

Abstract
Hydroxylysine aldehyde-derived collagen cross-links (HLCCs) accumulate in fibrotic tissues and certain types of cancer and are thought to drive the progression of these diseases. HLCC formation is initiated by lysyl hydroxylase 2 (LH2), an Fe(II) and α-ketoglutarate (αKG)-dependent oxygenase that hydroxylates telopeptidyl lysine residues on collagen. Development of LH2 antagonists for the treatment of these diseases will require a reliable source of recombinant LH2 protein and a non-radioactive LH2 enzymatic activity assay that is amenable to high throughput screens of small molecule libraries. However, LH2 protein generated previously using E coli– or insect-based expression systems was either insoluble or enzymatically unstable, and LH2 enzymatic activity assays have measured radioactive CO2 released from 14C-labeled αKG during its conversion to succinate. To address these deficiencies, we have developed a scalable process to purify human LH2 protein from Chinese hamster ovary cell-derived conditioned media samples and a luciferase-based assay that quantifies LH2-dependent conversion of αKG to succinate. These methodologies may be applicable to other Fe(II) and αKG-dependent oxygenase systems.
Keywords: lysyl hydroxylase 2, collagen, high-throughput assay, succinate detection, Chinese hamster ovary cell, oxygenase
1. Introduction
The collagen lysyl hydroxylation catalyzed by lysyl hydroxylases (LHs; EC 1.14.11.4) is critical to the formation of the collagen cross-links that covalently connect collagen molecules and stabilize the extracellular matrix [1]. LHs belong to a superfamily of more than 60 oxygenases that use Fe(II) as a cofactor and α-ketoglutarate (αKG) and oxygen as co-substrates; these oxygenases include, among others, the collagen prolyl hydroxylases, the jumonji family of histone demethylases, hypoxia-inducible factor (HIF) prolyl hydroxylase domain-2, and asparaginyl hydroxylase factor inhibiting HIF-1α [2]. Three LH family members—LH1, 2, and 3—are found in vertebrate genomes, whereas only one LH family member that is homologous to vertebrate LH3 has been identified in invertebrates such as Trichoplusia ni and Spodoptera frugiperda [3]. LH2 has 3 potential isoforms, two of which (LH2a and LH2b) have been shown to be functional enzymes [4]. LH2b is the major isoform and is 21 amino acids longer than LH2a owing to the inclusion of 63 nucleotides in exon 13A that are alternatively spliced [4]. LH2b, hereafter abbreviated to LH2, is unique because it is the only LH family member known to modify telopeptidyl lysine residues [5–7]. The hydroxylated telopeptidyl lysines that result from LH2 modification are converted by lysyl oxidases into hydroxylysine aldehydes, which subsequently condense with juxtaposed lysines or hydroxylysines to form hydroxylysine-derived collagen cross-links (HLCCs) [8]. HLCCs are resistant to collagenase cleavage, are more stable than lysine-derived collagen cross-links (LCCs) that form in the absence of LH2, and are particularly abundant in skeletal tissues such as cartilage and bone, which require high tensile strength. Genetic evidence supports a central role for LH2-mediated HLCC formation in normal bone function. Indeed, inactivating mutations in the procollagen-lysine,2-oxoglutarate 5-dioxygenase 2 gene, PLOD2, which encodes LH2, are found in patients with Bruck syndrome [9]; these individuals have deformed, fragile bones and are deficient in HLCCs.
Whereas LH2 plays a critical role in normal cartilage and bone development by forming HLCCs, the aberrant overexpression of LH2, leading to the accumulation of HLCCs, is a general feature of fibrotic diseases [6, 10]. The activation of the immune system after injury triggers tissue repair and collagen deposition as part of the natural repair progress. Collagen rich in LCCs can be resolved by collagenase action; however, the immune response often increases LH2 expression [11], which leads to the sustained formation of HLCCs that are resistant to removal, causing scarring or fibrosis under pathological conditions. LH2 levels are increased in multiple types of fibrosis [6, 10]. Moreover, in progressive fibrotic diseases such as idiopathic pulmonary fibrosis, the accumulation of collagen increases tissue stiffness and stimulates myofibroblast differentiation and additional collagen production, thereby activating a vicious cycle [12–16]. Thus, pharmacologic inhibitors of LH2 may stop or even reverse fibrotic disease progression.
