be more beneficial.
Our findings add to growing evidence that reduced PI3K/mTOR signaling can protect B-ALL cells from stress. Indeed, deletion or inhibition of PTEN phosphatase elevates PI3K/mTOR signaling and causes rapid death of B-ALL cells (45). Correspondingly, the data presented here demonstrate that inhibiting PI3K/mTOR can protect B-ALL cells from certain cytotoxic agents. Being a master regulator of diverse downstream pathways controlling cellular proliferation and survival, it is not surprising that inhibition of mTOR can have varying effects in different drug combinations. Further studies are needed to identify the best strategies for exploiting the chemo-sensitizing effects of mTOR inhibition in B-ALL and circumventing the chemo-protective effects.
Supplementary Material
Acknowledgments
We thank Dr. Kyoko Yokomori for advice on DNA damage.
Grant Support: This work was supported by NIH grants R01-CA158383 (to D.A. Fruman), R21-HD081319 (to D.A. Fruman and M. Konopleva), F32-CA189629-01 (to T.T. Vo), TL1-TR001415 (to J.S. Lee), and R35CA197628 (to M. Müschen).
Abbreviations
- mTOR
Mechanistic target of rapamycin
- B-ALL
B-cell acute lymphoblastic leukemia
- MTX
methotrexate
- 6-MP
6-mercaptopurine
- DNR
daunorubicin
- TKI
tyrosine kinase inhibitor
- Dox
doxycycline
- PDX
patient derived xenograft
References
- 1.Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology Am Soc Hematol Educ Program. 2014;2014:174–80. doi: 10.1182/asheducation-2014.1.174. [DOI] [PubMed] [Google Scholar]
- 2.Burmeister T, Schwartz S, Bartram CR, Gökbuget N, Hoelzer D, Thiel E, et al. Patients’ age and BCR-ABL frequency in adult B-precursor ALL: a retrospective analysis from the GMALL study group. Blood. 2008;112:918–9. doi: 10.1182/blood-2008-04-149286. [DOI] [PubMed] [Google Scholar]
- 3.Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang Y-L, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. 2014;371:1005–15. doi: 10.1056/NEJMoa1403088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JGCAM, Peters STCJM, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–34. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fielding AK, Rowe JM, Buck G, Foroni L, Gerrard G, Litzow MR, et al. UKALLXII/ECOG2993: addition of imatinib to a standard treatment regimen enhances long-term outcomes in Philadelphia positive acute lymphoblastic leukemia. Blood. 2014;123:843–50. doi: 10.1182/blood-2013-09-529008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foà R, Vitale A, Vignetti M, Meloni G, Guarini A, De Propris MS, et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 2011;118:6521–8. doi: 10.1182/blood-2011-05-351403. [DOI] [PubMed] [Google Scholar]
- 7.Rousselot P, Coudé MM, Gokbuget N, Gambacorti Passerini C, Hayette S, Cayuela J-M, et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome-positive ALL. Blood. 2016;128:774–82. doi: 10.1182/blood-2016-02-700153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ravandi F, O’Brien S, Thomas D, Faderl S, Jones D, Garris R, et al. First report of phase 2 study of dasatinib with hyper-CVAD for the frontline treatment of patients with Philadelphia chromosome-positive (Ph+) acute lymphoblastic leukemia. Blood. 2010;116:2070–7. doi: 10.1182/blood-2009-12-261586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quentmeier H, Eberth S, Romani J, Zaborski M, Drexler HG. BCR-ABL1-independent PI3Kinase activation causing imatinib-resistance. J Hematol Oncol. 2011;4:6. doi: 10.1186/1756-8722-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duy C, Hurtz C, Shojaee S, Cerchietti L, Geng H, Swaminathan S, et al. BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature. 2011;473:384–8. doi: 10.1038/nature09883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu P, Gan W, Chin YR, Ogura K, Guo J, Zhang J, et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015;5:1194–209. doi: 10.1158/2159-8290.CD-15-0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 13.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemes K, Sebestyén A, Márk A, Hajdu M, Kenessey I, Sticz T, et al. Mammalian target of rapamycin (mTOR) activity dependent phospho-protein expression in childhood acute lymphoblastic leukemia (ALL) PLoS ONE. 2013;8:e59335. doi: 10.1371/journal.pone.0059335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JS, Vo T-T, Fruman DA. Targeting mTOR for the treatment of B cell malignancies. Br J Clin Pharmacol. 2016 doi: 10.1111/bcp.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beagle BR, Nguyen DM, Mallya S, Tang SS, Lu M, Zeng Z, et al. mTOR kinase inhibitors synergize with histone deacetylase inhibitors to kill B-cell acute lymphoblastic leukemia cells. Oncotarget. 2015;6:2088–100. doi: 10.18632/oncotarget.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janes MR, Vu C, Mallya S, Shieh MP, Limon JJ, Li LS, et al. Efficacy of the investigational mTOR kinase inhibitor MLN0128/INK128 in models of B-cell acute lymphoblastic leukemia. Leukemia. 2013;27:586–94. doi: 10.1038/leu.2012.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janes MR, Limon JJ, So L, Chen J, Lim RJ, Chavez MA, et al. Effective and selective targeting of leukemia cells using a TORC1/2 kinase inhibitor. Nat Med. 2010;16:205–13. doi: 10.1038/nm.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saunders P, Cisterne A, Weiss J, Bradstock KF, Bendall LJ. The mammalian target of rapamycin inhibitor RAD001 (everolimus) synergizes with chemotherapeutic agents, ionizing radiation and proteasome inhibitors in pre-B acute lymphocytic leukemia. Haematologica. 2011;96:69–77. doi: 10.3324/haematol.2010.026997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teachey DT, Sheen C, Hall J, Ryan T, Brown VI, Fish J, et al. mTOR inhibitors are synergistic with methotrexate: an effective combination to treat acute lymphoblastic leukemia. Blood. 2008;112:2020–3. doi: 10.1182/blood-2008-02-137141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crazzolara R, Cisterne A, Thien M, Hewson J, Baraz R, Bradstock KF, et al. Potentiating effects of RAD001 (Everolimus) on vincristine therapy in childhood acute lymphoblastic leukemia. Blood. 2009;113:3297–306. doi: 10.1182/blood-2008-02-137752. [DOI] [PubMed] [Google Scholar]
