

A large-scale QTL mapping on 10 plant architecture traits across 10 RIL populations reveals the complex genetic basis of plant architecture in maize.
Abstract
Plant architecture is a key factor affecting planting density and grain yield in maize (Zea mays). However, the genetic mechanisms underlying plant architecture in diverse genetic backgrounds have not been fully addressed. Here, we performed a large-scale phenotyping of 10 plant architecture-related traits and dissected the genetic loci controlling these traits in 10 recombinant inbred line populations derived from 14 diverse genetic backgrounds. Nearly 800 quantitative trait loci (QTLs) with major and minor effects were identified as contributing to the phenotypic variation of plant architecture-related traits. Ninety-two percent of these QTLs were detected in only one population, confirming the diverse genetic backgrounds of the mapping populations and the prevalence of rare alleles in maize. The numbers and effects of QTLs are positively associated with the phenotypic variation in the population, which, in turn, correlates positively with parental phenotypic and genetic variations. A large proportion (38.5%) of QTLs was associated with at least two traits, suggestive of the frequent occurrence of pleiotropic loci or closely linked loci. Key developmental genes, which previously were shown to affect plant architecture in mutant studies, were found to colocalize with many QTLs. Five QTLs were further validated using the segregating populations developed from residual heterozygous lines present in the recombinant inbred line populations. Additionally, one new plant height QTL, qPH3, has been fine-mapped to a 600-kb genomic region where three candidate genes are located. These results provide insights into the genetic mechanisms controlling plant architecture and will benefit the selection of ideal plant architecture in maize breeding.
Maize (Zea mays) is the most widely grown grain crop worldwide and has become one of the most important crops for food, animal feed, and bioenergy production. Maize grain yield in the United States has increased 8-fold in the past 80 years, of which half was the result of breeder selection (Duvick, 2005). Although high grain yield per hectare is a primary breeding goal, these increases in grain yield are predominantly due to higher plant density (Duvick, 2005). In the United States, the average plant density of maize has increased from 30,000 plants per hectare in the 1930s to more than 80,000 plants per hectare currently, which has been accompanied by a change in maize plant aboveground architecture, especially maize leaf angle, which has become significantly more upright (Duvick, 2005). Ideal plant architecture in high-plant-density maize production can optimize canopy architecture, improve photosynthetic efficiency, and prevent lodging, thus resulting in overall high grain yield.
Here, plant architecture primarily refers to the aboveground parts of maize. It is a function of numerous specific traits, such as plant height (PH), ear height (EH), tassel branch number (TBN), tassel main axis length (TMAL), leaf length (LL), leaf width (LW), middle leaf angle (MLA), leaf number above ear (LNAE), leaf number blow ear (LNBE), and ear leaf number (LN), etc. As breeder selection for plant density leads to increased leaf angle, leaf size, and tassel size and angle, these traits have been optimized, allowing light to penetrate into the aboveground canopy with considerable yield advantages (Pepper et al., 1977; Fischer et al., 1987; Begna et al., 1999). All of the aboveground parts of maize plants are derived from the shoot apical meristem (SAM) and its derivative inflorescence meristem. The maintenance and differentiation of the SAM have been reported to determine the morphology of plant aboveground parts (Thompson et al., 2015). The developmental origination of all plant architecture traits explains their high correlations with each other. Previous mutant studies on the SAM indicate that multiple genes, especially homeobox genes, their interaction, as well as phytohormone genes, are involved in the regulation of meristem identity and differentiation (Barton, 2010). Therefore, plant architecture exhibits quantitative variation with complex genetic mechanisms. Although clarifying the genetic basis of these traits is still challenging, the genetic dissection of plant architecture will improve breeding for yield at high plant density and, thus, lead to continued maize productivity improvement.
Hundreds of quantitative trait loci (QTLs) related to plant architecture traits have been identified (http://www.maizegdb.org/data_center/qtl-loci-summary). Tian et al. (2011) performed a genome-wide association study (GWAS) of leaf architecture in the maize nested association mapping (NAM) population and demonstrated that the genetic basis of the leaf traits is dominated by small effects with little epistasis, environmental interaction, or pleiotropy. Peiffer et al. (2014) dissected the genetic basis of maize height in maize NAM populations and revealed that maize height is under strong genetic control and has a highly polygenic genetic basis. Li et al. (2016) employed a teosinte introgression population, conducted a comprehensive genetic dissection on leaf number and its genetic relationship to flowering time, and demonstrated that these traits are under relatively independent genetic control. Genes such as teosinte branched1, teosinte glume architecture1, dwarf plant3, dwarf plant8, dwarf plant9, nana plant1, and brachytic plant2 have been cloned, and their regulation of flowering time and plant height was explained at the molecular level (Winkler and Helentjaris, 1995; Doebley et al., 1997; Multani et al., 2003; Wang et al., 2005; Lawit et al., 2010; Hartwig et al., 2011). However, functional alleles of many of the genes associated with plant architecture have been already fixed in maize elite germplasm, limiting their use in maize improvement. Most previous genetic studies focused on single or a few plant architecture traits and lacked power to dissect plant architecture traits as a whole. Bouchet et al. (2017) assessed the genetic basis of 24 correlated maize traits and identified major pleioptropic effects and/or linkage for plant architecture traits in one population with 336 lines, which, however, only represents a small proportion of the genetic diversity in maize. Despite this progress, the genetics of plant architecture-related traits in maize have not been fully investigated in multiple diverse genetic backgrounds.
To this end, we constructed 10 recombinant inbred line (RIL) maize populations, designated as an random-open-parent association mapping (ROAM) population (Xiao et al., 2016, 2017), conducted large-scale phenotyping on 10 plant architecture traits, and performed a comprehensive genetic dissection of plant architecture in maize. Approximately 800 QTLs with major and minor effects were identified across 10 diverse genetic backgrounds in maize. Both pleiotropic and linked QTLs were detected, which, to a large extent, explains the high correlations between plant architecture traits. The phenotypic differences between the two parents for each of the 10 maize RIL populations were shown to be associated with the phenotypic diversity and the number of QTLs detected within populations, indicating that Mendelian effects contribute predominantly to phenotypic variation in the progeny. Additionally, residual heterozygous line (RHL) family analyses validated five QTLs and fine-mapped one plant height QTL to the 600-kb genomic region, confirming the robustness of our analysis. All of these results further our understanding of plant architecture variation and provide selection loci for further progress toward the ideal maize plant architecture.
RESULTS AND DISCUSSION
Large-Scale Phenotyping Reveals Complex Relationships among Plant Architecture-Related Traits in Maize
The ROAM population was developed, consisting of 10 RIL populations derived from 14 elite maize inbreds, for a total of 1,887 RIL lines (Pan et al., 2016; Xiao et al., 2016). These 14 elite maize inbreds were selected from a large association mapping panel and exhibited extensive genetic diversity (Yang et al., 2011; Supplemental Fig. S1). We phenotyped the ROAM population at five to 12 locations for 10 aboveground traits, PH, EH, TBN, TMAL, LL, LW, MLA, LNAE, LNBE, and LN, measured manually (Fig. 1A; see “Materials and Methods”).
Figure 1.

Extensive phenotypic variation of plant architecture in maize. A, Diagram of the measured 10 plant architecture traits in our study: PH, EH, TBN, TMAL, LNAE, LNBE, LN, LL, LW, and MLA. B, Phenotypic variation of five plant architecture traits. C, Relationships among 10 architecture traits. Red lines designate positive correlations between two traits, and blue lines designate negative correlations. The correlations with P < 1.0E-15 are shown. The circle size shows the number of correlated traits. D, Phenotypic tree of all 10 plant architecture traits. The scale represents Euclidean distance, which was calculated based on the standardized phenotypic data across 10 RIL populations.