We recently established a link between fibrosis and cancer progression and demonstrated a role of LH2 in lung cancer metastasis [17]. We showed that in patients with lung adenocarcinomas, intra-tumoral LH2 levels are prominently upregulated and predict a significantly shorter survival duration [17]. In addition, LH2 knockdown prominently reduces HLCC-to-LCC ratios without changing the total amount of collagen cross-links, decreases tumor stiffness, and abrogates tumor cell migration, invasion, and metastasis [17]. We also found that LH2 is secreted by many different types of lung cancer cells and can hydroxylate telopeptidyl lysines and switch the collagen crosslinking type from HLCC to LCC in the extracellular space [18]. This indicates that in tumor cells, LH2 overexpression functions as a regulatory switch that controls the relative abundance of distinct types of collagen cross-links that regulate stromal stiffness and influence lung cancer cells’ metastatic fate. LH2-driven metastasis has also been observed in other cancer types, including sarcoma [19] and breast cancer [20, 21], which suggests that LH2 promotes the progression of many cancer types and is a potential therapeutic target in cancer patients.
Therapeutically targeting LH2 requires a reliable and efficient LH2 expression and purification method that enables biochemical and crystallographic analysis, as well as a robust high throughput assay for large-scale LH2 antagonist screening. Previous studies have demonstrated that LH family members cannot be expressed as a soluble protein in E coli, and insect cell–based protein expression systems, such as Trichoplusia ni high-five cells and Spodoptera frugiperda Sf9 cells, produce LH2 protein with unstable enzymatic activity [22, 23], which may have resulted from the expression systems and/or purification procedures used [7]. However, several earlier studies successfully purified enzymatically active LH family members from chick embryos [24, 25], which suggests that a vertebrate host is important for the expression of vertebrate LHs.
To quantify LH2 enzymatic activity, investigators have implemented a method that detects radioactive CO2 released from 14C-labeled αKG [26, 27]. Although it produces a detectable signal from small amounts of LH enzyme, this assay is non-quantitative and therefore has no potential for high throughput screening. Non-radioactive methods that assess the enzymatic activity of Fe(II) and αKG-dependent oxygenases by measuring αKG diminution or succinate production have been developed [28, 29]. Compared with the assay measuring αKG diminution, succinate detection-based assays have shown superior sensitivity; however, the currently available assays still have limited sensitivity and thus require large amounts of recombinant protein to generate a reliable signal in a hydroxylation reaction, which makes them impractical for carrying out large-scale inhibitor screening.
To enable the characterization and therapeutic targeting of LH2, here we developed a scalable method of purifying LH2 protein from Chinese hamster ovary (CHO) cell–conditioned media, adapted a luciferase-based assay for the measurement of LH2’s enzymatic activity, and confirmed that the purified recombinant LH2 has stable enzymatic activity and that our assay has high signal-to-noise and minimal batch-to-batch variation.
2. Materials and methods
2.1. LH2 production and purification
LH2 was purified as described previously with minor modifications [30]. Briefly, human LH2 (residues 33–758, wild-type and inactive D689A mutant) recombinant proteins were produced from new Gibco™ ExpiCHO™ cells in suspension (Thermo Fisher Scientific, Waltham, MA) as a secreted protein with N-terminal His8 and human growth hormone (hGH) tags via large-scale transient transfection with polyethylenimine. The cells were transfected at a density of 2 × 106 cells/mL with 1 mg of DNA and 3 mg of polyethylenimine per liter of cells [30]. After 5 h, cells were split at a ratio of 2:9 and grown for 4 days. The LH2-containing conditioned media were then harvested by centrifugation at 7000 rpm for 10 min, filtered through 0.22 μm EMD Millipore Stericup™ Sterile Vacuum Filter Units (EMD Millipore, Billerica, MA), concentrated to 100 mL, and buffer-exchanged into Nickel-binding buffer (20 mM Tris, 200 mM NaCl, 15 mM imidazole, pH 8.0) using the Centramate™ & Centramate PE Lab Tangential Flow System (Pall Life Sciences, Ann Arbor, MI) at a flow rate of 100 ml per minute with a pressure of 20 to 30 psi. Using this system, a 15-Liter batch of conditioned medium is typically processed within 24 h. The recombinant LH2 proteins were then purified from CHO cell–conditioned media with immobilized metal affinity chromatography and anion exchange chromatography consecutively using NGC™ Medium-Pressure Liquid Chromatography Systems (Bio-Rad, Hercules, CA). CHO cell–conditioned media containing LH2 proteins were loaded into a Nickel column at a flow rate of 3 ml per min, washed with 10 bed volumes (50 mL) of Nickel-binding buffer followed by 6 bed volumes (30 mL) of nickel-binding buffer with 10 mM extra imidazole and then eluted with 400 mM imidazole in 200 mM NaCl, pH 8.0. The eluted LH2 proteins were diluted in water at a ratio of 1:4 and loaded into an anion exchange column at a flow rate of 3 ml per min. After the column was washed with 10 bed volumes of washing buffer (50 mM NaCl, 50 mM HEPES, pH 7.4), LH2 proteins were eluted with a linear gradient of up to 1 M NaCl, 50 mM HEPES, pH 7.4. The LH2 protein–containing fractions were collected, pooled, concentrated to 1 mg/mL with the Amicon Ultra-15 Centrifugal Filter Unit with Ultracel-30 membrane (EMD Millipore, Billerica, MA), buffer-exchanged into reaction buffer (150 mM NaCl, 50 mM HEPES, pH 7.4), snap-frozen in liquid nitrogen, and stored at −80°C. The protein concentration was determined with Nanodrop (Thermo Fisher Scientific) by measuring absorbance at 280 nm. The typical LH2 yield from a single 15-liter batch of CHO cell culture was approximately 2 mg/L.
