

Abstract
Objective
We previously demonstrated that diabetes exacerbates stroke-induced brain injury, and that this correlates with brain methylglyoxal (MG)-to-glutathione (GSH) status. Cerebral injury was reversed by N-acetylcysteine (NAC). Here we tested if the pro-thrombotic phenotype seen in the systemic circulation and brain during diabetes was associated with increased MG-glycation of proteins, and if NAC could reverse this.
Methods
The streptozotocin (STZ)-induced mouse model of type 1 diabetes was used. Thrombus formation in venules and arterioles (pial circulation) was determined by intravital videomicroscopy using the light-dye method. Circulating blood platelet-leukocyte aggregates (PLAs) were analyzed by flow cytometry 1 wk before other measurements. GSH and MG levels in platelets were measured by HPLC. MG-modified proteins, glutathione peroxidase-1 (GPx-1), and superoxide dismutase-1 (SOD1) levels were detected in platelets by western blot at 20 weeks. Proteins involved in coagulation were quantified by ELISA. NAC (2 mM) was given in drinking water for 3 weeks before the terminal experiment.
Results
Thrombus development was accelerated by diabetes in a time-dependent manner. % PLAs were significantly elevated by diabetes. Plasma activated plasminogen activator inhibitor type 1 levels were progressively increased with diabetes duration, with tail bleeding time reduced by 20 wks diabetes. Diabetes lowered platelet GSH levels, GPx-1 and SOD-1 expression. This was associated with higher MG levels, and increased MG-adduct formation in platelets. NAC treatment partly or completely reversed the effects of diabetes.
Conclusion
Collectively, these results show that the diabetic blood and brain become progressively more susceptible to platelet activation and thrombosis. NAC, given after the establishment of diabetes, may offer protection against the risk for stroke by altering both systemic and vascular prothrombotic responses via enhancing platelet GSH, and GSH-dependent MG elimination, as well as correcting levels of antioxidants such as SOD1 and GPx-1.
Abbreviations: ACD, citrate-dextrose solution; APC, activated protein C; ATIII, antithrombin III; FITC, fluorescein isothiocyanate; Glo, glyoxalase; GPx-1, glutathione peroxidase-1; GSH, glutathione; GSSG, glutathione disulfide; MG, methylglyoxal; NAC, N-acetylcysteine; PAI-1, activated plasminogen activator inhibitor type 1; PE, phycoerythrin; PLAs, platelet-leukocyte aggregates; Pl-Lymph, platelet-lymphocyte aggregates; Pl-mono, platelet-monocyte aggregates; Pl-neut, platelet-neutrophil aggregates; SOD1, superoxide dismutase-1; STZ, streptozotocin; TAT, thrombin-antithrombin; TIA, transient ischemic attack; tPA, tissue plasminogen activator; Veh, vehicle
Keywords: N-acetylcysteine, Diabetes, Thrombosis, Glutathione, Methylglyoxal
Graphical abstract
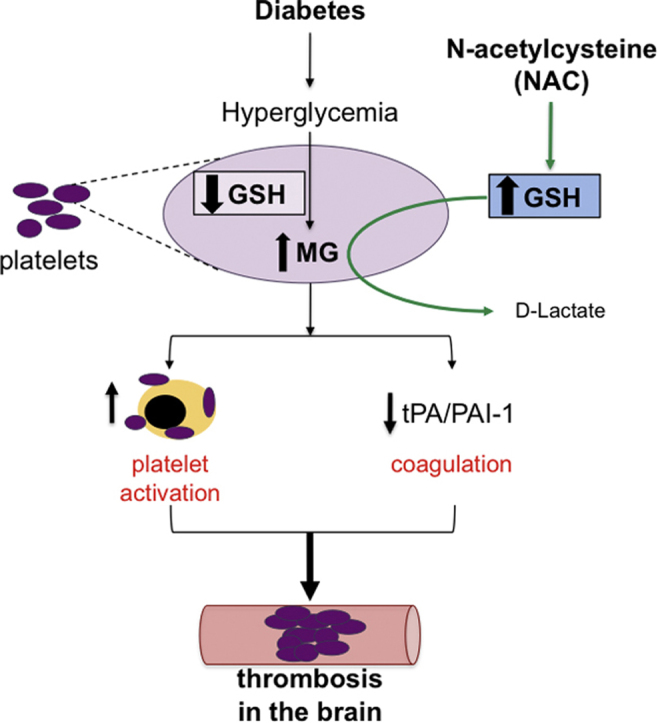
Highlights
-
•
Diabetes elevates dicarbonyl stress leading to enhanced thrombosis in the brain.
-
•
Glutathione levels decrease leading to impaired elimination of methylglyoxal in platelets during diabetes.
-
•
Platelet proteins are glycated and platelets form aggregates with leukocytes in diabetes.
-
•
Diabetes increases circulating levels of plasminogen activator inhibitor-1.
-
•
NAC, via GSH synthesis, reverses the platelet activation, protein glycation and pro-coagulation responses & protects against thrombosis in the diabetic brain.
1. Introduction
Diabetes affects 1 in 10 US adults and is associated with reduced longevity [1]. One of the reasons for this is that diabetes leads to a higher risk for stroke, transient ischemic attack (TIA), and cerebral small vessel disease. Stroke is a leading cause of mortality and disability worldwide [2], with approximately 795,000 people suffering a stroke each year in the US. In 2013, stroke was the second-leading global cause of death, and accounted for approximately 1 in 20 deaths in the US [1]. Importantly, diabetic stroke patients have a 3-fold higher mortality than their non-diabetic counterparts [3]. Ischemic stroke (thrombotic and embolic) accounts for approximately 85% of all strokes [1] and diabetic patients are at increased risk (3.35-fold greater) for both types of ischemic stroke [4]. Activation of platelets and the coagulation system are key events in the thrombotic event leading to ischemic stroke. The heightened platelet activation and hypercoagulable state found in the blood of diabetic patients suggests these systemic changes are at least in part involved in the enhanced stroke risk in this patient population. Furthermore, in thrombotic strokes, the thrombus can form in large or small arteries within the brain [5], indicating a prothrombotic environment in the brain vessels that may be exacerbated by diabetes.
