

Abstract
Heterotaxy results from a failure to establish normal left-right asymmetry early in embryonic development. By whole exome sequencing, whole genome sequencing and high-throughput cohort resequencing we identified recessive mutations in matrix metallopeptidase 21 (MMP21), in nine index cases with heterotaxy. In addition, Mmp21 mutant mice and morphant zebrafish display heterotaxy and abnormal cardiac looping, respectively, suggesting a novel role for extra-cellular remodeling in the establishment of laterality in vertebrates.
Initiation of left-right asymmetry involves the generation of leftward fluid flow by motile cilia of the embryonic node, leading to activation of the conserved NODAL gene expression cascade in the left lateral plate mesoderm, and ultimately to the asymmetric positioning of the heart and other visceral organs1. Heterotaxy (or situs ambiguus) comprises abnormal left-right positioning of visceral organs2, falls between the two extremes of situs solitus (normal) and situs inversus (complete mirror image version of normal) and is often associated with complex congenital heart defects (CHDs)1. Laterality defects (ie, heterotaxy and situs inversus) have an estimated prevalence of 1.1/10,000 live births3 and account for approximately 3% of all CHDs1. Situs inversus can occur in the context of primary ciliary dyskinesia (PCD; OMIM 244400), caused by mutations in structural components of motile cilia. Genes implicated in isolated heterotaxy include components of the NODAL pathway and ZIC3 (Supplementary Table 1), however these genes account for only a small proportion of cases.
We investigated two non-consanguineous families with recurrent heterotaxy including CHDs, but without any obvious symptoms of ciliopathy. Family 1 consists of affected dizygotic twins, one of whom died at one day of life due to haemodynamic failure, while family 2 consists of two affected brothers (Figure 1, Supplementary Table 2 and for family 2 see reference4). Whole exome sequencing (WES) of the affected twins of family 1 (Supplementary Table 3) led to the identification of rare, compound heterozygous mutations in matrix metallopeptidase 21 (MMP21, NM_147191.1): c.677T>C, p.(Ile226Thr) and c.1203G>A, p.(Trp401*); the latter corresponding to rs137955225 with Exome Aggregation Consortium (ExAC) allele frequency < 1/30,000. No rare, damaging variants were identified in genes previously implicated in isolated heterotaxy (Supplementary Table 1). Point mutations were verified by Sanger sequencing and segregation analysis indicated recessive inheritance (Figure 1). Based on previous whole genome sequencing (WGS) of the family 2 affected brothers and their healthy parents, compound heterozygous missense variants in BCL9L were suggested as potentially pathogenic4, however both BCL9L variants in that family are predicted benign and neutral by Polyphen2 and Provean programs respectively, and left-right patterning defects have not been reported in Bcl9l knockout mice or morphant zebrafish5,6. Re-analysis of the family 2 WGS data for identification of structural variants revealed compound heterozygosity in both brothers for a deletion of MMP21 exons 1-3 (chr10:127,460,915-127,466,819; Supplementary Figure 1) inherited from the mother and a frameshift mutation in MMP21 (c.365delT, p.(Met122Serfs*55)) inherited from the father. The heterozygous exonic deletion was validated by microarray analysis, and breakpoints were verified by long-range PCR and Sanger sequencing.
Figure 1.
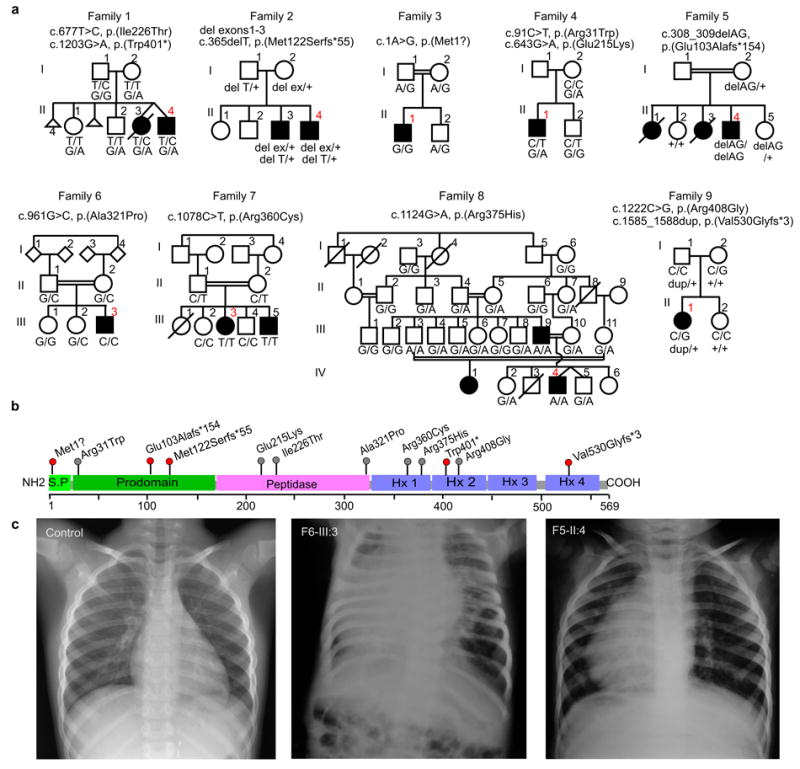
MMP21 mutations identified in 9 affected families. (a) Pedigrees of families 1-9. MMP21 variants refer to RefSeq transcript NM_147191.1. Index cases (including those submitted to HaloPlex sequencing) are indicated in red. (b) Domain structure of MMP21 with mutations identified. S.P., signal peptide; Hx, hemopexin repeat. (c), Radiography of a normal individual and of patients F6-III:3 and F5-II:4.