Best linear unbiased prediction (BLUP) analyses revealed extensive phenotypic variation (Fig. 1B; Supplemental Table S1). All 10 traits exhibited normal distributions, with a 2- to 10-fold difference between the smallest and largest lines in the RIL populations (Fig. 1B; Supplemental Table S1; Supplemental Fig. S2). TBN had the most dramatic variation, with a 10-fold change (the smallest and largest tassel numbers are two and 20, respectively; Fig. 1B; Supplemental Table S1). Tassel size is a key factor associated with maize grain yield (Duvick, 2005). A smaller tassel is frequently associated with higher yield; however, hybrid production would be affected if the tassel were too small (Hunter et al., 1969). The dramatic TBN variation in the ROAM population might allow a large flexible selection of optimal tassel size. And leaf angle had the second most extensive variation of ∼4-fold change, while the other eight traits had less than 3-fold phenotypic variations (Fig. 1B; Supplemental Table S1). Meanwhile, the broad sense heritability of all 10 plant architecture-related traits ranged from 0.9 to 0.95 (Table I), which is significantly higher than that (0.76–0.87) of grain yield-related traits (Xiao et al., 2016). The extensive phenotypic variation and high heritability suggest that these 10 RIL populations have a diverse genetic background and are suitable for dissecting the genetic factors underlying plant architecture-related traits in maize.
Table I. Summary of heritability and QTLs or SNPs identified by SLM, JLM, and GWAS methods for 10 plant architecture traits.
Plant architecture exhibits quantitative variation, which is sensitive to environment. Even for a specific genotype, plant individuals may show extensive phenotypic variations due to both genetic variation, such as new mutations, and environmental changes (Zhang, 2008; Fraser and Schadt, 2010). Although plant architecture traits had pretty high heritability, a fraction of phenotypic variations were derived from environmental effects. The environmental effects of phenotypic variations for all 10 plant architecture traits vary from 2.2% to 13.3% (Supplemental Table S2). Additionally, a significant effect of genotype by environment was observed (Supplemental Table S2).
Analyses showed that the 10 plant architecture-related traits are significantly correlated with each other, with each trait correlating with one to six other traits (Fig. 1C; Supplemental Table S3). We detected a total of 45 correlations among the 10 traits, the majority (43) of which are positive associations, except the two correlations between LL and LW or LNBE (Fig. 1C; Supplemental Table S3). We performed a phenotypic clustering analysis based on the phenotypic variation across the 10 RIL populations (Fig. 1D). A phenotypic tree of all 10 plant architecture traits was constructed based on the clustering analysis (see “Materials and Methods”) and demonstrated high correlations as well as an extensive phenotypic variation among plant architecture traits. This classified the 10 plant architecture traits into three unrooted groups (Fig. 1D). The biggest cluster includes eight measured traits, PH, EH, LL, LW, LN, LNAE, LNBE, and TMAL. All aboveground parts of maize plants are derived from SAM and its derivative inflorescence meristem (Barton, 2010). Recent studies have uncovered phenotypic correlations between meristem morphology and adult plant traits, revealing links between the undifferentiated and differentiated plant organs (Thompson et al., 2015). Additionally, Baute et al. (2015) revealed that the whole SAM traits are correlated with mature plant morphology-related traits. The similar origination of all aerial morphology traits may be the developmental factor that classifies these eight traits into one highly correlated phenotypic cluster.
Of particular interest, MLA and TBN are singletons in the clustering analysis, suggesting that these two traits may have a distinct genetic contribution to plant architecture compared with traits in the biggest cluster. Interestingly, these two traits also have the largest phenotypic variation in the mapping population. As plant density has increased, leaves have become more upright and TBN has been reduced dramatically to optimize light penetration into the plant canopy, suggesting that TBN and MLA are the important selection targets during maize improvement (Fischer et al., 1987; Russell, 1991; Andrade et al., 1993). Most of the parents of these 10 RIL populations are elite inbreds from different breeding programs. TBN and MLA, which have the most phenotypic variation, reflect the genetic diversity in our mapping populations. The extensive and continuous phenotypic variation of these two traits and their divergence from the major phenotypic cluster indicate that there were many different loci with rare alleles each contributing a small amount of variation for TBN and MLA in maize.
Genetic Dissection of Plant Architecture Uncovered Hundreds of QTLs with Major and Minor Effects That Contribute to Phenotypic Variation
To dissect the genetic mechanisms underlying plant architecture in maize, single linkage mapping (SLM), joint linkage mapping (JLM), and GWAS were employed to map the QTLs underlying phenotypic variation. While SLM identified QTLs in each RIL population, JLM and GWAS identified QTLs or single-nucleotide polymorphisms (SNPs) by a joint analysis of all 10 populations using an integrated high-density map. Hundreds of consistent QTLs were identified through all three genetic algorithms (Table I; Fig. 2; Supplemental Figs. S3–S12).
Figure 2.

Genome-wide landscape of genetic factors underlying plant architecture variation in maize. Genome-wide association mapping by the JLM was employed to detect the genetic factors underlying maize plant architecture variation. The x axis shows the genomic position (Mb), and the y axis shows the likelihood ratio (LTR). The gray section represents LTR values significantly less than the cutoff of 2.76, while the green and orange bars represents significant LTR values greater than the cutoff. Genes in blue were coincident with QTLs.
Through the SLM method, a large number of QTLs ranging from 64 to 83 were identified for each plant architecture trait. In total, 752 QTLs were uncovered for the 10 traits across 10 RIL populations, indicative of the complexity of maize plant development (Table I). Of these 752 QTLs, 23.2% have an effect of more than 10% of phenotypic variation (Supplemental Table S4; Supplemental Fig. S13). The proportion of QTLs with an additive effect greater than 10% ranged from 16% to 31% (Supplemental Fig. S14), suggesting that a few major genetic factors with large effects along with many genetic factors with marginal effects contribute to the phenotypic variation of maize plant architecture. QTL comparison among different RIL populations shows that a large proportion (ranging from 57 to 76) of QTLs could only be identified in one population, while a very small (from three to 12) proportion of QTLs can be uncovered simultaneously in at least two populations. This observation confirmed that our ROAM has an extensive genetic diversity. For example, out of the 83 QTLs associated with plant height across the 10 RIL populations, only seven could be detected in at least two populations. Interestingly, the number of QTLs detected in only a single population was significantly more than those detected in multiple populations (χ2 test, P = 1.1E-26), but there was no difference for QTL effects (ANOVA, P = 0.15). These results suggest that a large number of rare genetic factors play an important role in the phenotypic variation in maize.
Using the JLM method, we mapped 50 to 96 QTLs with R2 ranging from 0.31% to 4% for each trait. Concordant with the results of SJM, the majority QTLs detected by the JLM method are unique to a single population (Table I; Supplemental Table S5). Compared with SLM, the resolution of JLM reached up to 1.2 Mb, which was significantly smaller than the resolution of 3.4 Mb achieved by SLM (P < 2.0E-16; Supplemental Fig. S15). Through GWAS, we mapped nine to 43 significant SNPs associated with plant architecture traits (Table I). The effect of most loci was smaller than 1% (Supplemental Table S6). In total, these significant loci could explain 30.13% to 53.49% of the phenotype variation (Supplemental Fig. S16).
Different genetic algorithms identified different numbers of loci underlying plant architecture variation (Supplemental Tables S4–S6). Unlike NAM populations, the ROAM population was derived from random crosses of 14 diverse genetic backgrounds, which makes the whole population lack common variants. Therefore, GWAS was likely to detect fewer significant loci with smaller genetic effects conferring plant architecture variation. However, comparisons of the QTLs detected by the three methods still showed that a large proportion (ranging from 15.8% to 30.6% for each trait) of QTLs could be detected by all three methods (Supplemental Fig. S17). Additionally, a majority (60%–77.2% for SLM versus JLM, 26.6%–52.2% for SLM versus GWAS, and 6.5%–14.4% for JLM versus GWAS) of QTLs could be detected by two genetic algorithms, suggestive of the robustness of QTLs identified in our study.