Plasma glycemic levels serve as a predictor of increased vascular disease risk in diabetes, with stroke risk being exaggerated in diabetics who have poor glycemic control. However, an increased risk still remains, even in diabetic individuals with well-controlled blood glucose levels. This suggests the involvement of other factors. One candidate is the dicarbonyl metabolite of α-oxoaldehydes, methylglyoxal (MG) [6]. Because of its extremely high reactivity, and relatively high flux of formation, MG is the precursor of the most quantitatively and functionally significant advanced glycation endproducts [7]. MG levels are elevated in diabetes [8], [9], and have been implicated in diabetic complications. While there is a report showing that MG may inhibit platelet aggregation induced by several agonists, and ATP release induced by thrombin [10], most of the limited data available support the concept that MG is important in thrombosis during diabetes. In vitro incubation of platelets with MG has been shown to activate them, and promote platelet aggregation [11]. MG also promotes platelet-leukocyte aggregate formation, indicative of crosstalk between these two cell types, which leads to mutual activation of each other [12]. Although limited, there is evidence that MG can impair the activity of anticoagulants such as antithrombin III and plasminogen [13]. Furthermore, injection of MG into mice led to acceleration of ferric chloride-induced thrombosis in the carotid artery 30 min later [11]. While all of these studies focused on the acute effects of MG, the evidence nonetheless points to a possible role for MG in the enhanced risk for stroke in diabetic individuals, and the potential benefit for targeting MG therapeutically in diabetic individuals.
Glutathione (GSH) is the rate-limiting co-factor in the glyoxalase (Glo) elimination pathway for MG. GSH levels are decreased in diabetes, and there is some evidence that Glo enzymes may also be decreased/impaired [14], [15]. Thus, not only is more MG generated as part of glucose metabolism, but also the MG is not eliminated as efficiently. This creates a greater potential for glycation of proteins. One way to restore GSH levels is to provide N-acetylcysteine (NAC), a precursor of GSH synthesis. Within 2 h of treatment, NAC reduces platelet-monocyte aggregate formation and raises intra-platelet GSH levels in diabetics with depleted GSH [16]. Because GSH is also a major cellular antioxidant, the protection afforded by GSH or NAC in many studies has been attributed to the antioxidant capacity of the NAC-generated GSH. However, we have previously found that GSH protection against MG-induced brain endothelial barrier disruption [17] was due to GSH acting as a rate-limiting cofactor in MG metabolism [18], [19]. We went on to show that in a more chronic in vivo model, 3 wks NAC treatment of diabetic mice not only decreased brain injury following stroke, but also decreased the MG-to-GSH ratio in the brain and prevented the glycation of proteins, in particular the junctional protein occludin, in the brain [9]. Therefore, in this study we tested whether NAC could prevent the platelet activation, pro-coagulation and exacerbated thrombosis responses induced by diabetes, and if this is associated with correction of platelet GSH, and attenuation of diabetes-enhanced platelet MG levels and glycated protein content.
2. Materials and methods
The following reagents were purchased from Sigma: methylglyoxal, N-acetyl-cysteine, fluorescein isothiocyanate dextran (FITC-dextran; 150,000 molecular weight), citrate-dextrose solution (ACD) and 4% citrate solution. ELISAs were from Molecular Innovations Company (activated plasminogen activator inhibitor type 1 (PAI-1)), LifeSpan BioSciences company (activated protein C (APC) and tissue plasminogen activator (tPA)), and Abcam (thrombin-antithrombin (TAT)). The following antibodies were used: Rabbit anti-glutathione peroxidase-1 (GPx-1), rabbit anti-superoxide dismutase-1 (SOD1), HRP-conjugated goat-anti-rabbit IgG (Abcam), mouse anti-actin antibody, phycoerythrin (PE) anti-mouse Ly6G (BD company), HRP-conjugated sheep-anti-mouse (Amersham), fluorescein isothiocyanate (FITC) anti-mouse CD45.2, allophycocyanin anti-mouse CD41, pacific blue anti-mouse F4/80 (eBiosciences). ECL reagent was from BIO-RAD company. Fixative-free lysing solution, High yield lyse, was purchased from Invitrogen.
2.1. Animal preparation
Four-week-old male C57BL/6J mice, weighing 18–20 g, were purchased from Jackson Laboratory (Bar Habor, ME). All animal procedures were approved by the LSUHSC-S Institutional Animal Care and Use Committee and were in accordance with the US NIH guidelines in the Guide for the Care and Use of Laboratory Animals. The mice were divided randomly into: vehicle (Veh) 6 wk group, Veh 20 wk group, streptozotocin (STZ) 6wk group and STZ 20wk group. Experimental diabetes was achieved in STZ groups by injecting 50 mg/kg STZ for 5 consecutive days. Veh mice were injected with citrate buffer for 5 consecutive days and served as vehicle controls. On day 7, the mice whose plasma glucose was more than 300 mg/dl were deemed to be diabetic. Diabetic mice were further divided into two subgroups for treatment: 1) water ad libitum; 2) water ad libitum until 3 wks before the terminal experiment, at which time they were switched to 2 mmol/l of NAC in drinking water. This NAC dose took into account differences in rodent and human metabolism, and was chosen to give within the mouse equivalent dose range (326–570 mg/kg/day) for the human dose of 20–30 mg/kg/day. Based on body weight and water consumption our diabetic mice received 400 ± 70 mg/kg/day. Two endpoints, 6 wks and 20 wks, were used at which to measure tail bleed time and perform intravital videomicroscopy for observation of thrombosis, or to harvest blood for platelet isolation or plasma. 1 week before the experimental endpoint, blood was sampled from the tail to measure platelet-leukocyte aggregates (PLAs) by flow cytometry (denoted by 5wk or 19wk groups respectively in figures).
2.2. Flow cytometric analysis of PLAs
Blood from a tail clip was drawn into a heparinized capillary tube and incubated with Fc blocking antibody for 10 min at RT. The blood was then diluted 1:5 with phosphate buffered saline (PBS, PH 7.4) and incubated with anti-mouse CD45.2 FITC (1:200) to identify all leukocytes, anti-mouse F4/80 Pacific Blue (1:200) to identify monocytes, anti-mouse Ly6G PE (1:600) to label neutrophils, anti-mouse CD41 allophycocyanin (1:300) to identify platelets, or appropriate isotype controls for 45 min at RT in the dark. This was followed by red blood cell lysis, centrifugation at 300 g for 5 min, and a wash step using PBS before being resuspended in PBS for analysis by flow cytometry (n = 7–10/group). Total PLAs were expressed as a % of CD45.2+ cells, and separate leukocyte subpopulations forming aggregates were expressed as a % of CD45.2+ CD41+ positive cells.