Subsequently we sequenced MMP21 following HaloPlex target enrichment in 264 index cases with either heterotaxy (extra-cardiac and/or cardiac laterality defects such as dextrocardia or transposition of the great arteries (TGA), n=154) or non-heterotaxy CHDs such as isolated tetralogy of Fallot or common arterial trunk (n=110). The total included 25 syndromic cases in which heterotaxy or CHDs were associated with one or more other anomalies (for example, anal atresia, vertebral anomalies or cleft palate), and was comprised of 117 familial and 147 sporadic cases, including 34 index cases born to consanguineous parents. We identified nine cases (families 3-11) with rare, biallelic MMP21 variations predicted to affect protein function; all variations were confirmed by Sanger sequencing and segregated with heterotaxy or other laterality defects and complex CHDs (Figure 1, Supplementary Figures 2 and 3, Supplementary Tables 2 and 4). For families 10 and 11, one of the variants has been reported in the ExAC database in the homozygous state; the MMP21 variants are therefore unlikely to be causal in those cases. In families 3-9, all variants identified have ExAC allele frequencies <1/10,000 and have not been reported as homozygous. All missense variants (except p.(Arg408Gly)) were predicted probably damaging and deleterious by Polyphen2 and Provean, respectively (Supplementary Table 2). Parametric genome-wide linkage analysis performed on families 6 and 7 (analysed simultaneously) demonstrated a linkage peak with LOD score of 3.508 on a region of chromosome 10 containing MMP21 (Supplementary Figure 4); no other genes in the interval are known to cause laterality disorders (Supplementary Table 5). Also, homozygosity mapping in family 7 indicated that MMP21 fell within a large region of homozygosity shared by the two affected individuals (Supplementary Figure 5). Five families were consanguineous (families 3, 5-8). Of the three family 8 individuals with a homozygous MMP21 variant, F8-III:9 had situs inversus totalis while F8-III:3 is reported to be asymptomatic, suggesting possible incomplete penetrance in this family (Figure 1a). Overall, the penetrance of laterality defects in our series is very high compared to that observed in PCD (~50%). Based on the above findings, MMP21 mutations account for 5.9 % of non-syndromic heterotaxy cases.
MMP21 is predicted to be secreted and conforms to the basic domain structure of MMPs7,8: an N-terminal signal peptide, an auto-inhibitory pro-domain, a catalytic peptidase domain and four C-terminal hemopexin repeats that are involved in substrate recognition (Figure 1b). Mapping of the affected MMP21 peptidase domain residues Glu215, Ile226 and Ala321 to a model based on the crystal structure of the MMP11 peptidase domain indicated that all three fall within core secondary structure components (Supplementary Figure 6); their substitutions may therefore destabilize the domain. The variants p.(Arg360Cys), p.(Arg375His) and p.(Arg408Gly) map to the first and fourth hemopexin domains and therefore may influence substrate specificity.
Whole-mount in situ hybridization for mmp21 in zebrafish embryos at stages during which left-right asymmetry is established indicated that expression was restricted to Kupffer’s vesicle (the left-right organizer in fish; Figure 2a-e). Injection of splice-blocking (MO1; Supplementary Figure 7) or translation-blocking (MO-AUG) mmp21 morpholinos, but not standard control or 5 base mispair morpholinos, resulted in randomized heart looping in zebrafish at 48 hpf (Figure 2f,g). Defects in other organs known to be dependent on ciliary function were not observed (Supplementary Table 6), confirming a highly specialized role for mmp21 in the generation of left-right asymmetry.
Figure 2.