The density of genetic markers and the size of recombination blocks could allow us to map QTLs into 2-Mb genomic regions (Pan et al., 2016). We divided the maize genome into 2-Mb sliding window bins for the comparison of QTLs of different plant architecture traits across 10 RIL populations. Combining all three statistical methods, a total of 434 genomic recombinant bins with peaks significantly associated with phenotypic variation were identified. Colocalization of these genomic bins with well-known mutant genes indicates that 150 out of 416 plant architecture well-known genes were located within the confidence intervals of plant architecture QTLs for JLM and 250 well-known genes were coincident with detectable QTLs by SLM (Fig. 2). For example, brachytic2 is coincident with a QTL on chromosome 1 conferring plant height variation in the ZONG/YU87-1 population, which also was validated by QTL cloning (Multani et al., 2003; Xing et al., 2015; Fig. 3A). And liguleless1 (lg1) is colocalized with a QTL on chromosome 2 that controls leaf angle (Becraft et al., 1990; Fig. 3B). Maize plants without ligule have upright leaves, for which the leaf angle is nearly zero (Sylvester et al., 1990). Natural genomic variation in lg1 between inbreds is likely to quantitatively change leaf angle. Therefore, the identification of natural favorable alleles of lg1 would aid the target selection of leaf angle. Notably, based on the colocalization of well-known genes with QTLs by SLM, 39.9% of well-known plant architecture-related maize genes are not located in the QTL regions identified here as associated with plant architecture variation. The noncoincidence of well-known genes with detectable QTLs suggests either that most of these genes may have been fixed during the domestication and selection of maize or that the alleles present in the parents of the mapping population contribute little to the variation and thus were not detected, or that the power to detect these loci is still insufficient. A proportion (27.3%) of QTLs underlying plant architecture variation detected by SLM are not colocalized with any known functional mutant genes, implying that the genetic basis of plant architecture is complex and that many QTLs identified in this study would provide useful selection targets for the ideal plant architecture breeding in maize. New alleles at the loci lacking variation in the breeding germplasm may be introduced from the unadapted germplasm.
Figure 3.

Colocalization of two well-known genes with QTLs underlying plant architecture traits. A, brachytic2 is coincident with a QTL on chromosome 1 conferring plant height variation in the ZONG/YU87-1 population. B, lg1 is colocalized with a QTL on chromosome 2 that controls leaf angle in the SK/ZHENG58 population. LOD, Log of the odds.
To detect any epistatic interactions between the plant architecture QTLs, two-way ANOVA was employed and 100 epistatic pairs between QTLs were identified at a P value of 0.05. However, after Bonferroni correction, no significant epistatic interaction was retained. This is consistent with previous studies (Buckler et al., 2009; Tian et al., 2011), which also showed that the variation of plant architecture was rarely the result of epistatic effect.
Mendelian Effects Play an Important Role in the Phenotypic Variation of Progeny for Plant Architecture-Related Traits
Phenotypic variation between parental lines is a major consideration when developing a segregating population for QTL mapping on the assumption that large phenotypic variation can lead to the identification of large-effect QTLs and a large number of QTLs. The inclusion of multiple traits and multiple populations in our study allowed us to test this assumption directly. Surprisingly, our analysis indicated that neither the number nor the effect of QTLs is correlated significantly with parental phenotypic variation. However, they both showed significantly positive associations with phenotypic variation in the populations, which itself is weakly correlated to parental phenotypic variation (Fig. 4, A–E; Supplemental Fig. S18), suggesting the presence of parental Mendelian effects. Such Mendelian effects indicate that proper selection of parents is not only the basis of QTL mapping but also affects the efficiency of crop improvement. Interestingly, male parents provided significantly more additive alleles than female parents (55.8% versus 44.2%, P = 6.1E-3, ANOVA; Fig. 4F). However, the two parental alleles contributed equally to the additive phenotypic variations (P = 0.77; Fig. 4G). Taken together, these results indicate that parental Mendelian factors may play a critical role in the phenotypic variation of the progeny.
Figure 4.

Relationships between parental diversity and phenotypic variation in the progeny. A, Parental phenotypic variation (PPV) is significantly associated with sd of progeny phenotypic variation (STV). B, Parental genetic variance (PGV) is significantly associated with STV. C, STV is significantly associated with QTL number (QN). D, STV is significantly associated with QTL phenotypic variance rate (QPV). E, Model of relationships between STV, PGV, PPV, QN, and QPV. F, Proportion of QTLs with positive effects that were derived from male and female alleles. G, Distribution of QTL positive effects that were derived from male and female alleles.
Transcriptomic analyses of tens of thousands of expressed genes in SAMs of 105 RILs and their parents (B73 and Mo17) uncovered that a majority of maize genes exhibited Mendelian variation where the expression of genes in progeny is largely determined by parental variation, confirming the prevalence of Mendelian effects (Li et al., 2013b). Here, we analyzed 10 plant architecture-related traits across 10 RIL populations and observed that parental phenotypic variations are associated significantly with progeny phenotypic variation, the number of QTLs, and the effects of QTLs. Notably, although the number of QTLs with positive effects varies between male and female parents for plant architecture-related traits, the total positive contribution of alleles from two parents is equal. Consistent with previous studies, our results confirm that the parental Mendelian effects are widespread for many agronomic traits. Most of our elite parents are derived from conventional breeding programs, indicating that parental Mendelian effects may originate from the process of breeding selection. The phenomena of parental Mendelian effects and the QTLs associated with plant architecture traits may help breeders to select elite inbreds for the selection of breeding cycle and accelerate the breeding process.
A Substantial Number of Pleiotropic Loci or Linked QTLs Conferring Plant Architecture in Maize Are Likely to Be Key Developmental Genes
QTL mapping of different plant architecture traits showed that QTLs controlling different traits often overlap with each other. For any two plant architecture-related traits, the number of QTLs controlling both traits ranged from 4 to 19 (Fig. 5A). As the phenotypic distance between the two traits increased, the number of common QTLs for both traits was reduced (Fig. 5, B and C), which is consistent with the idea that phenotypic correlation among traits is due to the same or linked genetic factors controlling these traits. We identified 55 QTLs that are associated with at least three different traits (Fig. 5D). Notably, there are 11 QTLs that are associated significantly with at least five traits (Supplemental Fig. S19). Of all 434 2-Mb sliding genomic bins with detectable QTLs, 61.5% are associated with only a single plant morphology trait, while 38.5% are related to at least two traits.
Figure 5.

Genomic regions with pleiotropic effects contribute largely to the genetic relationships within plant architecture traits in maize. A, Summary of overlapped QTLs between 10 plant architecture traits. The bottom left part shows the –log10 values of P, showing the extent of genetic overlap between plant architecture traits. The top right part illustrates the overlapped QN for different pairs of architecture traits. B, Relationship between the proportion of pleiotropic QTLs and phenotypic correlation between all pairs of architecture traits. PQR, Pleiotropic QTL rate. C, Relationship between the proportion of phenotypic variation explained by pleiotropic QTL (PPR) and phenotypic correlation. D, Genomic region rate with different number traits of mapping. E, Pleiotropic QTL among different clusters. The clusters are defined as described in Figure 1D. F, Colocalization of well-known genes with pleiotropic QTLs, showing the distribution of well-known genes in different genomic regions.
Consistent with the phenotypic clustering tree of all 10 plant morphology traits, a large number (275) of pleiotropic loci have been detected for any pair of morphology traits within the biggest phenotypic cluster, significantly more (P = 9.0E-3) than that for phenotypic pairs between TBN, MLA, and the eight traits within the biggest cluster (Fig. 5E). TBN and MLA, which are two singletons in the phenotypic clustering tree, showed the lowest genetic overlap as compared with any phenotypic pairs of all 10 plant morphology traits. Based on the significant P values of the pleiotropic QTL matrix of any pair of plant architecture traits, the genetic distances of any two plant architecture traits were obtained. Furthermore, we constructed a phylogenetic tree of plant architecture traits, which is largely consistent with a phenotypic clustering tree (Supplemental Fig. S20). Both phylogenetic and phenotypic clustering trees intuitively represent the divergence and relationships among plant architecture traits in maize.