2.3. Intravital videomicroscopy and light/dye induced thrombosis
As previously described [20], the mice were anesthetized with ketamine (125 mg/kg) and xylazine (6.25 mg/kg) IP. The jugular vein was canulated, and a craniectomy (3 mm diameter; 1 mm posterior, 4 mm lateral from the bregma) was performed on the opposite side and covered with a cover-glass. The space between cover-glass and pia mater was filled with artificial cerebrospinal fluid. The mouse was fixed on the platform of an upright fluorescent microscope (BX51WI; Olympus, Japan) and equilibrated for 20 min. 5% FITC-dextran (10 mL/kg BW) was injected into the jugular vein and allowed to circulate for 10 mins. Cerebral microvessels were visualized using a 40X water immersion objective lens (E Plan FI/IR 40×/0.65×; Olympus), and videos were recorded on DVD. FITC photoactivation (excitation, 495 nm; emission, 519 nm) was induced by exposing 100 µm vessels to epi-illumination with a xenon lamp (LB-LS/17, Novato, CA, US) coupled with a fluorescein filter cube (HQ-FITC; Chroma Technology, US). The excitation power density was calibrated daily and maintained within 1% of 0.17 W/cm2, as previously described [20]. This light/dye model of inducing thrombosis is a widely used model of thrombosis, with acceptable limitations addressed elsewhere [21], [22]. During continuous epi-illumination, thrombus formation was quantified in both venules and arterioles (diameters: 20–40 µm) by determining the time of onset of platelet deposition/aggregation on the vessel wall (onset time) and the time to complete blood flow cessation for >30 s (cessation time) (n = 6–11/group). Onset and cessation times were averaged from 2 to 3 thrombi formed in both venules and arterioles in each mouse.
2.4. Tail bleeding time
As a measure of the hemostatic function of platelets, tail bleeding time was measured by placing the anesthetized mouse on a warm pad, transecting the tail 5 mm from the tip and immediately immersing it into a 15 mL falcon tube filled with 14 mL saline (37 °C) (n = 6–8/group). The total time for cessation of bleeding was recorded, and if necessary the bleeding was stopped by cauterization at 10 mins.
2.5. Separation of platelets for assay of GSH, GSSG and MG levels
Mice were anesthetized with ketamine (125 mg/kg) and xylazine (6.25 mg/kg) IP. The carotid artery was canulated and blood was collected in citrate-dextrose solution at a ratio of 9:1. The blood was centrifuged at 1200 rpm for 8 min, and the platelet-rich plasma was further centrifuged at 1200 rpm for 3 min to remove contaminating leukocytes. The platelet-rich plasma was then centrifuged at 3000 rpm for 10 min, and the platelet pellet collected and stored at −80 °C for measuring GSH, GSSG and MG levels (n = 5–8/group), and to perform western blots (n = 5/group for MG; n = 6/group for SOD1 and GPx-1).
2.6. Quantification of GSH, GSSG and MG in platelets
Determination of GSH and GSSG was performed as previously described [23]. Trichloroacetic acid-soluble supernatants of platelets were derivatized with 6 mmol/l iodoacetic acid and 1% 2,4-dinitrophenyl fluorobenzene (pH adjusted to 7–8 and 7.0, respectively) to yield the S-carboxymethyl and 2,4-dinitrophenyl derivatives, respectively. GSH and glutathione disulfide (GSSG) derivatives were separated on a 250 × 4.6-mm Alltech Lichrosorb NH2 10 µm anion-exchange column and detected at 365 nm. The platelet GSH and GSSG contents, expressed as nmol/mg protein, were quantified by comparison to standards derivatized in the same manner.
Determination of MG was as previously described [24]. Platelet homogenates in PBS were treated with 60% perchloric acid (29:1 v/v) and the acid supernatants were derivatized with 0.1 mol/l of o-phenylenediamine (100:1 v/v) for 24 h. MG derivatives were separated on a 250 × 4.6-mm Chromegabond Ultra C-18 reversed phase column, and detected at 315 nm. Platelet MG contents, expressed as nmol/mg protein, were quantified by comparison to MG standards derivatized with o-phenylenediamine.
2.7. ELISAs for proteins involved in coagulation
Mice were anesthetized with ketamine (125 mg/kg) and xylazine (6.25 mg/kg) IP. Blood was collected via a carotid artery canula into 4% citrate concentrated solution, at a ratio of 9:1. The plasma was separated as per the ELISA manufacturers recommendations, and stored at −80 °C for analysis later (n = 6/group). ELISAs for APC, TAT, activated PAI-1 and tPA were performed according to the manufacturers instructions. All samples were performed in triplicate, and the minimal detectable concentrations were 0.05 pg/mL for PAI-1 and tPA, 78 pg/mL for APC, and 0.01 ng/mL for TAT.
2.8. Western blot assay for GPx-1, SOD1 and MG-modified protein
Platelets were homogenized with RIPA lysis buffer containing 50 mM Tris, 150 mM sodium chloride, 0.5% sodium deoxycholate, 0.1% SDS and protease inhibitor cocktail, pH 8.0. 20 μg protein per sample was loaded on 15% SDS-polyacrylamide gels. Gel electrophoresis was performed @120 V, 140 min followed by transfer onto a PVDF membrane, @100 V for 1 h. The membranes were blocked in 5% nonfat milk in TBST buffer containing 20 mM Tris, 137 mM NaCl, 0.1% Tween20, pH 7.6 for 1 h @RT. The membranes were then probed with rabbit anti-GPx-1 monoclonal antibody (1:2000), rabbit anti-SOD1 polyclonal antibody (1:2000) or mouse anti-MG polyclonal antibody (1:2000) at 4 °C overnight, followed by HRP-conjugated goat-anti-rabbit or sheep-anti-mouse secondary antibodies (1:5000) as appropriate, for 2 h at RT. Protein expression was detected using enhanced chemiluminescence (BIO-RAD) as per the manufacturer's instructions. Protein expression was normalized to β-actin.
2.9. Statistical analysis
All data are mean±SEM. The significance of difference was assessed by Student t-test (single comparisons) or by one-way ANOVA with Newman-Keuls post hoc tests (multiple comparisons). Differences were considered significant at P < 0.05.
3. Results
3.1. NAC partly reverses the accelerating effect of diabetes on thrombus development in cerebral microvessels at 6 weeks
Six weeks after inducing diabetes, the time to thrombus onset was shortened in both venules and arterioles. Time to complete cessation of blood flow in arterioles was also reduced in the STZ 6wk group compared with Veh controls. NAC significantly prolonged the onset times and the arteriolar cessation time in the diabetic mice. These data are consistent with an anti-thrombosis potential for NAC (Fig. 1).
Fig. 1.

Effect of NAC on acceleration of thrombosis during early diabetes: Thrombosis in blood vessels of the mouse brain at 6 wks after injection with vehicle (Veh), or streptozotocin to induce diabetes (STZ 6wk). One STZ 6wk group was treated with N-acetylcysteine in drinking water for 3 wks before observation (STZ 6wk +NAC). Times to thrombus onset and cessation are shown in postcapillary venules (A&B) and arterioles (C&D). * P < 0.01 vs. Veh; # P < 0.05 vs. STZ 6wk.