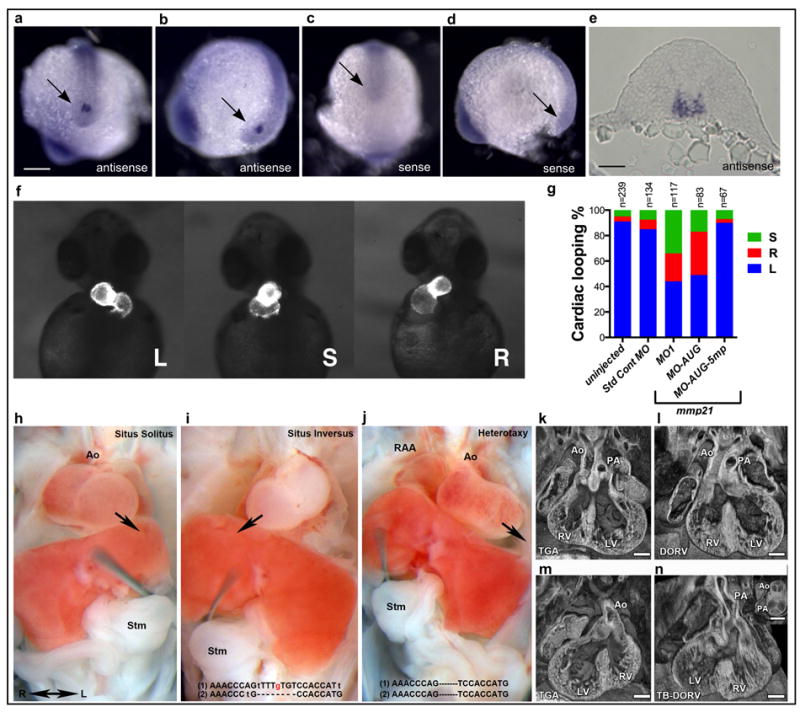
mmp21 expression and knock-down in zebrafish and CRISPR/Cas9 genome editing of Mmp21 in mice. (a-d) Whole-mount in situ hybridization (WISH) for zebrafish mmp21 at 12 hpf. (a, b) antisense probe; (c, d) sense probe. (a, c) caudal views; (b, d) lateral views. An arrow indicates Kupffer’s vesicle. Scale bar = 200 μm. (e) Transverse cryosection through tail-bud of an mmp21 WISH-stained embryo. Scale bar = 60 μm. (f, g) Cardiac looping in zebrafish mmp21 morphants. Wildtype zebrafish embryos or embryos from a transgenic line expressing GFP under the control of myosin light chain 7 (myl7 or cmlc2) regulatory elements were injected with a standard control morpholino (Std Cont MO), a splice blocking morpholino (MO1), a translation-blocking morpholino (MO-AUG) or a 5 bp-mispair control morpholino (MO-AUG-5mp). L, left (normal); S, straight; R, right; n, number of embryos injected. (h-j) Mouse embryos generated by Mmp21 CRISPR/Cas9 genome editing exhibit CHDs and laterality phenotypes. Situs solitus (left sided heart and stomach (stm)) (h), situs inversus with right sided heart and stomach (i), or heterotaxy with discordant stomach/heart situs (j). Arrows indicate orientation of the heart. Genotyping indicated (i) compound heterozygosity for a null allele and a mutation causing the desired p.(Ile226Thr) substitution or (j) homozygous null. (k-n). CHDs observed in CRISPR targeted embryos included transposition of the great arteries (TGA) (k), double outlet right ventricle (DORV) (l), TGA with dextrocardia (m), and Taussig-Bing sub-type DORV with hypoplastic aortic valve and dextrocardia (n). Scale bars = 0.2mm. RAA, right aortic arch; Ao, aorta; PA, pulmonary artery; RV, right ventricle; LV, left ventricle.
Consistent with the above findings, two ENU-induced Mmp21 missense mutations cause laterality defects and complex CHDs in mice9 (Supplementary Tables 7,8,9 and accompanying manuscript by Akawi et al). We performed CRISPR/Cas9 mediated genome-editing in mouse zygotes to knock-in the MMP21 p.(Ile226Thr) or p.(Ala321Pro) missense mutations from family 1 and 6. We harvested 49 F0 embryos at E13.5 and observed mutations (including deletions and insertions) in Mmp21 in 15/15 (100%) embryos and 20/34 (59%) embryos injected with p.(Ile226Thr) or p.(Ala321Pro) oligos, respectively (Supplementary Figure 8 and Supplementary Tables 10 and 11). Phenotyping showed 97% of the mutation-positive embryos (32/33; not included are two mutation-positive dead embryos whose phenotype could not be analysed) had CHDs including TGA, with 20 (61%) exhibiting situs inversus or heterotaxy and 13 (39%) exhibiting situs solitus (Figure 2 and Supplementary Table 10). No defects were observed in 14 embryos with wildtype genotypes and in one mutation-positive embryo (A321P-20). Subcloning of PCR products amplified from yolk sac genomic DNA revealed that embryo I226T-1 harboured a loss of function deletion on one allele and on the other the desired A to G mutation causing the homologous p.(Ile226Thr) substitution (Supplementary Figure 8 and Supplementary Table 11). This embryo exhibited situs inversus with CHD (Figure 2 and Supplementary Table 10), while embryos I226T-4 and -5 also harboured laterality defects and CHDs and had a high abundance of subclones with the knock-in allele (Supplementary Table 11), supporting the pathogenicity of the p.(Ile226Thr) missense mutation. No p.(Ala321Pro) knock-in alleles were identified. The above findings show that Mmp21 loss of function can cause the same spectrum of phenotypes seen in mice with ENU-induced missense mutations, suggesting that the latter are loss of function.