Colocalization analysis between pleiotropic loci and well-known genes, of which most have been cloned through mutant studies (Schnable and Freeling, 2011), suggested that many mutant genes (166) are not located in any QTL regions, such as tassel branch1, rough sheath1, rough sheath2, and tassel seed1 (Doebley et al., 1995; Schneeberger et al., 1995; Timmermans et al., 1999; Theodoris et al., 2003; Acosta et al., 2009). This indicates that there is little phenotypically relevant genetic diversity for these genes among our parents or, perhaps, in the maize breeding germplasm. However, we did observe that a large number of 250 well-known genes, especially the key developmental genes, are coincident with QTLs by SLM, and 119 are in the genomic regions with pleiotropic effects for plant morphology traits, which is significantly higher (χ2 test, P < 0.01) than expectation (Supplemental Table S7).
This biggest phenotypic cluster contains mainly SAM-derived or inflorescence meristem-derived traits, which may be controlled by developmental genetic mechanisms (Sheridan, 1988). Previous studies have identified and cloned many developmental genes controlling SAM identity and differentiation (Barton, 2010; Schnable and Freeling, 2011). Interestingly, these well-known cloned developmental genes (416) are frequently colocated with plant architecture QTL regions, especially the pleiotropic or linked genomic regions (Supplemental Table S7). Zea floricaula leafy2 (Zfl2), which is a homolog of the Arabidopsis (Arabidopsis thaliana) mutant gene LEAFY, which generates more rosette leaves, functions by promoting the transition from inflorescence to floral meristem and controls quantitative aspects of inflorescence phyllotaxy in maize (Weigel et al., 1992; Bomblies et al., 2003). As expected, Zfl2 is coincident with a pleiotropic genomic region on chromosome 2, which controls LNBE, LN, and LW at the same time. This coincidence makes Zfl2 a strong candidate for this pleiotropic locus for future study (Fig. 6A). A pleiotropic locus on chromosome 8, controlling PH and LL, is coincident with Zmhomeobox1a (Zmhox1a), which is a homeobox gene controlling tissue differentiation and development (Bellmann and Werr, 1992; Fig. 6B).
Figure 6.

Colocalization of two well-known developmental genes with the pleiotropic loci conferring plant architecture in maize. A, Zfl2 is coincident with a pleiotropic genomic region on chromosome 2 that controls LNBE, LN, and LW. B, Zmhox1a is coincident with a pleiotropic genomic region on chromosome 8 that controls PH and LL.
Although TBN and MLA were divergent from the biggest phenotypic cluster, there are a few pleiotropic loci that have been colocalized with well-known developmental genes. The dissection of the genetic basis of floral branch systems in maize and related grasses suggested a model in which ramosa1, ramosa2, barren stalks, and tassel branch1 modulate plant inflorescence and plant architecture (Vollbrecht et al., 2005). Except for tassel branch1, the other three key genes were colocalized in the pleiotropic loci, which are associated with TBN, TMAL, LL, LW, and EH. Notably, lg1, which was reported previously to induce ligules and auricles during maize leaf organogenesis and further affect proximal-distal signaling and leaf growth, is coincident with a pleiotropic locus affecting MLA, LL, LW, and TBN as well (Moreno et al., 1997; Moon et al., 2013). These results indicate that developmentally associated loci contribute to more correlations between plant architecture-related traits than expected by chance (P = 1E-3, Monte Carlo 1,000 random resample), suggesting that key developmental genes may be the source of the genetic overlap of plant architecture in maize (Supplemental Table S7).
Validation of Five QTLs Using RHLs
Although fine-mapping using near isogenic lines is an accepted standard way to validate a locus that controls a phenotypic variation, the process of developing near isogenic lines is time and labor consuming. RHLs, which harbor heterozygous regions where the phenotypic QTL of interest is located, may be available from an RIL population (Yamanaka et al., 2001; McMullen et al., 2009). The use of RHLs to validate and fine-map QTLs has proven to be an efficient method (Yamanaka et al., 2001; Watanabe et al., 2011). To validate the QTL mapping results of plant architecture QTLs identified in this study, RHLs were obtained for five randomly selected QTLs of plant height, TMAL, and TBN. As expected, all five QTLs could be detected with genetic effects consistent with whole-genome QTL scanning (Fig. 7). This implies the robustness of our genetic dissection and provides candidate fine-mapping regions for the future improvement of plant morphology in maize.
Figure 7.

Map-based validation using RHLs for five QTLs. RHL family validation is shown for qPH3, qTMAL3, qPH1, qPH7, and qTBN3. For each trait, the image at top shows the phenotypic variation between the two parents, while the graph at bottom shows the phenotypic variation between two different homozygous genotypes of the target QTL region.
We elected to focus on the fine-mapping of qPH3, which is a major QTL with an additive effect of ∼8 cm and which explains 17% of the plant height variation in the DE3×BY815 RIL population (Fig. 8A). Primary QTL mapping has anchored qPH3 into a genomic region ranging from 162.2 to 166.7 Mb on chromosome 3 (Fig. 8B). We obtained the segregating RHLs and developed nine competitive allele-specific PCR (KASP) markers in the target region of qPH3. Phenotypic Student’s t test of the progeny of these segregating RHLs narrowed down the target region to between KASP markers K4 and K8, which spans a 1-Mb (from 165.72 to 166.59 Mb) genomic region according to the B73 reference genome (Fig. 8C). RHL subfamilies with segregating genomic substitution were planted and subjected to phenotyping and genotyping with KASP markers M1 to M5. The progeny phenotypic test indicates that the causal gene of qPH3 is located in the ∼600-kb genomic region from M1 to M5 (Fig. 8D).
Figure 8.

Fine-mapping of qPH3 using RHLs in the DE3/BY815 RIL population. A, Primary QTL mapping of qPH3 in the DE3/BY815 population. B, RHL screening of qPH3. C and D, ANOVA based on the progeny genotypic and phenotypic variation in the RHL families in Wuhan (C) and Hainan (D).
The 600-kb genomic region has 16 annotated genes according to the maize B73 reference genome (Schnable et al., 2009). By integrating the genome-wide association mapping results of plant height in a Chinese association panel consisting of 505 diverse inbred lines (Yang et al., 2011), eight SNPs derived from four genes in the 1-Mb target genomic region were identified to be significantly associated with plant height in maize (Fig. 9A). Subsequent RNA-seq analysis on the leaves of the segregating lines of RHLs identified 146 differentially expressed genes genome wide. Gene Ontology enrichment showed that these differentially expressed genes were enriched in the category of developmental growth, response to biotic stimulus, etc. Of these differentially expressed genes between progeny lines with BY815 alleles and the ones with DE3 alleles, only two genes (GRMZM2G058250 and GRMZM2G177227) are located in the target genomic region, providing targets of the cloning of qPH3 (Fig. 9B). For a causal gene of the QTL of interest, the parents of the mapping population should have different haplotypes. We analyzed the haplotype variation at all 16 genes in the target genomic region, 13 of which have alternative haplotypes between parents of five RIL populations (K22×CI7, K22×DAN340, K22×BY815, BY815×KUI3, and KUI3×B77). However, qPH3 was not detected in all five RIL populations. Resequencing of these 16 candidate genes in the parents also revealed that three genes had dramatic genomic variations, which could result in nonsynonymous mutations, early translation termination, etc., between parents of all five RIL populations (Supplemental Tables S8 and S9). However, these three genes (GRMZM2G058250, GRMZM2G177227, and GRMZM2G177220) within the target region, two of which were differentially expressed, have identical haplotypes for the parents of these five RIL populations but different haplotypes between DE3 and BY815. qPH3 has been identified in the DE3×BY815 RIL population, implying that these three genes are likely to be the causal candidates of qPH3 (Fig. 9C). Of particular interest, GRMZM2G177220 has a nucleotide mutation between DE3 and BY815, which could cause an early stop codon. This nucleotide mutation does not exist in the parents of the other five RIL populations but solely between DE3 and BY815 (Fig. 9D).