2.2. Protein gel electrophoresis and Western blotting
Protein purity was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). LH2 proteins were visualized by staining the gels with SimplyBlue™ Safe Stain (Thermo Fisher Scientific). To confirm that the protein was LH2, we transferred the protein in the SDS-PAGE gel to polyvinylidene fluoride (PVDF) membrane using the Trans-Blot Turbo Transfer System and a Trans-Blot Turbo Mini PVDF Transfer Pack (Bio-Rad). The membrane was blocked with 5% milk in phosphate-buffered saline–Tween 20 (PBS-T; 0.01 M PBS, 0.1% Tween 20, pH 7.4) for 1 hour at room temperature and incubated with antibodies against His6, hGH (sc-8036 and sc-10365, respectively, Santa Cruz, Dallas, Texas), or human LH2 (21214-1-AP, Proteintech, Rosemont, Illinois) at a 1:1000 dilution in PBS-T with 1% milk overnight at 4°C. The membrane was then incubated with sheep anti-mouse (ECL™ Anti-Mouse IgG, GE Healthcare Life Sciences, Pittsburgh, PA) or rabbit anti-goat (FKA1407041, R&D, Minneapolis, MN) horseradish peroxidase–conjugated secondary antibodies at a 1:3000 dilution in PBS-T for 1 hour at room temperature. The Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific) was used for the detection of immunoreactivity on X-ray films (HyBlot CL; Denville Scientific, Inc., Metuchen, NJ).
2.3. Identification of LH2 glycosylation by NanoLC MS/MS Analysis
Approximately 10 μg of LH2 recombinant protein was diluted to 1μg/μL in 50 mM Ammonium Bicarbonate, 10 mM DTT at pH = 7.6 and incubated at 95°C for 10 min. The sample was then cooled to RT and 3.75 μL of 1 M iodoacetic acid added and allowed to react for 20 minutes in the dark, after which 0.5 μL of 2 M DTT was added to quench the reaction. Trypsin was added to the protein mixture in a ratio of 1:100, vortexed and incubated at 37°C overnight. Trifluoracetic acid was then added to a final concentration of 0.1% TFA to quench trypsinolysis.
Peptide mixtures were analyzed by nanoflow liquid chromatography-tandem mass spectrometry (nanoLC-MS/MS) using a nano-LC chromatography system (UltiMate 3000 RSLCnano, Dionex), coupled on-line to a Thermo Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific, San Jose, CA) through a nanospray ion source (Thermo Scientific). A trap and elute method was used. The trap column was a C18 PepMap100 (300 μm X 5mm, 5um particle size) from ThermoScientific. The analytical column was an Acclaim PepMap 100 (75 μm X 15 cm) from Thermo Scientific. After equilibrating the column in 98% solvent A (0.1% formic acid in water) and 2% solvent B (0.1% formic acid in acetonitrile (ACN)), the samples (1 μL in solvent A) were injected onto the trap column and subsequently eluted (400 nL/min) by gradient elution onto the C18 column as follows: isocratic at 2% B, 0–5 min; 2% to 45% B, 2–37 min; 45% to 90% B, 37–40 min; isocratic at 90% B, 40–45 min; 90% to 2%, 45–47 min; and isocratic at 2% B, 47–60 min.
All LC-MS/MS data were acquired using XCalibur, version 2.1.0 (Thermo Fisher Scientific) in positive ion mode using a top speed data-dependent acquisition (DDA) method with a 3 sec cycle time. The survey scans (m/z 400–1600) were acquired in the Orbitrap at 120,000 resolution (at m/z = 400) in profile mode, with a maximum injection time of 50 msec and an AGC target of 200,000 ions. The S-lens RF level was set to 60. Isolation was performed in the quadrupole with a 2.0 Da isolation window, and HCD MS/MS acquisition was performed in profile mode using rapid scan rate with detection in the ion trap, with the following settings: parent threshold = 5,000; collision energy = 28%; maximum injection time 250 msec; AGC target 20,000 ions. Monoisotopic precursor selection (MIPS) and charge state filtering were on, with charge states 2–6 included. Dynamic exclusion was used to remove selected precursor ions, with a +/− 10 ppm mass tolerance, for 60 sec after acquisition of one MS/MS spectrum.