Specific ECM molecules play various roles at the node during left-right patterning10, suggesting candidate MMP21 cleavage targets. Also, relevant to the central role of TGFβ family members (e.g. NODAL, LEFTY) in generation of left-right asymmetry, MMPs can activate latent TGFβ factors11. Important future work will involve investigation of MMP21 cleavage targets at the node. Interestingly, an MMP21 orthologue appears to be either absent or undergoing decay in genomes of birds, reptiles and cetartiodactyl mammals (Supplementary Figures 9-11). It has been reported that in chick and pig (a cetartiodactyl), cells of the embryonic node are covered by endoderm, such that asymmetry would be established by rotational cell movements around the node rather than by cilia-driven fluid flow12. We speculate that MMP21 is only required in those species in which asymmetry depends on ciliated nodal cells exposed to an extracellular cavity.
In conclusion, our results suggest that MMP21 plays an essential role in the pathway specifying left-right asymmetry by providing a highly specific cleavage activity at the embryonic node, and that MMP21 mutations are a relatively frequent cause of heterotaxy in humans.
Online Methods
Whole exome sequencing
For WES of family 1 individuals F1-II:3 and F1-II:4, Agilent SureSelect libraries were prepared from 3 μg of genomic DNA sheared with a Covaris S2 Ultrasonicator as recommended by the manufacturer. Exome capture was performed with the 51 Mb Agilent SureSelect Human All Exon kit V5. Paired-end sequencing (75 bp + 75 bp reads) was carried out on a pool of barcoded exome librairies using a HiSeq2500 (Illumina). After demultiplexing, paired-end sequences were mapped to the human genome (NCBI build37/hg19) using Burrows-Wheeler Aligner. The mean depth of coverage obtained for the two family 1 samples was 107 and 110 fold, with 98 % of the exome covered at least 15 fold. Downstream processing was performed using Genome Analysis Toolkit (GATK), SAMtools and Picard, and variant calls were made with the GATK Unified Genotyper. We removed from consideration all calls with read coverage 2 fold or less or a Phred-scaled SNP quality score of 20 fold or less. Variant annotation was based on Ensembl release 71, and filtering was performed with an in-house software, using the following SNP databases for filtering: dbSNP (build 135), Exome Variant Server (release ESP6500SI-V2), 1000 Genomes (release date May 21, 2011) and variants from more than 5,000 in-house exomes. WES was performed in accordance with approved institutional protocols (Comité de protection des personnes Ile-de-France II), and informed consent was obtained for genetic testing.
Whole genome sequencing
Study Participants
Family 2 individuals F2-I:1, F2-I:2, F2-II:3 and F2-II:4 were enrolled in the Center for Pediatric Genomic Medicine research repository and received WGS to diagnose suspected monogenic diseases of unknown etiology in affected children4,13. The study was approved by the institutional review board at Children’s Mercy Hospital Kansas City, MO.
Library preparation and sequencing
Isolated genomic DNA was prepared for WGS with a modification of the Illumina TruSeq sample preparation4,13. Briefly, 500 ng of DNA was sheared with a Covaris S2 Biodisruptor, end-repaired, A-tailed, and adaptor-ligated. Polymerase chain reaction (PCR) was omitted. Libraries were purified with SPRI beads (Beckman Coulter). Quantitation was carried out by real-time PCR. Libraries were denatured with 0.1 M NaOH and diluted to 2.8 pM in hybridization buffer. Samples were each loaded onto two flowcells, followed by 2 × 100 cycle sequencing on Illumina HiSeq 2500 instruments.