Figure 9.

Identification of candidate genes for qPH3. A, Haplotype association mapping of qPH3. The colors of the points indicate minor allele frequency, yellow for 0.05 to 0.1, purple for 0.1 to 0.2, red for 0.2 to 0.3, blue for 0.3 to 0.4, and dark blue for 0.4 to 0.5. B, Expression-level variation for all candidate genes in the qPH3 region between RHLs with and without 600-kb target genomic substitutions. C, Haplotype comparison of all candidate genes in the qPH3 region in different elite inbred lines. Only the DE3/BY815 RIL population, of which the two parents had haplotype divergence for three genes, had qPH3 detected. D, GRMZM2G177220 has a nucleotide mutation detected only between DE3 and BY815, which could cause an early stop codon.
Taking the results from association mapping, transcriptome analysis, and haplotype diversity based on candidate gene resequencing together, GRMZM2G058250, GRMZM2G177227, and GRMZM2G177220 are considered to be the candidate genes underlying qPH3. These three genes encode plastid-specific ribosomal protein4, exocyst subunit exo70 family protein F1, and a transcription factor with Myb-like domain and homeodomain, respectively. Of particular interest, GRMZM2G177227 has been shown to be associated with plant height variation in a U.S. association mapping panel (Wallace et al., 2014). The Arabidopsis homologous gene GRMZM2G177220 contains Myb-like, homeodomain-like, and signal transduction response domains, and its mutants showed reduced sensitivity to cytokinin inhibition on root elongation and lateral root formation, suggestive of a potential role in the regulation of plant height (Riechmann et al., 2000). The identification of these candidate genes provides critical information for future cloning of qPH3.
MATERIALS AND METHODS
Populations and Phenotypes
In previous studies, we developed a ROAM population, consisting of 10 RIL populations (B73×BY804, BY815×KUI3, DAN340×K22, DE3×BY815, K22×BY815, K22×CI7, KUI3×B77, YU87-1×BK, ZHENG58×SK, and ZONG3×YU87-1), derived from 14 diverse elite inbred lines of maize (Zea mays; Pan et al., 2016; Xiao et al., 2016). Populations of B73×BY804, KUI3×B77, K22×CI7, DAN340×K22, ZHENG58×SK, YU87-1×BK, and ZONG3×YU87-1 were planted in 12 locations, including Hubei (E114°17, N30°35), Sichuan (E104°04, N30°40), Guangxi (E108°19, N22°48), Chongqing (E106°33, N29°35), Henan (E113°40, N34°46), Yunnan (E102°42, N25°04), and Hainan (E109°31, N18°14) province in 2011 and Hubei (E114°7, N30°35), Chongqing (E106°33, N29°35), Henan (E113°40, N34°46), Yunnan (E102°42, N25°04), and Hainan (E109°31, N18°14) province in 2012. The other three populations, DE3×BY815, K22×BY815, and BY815×KUI3, were planted in five locations in 2012: Hubei, Chongqing, Henan, Yunnan, and Hainan province. In each location, RILs from a RIL family were randomly placed, with 12 plants of one RIL planted in a row. Five plants in the middle of the row were selected for the phenotyping of each RIL.
All populations were subject to phenotyping in 2011 and 2012. In total, 10 plant architecture-related traits, PH (cm), EH (cm), TBN (number), TMAL (cm), LNAE (number), LNBE (number), LN (number), LL (cm), LW (cm), and MLA (°), were measured manually (Supplemental Fig. S2). For the measurement of LNAE and LNBE, we considered the topmost ear as the dividing point regardless of how many ears were on the main stalk. We counted the leaf number above and below the topmost ear as LNAE and LNBE, respectively. To accurately measure phenotypic variations, we transformed the raw phenotypic data from all 13 and five locations of 2 years into BLUP values for all 10 plant architecture traits using the R package lme4 of lme function. The heritability of 10 plant architecture traits also was calculated using the R package lme4 of lme function. All analyses, including phenotypic clustering, correlation, and ANOVA, were based on BLUP phenotype data values.
Genotyping and Mapping
The Illumina MaizeSNP50 BeadChip of 56,110 SNPs was employed to identify the recombination bins, and ultra-high-resolution genetic maps were constructed for all 10 RIL populations (Pan et al., 2016). Additionally, 14 diverse elite inbred lines were selected from a diverse association mapping panel (Yang et al., 2010, 2011) and were subject to RNA-seq experiments on kernels at 15 d after pollination (Fu et al., 2013), generating hundreds of thousands of SNP markers. Recombination bins and ultra-high-density linkage maps of the ROAM population were used to dissect the genetic factors underlying maize plant architecture as follows.
(1) SLM was employed to detect QTLs in each RIL population. Winqtlcart 2.5 software along with linkage map and BLUP phenotypic data were used for QTL mapping (Wang et al., 2006). The mapping function cim was used. The 95% LOD values for 10 traits across 10 RIL populations were obtained through 1,000 permutations. The LOD thresholds ranged from 2.89 to 3.53, with a mean value of 3. If the genomic region had a detectable LOD value beyond 3, we defined it as one QTL, and the QTL confidence interval spanned the genomic regions corresponding to one LOD drop from the peak.
(2) JLM was performed based on the combined 10 RIL populations and the BLUP phenotypic data. We used the following formula to estimate the relationship between ROAM genotype and phenotype.
where y is the phenotype of ROAM populations with 1,887 individuals; X is the populations mean value of the phenotype; Z is an N × P matrix for the genotype of the haplotype (N is 1,887 and P = 14 is the parent number); and γ is a vector of genetic effects for the SNP of interest. The likelihood ratio test (LRT) was employed to test the phenotypic association significance of each haplotype bin. The permutation test was conducted 500 times to determine the threshold of the LRT value, and the 99.5 percentile of the LRT value was defined as the cutoff. The final threshold of LRT was 2.76 (Xiao et al., 2016).
(3) GWAS based on ultra-high-density genotyping data across 10 RIL populations was performed. High-density SNP genotypes of parents derived from RNA-seq were projected to offspring based on the recombination/haplotype bins of 1,887 lines, which generated the ultra-high-density genotyping data for subsequent GWAS with plant architecture data. The main step of this method was stepwise regression. Briefly, a subsample composed of 80% of lines randomly selected from the whole ROAM population without replacement was used for GWAS scans, and the process was repeated 100 times. Resample model inclusion probability was used to evaluate the robustness of the SNPs included in the model. The resample model inclusion P cutoff was defined as 0.05.
Detailed descriptions of all three methods can be found in a previous study (Xiao et al., 2016).
Analysis of Epistasis
Based on all significant QTLs or loci obtained as described above by SLM, JLM, and GWAS, we extracted loci with the peak value LOD score for each QTL. For the single locus, we performed epistatic analysis for every pair of peak loci using two-way ANOVA in the R environment. P < 0.05 was adjusted by the total number of tests for the detection of significant epistatic interaction. Combined with the genotypic information of all significant single loci and two-locus interactions, we used the lm of the R language to estimate their contributions to the phenotypic variation (Yu et al., 1997).
Estimation of Correlations between Parental Variation and Progeny Diversity
To understand the source of genetic diversity of plant architecture traits, we conducted a comprehensive correlation analysis of PPV, PGV, STV, QN, and QPV for all 10 plant architecture traits across 10 maize RIL populations. PPVs of all 10 aboveground traits were the absolute difference between two parents of a population. The formula used was:
where P1 is the one parent phenotypic value, P2 is the other parent phenotypic value, and P1 and P2 were the male and female parents of the RIL population. PGVs of all pairs of parents were calculated based on genetic diversity estimated with high-density markers. We evaluated the genetic relationship between the pairs of parents based on the high-density markers generated by our laboratory previously (Yang et al., 2014). QN was obtained based on the results of the SLM method (Supplemental Table S3). QPV was calculated by integrating all significant QTLs of SLM together with the lm function to represent the proportion of phenotypic variation that a QTL could explain for a specific trait. In order to make all different variables comparable, we adjusted each variable using this formula:
to compare the relationship for PPV, PGV, STV, QN, and QPV, where X is the original value of PPV, PGV, STV, QN, and QPV, Y is the adjusted value for comparison, and mean is the average of X. Furthermore, for the 10 RIL populations, the QN and phenotypic effect to which male and female parents contributed positively also were summarized based on the SLM results. All statistical analyses of this section were performed using R software (https://www.r-project.org/).