3.2. NAC protects against systemic increases in platelet-leukocyte aggregates and PAI-1 levels at early stage diabetes
Circulating % of blood leukocytes forming aggregates with platelets was significantly increased in STZ 6wk mice compared with the Veh group (Fig. 2A). This was primarily due to an elevation in platelet-lymphocyte aggregates (Pl-Lymph). NAC treatment for 2 weeks significantly decreased the levels of total PLAs and Pl-Lymph aggregates at 5 weeks of diabetes compared with levels in untreated diabetic mice. The number of platelets in the aggregates, as indicated by mean CD41 fluorescence per aggregate, was not different between the groups for total or individual leukocyte subpopulations (Fig. 2B).
Fig. 2.

Impact of NAC on systemic platelet and coagulation changes induced by early diabetes: Mice were injected with vehicle (Veh) or streptozotocin (STZ) to induce diabetes. A third group of mice was injected with STZ, and 3 weeks later started on NAC drinking water. 5 wks after injections, blood was drawn for flow cytometry to measure (A) the % of leukocytes and leukocyte subsets forming aggregates with platelets, and (B) CD41 (platelet marker) expression on the aggregates. One week later, at 6 wks post-injection, (C-E) blood was drawn to measure circulating plasma levels of PAI-1, tPA and the ratio of tPA/PAI-1, and (F) tail bleed time was determined. * P < 0.05 vs. Veh; # P < 0.05 vs. STZ.
Plasma activated PAI-1 levels were also elevated by diabetes at 6 wks (Fig. 2C). There were no changes observed between groups for plasma tPA levels (Fig. 2D), but the tPA/PAI-1 ratio was significantly decreased in diabetic mice (Fig. 2E). Treatment with NAC for 3 wks increased the ratio by attenuating the PAI-1 levels. There was a slight reduction in tail bleeding time in diabetic mice compared with Veh mice. This was reversed by NAC (Fig. 2F).
3.3. Thrombus formation is further accelerated at 20 weeks diabetes, and NAC protects against the thrombotic phenotype
The onset and cessation times of venules and arterioles were all reduced at 20 wks in diabetic mice compared with those of Veh mice (Fig. 3A). Diabetes duration was found to be an important factor affecting thrombus formation, in that the cessation time in venules and both the onset and cessation times in arterioles were shorter at 20 wks versus 6 wks of diabetes (Fig. 3B). Despite the further acceleration of thrombosis times at 20 wks of diabetes, just 3 weeks of NAC treatment was sufficient to reverse the faster thrombus formation in venules, and offer partial protection in the arterioles (Fig. 3A).
Fig. 3.

Outcome of NAC treatment for the acceleration of thrombosis at 5 months diabetes. (A) Thrombosis in blood vessels of the mouse brain at 20 wks after injection with vehicle (Veh), or streptozotocin to induce diabetes (STZ 20wk). One STZ 20wk group was treated with N-acetylcysteine in drinking water for 3 wks before observation (STZ 20wk +NAC). (B) Comparison of thrombosis times between STZ groups 6wk and 20wk after induction of diabetes. Times to thrombus onset and cessation are shown in postcapillary venules and arterioles in both panels. * P < 0.05 vs. Veh; # P < 0.05 vs. STZ 20wk; ^ P < 0.05 vs. STZ 6wk.
3.4. Diabetes causes activation of systemic platelets, and coagulation, and NAC affords protection
At 1 wk before the observation of thrombosis (i.e. 19 wks diabetes), the % of circulating PLAs were elevated in STZ mice versus Veh controls. At this time, not only were Pl-Lymph aggregates increased, but also platelet-monocyte (Pl-Mono) and platelet-neutrophil (Pl-Neut) aggregates were both higher than seen in controls (Fig. 4A and Supplementary Fig. 1). Although the %’s of total PLAs were comparable between the short and long duration of diabetes (Fig. 4B), the type of aggregates that formed differed between the two diabetic time points. Furthermore, the CD41 mean fluorescence intensity per aggregate was significantly increased at 19 wks diabetes, both versus corresponding controls (Fig. 4C) and compared with the 5 wks diabetic mice (Fig. 4D). This suggests that, on average, the aggregates at 19wks diabetes contained more platelets. The higher CD41 expression was observed in all aggregate subtypes. NAC attenuated Pl-Neut and Pl-Mono aggregates back to non-diabetic levels, and partly decreased Pl-Lymph aggregates (Fig. 4A). NAC treatment also reduced the CD41 expression in each aggregate subtype to control levels (Fig. 4C).
Fig. 4.

Effect of NAC on systemic platelet activation and coagulation during diabetes: Mice were injected with vehicle (Veh) or streptozotocin (STZ) to induce diabetes. A third group of mice was injected with STZ, and 3 or 17 wks later started on NAC drinking water. At 2 wks of NAC, blood was drawn for flow cytometry to measure (A-B) the % of leukocytes and leukocyte subsets forming aggregates with platelets, and (C-D) CD41 (platelet marker) expression on the aggregates. One week later, at 6 or 20 wks post-injection, (E-I) blood was drawn to measure circulating plasma levels of PAI-1, tPA and the ratio of tPA/PAI-1, and (J-K) tail bleed time was determined. * P < 0.05 vs. Veh; # P < 0.05 vs. STZ; ^ P < 0.05 vs. STZ 6wk.
Plasma activated PAI-1 levels were elevated in STZ 20wk mice compared with corresponding Veh controls (Fig. 4E). NAC alleviated this response by approximately one third. The tPA levels were only slightly decreased by diabetes (Fig. 4F), therefore the observed drop in the ratio of tPA/PAI-1 in STZ 20wk mice was primarily attributed to the elevated PAI-1 levels (Fig. 4G). NAC partially restored the tPA/PAI-1 ratio towards normal. When PAI-1 and tPA levels were compared among diabetic mice at 6 wk and 20 wk, it was found that the PAI-1 levels increased progressively with diabetic duration, which contributed to a more severe fall in the tPA/PAI-1 ratio in STZ 20wk versus STZ 6wk mice (Figs. 4H, 4I). TAT and APC levels were unchanged by diabetes (data not shown).
The tail bleeding time was significantly decreased by 20 wk diabetes, and NAC completely reversed the procoagulation effect of diabetes. (Fig. 4J). The tail bleeding time was shorter in the STZ 20wk mice versus STZ 6wk mice, suggesting a progressive deterioration of hemostatic function (Fig. 4K).