Next Generation Sequencing Analysis
Sequence data was generated with Illumina RTA 1.12.4.2 & CASAVA-1.8.2, aligned to the human reference GRCh37.p5 using Genomic Short Read Nucleotide Alignment Program (GSNAP). Deletion structural variants (SVs) were identified with a novel computational pipeline (SKALD, Screening Konsensus and Annotation of Large Deletions; unpublished data, A.N. et al). Briefly, SKALD starts from a GSNAP-derived bam file, executes the SV detection tools Breakdancer14 and GenomeStrip15 concurrently, and applies filters (depth ratio within SV to region flanking SV ≤0.7, GenomeStrip confidence interval <40, Breakdancer score ≥30, deletion size ≤0.5Mb, supporting “stretched” read pairs ≥2, overlapping repeat features ≤1500) to yield a high confidence deletion variant set. Since genes that are commonly deleted are unlikely to be deleterious, only SV deletions harboring rarely deleted genes were retained. Nucleotide variants were detected and genotyped with the GATK v. 1.4, and annotated with RUNES4,13. Likely pathogenic hemizygous nucleotide variants were sought within SV deletion boundaries by limitation to rare coding and splice variants with allele frequency <1% in an internal database4,13, and were included as part of the SKALD output in the form of a tab separated text file. Variant analysis programs were either written in Perl, R, Make or the Linux shell scripting language. Variant statistics for WGS of family 2 have been previously published4; see data for individuals CMH184, CMH185, CMH186 and CMH202 in that reference.
Microarray analysis
For validation of the deletion identified by WGS in family 2, isolated genomic DNA was prepared using the Gentra Puregene Kit for whole blood. Test sample DNA was hybridized to Affymetrix CytoScan HD CN+SNP microarray chips. The chips were washed, stained and scanned. Raw .cel and .dat files were converted to .cychp files using CytoScan® HD Array. Chromosome Analysis Suite 2.0 NetAffx 32.3 (hg19) was used for final analysis and exporting of deletion calls.
HaloPlex sequencing
DNA extraction, quantification, quality control and pooling
The study protocol was approved by the local institutional review board (Comité de protection des personnes Sud-Est II) and a signed informed consent was obtained from all participants (or parents of minors or fetuses). DNA was extracted from either whole blood or fetal tissue using a variety of extraction protocols. DNA concentration and quality were assessed using NanoDrop (Thermo Scientific) and Qubit (Life technologies) fluorometers. A260/A280 ratios of 1.8 to 2.0 and A260/A230 ratios >1.5 were accepted. DNA fragmentation was assessed using agarose gel (0.8%) electrophoresis. Pools of six DNA samples were prepared by pooling together DNA extracted with the same protocol.
HaloPlex design
The seven exons and 25 intronic nucleotides on each side of each exon of the unique MMP21 transcript (ENST00000368808, NM_147191.1, GRCh37) and of two other genes (total target region: 5,624 nucleotides, total target on MMP21: 2060 nucleotides) were selected to generate probes according to SureDesign (https://earray.chem.agilent.com/suredesign/index.htm) with a predicted MMP21 sequencing coverage of 99.47%.
HaloPlex library preparation
Amplicon libraries were prepared from genomic DNA of each pool using the HaloPlex PCR target enrichment system according to the manufacturer’s recommendations. In brief, 200 ng of pooled DNA (33 ng of each individual DNA) was used for restriction reactions. Successful digestion was verified on a Bioanalyzer 2100 with a high-sensitivity chip (Agilent Technologies). DNA pools were then hybridized for 3 hours at 54°C to customized, biotinylated probes in a Biometra T Professional thermocycler (Analytik Jena). Hybridized probes in presence of the primer barcode cassette were captured with magnetic beads and target fragments were ligated to create circular DNA molecules. Libraries were then amplified by PCR using HaloPlex Ion primers. Libraries were quantified using a Bioanalyzer 2100 with a high-sensitivity chip. The same molar amount of each library was used to make pools of 16 libraries (6×16 = 96 DNA samples) at a final concentration of 20 pM.
Sequencing, quality controls and variant calling
Sequencing was performed using an Ion PGM sequencer (Life Technologies) with Ion 318™ Chip v2. 25 microlitres of a pool of DNA at 20 pM was used for emulsion PCR. Quality of sequencing was assessed by the PGM sequencer by providing the Ion Sphere Particle (ISP) density, number of total reads, percentage of usable reads, percentages of monoclonal and polyclonal reads, mean read length (AQ17, AQ20 and perfect), percentages of low quality sequences, adapter dimers and aligned bases. Bam files were loaded on Alamut Visual 2.5 (Interactive Biosoftware) and variant detection was performed manually by setting the variant detection threshold to 0.04. Gained stop, splice site, frameshift and rare missense variants (at a frequency of less than 0.1% in the Exome Variant Server http://evs.gs.washington.edu/EVS/) were selected for Sanger sequencing of the six individual DNAs composing the pool carrying each selected variant. Variants of interest were confirmed on a second dilution of DNA and on an independent sample. Subsequently, relatives were tested for variant segregation analysis.