The Dissection of QTL Overlap of Plant Architecture Traits
In order to detect pleiotropic QTLs, two methods were used. (1) Using SLM, we compared the QTL confidence intervals to calculate the number of overlapping QTLs for different traits. (2) We split the genome into sliding 2-Mb bins, and if a specific genomic bin had at least two SLM QTL peaks for different traits, it was defined as a pleiotropic locus.
To test the pleiotropic effect between different traits, we used the following formula based on SLM QTL information to estimate the relationships (Li et al., 2016):
 |
where n is the number of QTLs, which is calculated as the total physical genome length divided by the average QTL interval (in this study, the average QTL confidence interval was 2 Mb, so n was equal to 1,031); m is the number of overlapping QTLs between two traits of method (2) in this section; l is the QN for the trait with more detectable QTLs; and s is the QN for the trait with fewer detectable QTLs by SLM.
To systematically dissect the genetic overlap among different plant architecture traits, a phylogenetic tree of plant architecture traits was constructed based on the converted distances of P values described above. Briefly, we used the R packages ape and phangorn to do the phylogenetic tree analysis. The nj function was used to reestimate the genetic relationships by the neighbor-joining method.
Construction of a Phenotypic Clustering Tree of All Maize Plant Architecture Traits
For all 1,887 progeny families of 10 phenotypic traits, we first used the following formula to adjust the phenotypic ranges of different traits to make the different traits comparable:
where X is original phenotypic value of 10 traits and Y is the transformed value, which was used for further analysis. The correlation analyses of 10 plant architecture traits were implemented based on the transformed Y values. Second, we used the dist function of the euclidean method in the R environment with transformed phenotypic values to calculate their distance. The Euclidean distance was calculated between all pairs of traits. The formula used is:
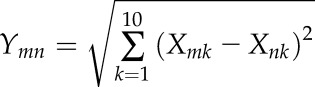 |
Where Ymn is the Euclidean distance value between traits m and n (where m and n are traits selected from the 10 measured traits) and X is the transformed phenotypic value. Based on the phenotypic distance of all pairs of 10 traits, we used the hclust function to construct the hierarchical cluster, which we called a phenotypic clustering tree relationship. Furthermore, in order to dissect the genetic basis that underlies the phenotypic clustering tree relationship of all 10 plant architecture traits, we analyzed the relationships between phenotypic distances of all 10 plant architecture traits, the number of pleiotropic QTLs (see above), and the effect of pleiotropic QTLs (the proportion of phenotypic variation that pleiotropic QTLs could explain). The effect of pleiotropic QTLs was calculated as pleiotropic QTLs divided by the total QTL effect value. Based on phenotypic distance, pleiotropic QTLs, and pleiotropic QTL effect, we used the lm function of R to estimate the relationship using the following formula:
where Y is the phenotypic distance, X is the pleiotropic QTLs or pleiotropic QTL effect, and a and b are the evaluation parameters. For the regression formula, the regression coefficient and P value also were calculated.
QTL Validation Using RHLs
Five QTLs were selected arbitrarily to verify the mapping results. They were qPH3 on chr3:161959511-166378535 affecting PH in the DE3×BY815 populations, qTMAL3 on chr3:172477014-176570244 affecting TMAL in the DE3×BY815 populations, qPH1 on chr1:98297572-116455713 affecting PH in the SK×ZHENG58 populations, qPH7 on chr7:126535776-130704819 affecting PH in the K22×BY815 populations, and qTBN3 on chr3:173803352-178006685 affecting TBN in the K22×BY815 populations. Based on the high-density linkage map, we identified the RHLs from the database (Liu et al., 2016) and planted them in Wuhan, China, in 2015. KASP markers were designed and employed for the genotyping of RHLs. We identified five lines homozygous for each parental allele in the region of the QTL of interest. Accurate phenotyping was performed in each RHL family. Phenotypic differences between the lines homozygous for different alleles at the target QTL regions were calculated and compared using ANOVA. QTLs were validated preliminarily if phenotypic variation between two different homozygous genotypes of the target QTL region was significant (P ≤ 0.05). Primer information is provided in Supplemental Table S10.
Fine-Mapping of qPH3 and Candidate Gene Mining Using Omics
A major QTL affecting PH, qPH3, with R2 > 15% was identified on chromosome 3 in the DE3/BY815 population. RHLs were selected to fine-map this QTL. First, we developed new markers to validate the QTL’s existence as described above. Then, more KASP markers were developed, and additional RHLs with recombination crossovers in the region were identified and tested in 2015 and 2016. This narrowed down the target genomic region to 600 kb. RNA-seq was conducted on the seedling leaves of RHLs with and without 600-kb genomic substitutions. RNA-seq reads were mapped on the maize reference genome AGPv2 (www.maizesequence.org; Schnable et al., 2009) using Tophat 2.0 (Kim et al., 2013) with default parameters, and the reads per kilobase of exon model per million mapped reads of all detectable genes in RHLs with and without 600-kb target genomic substitutions were calculated using Cufflinks (Trapnell et al., 2010). Differentially expressed genes were identified between RHLs with and without 600-kb target genomic substitutions. Additionally, association mapping of the target genomic region of qPH3 was performed using 1.03 million SNPs genotyped in a diverse Chinese association mapping panel with 505 inbred lines (Yang et al., 2010, 2011, 2014; Li et al., 2013a). Furthermore, the haplotypes of all candidate genes in the 600-kb genomic region in all 14 parents were analyzed and compared. Additionally, we designed primers for the amplifications of genic coding sequence regions, 5′ 2 kb upstream and 3′ 1 kb downstream, of all 16 candidate genes in all 14 parents of the ROAM population. The PCR products were subjected to Sanger sequencing for genomic variant identification followed by haplotype analysis for the determination of causal gene mining. Taking account of the results from fine-mapping, RNA-seq analysis, candidate association mapping, and haplotype comparison together, we proposed the candidate genes. Primer information of fine-mapping is provided in Supplemental Table S11.
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Genetic diversity of 14 parents of our ROAM population.
Supplemental Figure S2. Phenotypic variation of 10 plant architecture traits across 10 RIL populations in maize.
Supplemental Figure S3. Overview of QTL results for PH in maize.
Supplemental Figure S4. Overview of QTL results for EH in maize.
Supplemental Figure S5. Overview of QTL results for TMAL in maize.
Supplemental Figure S6. Overview of QTL results for TBN in maize.
Supplemental Figure S7. Overview of QTL results for LNAE in maize.
Supplemental Figure S8. Overview of QTL results for LNBE in maize.
Supplemental Figure S9. Overview of QTL results for LN in maize.
Supplemental Figure S10. Overview of QTL results for LL in maize.
Supplemental Figure S11. Overview of QTL results for LW in maize.
Supplemental Figure S12. Overview of QTL results for MLA in maize.
Supplemental Figure S13. Distribution of QTL effects (R2) for all 10 plant architecture traits by the SLM method.
Supplemental Figure S14. Number of QTLs with R2 > 10% for all 10 plant architecture traits in maize.
Supplemental Figure S15. Comparison of mapping resolution for SLM, JLM, and GWAS methods.
Supplemental Figure S16. Phenotypic variance explained by all significant GWAS SNPs.
Supplemental Figure S17. Number of consistent QTLs identified by SLM, JLM, and GWAS methods.
Supplemental Figure S18. Relationships between PPV, PGV, progeny phenotypic average value variation, STV, QN, and QPV.
Supplemental Figure S19. Distribution of pleiotropic QTLs.