Tandem mass spectra were extracted and charge state deconvoluted by Proteome Discoverer (Thermo Fisher, version 1.4.1.14). Deisotoping was not performed. All MS/MS spectra were searched against a Uniprot Murine database (version 08-11-2014) using Sequest. Searches were performed with a parent ion tolerance of 5 ppm and a fragment ion tolerance of 0.60 Da. Trypsin was specified as the enzyme, allowing for two missed cleavages. Fixed modification of carbamidomethyl (C) and variable modifications of oxidation (M) and glycosylation were specified in Sequest. Glycopeptides were discovered with the aid of the Pinnacle software (http://www.optystech.com).
2.4. LH2 enzymatic activity assay
The collagen helical peptide substrate IKGIKGIKG ([IKG]3) was synthesized using solid phase synthesis and purified with reverse-phase high-performance liquid chromatography (HPLC) to >98% purity using an Ascentis® Express C18 column (LifeTein, Hillsborough, NJ) [31]. The standard LH2 enzymatic activity assay was performed in reaction buffer (50 mM HEPES buffer pH 7.4, 150 mM NaCl) at 37°C for 1 h with, unless otherwise stated, 1 μM LH2 enzyme, 50 μM FeSO4, 100 μM αKG, 500 μM ascorbate, 1 mM (IKG)3, and 1.5 μM catalase. Reactions were carried out under different conditions to determine enzyme kinetic properties (1 mM αKG and 0.1 μM LH2) and to compete αKG (10 μM αKG and 0.1 μM LH2). The reactions were stopped by boiling the samples for 2 min. The samples were chilled on ice for additional 2 min before performing the succinate detection assay. Data from enzyme kinetic and inhibition experiments were plotted and fitted (GraphPad Prism 6.0h software). With the exception of LH2 recombinant protein, all reagents were prepared immediately before use. All of these reagents were dissolved in reaction buffer with the exception of FeSO4, which was prepared in 10 mM HCl to keep iron in its reduced state, and the pH of the reaction mixture was checked with pH papers to ensure that HCl did not change the overall sample pH. LH2 activity was measured by detecting succinate production with a nicotinamide adenine dinucleotide (NADH)-based succinate detection assay (Succinate Colorimetric Assay Kit, BioVision, Milpitas, CA) or an adenosine triphosphate (ATP)-based luciferase assay (Succinate-Glo™ Assay, Promega, Madison, WI) according to manufacturers’ instructions. NADH-based succinate detection assay was measured either by detecting NADH fluorescence at 450 nm after exciting it at 340 nm or by monitoring light absorbance at 450 nm after converting NADH to a colored product with the reagents provided by the Succinate Colorimetric Assay Kit. Experiments were performed in triplicate, and an unpaired t-test was used to compare the enzymatic activity of different samples. The findings from studies on enzyme kinetics and the effects of αKG competitive inhibitors were reported as the mean ± standard error.
3. Results and discussion
We previously sought to overexpress LH2 protein in multiple cancer cell lines and CHO cells and found that varying amounts of LH2 remain inside the cells depending on the cell type [18]. We used CHO cells as host based on the reasoning that a mammalian cell would generate post-translational modifications that enhance the solubility and stability of recombinant human LH2 and thereby maximize activity. Recombinant LH2 was expressed as a fusion protein with N-terminal hGH and His8 tags (hGH-His8-LH2), which allowed recombinant LH2 to be secreted into, and affinity-purified from, conditioned media samples, respectively. The concentrated and buffer-exchanged conditioned media samples were subjected to immobilized metal affinity chromatography (Fig 1A) and anion exchange chromatography (Fig 1B). This two-step purification procedure allowed the removal of contaminants and the elution of recombinant LH2 as a single peak with more than 90% purity (Fig 1B and 1C). In contrast, a one-step purification using immobilized metal affinity chromatography alone generated a protein product of similar purity to that isolated from the two-step procedure but had minimal enzymatic activity when subjected to the detection assays described below (data not shown). Although the reason for the low activity of the LH2 isolated from the single-step procedure is unclear, it may be related to imidazole, which inhibits the activity of recombinant LHs [32] and is removed during the second step of the purification process. The two-step purification method was used for all subsequent experiments.
Figure 1.

Two step LH2 protein purification (A) LH2 was purified by immobilized metal ion affinity chromatograph (IMAC) of concentrated, buffer-exchanged conditioned medium samples taken from CHO cells transiently transfected with a vector expressing human LH2 tagged with His8 and human growth hormone (hGH). The protein was eluted with 400 mM imidazole as a single peak. (B) Anion exchange chromatography purification of LH2 recombinant protein. (C) SDS-polyacrylamide gel electrophoresis of LH2 protein after IMAC (N) and anion exchange chromatography (Q) purification. LH2 protein isolated from Kifunesine treated cells (K) showed a narrower band, which shifted down by ~25 kDa after EndoH treatment (E), suggesting LH2 is glycosylated. The positions and sizes of molecular weight markers (MW) are indicated. (D) The identity of LH2 protein was confirmed by Western blotting with anti-His6, anti-hGH and anti-human LH2 antibodies.