Genome-wide linkage analysis and homozygosity mapping
DNA was extracted from either whole blood or fetal tissue using a variety of extraction protocols. DNA concentration and quality were assessed using NanoDrop (Thermo Scientific) and Qubit (Life technologies) fluorometers. A260/A280 ratios of 1.8 to 2.0 and A260/A230 ratios > 1.5 were accepted. DNA fragmentation was assessed using agarose gel (0.8%) electrophoresis.
A minimal amount of 1 μg at a minimal concentration of 50 ng/μL of each DNA was used for genotyping on a HumanCoreExome-12 v1.0 (WG-353-1104) cell (Illumina) according to the Illumina protocol ‘Infinium HD Ultra Assay Automated EUC (11328108 B)’ (Illumina). Cells were scanned on an IScan+.
Genotypes were obtained using GenomeStudio (Illumina). Quality controls were performed on in-house controls and control DNA. Genotyping and pedigree information were compared (sex, familial relationship, Mendelian allele segregation) with Linkdatagen16,17, a PERL script that generates datasets for linkage analysis, relatedness checking, IBD and HBD inference. Parental relatedness was also checked with Graphical Representation of Relationships (http://csg.sph.umich.edu/abecasis/GRR/index.html). Linkdatagen created output files for MERLIN18. Parametric (disease allele frequency of 0.0001, a fully penetrant disease, a 0% phenocopy rate, autosomal recessive inheritance) and non-parametric analyses were performed.
Homozygosity mapping was performed using FSuite version 1.0.319. Map and ped files in PLINK format derived from genotyping files were prepared with Linkdatagen. Minor allele frequencies were obtained from the Ensembl database in order to prepare the freq file. One hundred random submaps were generated. The inbreeding coefficient of each individual was estimated by running FEstim on the consecutive submaps. HBD (Homozygous-by-descent) posterior probabilities, FLOD and HFLOD of inbred cases were calculated on the consecutive submaps. Finally, the results were plotted. FSuite calls R, MERLIN, and plink2.
Homology modeling of the MMP21 peptidase domain
Homology modeling of the MMP21 peptidase domain (amino acids 170-327; NP_671724.1) was performed via http://swissmodel.expasy.org. The Swiss Model Template Library (SMTL) template used for modeling was 1hv5.6.A (stromelysin-3, ie, MMP11)20. This template was selected because it was one of the most highly ranked templates for sequence identity to MMP21 (43.33%) and for Global Model Quality Estimation (GQME) score (0.72; possible scores between 0 and 1). The model was visualized using UCSF Chimera (http://www.cgl.ucsf.edu/chimera/).
Zebrafish in situ hybridization
We were unable to identify an mRNA or EST clone corresponding to zebrafish mmp21 in public databases. In order to generate probes for whole-mount in situ hybridization (WISH), we designed primers based on the exon-intron structure predicted by Genscan in the UCSC browser (Zv9/danRer7 assembly) and by visual comparison with the structure of the human gene. cDNA was generated from wildtype (AB strain) whole embryo RNA by RT using GeneAmp RNA PCR Core Kit (Applied Biosystems, Roche). Primers used for PCR amplification of mmp21 5’ and 3’ probes are listed in Supplementary Table 12 (primers 5’F, 5’R, 3’F, 3’R). The two probes are non-overlapping, with the 5’ probe (654 bp) spanning exons 1-4 and the 3’ probe (522 bp) spanning the final two exons (see Supplementary Figure 7). Amplicons were cloned into the pCRII-TOPO vector (Invitrogen). Positive clones confirmed by DNA sequencing were used to generate digoxigenin-AP labeled T7 antisense and SP6 sense probes. WISH was performed as previously described21. Similar WISH results were obtained with both mmp21 probes. Stained embryos were photographed in 30% sucrose and then cryosectioned. An mmp21 cDNA containing the entire ORF was also amplified using the primers 5’F and 3’R above. This sequence has been deposited in GenBank, with accession number KT207790.
Zebrafish mmp21 knock-down
Zebrafish knock-down experiments were carried out by the Zebrafish Core of the Cardiovascular Development Consortium under approved IACUC protocols of the University of Utah and at the Institut Imagine and University of Pittsburgh with locally approved IACUC protocols. Antisense morpholino oligos (MOs) were designed to suppress mmp21 splicing or translation and purchased from Gene Tools (www.gene-tools.com), along with control MOs: MO1 targeting the exon 4-intron 4 junction (exon numbering corresponding to the transcript described above), MO2 targeting the intron 4-exon 5 junction, MO-AUG, MO-AUG-5mp and the standard control MO (for sequences, see Supplementary Table 12). For the experiments presented in Figure 2f-g, 3-8 ng of MO was injected at the 1 cell stage by standard techniques into wildtype AB strain embryos or a transgenic line expressing GFP under the control of myosin light chain 7 (myl7 also known as cmlc2) regulatory elements22. For the experiments presented in Supplementary Table 6, 12 or 16 ng of MO1 was injected. The ability of MO1 and MO2 to induce aberrant splicing was assessed by RT-PCR at 24 hpf (Supplementary Figure 7), using primers within exon 3 and exon 6 (for sequences, see Supplementary Table 12). Only MO1 was effective at blocking splicing. Cardiac looping was scored at 48 hpf.