Supplemental Figure S20. Phylogenetic tree of different plant architecture traits based on the QTL mapping results.
Supplemental Table S1. Phenotypic variation of 10 plant architecture traits across 10 RIL populations in maize.
Supplemental Table S2. Effects of environment and genotype by environment in the phenotypic variation of plant architecture traits in maize.
Supplemental Table S3. Correlations between 10 plant architecture traits of the ROAM population in maize.
Supplemental Table S4. Detailed QTL information identified by single population linkage analyses for all 10 plant architecture traits.
Supplemental Table S5. Detailed QTL information identified by joint population linkage analyses for all 10 plant architecture traits.
Supplemental Table S6. Significant SNPs identified by genome-wide association analyses for all 10 plant architecture traits.
Supplemental Table S7. Coincidence between QTLs and well-known functional genes.
Supplemental Table S8. Detailed polymorphism information of three candidate genes, GRMZM2G058250, GRMZM2G177227, and GRMZM2G177220, for the fine-mapping of QTL qPH3.
Supplemental Table S9. Detailed polymorphism information of other candidate genes in the 600-kb region for the fine-mapping of QTL qPH3.
Supplemental Table S10. Detailed maker information for the validation of five QTLs in RHLs.
Supplemental Table S11. Detailed marker information for the fine-mapping of QTL qPH3.
Acknowledgments
We thank all the students and colleagues from J.Y.’s present and former laboratories at China Agricultural University and from X.Y.’s and J.L.’s laboratories at China Agricultural University for help with the phenotype survey.
Glossary
- PH
plant height
- EH
ear height
- TBN
tassel branch number
- TMAL
tassel main axis length
- LL
leaf length
- LW
leaf width
- MLA
middle leaf angle
- LNAE
leaf number above ear
- LNBE
leaf number blow ear
- LN
ear leaf number
- SAM
shoot apical meristem
- QTL
quantitative trait locus
- NAM
nested association mapping
- RIL
recombinant inbred line
- ROAM
random-open-parent association mapping
- RHL
residual heterozygous line
- BLUP
Best linear unbiased prediction
- SLM
single linkage mapping
- JLM
joint linkage mapping
- GWAS
genome-wide association study
- SNP
single-nucleotide polymorphism
- KASP
competitive allele-specific PCR
- LOD
log of the odds
- LRT
likelihood ratio test
- PPV
parental phenotypic variation
- PPV
parental phenotypic variation
- PGV
parental genomic variation
- STV
sd of phenotypic variation in the progeny
- QN
quantitative trait locus number
- QPV
quantitative trait locus effect
Footnotes
This research was supported by the National Natural Science Foundation of China (31501315 and 91635303), the National Key Research and Development Program of China (2016YFD0101003 and 2016YFD0100404), and Huazhong Agricultural University Scientific & Technological Self-Innovation Foundation (2015RC016).
Articles can be viewed without a subscription.
References
- Acosta IF, Laparra H, Romero SP, Schmelz E, Hamberg M, Mottinger JP, Moreno MA, Dellaporta SL (2009) tasselseed1 is a lipoxygenase affecting jasmonic acid signaling in sex determination of maize. Science 323: 262–265 [DOI] [PubMed] [Google Scholar]
- Andrade FH, Uhart SA, Frugone MI (1993) Intercepted radiation at flowering and kernel number in maize: shade versus plant density effects. Crop Sci 33: 482–485 [Google Scholar]
- Barton MK. (2010) Twenty years on: the inner workings of the shoot apical meristem, a developmental dynamo. Dev Biol 341: 95–113 [DOI] [PubMed] [Google Scholar]
- Baute J, Herman D, Coppens F, De Block J, Slabbinck B, Dell’Acqua M, Pè ME, Maere S, Nelissen H, Inzé D (2015) Correlation analysis of the transcriptome of growing leaves with mature leaf parameters in a maize RIL population. Genome Biol 16: 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becraft PW, Bongard-Pierce DK, Sylvester AW, Poethig RS, Freeling M (1990) The liguleless-1 gene acts tissue specifically in maize leaf development. Dev Biol 141: 220–232 [DOI] [PubMed] [Google Scholar]
- Begna HS, Hamilton RI, Dwyer LM, Stewart DW, Smith DL (1999) Effects of population density on the vegetative growth of leafy reduced-stature maize in short-season areas. J Agron Crop Sci 182: 49–55 [Google Scholar]
- Bellmann R, Werr W (1992) Zmhox1a, the product of a novel maize homeobox gene, interacts with the Shrunken 26 bp feedback control element. EMBO J 11: 3367–3374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomblies K, Wang RL, Ambrose BA, Schmidt RJ, Meeley RB, Doebley J (2003) Duplicate FLORICAULA/LEAFY homologs zfl1 and zfl2 control inflorescence architecture and flower patterning in maize. Development 130: 2385–2395 [DOI] [PubMed] [Google Scholar]
- Bouchet S, Bertin P, Presterl T, Jamin P, Coubriche D, Gouesnard B, Laborde J, Charcosset A (2017) Association mapping for phenology and plant architecture in maize shows higher power for developmental traits compared with growth influenced traits. Heredity (Edinb) 118: 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler ES, Holland JB, Bradbury PJ, Acharya CB, Brown PJ, Browne C, Ersoz E, Flint-Garcia S, Garcia A, Glaubitz JC, et al. (2009) The genetic architecture of maize flowering time. Science 325: 714–718 [DOI] [PubMed] [Google Scholar]
- Doebley J, Stec A, Gustus C (1995) teosinte branched1 and the origin of maize: evidence for epistasis and the evolution of dominance. Genetics 141: 333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doebley J, Stec A, Hubbard L (1997) The evolution of apical dominance in maize. Nature 386: 485–488 [DOI] [PubMed] [Google Scholar]
- Duvick DN. (2005) Genetic progress in yield of United States maize (Zea mays L.). Maydica 50: 193–202 [Google Scholar]
- Fischer K, Edmeades G, Johnson E (1987) Recurrent selection for reduced tassel branch number and reduced leaf area density above the ear in tropical maize populations. Crop Sci 27: 1150–1156 [Google Scholar]
- Fraser HB, Schadt EE (2010) The quantitative genetics of phenotypic robustness. PLoS ONE 5: e8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu J, Cheng Y, Linghu J, Yang X, Kang L, Zhang Z, Zhang J, He C, Du X, Peng Z, et al. (2013) RNA sequencing reveals the complex regulatory network in the maize kernel. Nat Commun 4: 2832. [DOI] [PubMed] [Google Scholar]
- Hartwig T, Chuck GS, Fujioka S, Klempien A, Weizbauer R, Potluri DP, Choe S, Johal GS, Schulz B (2011) Brassinosteroid control of sex determination in maize. Proc Natl Acad Sci USA 108: 19814–19819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter RB, Daynard TB, Hume DJ, Tanner JW, Curtis JD, Kannenberg LW (1969) Effect of tassel removal on grain yield of corn (Zea mays L.). Crop Sci 9: 405–406 [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawit SJ, Wych HM, Xu D, Kundu S, Tomes DT (2010) Maize DELLA proteins dwarf plant8 and dwarf plant9 as modulators of plant development. Plant Cell Physiol 51: 1854–1868 [DOI] [PubMed] [Google Scholar]
- Li D, Wang X, Zhang X, Chen Q, Xu G, Xu D, Wang C, Liang Y, Wu L, Huang C, et al. (2016) The genetic architecture of leaf number and its genetic relationship to flowering time in maize. New Phytol 210: 256–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Peng Z, Yang X, Wang W, Fu J, Wang J, Han Y, Chai Y, Guo T, Yang N, et al. (2013a) Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels. Nat Genet 45: 43–50 [DOI] [PubMed] [Google Scholar]