Western blotting of the recombinant LH2 protein using anti-hGH, anti-His8, or anti-LH2 antibodies identified a single band with a molecular weight identical to that of the protein identified by SDS-PAGE (Fig 1C and 1D). Although the predicted molecular weight of the LH2b recombinant protein is 110 kDa (87 kDa LH2b plus 25 kDa hGH tag), the apparent molecular weight was approximately 135 kDa, nearly 25 kDa larger than predicted, suggesting that the recombinant protein is glycosylated. Supporting this conclusion, we purified LH2 from CHO cells that had been treated with the mannosidase I inhibitor kifunensine, and the recombinant LH2 was then treated with EndoH, which shifted the protein’s molecular weight from 135 kDa to approximately 110 kDa (Fig 1C). Using a prediction algorithm (NetNGlyc 1.0 Server), we identified 7 potential glycosylation sites (N63, N209, N297, N365, N543, N717 and N746). Mass spectrometry analysis identified N-linked glycosylation on N63, N209, N543, N717 and N746 (Supplemental Table 1). These results suggest that the human recombinant LH2 protein isolated from CHO cell–conditioned media is highly pure and glycosylated.
LH2 catalyzes the hydroxylation of collagen telopeptidyl and helical lysines by using oxygen and αKG as co-substrates and Fe2+ and ascorbic acid as co-factors to produce succinate and CO2 (Fig 2A). Historically, this reaction was monitored by using radioisotope-labeled αKG and measuring the release of the CO2 (Fig 2A). This is a highly sensitive method suitable for studying small amounts of enzyme with low activity [26, 27]. We previously confirmed the activity of LH2 isolated from CHO cell–conditioned media by performing the hydroxylation reaction using (IKG)3 as a collagen helical peptide mimic and detecting hydroxylated lysine using an HPLC-based amino acid analysis [18]. Because this HPLC-based assay, like the conventional radioactive CO2 detection method, cannot be used to identify LH2 antagonists from high-throughput small molecule screens, we sought to develop a new assay that addresses this deficiency.
Figure 2.

Current detection strategies of LH2-catalyzed lysine hydroxylation. (A) Schematic illustration of a LH2-catalyzed lysine hydroxylation reaction. (B) Schematic illustration of a LH2 enzymatic detection assay that couples succinate production to NADH consumption. Abbreviations not included in text: Coenzyme A (CoA), Succinyl coA synthetase (SCS), phosphoenolpyruvate (PEP), pyruvate kinase (PK), and lactate dehydrogenase (LDH). (C) Activity of wild-type (WT) and mutant (D689A) LH2 was measured based on the disappearance of NADH fluorescence. p value, t-test.
Recently, an assay coupling succinate production to nicotinamide adenine dinucleotide (NADH) consumption (Fig. 2B) was shown to have better sensitivity than assays measuring αKG diminution [28, 29]. Using NADH consumption as a readout, wild-type LH2 (LH2WT) or mutant LH2 containing an inactivating mutation (LH2D689A) was reacted with (IKG)3 and co-factors, and NADH was quantified on the basis of fluorescence (NADH excitation/emission of 340/450 nm). NADH consumption by wild-type LH2 was greater than that by mutant LH2 (mean NADH fluorescence 837 vs. 792, p=0.009, t-test), but the signal-to-background (S/B) ratio was rather limited (S/N ratio=1.06, Fig. 2C). To address this issue, we converted NADH to a colored product with absorbance at 450 nm; however, no absorbance signal was detected even with 10 μM LH2 (data not shown). Thus, coupling succinate detection to NADH consumption was not sufficiently sensitive to detect LH2 enzymatic activity.
As an alternative to monitoring NADH consumption, we quantified succinate production by using a luminescence-based approach in which succinate and adenosine triphosphate (ATP) production are coupled with luciferase to generate luminescence from a series of succinate-to-ATP conversions (Fig. 3A). Luciferase activity was detected over a wide range of concentrations of LH2, and the luminescence signal was proportional to the concentration of LH2 (Fig. 3B). This luminescence signal was further confirmed to be specific to LH2, since inactive mutant D689A produced little luminescence signal and generated a wild-type-to-mutant signal ratio of over 15 when 1 μM LH2 recombinant enzymes were assayed (Fig. 3C). Of note, the reaction mixture without the substrate peptide produced a stronger signal than inactive mutant D689A did, suggesting that LH2 is able to modify itself and/or catalase and other peptide contaminants. Furthermore, compared to the reaction mixture without any LH2 enzyme, the inactive mutant D689A sample produced a slightly higher luminescence signal, which could have come from remnant activity of the D689A mutant or endogenous LH2 produced by CHO cells that forms dimeric complexes [33] with the recombinant human LH2 and are not removed by the two-step purification.