Mouse Mmp21 CRISPR/Cas9 Genome Editing
Design of guides
Guides for CRISPR/Cas9 gene editing were identified utilizing http://crispr.mit.edu and efficiency scores were calculated using http://www.broadinstitute.org/rnai/public/analysis-tools/sgrna-design. Guides were selected based on the least number of off-target genes, efficiency scores, as well as proximity to the orthologous human mutation site. PAGE-purified, 150bp donor oligonucleotides for the p.(Ile226Thr) mutation and the p.(Ala321Pro) mutation were designed to create the mutation of interest (underlined nucleotide in the sequences in Supplementary Table 12).
Generation of sgRNA
T7 promoter was added to sgRNA template via PCR amplification using the Mmp21 primers (Supplementary Table 12): p(Ile226Thr)-T7-forward (near orthologous p.(Ile226Thr)) and p(Ala321Pro)-T7-forward (near orthologous p.(Ala321Pro)) and common reverse primer containing the stemloop,. The PCR product was purified with a column PCR purification kit (Qiagen). In vitro transcription was then performed with an Ambion T7 MEGAshortscript kit (Life Technologies) and RNA was recovered using an Ambion MEGAclear kit (Life Technologies). The concentration of RNA was measure by a NanoDrop instrument (Thermo Scientific) and Agilent Bioanalyzer (Agilent Technologies, Inc).
Microinjection
All animal procedures were conducted according to relevant national and international guidelines (AALAC and IACUC) and have been approved by the Jackson Laboratory Animal Care and Use Committee (Protocol #99066). C57BL/6J and CB6F1/J mouse strains were used as embryo donors and pseudopregnant recipient dams, respectively. Cas9 RNA (100 ng/μl) (TriLink Biotechnologies), sgRNA (50 ng/μl), and oligonucleotide donor DNA (20ng/μl) were injected into the pronuclei of zygotes. Oviduct transfers were performed on the same day into pseudopregnant dams. Embryos were harvested at embryonic day 13.5, denoting the injection day as 0.5, and fixed in 4% paraformaldehyde.
Genotyping of CRISPR F0 embryos
Genomic DNA was extracted with 50 mM NaOH from yolk sacs and PCR-amplified using forward and reverse primers (Supplementary Table 12): p.(Ile226Thr)-F, p.(Ile226Thr)-R, p.(Ala321Pro)-F, p.(Ala321Pro)-R, with the expected product sizes of 455 bp for the p.(Ile226Thr) amplicon and 422 bp for the p.(Ala321Pro) amplicon. PCR products were then submitted for Sanger sequencing. For some embryos, the PCR products were subcloned using TOPO PCR cloning (Life Technologies, Inc.), with multiple clones analyzed per embryo by Sanger sequencing.
Mouse Mutant Phenotyping Analysis
Mouse mutants were analyzed by necropsy to examine overall visceral laterality. To examine intracardiac anatomy, serial section histopathology was carried out using episcopic confocal microscopy to generate 3D reconstructions.
Supplementary Material
Acknowledgments
This work was supported by grants from the Fondation pour la Recherche Médicale (HeartGenomics), the Agence Nationale de la Recherche (ANR-10-IAHU-01), the Programme Hospitalier de Recherche Clinique (PHRC, 2008), the fondation Renaud Fèbvre, NICHD and NHGRI (U19HD077693), NCATS (CTSA grant TL1TR000120), NHLBI Bench to Bassinet grant U01-HL098180 and the NIH (HL098180). We thank the Fédération Française de Cardiologie, the Société Française de Cardiologie, the Centre de Ressources Biologiques (CRB-ADN; BB-033-00065) at the Institut Imagine, the zebrafish core facility of the Cardiovascular Development Consortium (CvDC), the divisions of Clinical Genetics and Neonatology at Children’s Mercy-Kansas City, the families for their participation, Virginie Salle, Christophe Ollagnier and Brigitte Aime for excellent technical assistance, Xiaoqin Liu and William Devine for diagnosis of CHDs in mutant mouse embryos and Hugues Roest Crollius and Marion Delous for discussions.