3.5. Diabetes decreases GSH and increases MG and MG-modified protein levels; NAC blunts these effects
Platelet GSH and MG levels were unaltered early during diabetes (STZ 6wk versus Veh controls) (Figs. 5A, 5B). However, as the disease progressed, GSH levels in platelets declined alongside a rise in MG levels and significant differences were found between STZ and Veh groups at the 20 wk timepoint (Figs. 5C, 5D). After treatment with NAC for 3 weeks, platelet GSH was restored to normal levels (Fig. 5C). Interestingly, while not complete, platelet MG levels were decreased in diabetic mice treated with NAC compared with untreated diabetic counterparts (Fig. 5D). NAC treatment resulted in an almost complete normalization of MG-to-GSH ratio. Furthermore, GSSG levels were comparable between Veh, STZ and STZ+NAC groups (Figs. 5E, 5F), and did not change with time (i.e. between 6 and 20 wks diabetes) in any of the groups. Taken together, these data suggest that NAC-derived GSH did not function primarily as an antioxidant, but instead NAC promoted GSH-catalyzed MG elimination. Additionally, MG-adduct formation in platelets was significantly elevated by 20 wks diabetes (Fig. 5G). The two major MG-modified proteins had Mws of almost 24 kD and 54 kD. The glycation of the 54kD protein was prevented by NAC treatment. NAC also decreased the glycation of the 24 kD protein to levels comparable to the Veh group. Further work is required to identify these MG-modified proteins.
Fig. 5.

Impact of NAC on diabetes-induced alterations in GSH and MG: HPLC was used to measure GSH, MG or GSSG (A-F) and western blot was used to probe for MG-adducts (G) in platelets isolated from mice 6 or 20 wks after injection with vehicle (Veh), or streptozotocin to induce diabetes (STZ). Separate STZ groups were treated with N-acetylcysteine in drinking water for 3 wks before obtaining platelets (STZ +NAC). In (G), a sample blot and the quantification of 24kD and 54kD protein band intensities normalized to β-actin are shown. * P < 0.01 vs. Veh; # P < 0.01 vs. STZ.
3.6. NAC restores the antioxidant reserve of platelets during diabetes
There is evidence that MG not only acts as a glycating agent, but also may enhance reactive oxygen species (most likely H2O2) generation in platelets [10]. Therefore we tested if diabetes altered levels of antioxidants important in the generation and detoxification of H2O2. The expression of the antioxidant enzymes, SOD1 (Fig. 6A) and GPx-1 (Fig. 6B), in platelets at 20 wks diabetes was lower than levels in non-diabetic mice. Notably, NAC treatment for 3 weeks prevented the changes to SOD1 and GPx-1 induced by diabetes, suggesting NAC restored the antioxidant reserve of the platelets.
Fig. 6.

Expression of antioxidant enzymes in platelets from diabetic mice without and with NAC treatment. Western immunoblotting was employed to measure levels of (A) SOD-1 and (B) GPx-1 in platelets isolated from mice 20 wks after injection with vehicle (Veh), or streptozotocin to induce diabetes (STZ 20wk). One STZ 20wk group was treated with N-acetylcysteine in drinking water for 3 wks before obtaining platelets (STZ 20wk +NAC). For both, sample immunoblots are shown at the top, and the bar graphs show the quantification of protein band intensities normalized to β-actin. * P < 0.05 vs. Veh; # P < 0.05 vs. STZ 20wk.
4. Discussion
Diabetics are at increased risk for stroke and TIAs compared with the non-diabetic population. Even diabetic patients with well-controlled glucose levels remain at higher risk for cardiovascular disease, suggesting there are factors other than hyperglycemia that also play a role. A candidate molecule is MG, which is not only increased in diabetes, but has been implicated in diabetic complications such as nephropathy [25]. More recently our group showed that MG interrupts vascular endothelial integrity [17], [26], and is associated with worse stroke outcome in diabetes [9]. In the current study we found that MG contributed to enhanced thrombosis during diabetes. Providing N-acetylcysteine (NAC), a precursor of GSH, which is the rate-limiting factor in the MG elimination pathway, attenuated platelet activation, PAI-1 generation and thrombus formation in a mouse model of diabetes. NAC administration was associated with decreased MG levels and MG-adduct formation in platelets. Thus our findings suggest that MG may also be important in the increased risk for stroke in diabetes.
Stroke or TIA occur when thrombosis interrupts blood flow to an area of the brain. The thrombi often occur in arteries of, or supplying, the brain. In order to test if the vasculature of the brain during diabetes is more vulnerable to thrombus formation, we induced thrombi in pial vessels of the brain. To our knowledge, this is the first time this has been assessed in the diabetic brain. Both arterioles and venules exhibited accelerated thrombosis early during diabetes and this worsened as diabetes progressed (Fig. 1, Fig. 3). In our study, NAC afforded protection against thrombosis in both vessel types and at both timepoints, and was particularly effective in the venules. We previously found that diabetes-induced formation of MG-adducts are observed primarily in microvessels [17]. The venules may be particularly vulnerable to MG-glycation, in that our diabetes model causes increased blood brain barrier permeability, a response that is typically seen in venules, and is associated with glycation of an endothelial junctional protein, occludin. We found that NAC protected against the permeability and increased MG-glycation [9], suggesting venular endothelium may at least be one of the targets of MG during diabetes. Taken together, our previous and current findings are in line with the idea that during diabetes, MG makes the vasculature more susceptible to thrombosis by targeting the endothelium. We found a single report showing that MG accelerates thrombosis [11], and several studies showing MG affects key processes of the thrombotic process, namely the coagulation/fibrinolytic pathways and platelet activation. Therefore we also tested whether NAC could protect against these responses in our diabetic model.
In diabetic patients, plasma levels of PAI-1 [27], [28], factor VII and fibrinogen are elevated, and anti-coagulant proteins (protein C, antithrombin III (ATIII) and plasminogen) are decreased [29], [30]. There are several studies consistent with the concept that MG may be involved in this dysregulation. For instance, MG increases the expression of tissue factor (part of the extrinsic pathway) by isolated monocytes [12], and can glycate fibrinogen in vitro, leading to enhanced polymerization and decreased susceptibility to fibrinolysis [31]. In contrast, activities of ATIII and plasminogen, are decreased by MG [13]. Although TAT, tPA and APC levels were unchanged in our diabetes model, PAI-1 levels were increased, a finding consistent with others [32]. High plasma PAI-1 is an independent predictor of atherothrombotic ischemic stroke in diabetics [33]. Because PAI-1 forms a complex with tPA to inhibit the fibrinolytic activity of tPA, elevated PAI-1 levels may also be important in efficacy of stroke therapy. In line with this, exogenous tPA has been shown to be less effective in diabetic humans and rats following stroke [34], [35]. NAC has previously been shown to decrease hyperglycemia-induced PAI-1 expression in cultured endothelial cells [36], and lower PAI-1 levels in diabetic patients [27]. Here we showed that NAC decreased PAI-1 levels in diabetic mice, offering promise for reducing risk for stroke, as well as improving the resolution of a thrombus in a diabetic individual. Importantly, NAC restored the tail bleed time to normal, which suggests that although NAC exhibits anticoagulant properties, it is not associated with disruption of hemostasis. Further work is required to determine if NAC also decreases glycation of fibrinogen or other key proteins in the coagulation/fibrinolytic processes.