Figure 3.

Multiwell-based LH2 enzymatic detection assay that detects succinate production from αKG on the basis of a luminescence signal generated by the conversion of succinate to ATP. (A) Schematic illustration of succinate detection by a luciferase-induced light emission. (B) Enzymatic activities from reactions containing different LH2 protein concentrations with or without substrate peptide (IKG)3. (C) Enzymatic activities of wild-type (WT) and mutant (D689A) LH2.
To determine the kinetic properties of the recombinant LH2 protein, we first established a standard curve (Supplemental Fig. 1) and lowered the concentration of LH2 used in the reaction to ensure that the consumption of substrates during the reaction is less than 5% of those initially added. We then collected velocity data as a function of varied αKG concentrations using saturating concentrations of FeSO4 (50 μM), ascorbate (500 μM), (IKG)3 (1 mM), and oxygen (ambient levels). Over a range of αKG concentrations (4 to 250 μM), LH2 showed simple saturation kinetics with a Km value of 11 ± 2 μM (Fig. 4A). Next, we collected velocity data as a function of varied peptide substrate (IKG)3 concentrations using saturating concentrations of FeSO4 (50 μM), ascorbate (500 μM), αKG (1 mM), and oxygen (ambient levels), which generated a Km value of 169 ± 39 μM (Fig. 4B). The Kcat and Kcat/Km values were calculated from the velocity data (Fig. 4A and 4B).
Figure 4.

Steady state kinetics of LH2 with varied concentrations of αKG (A) and substrate peptide (IKG)3 (B) and fixed concentrations of LH2 (0.1 μM), FeSO4 (50 μM), ascorbate (500 μM), and oxygen (ambient levels).
To determine whether the assay is sufficiently robust for use in LH2 antagonist screens, we performed the assay in the presence of a competitive inhibitor of αKG. We lowered the concentrations of enzyme and αKG (0.1 μM and 10 μM, respectively) to ensure that LH2 is sensitive to αKG competitive inhibition. LH2 activity was decreased by L-α-Hydroxyglutarate (IC50 25 ± 4 μM) but not its enantiomer D-α-Hydroxyglutarate (Fig. 5), providing proof-of-principle that the assay is sufficiently robust for application to small molecule screens.
Figure 5.
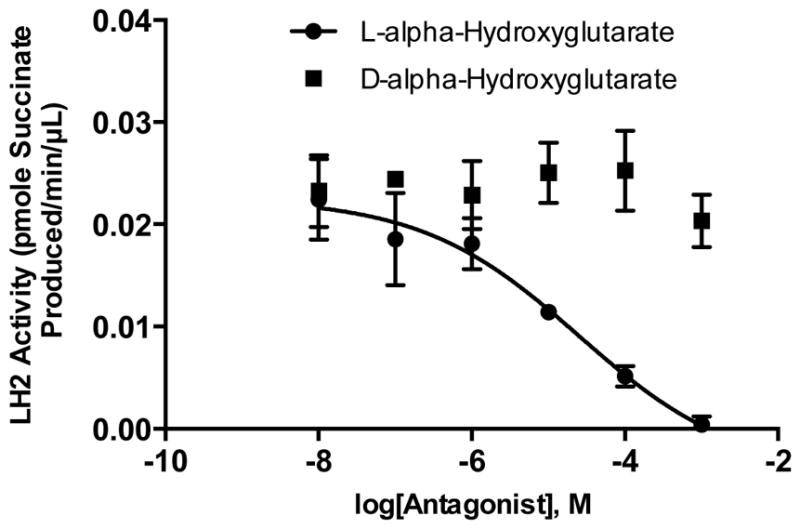
LH2 enzymatic activity in the presence of an αKG competitive inhibitor. Activity was inhibited by L-α-Hydroxyglutarate but not its enantiomer D-α-Hydroxyglutarate.
Other groups’ work has shown that controlling batch-to-batch variations in the enzymatic activity of LH2 preparations is challenging [7, 22], and this presents a technical hurdle in carrying out high-throughput inhibitor screens. To overcome this obstacle, we have developed standardized operating procedures to improve consistency. We have obtained more than 200 mg of recombinant human LH2 protein by purifying from seven separate batches of 15-Liter CHO cell cultures (Fig. 6A). The yield per batch was typically 2 mg protein per liter. To determine the reproducibility of the enzymatic activity assay, we measured the activities of seven independently prepared LH2 protein batches; the mean values were not significantly different (Fig 6A). The mean activity (9,235 ± 382) of replicate aliquots (n=9) from batch #1 was not significantly different from the mean activity (9,631 ± 549) of replicate aliquots from all seven batches (n=3 per batch, n=21 total aliquots) (Fig 6B). Furthermore, enzymatic activity was stable after leaving the recombinant LH2 protein at room temperature for up to 2 hours (Supplemental Fig 2).