Footnotes
Author Contributions
A.G. analyzed WES data, performed mutation validation, segregation studies and zebrafish experiments and wrote the paper. G.C.G., M.S, N.T.K. and H.Y. performed and analyzed CRISPR/Cas9 and ENU experiments. F.B., L.D.S., S.D.F., P.S., S.L., L.d.P. and D.B. recruited patients. M.T. performed zebrafish experiments. H.L., R.E.M., G.J. and A.M.d.B. performed HaloPlex sequencing, mutation validation and segregation studies. A.N., N.A.M., C.J.S., I.T., L.D.C., E.G.F. and S.F.K. performed and analyzed WGS data and deletion validation studies of family 2. M.O. performed mutation validation and segregation studies. C.N.B, K.A.P. and S.A.M. performed and analyzed CRISPR/Cas9 experiments. C.M., C.B.-F. and P.N. performed WES. J.-F.D. performed linkage analysis. J.A. recruited patients, supervised genetic studies and wrote the paper. P.B. recruited patients, supervised HaloPlex studies, performed linkage analysis and homozygosity mapping and wrote the paper. C.W.L. supervised CRISPR/Cas9 and ENU experiments and wrote the paper. C.T.G. analyzed WES and CRISPR/Cas9 data, performed homology modeling, evolutionary analysis and zebrafish experiments and wrote the paper.
Competing Financial Interests
The authors declare no competing financial interests.
URLs
ENU-induced mutant Mmp21 mice, http://www.informatics.jax.org/allele/MGI:5311376 and http://www.informatics.jax.org/allele/MGI:5554438. Online Mendelian Inheritance in Man, http://www.omim.org. ExAC browser, http://exac.broadinstitute.org. UCSC genome browser, http://genome.ucsc.edu. Polyphen2, http://genetics.bwh.harvard.edu/pph2/index.shtml. Provean, http://provean.jcvi.org/index.php. MMP21 variants, http://databases.lovd.nl/shared/view/MMP21.
Accession codes
GenBank accession number for zebrafish mmp21: KT207790.
References
- 1.Sutherland MJ, Ware SM. Am J Med Genet C Semin Med Genet. 2009;151C:307–17. doi: 10.1002/ajmg.c.30228. [DOI] [PubMed] [Google Scholar]
- 2.Jacobs JP, et al. Cardiol Young. 2007;17(Suppl 2):1–28. doi: 10.1017/S1047951107001138. [DOI] [PubMed] [Google Scholar]
- 3.Lin AE, et al. Am J Med Genet A. 2014;164A:2581–91. doi: 10.1002/ajmg.a.36695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Saunders CJ, et al. Sci Transl Med. 2012;4:154ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brembeck FH, et al. Genes Dev. 2004;18:2225–30. doi: 10.1101/gad.317604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsuura K, et al. Nat Commun. 2011;2:548. doi: 10.1038/ncomms1551. [DOI] [PubMed] [Google Scholar]
- 7.Ahokas K, et al. Gene. 2002;301:31–41. doi: 10.1016/s0378-1119(02)01088-0. [DOI] [PubMed] [Google Scholar]
- 8.Marchenko GN, Marchenko ND, Strongin AY. Biochem J. 2003;372:503–15. doi: 10.1042/BJ20030174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, et al. Nature. 2015;521:520–4. doi: 10.1038/nature14269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Collins MM, Ryan AK. Genesis. 2014;52:488–502. doi: 10.1002/dvg.22774. [DOI] [PubMed] [Google Scholar]
- 11.Yu Q, Stamenkovic I. Genes Dev. 2000;14:163–76. [PMC free article] [PubMed] [Google Scholar]
- 12.Gros J, Feistel K, Viebahn C, Blum M, Tabin CJ. Science. 2009;324:941–4. doi: 10.1126/science.1172478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soden SE, et al. Sci Transl Med. 2014;6:265ra168. doi: 10.1126/scitranslmed.3010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan X, Abbott TE, Larson D, Chen K. Curr Protoc Bioinformatics. 2014;2014 doi: 10.1002/0471250953.bi1506s45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handsaker RE, Korn JM, Nemesh J, McCarroll SA. Nat Genet. 2011;43:269–76. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bahlo M, Bromhead CJ. Bioinformatics. 2009;25:1961–2. doi: 10.1093/bioinformatics/btp313. [DOI] [PubMed] [Google Scholar]
- 17.Smith KR, et al. Genome Biol. 2011;12:R85. doi: 10.1186/gb-2011-12-9-r85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 19.Gazal S, Sahbatou M, Babron MC, Genin E, Leutenegger AL. Bioinformatics. 2014;30:1940–1. doi: 10.1093/bioinformatics/btu149. [DOI] [PubMed] [Google Scholar]
- 20.Gall AL, et al. J Mol Biol. 2001;307:577–86. doi: 10.1006/jmbi.2001.4493. [DOI] [PubMed] [Google Scholar]
- 21.Thisse C, Thisse B. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- 22.Huang CJ, Tu CT, Hsiao CD, Hsieh FJ, Tsai HJ. Dev Dyn. 2003;228:30–40. doi: 10.1002/dvdy.10356. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.