Platelets in diabetic patients and animal models exhibit heightened aggregability, not only with each other, but also with leukocytes [37], [38], [39]. Incubation of whole blood with MG results in platelet activation and platelet-leukocyte interactions [12]. We observed increased PLA formation as early as 6 wks of diabetes, made up primarily of lymphocytes. As diabetes progressed, the type of PLAs forming evolved, such that Pl-mono, Pl-neut and Pl-lymph aggregates were all increased by 20 wks diabetes. Our CD41 expression data also suggested the PLAs at 20 wks diabetes contained more platelets, perhaps reflecting a more activated platelet state versus 6 wks diabetes. NAC decreased the PLA formation towards control levels at both timepoints, and was more effective against the Pl-mono and Pl-neut aggregation. This could have particular significance for diabetic patients in that Pl-neut and Pl-mono aggregates are elevated in both type 1 and 2 diabetic humans, and are both associated with the incidence of microvascular complications [38]. Increased Pl-mono aggregation in individuals with well-controlled diabetes is inversely proportional to intraplatelet GSH, suggesting that platelet GSH plays a key role in limiting the formation of PLAs. NAC, given for just 7 days decreased Pl-mono aggregates in these patients, and increased GSH levels in platelets with low baseline GSH content [16]. Our findings are in agreement with NAC as a potential anti-platelet therapy in diabetes, in that platelet GSH levels were decreased in our diabetic mice and NAC corrected not only the platelet GSH levels, but also the Pl-mono and Pl-neut aggregate formation.
Our previous findings in endothelial cells show that the protection afforded by NAC against MG-induced permeability was blocked by inhibiting GSH synthesis using buthionine sulfoximine, suggesting that NAC is primarily functioning through the generation of GSH. Here, we found that NAC also restored GSH in platelets from diabetic mice. While this GSH could be used as an antioxidant and/or in the MG elimination pathway, our data supports the later possibility. First, NAC decreased the high levels of MG in platelets, and prevented MG-glycation of platelet proteins in diabetic mice. Second, GSSG levels in platelets were unchanged by diabetes, even at the 20 wk time point when significant changes in platelet GSH and MG were observed. This suggests there was no significant oxidative stress in these cells. Third, NAC did not significantly alter GSSG levels. The fact that GPx-1 levels were decreased in the diabetic group further suggests this antioxidant pathway was not primarily responsible for the fall in platelet GSH levels during diabetes. Nonetheless, it should be noted that the drop in GPx-1 and SOD1 levels in diabetic platelets was reversed by NAC. While this indicates that NAC restored the antioxidant reserve of the platelets, which could become important as diabetes progresses, we acknowledge that increasing the expression of antioxidant enzymes does not necessarily reflect enhancement of their activity. However, there is evidence that both SOD and GPx-1 are not only susceptible to glycation by MG, but that incubation with MG decreases the activity of both of these enzymes [40], [41]. Therefore, NAC may not only preserve the expression of antioxidant enzymes, but may also protect their activity by preventing their glycation by MG, although this requires further study. Overall, our previous findings in endothelial cells are in agreement with events in platelets during diabetes in that GSH supplied by NAC is being primarily used to eliminate MG, and together this protects against thrombosis and platelet activation.
5. Conclusions
In summary, our study demonstrated that diabetes aggravates thrombosis by activating the coagulation system and platelets. These responses were progressive, becoming more robust as the length of diabetes increased. Furthermore, the phenotype of platelet-leukocyte aggregates changed with diabetes duration, and aggregates began involving more platelets. The platelets were undergoing dicarbonyl stress at this time, characterized by decreased GSH levels, increased platelet MG levels, and MG-adduct formation, and decreased GPx-1 and SOD1 expression. NAC was able to reverse all of the thrombosis-associated changes in the platelet. By supplying GSH as a co-factor in MG elimination, NAC protected against diabetes induced platelet and coagulation activation, and thrombosis. Together with our previous work showing NAC improves post-stroke outcome, we have now shown that NAC, given after the establishment of diabetes, may offer protection against the risk for stroke by altering both systemic and vascular prothrombotic responses. Since Type 2 diabetics also exhibit dicarbonyl stress, increased MG levels and decreased glyoxalase-catalyzed MG elimination, our current findings could apply to this larger diabetic population, and indicate NAC as a prophylactic approach to diminish dicarbonyl stress and offer protection against the thrombotic ischemic strokes seen in diabetics.
Funding
This work was supported by a Malcolm Feist Cardiovascular Research Fellowship, LSUHSC-CCDS (BW), and an LSU LIFT grant (KYS).
Acknowledgments
Contribution statement
BW generated most of the data for this study and was involved in data analyses, interpretation and writing the manuscript. TYA was responsible for the overall design and supervision of the study, including data analyses and manuscript preparation. KYS performed some of the experiments, had intellectual input in the study design and part supervision of the study, participated in manuscript preparation and is the guarantor of this work. We acknowledge that no conflicts exist.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2017.09.005.
Appendix A. Supplementary material
Fig. S1.

Examples of flow cytometry analysis for platelet-leukocyte aggregates (PLAs). Left: CD45 (leukocytes), CD41 (platelets) double positive events identified platelet-leukocyte aggregates. Right: This population was further divided into F4/80+ (Pl-Mono aggregates), Ly-6 G+ (Pl-Neut aggregates) or F4/80-negative, Ly-6 G-negative (Pl-Lymph aggregates). Examples are provided from the three experimental groups: mice injected with vehicle (Veh 19 week), mice injected with STZ to induce diabetes (STZ 19 week) and STZ-treated mice given NAC in drinking water for 2 wks before observation (STZ 19 week+NAC 2wk).