Figure 6.

Batch-to-batch variations in LH2 enzymatic activity detected by succinate production. (A) Activities of separate batches of recombinant LH2 protein. Mean values determined from replicate aliquots (n=3) taken from each batch. (B) “Intra-batch” is the mean value of replicate aliquots (n=9) taken from batch #1 in (A). “Inter-batch” is the mean value of replicate aliquots taken from all seven batches (n=3 aliquots per batch, n=21 total aliquots) in (A).
4. Conclusions
In summary, we have established a scalable method for purifying enzymatically active human LH2 protein from a mammalian expression system. Our purification procedure involves two chromatography steps to obtain protein with >90% purity, and the resultant LH2 isolated from CHO-conditioned medium is highly soluble and has stable enzymatic activity with little batch-to-batch variation. We compared 3 different detection methods of succinate production and found that the Succinate-Glo assay, which employs a luminescence detection system that is readily available in most laboratories and was more robust and sensitive and had a greater signal-to-background ratio than the NADH-based assay reported previously. The stability of this assay may be attributed to the utilization of a mammalian expression system that provides chaperones and post-translational modifications that stabilize the recombinant LH2 protein. The methodologies established here will be amenable to high-throughput screening of LH2 small molecule antagonists and might be applicable to other α-ketoglutarate– and Fe2+–dependent oxygenases.
Supplementary Material
{kind=link}
Highlights.
A scalable method for expressing and purifying recombinant human LH2 with robust enzymatic activity.
A convenient and sensitive biochemical assay for Fe(II) and α-ketoglutarate dependent oxygenases.
Acknowledgments
We thank Dr. Craig W. Vander Kooi at the University of Kentucky for helpful discussions and technical assistance.
Funding:
This work was supported by P50-CA70907-15 (JMK), R01 CA105155 (JMK, MY), the Welch Foundation (F-1390) (KND), and CPRIT grants RP160652 (JMK, GNP), RP160657 (KND), and RP110532-P1 (KND).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yamauchi M, Sricholpech M. Lysine post-translational modifications of collagen. Essays Biochem. 2012;52:113–33. doi: 10.1042/bse0520113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4(3):152–6. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- 3.Myllyharju J, Kivirikko KI. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004;20(1):33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Yeowell HN, Walker LC. Tissue specificity of a new splice form of the human lysyl hydroxylase 2 gene. Matrix Biol. 1999;18(2):179–87. doi: 10.1016/s0945-053x(99)00013-x. [DOI] [PubMed] [Google Scholar]
- 5.Uzawa K, et al. Differential expression of human lysyl hydroxylase genes, lysine hydroxylation, and cross-linking of type I collagen during osteoblastic differentiation in vitro. J Bone Miner Res. 1999;14(8):1272–80. doi: 10.1359/jbmr.1999.14.8.1272. [DOI] [PubMed] [Google Scholar]
- 6.van der Slot AJ, et al. Identification of PLOD2 as telopeptide lysyl hydroxylase, an important enzyme in fibrosis. J Biol Chem. 2003;278(42):40967–72. doi: 10.1074/jbc.M307380200. [DOI] [PubMed] [Google Scholar]
- 7.Takaluoma K, Lantto J, Myllyharju J. Lysyl hydroxylase 2 is a specific telopeptide hydroxylase, while all three isoenzymes hydroxylate collagenous sequences. Matrix Biol. 2007;26(5):396–403. doi: 10.1016/j.matbio.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Pornprasertsuk S, et al. Lysyl hydroxylase-2b directs collagen cross-linking pathways in MC3T3-E1 cells. J Bone Miner Res. 2004;19(8):1349–55. doi: 10.1359/JBMR.040323. [DOI] [PubMed] [Google Scholar]
- 9.Ha-Vinh R, et al. Phenotypic and molecular characterization of Bruck syndrome (osteogenesis imperfecta with contractures of the large joints) caused by a recessive mutation in PLOD2. Am J Med Genet A. 2004;131(2):115–20. doi: 10.1002/ajmg.a.30231. [DOI] [PubMed] [Google Scholar]