- 22.Daver N, Boumber Y, Kantarjian H, Ravandi F, Cortes J, Rytting ME, et al. A Phase I/II Study of the mTOR Inhibitor Everolimus in Combination with HyperCVAD Chemotherapy in Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. Clin Cancer Res. 2015;21:2704–14. doi: 10.1158/1078-0432.CCR-14-2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parameswaran R, Müschen M, Kim Y-M, Groffen J, Heisterkamp N. A functional receptor for B-cell-activating factor is expressed on human acute lymphoblastic leukemias. Cancer Res. 2010;70:4346–56. doi: 10.1158/0008-5472.CAN-10-0300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khoury MK, Parker I, Aswad DW. Acquisition of chemiluminescent signals from immunoblots with a digital single-lens reflex camera. Anal Biochem. 2010;397:129–31. doi: 10.1016/j.ab.2009.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.So L, Lee J, Palafox M, Mallya S, Woxland CG, Arguello M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Sci Signal. 2016;9:ra57. doi: 10.1126/scisignal.aad8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kharas MG, Janes MR, Scarfone VM, Lilly MB, Knight ZA, Shokat KM, et al. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest. 2008;118:3038–50. doi: 10.1172/JCI33337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485:55–61. doi: 10.1038/nature10912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70:288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 29.Fransecky L, Mochmann LH, Baldus CD. Outlook on PI3K/AKT/mTOR inhibition in acute leukemia. Molecular and cellular therapies. 2015;3:2. doi: 10.1186/s40591-015-0040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, et al. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–4. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 31.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–32. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 32.Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–406. doi: 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- 33.Bucknall RA, Moores H, Simms R, Hesp B. Antiviral effects of aphidicolin, a new antibiotic produced by Cephalosporium aphidicola. Antimicrob Agents Chemother. 1973;4:294–8. doi: 10.1128/aac.4.3.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bhat M, Robichaud N, Hulea L, Sonenberg N, Pelletier J, Topisirovic I. Targeting the translation machinery in cancer. Nat Rev Drug Discov. 2015;14:261–78. doi: 10.1038/nrd4505. [DOI] [PubMed] [Google Scholar]
- 35.Dowling RJO, Topisirovic I, Alain T, Bidinosti M, Fonseca BD, Petroulakis E, et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science. 2010;328:1172–6. doi: 10.1126/science.1187532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mothe-Satney I, Yang D, Fadden P, Haystead TA, Lawrence JC. Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol Cell Biol. 2000;20:3558–67. doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013;110:11988–93. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li S, Ilaria RL, Million RP, Daley GQ, Van Etten RA. The P190, P210, and P230 forms of the BCR/ABL oncogene induce a similar chronic myeloid leukemia-like syndrome in mice but have different lymphoid leukemogenic activity. J Exp Med. 1999;189:1399–412. doi: 10.1084/jem.189.9.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Anta JM, Pérez-Castro AJ, Freire R, Mayol X. The DNA damage checkpoint is activated during residual tumour cell survival to methotrexate treatment as an initial step of acquired drug resistance. Anticancer Drugs. 2006;17:1171–7. doi: 10.1097/01.cad.0000236311.73703.d2. [DOI] [PubMed] [Google Scholar]
- 40.Karran P, Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat Rev Cancer. 2008;8:24–36. doi: 10.1038/nrc2292. [DOI] [PubMed] [Google Scholar]
- 41.Johnson SM, Torrice CD, Bell JF, Monahan KB, Jiang Q, Wang Y, et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J Clin Invest. 2010;120:2528–36. doi: 10.1172/JCI41402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorlick R, Goker E, Trippett T, Waltham M, Banerjee D, Bertino JR. Intrinsic and acquired resistance to methotrexate in acute leukemia. N Engl J Med. 1996;335:1041–8. doi: 10.1056/NEJM199610033351408. [DOI] [PubMed] [Google Scholar]
- 43.Li JC, Kaminskas E. Accumulation of DNA strand breaks and methotrexate cytotoxicity. Proc Natl Acad Sci U S A. 1984;81:5694–8. doi: 10.1073/pnas.81.18.5694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kano Y, Akutsu M, Tsunoda S, Mano H, Sato Y, Honma Y, et al. In vitro cytotoxic effects of a tyrosine kinase inhibitor STI571 in combination with commonly used antileukemic agents. Blood. 2001;97:1999–2007. doi: 10.1182/blood.v97.7.1999. [DOI] [PubMed] [Google Scholar]
- 45.Shojaee S, Chan LN, Buchner M, Cazzaniga V, Cosgun KN, Geng H, et al. PTEN opposes negative selection and enables oncogenic transformation of pre-B cells. Nat Med. 2016;22:379–87. doi: 10.1038/nm.4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.