References
- 1.Writing Group M., Mozaffarian D., Benjamin E.J., Go A.S., Arnett D.K., Blaha M.J., Cushman M., Das S.R., de Ferranti S., Despres J.P., Fullerton H.J., Howard V.J., Huffman M.D., Isasi C.R., Jimenez M.C., Judd S.E., Kissela B.M., Lichtman J.H., Lisabeth L.D., Liu S., Mackey R.H., Magid D.J., McGuire D.K., Mohler E.R., 3rd, Moy C.S., Muntner P., Mussolino M.E., Nasir K., Neumar R.W., Nichol G., Palaniappan L., Pandey D.K., Reeves M.J., Rodriguez C.J., Rosamond W., Sorlie P.D., Stein J., Towfighi A., Turan T.N., Virani S.S., Woo D., Yeh R.W., Turner M.B. American heart association statistics C, stroke statistics S. Executive summary: heart disease and stroke statistics-−2016 update: a report from the american heart association. Circulation. 2016;133:447–454. doi: 10.1161/CIR.0000000000000366. [DOI] [PubMed] [Google Scholar]
- 2.O'Donnell M.J., Xavier D., Liu L., Zhang H., Chin S.L., Rao-Melacini P., Rangarajan S., Islam S., Pais P., McQueen M.J., Mondo C., Damasceno A., Lopez-Jaramillo P., Hankey G.J., Dans A.L., Yusoff K., Truelsen T., Diener H.C., Sacco R.L., Ryglewicz D., Czlonkowska A., Weimar C., Wang X., Yusuf S., investigators I. Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the interstroke study): a case-control study. Lancet. 2010;376:112–123. doi: 10.1016/S0140-6736(10)60834-3. [DOI] [PubMed] [Google Scholar]
- 3.Singer D.E., Moulton A.W., Nathan D.M. Diabetic myocardial infarction. Interaction of diabetes with other preinfarction risk factors. Diabetes. 1989;38:350–357. doi: 10.2337/diab.38.3.350. [DOI] [PubMed] [Google Scholar]
- 4.McCarron P., Greenwood R., Elwood P., Shlomo Y.B., Bayer A., Baker I., Frankel S., Ebrahim S., Murray L., Smith G.D. The incidence and aetiology of stroke in the caerphilly and speedwell collaborative studies ii: risk factors for ischaemic stroke. Public Health. 2001;115:12–20. doi: 10.1038/sj.ph.1900724. [DOI] [PubMed] [Google Scholar]
- 5.Li H.F., Zhang X., Zhang Y., Pan X.D., Zhao H.Q., Li H. Clinical and neuroradiological features of internal watershed infarction and the occlusive diseases of carotid artery system. Neurol. Res. 2010;32:1090–1096. doi: 10.1179/016164110X12681290831324. [DOI] [PubMed] [Google Scholar]
- 6.Turk Z. Glycotoxines, carbonyl stress and relevance to diabetes and its complications. Physiol. Res. 2010;59:147–156. doi: 10.33549/physiolres.931585. [DOI] [PubMed] [Google Scholar]
- 7.Rabbani N., Xue M., Thornalley P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016;33:513–525. doi: 10.1007/s10719-016-9705-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemet I., Turk Z., Duvnjak L., Car N., Varga-Defterdarovic L. Humoral methylglyoxal level reflects glycemic fluctuation. Clin. Biochem. 2005;38:379–383. doi: 10.1016/j.clinbiochem.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 9.Wang B., Aw T.Y., Stokes K.Y. The protection conferred against ischemia-reperfusion injury in the diabetic brain by n-acetylcysteine is associated with decreased dicarbonyl stress. Free Radic. Biol. Med. 2016;96:89–98. doi: 10.1016/j.freeradbiomed.2016.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leoncini G., Poggi M. Effects of methylglyoxal on platelet hydrogen peroxide accumulation, aggregation and release reaction. Cell Biochem. Funct. 1996;14:89–95. doi: 10.1002/cbf.655. [DOI] [PubMed] [Google Scholar]
- 11.Hadas K., Randriamboavonjy V., Elgheznawy A., Mann A., Fleming I. Methylglyoxal induces platelet hyperaggregation and reduces thrombus stability by activating pkc and inhibiting pi3k/akt pathway. PLoS One. 2013;8:e74401. doi: 10.1371/journal.pone.0074401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gawlowski T., Stratmann B., Stirban A.O., Negrean M., Tschoepe D. AGEs and methylglyoxal induce apoptosis and expression of mac-1 on neutrophils resulting in platelet-neutrophil aggregation. Thromb. Res. 2007;121:117–126. doi: 10.1016/j.thromres.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Gugliucci A. A practical method to study functional impairment of proteins by glycation and effects of inhibitors using current coagulation/fibrinolysis reagent kits. Clin. Biochem. 2003;36:155–158. doi: 10.1016/s0009-9120(02)00442-3. [DOI] [PubMed] [Google Scholar]
- 14.Reiniger N., Lau K., McCalla D., Eby B., Cheng B., Lu Y., Qu W., Quadri N., Ananthakrishnan R., Furmansky M., Rosario R., Song F., Rai V., Weinberg A., Friedman R., Ramasamy R., D'Agati V., Schmidt A.M. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic ove26 mouse. Diabetes. 2010;59:2043–2054. doi: 10.2337/db09-1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skapare E., Konrade I., Liepinsh E., Makrecka M., Zvejniece L., Svalbe B., Vilskersts R., Dambrova M. Glyoxalase 1 and glyoxalase 2 activities in blood and neuronal tissue samples from experimental animal models of obesity and type 2 diabetes mellitus. J. Physiol. Sci. 2012;62:469–478. doi: 10.1007/s12576-012-0224-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Treweeke A.T., Winterburn T.J., Mackenzie I., Barrett F., Barr C., Rushworth G.F., Dransfield I., MacRury S.M., Megson I.L. N-acetylcysteine inhibits platelet-monocyte conjugation in patients with type 2 diabetes with depleted intraplatelet glutathione: a randomised controlled trial. Diabetologia. 2012;55:2920–2928. doi: 10.1007/s00125-012-2685-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li W., Maloney R.E., Circu M.L., Alexander J.S., Aw T.Y. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: role for glutathione-dependent metabolism of methylglyoxal. Free Radic. Biol. Med. 2013;54:51–61. doi: 10.1016/j.freeradbiomed.2012.10.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thornalley P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988;254:751–755. doi: 10.1042/bj2540751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thornalley P.J. The glyoxalase system in health and disease. Mol. Asp. Med. 1993;14:287–371. doi: 10.1016/0098-2997(93)90002-u. [DOI] [PubMed] [Google Scholar]
- 20.Gavins F.N., Russell J., Senchenkova E.L., De Almeida Paula L., Damazo A.S., Esmon C.T., Kirchhofer D., Hebbel R.P., Granger D.N. Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-s. Blood. 2011;117:4125–4133. doi: 10.1182/blood-2010-08-301366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rumbaut R.E., Randhawa J.K., Smith C.W., Burns A.R. Mouse cremaster venules are predisposed to light/dye-induced thrombosis independent of wall shear rate, cd18, icam-1, or p-selectin. Microcirculation. 2004;11:239–247. doi: 10.1080/10739680490425949. [DOI] [PubMed] [Google Scholar]