- Li L, Petsch K, Shimizu R, Liu S, Xu WW, Ying K, Yu J, Scanlon MJ, Schnable PS, Timmermans MC, et al. (2013b) Mendelian and non-Mendelian regulation of gene expression in maize. PLoS Genet 9: e1003202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Wang F, Xiao Y, Tian Z, Wen W, Zhang X, Chen X, Liu N, Li W, Liu L, et al. (2016) MODEM: multi-omics data envelopment and mining in maize. Database (Oxford) 2016: baw117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullen MD, Kresovich S, Villeda HS, Bradbury P, Li H, Sun Q, Flint-Garcia S, Thornsberry J, Acharya C, Bottoms C, et al. (2009) Genetic properties of the maize nested association mapping population. Science 325: 737–740 [DOI] [PubMed] [Google Scholar]
- Moon J, Candela H, Hake S (2013) The Liguleless narrow mutation affects proximal-distal signaling and leaf growth. Development 140: 405–412 [DOI] [PubMed] [Google Scholar]
- Moreno MA, Harper LC, Krueger RW, Dellaporta SL, Freeling M (1997) liguleless1 encodes a nuclear-localized protein required for induction of ligules and auricles during maize leaf organogenesis. Genes Dev 11: 616–628 [DOI] [PubMed] [Google Scholar]
- Multani DS, Briggs SP, Chamberlin MA, Blakeslee JJ, Murphy AS, Johal GS (2003) Loss of an MDR transporter in compact stalks of maize br2 and sorghum dw3 mutants. Science 302: 81–84 [DOI] [PubMed] [Google Scholar]
- Pan Q, Li L, Yang X, Tong H, Xu S, Li Z, Li W, Muehlbauer GJ, Li J, Yan J (2016) Genome-wide recombination dynamics are associated with phenotypic variation in maize. New Phytol 210: 1083–1094 [DOI] [PubMed] [Google Scholar]
- Peiffer JA, Romay MC, Gore MA, Flint-Garcia SA, Zhang Z, Millard MJ, Gardner CA, McMullen MD, Holland JB, Bradbury PJ, et al. (2014) The genetic architecture of maize height. Genetics 196: 1337–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepper GE, Pearce RB, Mock JJ (1977) Leaf orientation and yield of maize. Crop Sci 17: 883–886 [Google Scholar]
- Riechmann JL, Heard J, Martin G, Reuber L, Jiang C, Keddie J, Adam L, Pineda O, Ratcliffe OJ, Samaha RR, et al. (2000) Arabidopsis transcription factors: genome-wide comparative analysis among eukaryotes. Science 290: 2105–2110 [DOI] [PubMed] [Google Scholar]
- Russell W. (1991) Genetic improvement of maize yields. Adv Agron 46: 245–298 [Google Scholar]
- Schnable JC, Freeling M (2011) Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS ONE 6: e17855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115 [DOI] [PubMed] [Google Scholar]
- Schneeberger RG, Becraft PW, Hake S, Freeling M (1995) Ectopic expression of the knox homeo box gene rough sheath1 alters cell fate in the maize leaf. Genes Dev 9: 2292–2304 [DOI] [PubMed] [Google Scholar]
- Sheridan WF. (1988) Maize developmental genetics: genes of morphogenesis. Annu Rev Genet 22: 353–385 [DOI] [PubMed] [Google Scholar]
- Sylvester AW, Cande WZ, Freeling M (1990) Division and differentiation during normal and liguleless-1 maize leaf development. Development 110: 985–1000 [DOI] [PubMed] [Google Scholar]
- Theodoris G, Inada N, Freeling M (2003) Conservation and molecular dissection of ROUGH SHEATH2 and ASYMMETRIC LEAVES1 function in leaf development. Proc Natl Acad Sci USA 100: 6837–6842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson AM, Yu J, Timmermans MC, Schnable P, Crants JC, Scanlon MJ, Muehlbauer GJ (2015) Diversity of maize shoot apical meristem architecture and its relationship to plant morphology. G3 (Bethesda) 5: 819–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Bradbury PJ, Brown PJ, Hung H, Sun Q, Flint-Garcia S, Rocheford TR, McMullen MD, Holland JB, Buckler ES (2011) Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat Genet 43: 159–162 [DOI] [PubMed] [Google Scholar]
- Timmermans MC, Hudson A, Becraft PW, Nelson T (1999) ROUGH SHEATH2: a Myb protein that represses knox homeobox genes in maize lateral organ primordia. Science 284: 151–153 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L (2010) Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollbrecht E, Springer PS, Goh L, Buckler ES IV, Martienssen R (2005) Architecture of floral branch systems in maize and related grasses. Nature 436: 1119–1126 [DOI] [PubMed] [Google Scholar]
- Wallace JG, Bradbury PJ, Zhang N, Gibon Y, Stitt M, Buckler ES (2014) Association mapping across numerous traits reveals patterns of functional variation in maize. PLoS Genet 10: e1004845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Nussbaum-Wagler T, Li B, Zhao Q, Vigouroux Y, Faller M, Bomblies K, Lukens L, Doebley JF (2005) The origin of the naked grains of maize. Nature 436: 714–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Basten CJ, Zeng Z (2006) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC [Google Scholar]
- Watanabe S, Xia Z, Hideshima R, Tsubokura Y, Sato S, Yamanaka N, Takahashi R, Anai T, Tabata S, Kitamura K, et al. (2011) A map-based cloning strategy employing a residual heterozygous line reveals that the GIGANTEA gene is involved in soybean maturity and flowering. Genetics 188: 395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel D, Alvarez J, Smyth DR, Yanofsky MF, Meyerowitz EM (1992) LEAFY controls floral meristem identity in Arabidopsis. Cell 69: 843–859 [DOI] [PubMed] [Google Scholar]
- Winkler RG, Helentjaris T (1995) The maize Dwarf3 gene encodes a cytochrome P450-mediated early step in gibberellin biosynthesis. Plant Cell 7: 1307–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Liu H, Wu L, Warburton M, Yan J (2017) Genome-wide association studies in maize: praise and stargaze. Mol Plant 10: 359–374 [DOI] [PubMed] [Google Scholar]
- Xiao Y, Tong H, Yang X, Xu S, Pan Q, Qiao F, Raihan MS, Luo Y, Liu H, Zhang X, et al. (2016) Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol 210: 1095–1106 [DOI] [PubMed] [Google Scholar]
- Xing A, Gao Y, Ye L, Zhang W, Cai L, Ching A, Llaca V, Johnson B, Liu L, Yang X, et al. (2015) A rare SNP mutation in Brachytic2 moderately reduces plant height and increases yield potential in maize. J Exp Bot 66: 3791–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka N, Ninomiya S, Hoshi M, Tsubokura Y, Yano M, Nagamura Y, Sasaki T, Harada K (2001) An informative linkage map of soybean reveals QTLs for flowering time, leaflet morphology and regions of segregation distortion. DNA Res 8: 61–72 [DOI] [PubMed] [Google Scholar]
- Yang N, Lu Y, Yang X, Huang J, Zhou Y, Ali F, Wen W, Liu J, Li J, Yan J (2014) Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet 10: e1004573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Gao S, Xu S, Zhang Z, Prasanna B, Li L, Li JS, Yan J (2011) Characterization of a global germplasm collection and its potential utilization for analysis of complex quantitative traits in maize. Mol Breed 28: 511–526 [Google Scholar]
- Yang X, Yan J, Shah T, Warburton ML, Li Q, Li L, Gao Y, Chai Y, Fu Z, Zhou Y, et al. (2010) Genetic analysis and characterization of a new maize association mapping panel for quantitative trait loci dissection. Theor Appl Genet 121: 417–431 [DOI] [PubMed] [Google Scholar]
- Yu SB, Li JX, Xu CG, Tan YF, Gao YJ, Li XH, Zhang Q, Saghai Maroof MA (1997) Importance of epistasis as the genetic basis of heterosis in an eliterice hybrid. Proc Natl Acad Sci USA 94: 9226–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XS. (2008) Increase in quantitative variation after exposure to environmental stresses and/or introduction of a major mutation: G x E interaction and epistasis or canalization? Genetics 180: 687–695 [DOI] [PMC free article] [PubMed] [Google Scholar]