- 10.van der Slot AJ, et al. Increased formation of pyridinoline cross-links due to higher telopeptide lysyl hydroxylase levels is a general fibrotic phenomenon. Matrix Biol. 2004;23(4):251–7. doi: 10.1016/j.matbio.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Knipper JA, et al. Interleukin-4 Receptor alpha Signaling in Myeloid Cells Controls Collagen Fibril Assembly in Skin Repair. Immunity. 2015;43(4):803–16. doi: 10.1016/j.immuni.2015.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellaye PS, Kolb M. Why do patients get idiopathic pulmonary fibrosis? Current concepts in the pathogenesis of pulmonary fibrosis. BMC Med. 2015;13:176. doi: 10.1186/s12916-015-0412-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu F, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015;308(4):L344–57. doi: 10.1152/ajplung.00300.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Southern BD, et al. Matrix-driven Myosin II Mediates the Pro-fibrotic Fibroblast Phenotype. J Biol Chem. 2016;291(12):6083–95. doi: 10.1074/jbc.M115.712380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15(12):786–801. doi: 10.1038/nrm3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang X, et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012;47(3):340–8. doi: 10.1165/rcmb.2012-0050OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, et al. Hydroxylysine aldehydre-derived collagen cross-links promote lung cancer metastasis. The Journal of Clinical Investigation. 2015 [Google Scholar]
- 18.Chen Y, et al. Lysyl Hydroxylase 2 Is Secreted By Tumor Cells and Can Modify Collagen in the Extracellular Space. J Biol Chem. 2016 doi: 10.1074/jbc.M116.759803. [DOI] [PMC free article] [PubMed]
- 19.Eisinger-Mathason TS, et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discov. 2013;3(10):1190–205. doi: 10.1158/2159-8290.CD-13-0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gilkes DM, et al. Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol Cancer Res. 2013;11(5):456–66. doi: 10.1158/1541-7786.MCR-12-0629. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Gilkes DM, et al. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J Biol Chem. 2013;288(15):10819–29. doi: 10.1074/jbc.M112.442939. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Pirskanen A, et al. Site-directed mutagenesis of human lysyl hydroxylase expressed in insect cells. Identification of histidine residues and an aspartic acid residue critical for catalytic activity. J Biol Chem. 1996;271(16):9398–402. doi: 10.1074/jbc.271.16.9398. [DOI] [PubMed] [Google Scholar]
- 23.Krol BJ, et al. The expression of a functional, secreted human lysyl hydroxylase in a baculovirus system. J Invest Dermatol. 1996;106(1):11–6. doi: 10.1111/1523-1747.ep12326956. [DOI] [PubMed] [Google Scholar]
- 24.Puistola U, et al. Studies on the lysyl hydroxylase reaction. I. Initial velocity kinetics and related aspects. Biochim Biophys Acta. 1980;611(1):40–50. doi: 10.1016/0005-2744(80)90040-6. [DOI] [PubMed] [Google Scholar]
- 25.Puistola U, et al. Studies on the lysyl hydroxylase reaction. II. Inhibition kinetics and the reaction mechanism. Biochim Biophys Acta. 1980;611(1):51–60. doi: 10.1016/0005-2744(80)90041-8. [DOI] [PubMed] [Google Scholar]
- 26.Kivirikko KI, et al. Studies on protocollagen lysine hydroxylase. Hydroxylation of synthetic peptides and the stoichiometric decarboxylation of -ketoglutarate. Biochemistry. 1972;11(1):122–9. doi: 10.1021/bi00751a021. [DOI] [PubMed] [Google Scholar]
- 27.Wang C, et al. Lysyl hydroxylase 3 is secreted from cells by two pathways. J Cell Physiol. 2012;227(2):668–75. doi: 10.1002/jcp.22774. [DOI] [PubMed] [Google Scholar]
- 28.Luo L, et al. An assay for Fe(II)/2-oxoglutarate-dependent dioxygenases by enzyme-coupled detection of succinate formation. Anal Biochem. 2006;353(1):69–74. doi: 10.1016/j.ab.2006.03.033. [DOI] [PubMed] [Google Scholar]
- 29.McNeill LA, et al. A fluorescence-based assay for 2-oxoglutarate-dependent oxygenases. Anal Biochem. 2005;336(1):125–31. doi: 10.1016/j.ab.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Longo PA, et al. Transient mammalian cell transfection with polyethylenimine (PEI) Methods Enzymol. 2013;529:227–40. doi: 10.1016/B978-0-12-418687-3.00018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo HF, et al. Mechanistic basis for the potent anti-angiogenic activity of semaphorin 3F. Biochemistry. 2013;52(43):7551–8. doi: 10.1021/bi401034q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heikkinen J, et al. Lysyl hydroxylase 3 is a multifunctional protein possessing collagen glucosyltransferase activity. J Biol Chem. 2000;275(46):36158–63. doi: 10.1074/jbc.M006203200. [DOI] [PubMed] [Google Scholar]
- 33.Heikkinen J, et al. Dimerization of human lysyl hydroxylase 3 (LH3) is mediated by the amino acids 541–547. Matrix Biol. 2011;30(1):27–33. doi: 10.1016/j.matbio.2010.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.