- 22.Rumbaut R.E., Slaff D.W., Burns A.R. Microvascular thrombosis models in venules and arterioles in vivo. Microcirculation. 2005;12:259–274. doi: 10.1080/10739680590925664. [DOI] [PubMed] [Google Scholar]
- 23.Reed D.J., Babson J.R., Beatty P.W., Brodie A.E., Ellis W.W., Potter D.W. High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Anal. Biochem. 1980;106:55–62. doi: 10.1016/0003-2697(80)90118-9. [DOI] [PubMed] [Google Scholar]
- 24.Dhar A., Desai K., Liu J., Wu L. Methylglyoxal, protein binding and biological samples: are we getting the true measure? J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009;877:1093–1100. doi: 10.1016/j.jchromb.2009.02.055. [DOI] [PubMed] [Google Scholar]
- 25.Giacco F., Du X., D'Agati V.D., Milne R., Sui G., Geoffrion M., Brownlee M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes. 2014;63:291–299. doi: 10.2337/db13-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li W., Maloney R.E., Aw T.Y. High glucose, glucose fluctuation and carbonyl stress enhance brain microvascular endothelial barrier dysfunction: implications for diabetic cerebral microvasculature. Redox Biol. 2015;5:80–90. doi: 10.1016/j.redox.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martina V., Bruno G.A., Zumpano E., Origlia C., Quaranta L., Pescarmona G.P. Administration of glutathione in patients with type 2 diabetes mellitus increases the platelet constitutive nitric oxide synthase activity and reduces pai-1. J. Endocrinol. Investig. 2001;24:37–41. doi: 10.1007/BF03343806. [DOI] [PubMed] [Google Scholar]
- 28.Nemmar A., Subramaniyan D., Yasin J., Ali B.H. Impact of experimental type 1 diabetes mellitus on systemic and coagulation vulnerability in mice acutely exposed to diesel exhaust particles. Part. Fibre Toxicol. 2013;10:14. doi: 10.1186/1743-8977-10-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ElGendy A.A., Abbas A.M. Effects of warfarin and l-carnitine on hemostatic function and oxidative stress in streptozotocin-induced diabetic rats. J. Physiol. Biochem. 2014;70:535–546. doi: 10.1007/s13105-014-0333-4. [DOI] [PubMed] [Google Scholar]
- 30.Okazaki M., Zhang H., Tsuji M., Morio Y., Oguchi K. Blood coagulability and fibrinolysis in streptozotocin-induced diabetic rats. J. Atheroscler. Thromb. 1997;4:27–33. doi: 10.5551/jat1994.4.27. [DOI] [PubMed] [Google Scholar]
- 31.Krantz S., Lober M., Thiele M., Teuscher E. Properties of in vitro nonenzymatically glycated plasma fibrinogens. Exp. Clin. Endocrinol. 1987;90:37–45. doi: 10.1055/s-0029-1210670. [DOI] [PubMed] [Google Scholar]
- 32.Guldiken S., Turgut B., Demir M., Arikan E., Kara M., Vural O., Tugrul A., Fareed J. The effects of rosiglitazone treatment on the fibrinolytic system in patients with type 2 diabetes mellitus. Clin. Appl. Thromb. Hemost. 2006;12:55–60. doi: 10.1177/107602960601200109. [DOI] [PubMed] [Google Scholar]
- 33.Jotic A., Milicic T., Covickovic Sternic N., Kostic V.S., Lalic K., Jeremic V., Mijajlovic M., Lukic L., Rajkovic N., Civcic M., Macesic M., Seferovic J.P., Stanarcic J., Aleksic S., Lalic N.M. Decreased insulin sensitivity and impaired fibrinolytic activity in type 2 diabetes patients and nondiabetics with ischemic stroke. Int. J. Endocrinol. 2015;2015:934791. doi: 10.1155/2015/934791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Masrur S., Cox M., Bhatt D.L., Smith E.E., Ellrodt G., Fonarow G.C., Schwamm L. Association of acute and chronic hyperglycemia with acute ischemic stroke outcomes post-thrombolysis: findings from get with the guidelines-stroke. J. Am. Heart Assoc. 2015;4:e002193. doi: 10.1161/JAHA.115.002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ning R., Chopp M., Yan T., Zacharek A., Zhang C., Roberts C., Cui X., Lu M., Chen J. Tissue plasminogen activator treatment of stroke in type-1 diabetes rats. Neuroscience. 2012;222:326–332. doi: 10.1016/j.neuroscience.2012.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rikitake Y., Liao J.K. Rho-kinase mediates hyperglycemia-induced plasminogen activator inhibitor-1 expression in vascular endothelial cells. Circulation. 2005;111:3261–3268. doi: 10.1161/CIRCULATIONAHA.105.534024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Angiolillo D.J., Fernandez-Ortiz A., Bernardo E., Ramirez C., Sabate M., Jimenez-Quevedo P., Hernandez R., Moreno R., Escaned J., Alfonso F., Banuelos C., Costa M.A., Bass T.A., Macaya C. Platelet function profiles in patients with type 2 diabetes and coronary artery disease on combined aspirin and clopidogrel treatment. Diabetes. 2005;54:2430–2435. doi: 10.2337/diabetes.54.8.2430. [DOI] [PubMed] [Google Scholar]
- 38.Elalamy I., Chakroun T., Gerotziafas G.T., Petropoulou A., Robert F., Karroum A., Elgrably F., Samama M.M., Hatmi M. Circulating platelet-leukocyte aggregates: a marker of microvascular injury in diabetic patients. Thromb. Res. 2008;121:843–848. doi: 10.1016/j.thromres.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 39.Schmatz R., Mann T.R., Spanevello R., Machado M.M., Zanini D., Pimentel V.C., Stefanello N., Martins C.C., Cardoso A.M., Bagatini M., Gutierres J., Leal C.A., Pereira L.B., Mazzanti C., Schetinger M.R., Morsch V.M. Moderate red wine and grape juice consumption modulates the hydrolysis of the adenine nucleotides and decreases platelet aggregation in streptozotocin-induced diabetic rats. Cell Biochem. Biophys. 2013;65:129–143. doi: 10.1007/s12013-012-9407-5. [DOI] [PubMed] [Google Scholar]
- 40.Khan M.A., Anwar S., Aljarbou A.N., Al-Orainy M., Aldebasi Y.H., Islam S., Younus H. Protective effect of thymoquinone on glucose or methylglyoxal-induced glycation of superoxide dismutase. Int. J. Biol. Macromol. 2014;65:16–20. doi: 10.1016/j.ijbiomac.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Suravajjala S., Cohenford M., Frost L.R., Pampati P.K., Dain J.A. Glycation of human erythrocyte glutathione peroxidase: effect on the physical and kinetic properties. Clin. Chim. Acta. 2013;421:170–176. doi: 10.1016/j.cca.2013.02.032. [DOI] [PubMed] [Google Scholar]