

Abstract
Bacteria downregulate their ribosomal activity through dimerization of 70S ribosomes, yielding inactive 100S complexes. In Escherichia coli, dimerization is mediated by the hibernation promotion factor (HPF) and ribosome modulation factor. Here we report the cryo-electron microscopy study on 100S ribosomes from Lactococcus lactis and a dimerization mechanism involving a single protein: HPFlong. The N-terminal domain of HPFlong binds at the same site as HPF in Escherichia coli 100S ribosomes. Contrary to ribosome modulation factor, the C-terminal domain of HPFlong binds exactly at the dimer interface. Furthermore, ribosomes from Lactococcus lactis do not undergo conformational changes in the 30S head domains upon binding of HPFlong, and the Shine–Dalgarno sequence and mRNA entrance tunnel remain accessible. Ribosome activity is blocked by HPFlong due to the inhibition of mRNA recognition by the platform binding center. Phylogenetic analysis of HPF proteins suggests that HPFlong-mediated dimerization is a widespread mechanism of ribosome hibernation in bacteria.
When bacteria enter the stationary growth phase, protein translation is suppressed via the dimerization of 70S ribosomes into inactive complexes. Here the authors provide a structural basis for how the dual domain hibernation promotion factor promotes ribosome dimerization and hibernation in bacteria.
Introduction
Ribosomes are responsible for peptide-bond synthesis and are a main target for antibiotics1. They translate the genetic information from mRNA into a defined sequence of amino-acid residues, which eventually fold into mature proteins. When bacteria enter the stationary growth phase, the expression levels of hundreds of genes are changed and so is the synthesis of proteins2–4. The ability of a cell to control protein synthesis is essential for life. If ribosomal activity is not well controlled, over-activity or over-abundance of ribosomes can lead to serious problems, including stalling of ribosomes due to amino acid scarcity5. Ribosomes can be degraded to lower the translation activity but this is a costly strategy, both in terms of resources and competitive strength5, 6. A more efficient and faster response involves downregulation of ribosome activity.
Cells can do this via dimerization of 70S ribosomes into inactive 100S complexes4, a process that has been observed in bacteria7 and mammalian cells8. Ribosome dimerization is best documented for the Gram-negative bacteria Escherichia coli 5, 9, 10 and Thermus thermophilus 11. Ribosome dimers of E. coli, connected through their 30S subunits, were first observed by electron microscopy in 196012, and proposed to be translationally inactive13. The mechanism in E. coli is mediated by the ribosome modulation factor (RMF)10, which binds to a site in the 30S and interferes with the Shine–Dalgarno (SD) sequence. This prevents the interaction between mRNA and the 16S rRNA and leads to the formation of 90S dimers11. Subsequently, the hibernation promotion factor (HPFshort, YhbH) binds to a site that overlaps with that of mRNA, tRNA and initiation factors. Binding of HPFshort modifies the structure of the 90S dimer to form the 100S complex11. Alternatively, a competing homolog of HPFshort, named YfiA (protein Y, pY or earlier termed RaiA or the ribosome-associated inhibitor A), inhibits ribosome activity by binding to the same location as HPF, but its C terminus protrudes into the binding site of RMF preventing 100S formation. Only the binding of RMF induces the conformational change in the 30S head domains, which has been proposed to directly take part in 100S formation11. Although rmf may be specific for γ-proteobacteria14, most bacteria and some plant plastids15, 16 carry a gene homologous to hpf short, here referred to as hpf long, and form 100S particles through a different mechanism, which we describe in this paper.
This second mechanism of dimerization exists in the majority of bacteria7 and is mediated by a long version of HPF alone6, 17–19. This homolog of YfiA and HPFshort is about twice as long and its C-terminal domain has no counterpart in E. coli. In a previous study6, we demonstrated that deletion of the C-terminal part of this dual-domain protein (hereafter named HPFlong) in Lactococcus lactis results in the loss of dimerization. Thus, whereas the N-terminal domain may have a similar role and binding site as YfiA and HPFshort 20, the C-terminal domain of HPFlong is necessary for dimer formation. Although the C-terminal domain differs from RMF, HPFlong may induce a similar conformational change in the head region of the 30S subunit to facilitate the formation of 100S dimers4, 6. Alternatively, HPFlong may directly trigger dimer formation upon binding at the interface, as has been suggested by Khusainov et al.20. To determine the binding site of HPFlong and the actual mechanism of HPFlong-mediated dimerization, higher resolution structures are required6, 7, 20, which we provide in this study.
We obtained by single particle cryo-transmission electron microscopy (cryo-TEM) two overall 100S maps of L. lactis ribosomes in different conformations at 19 Å resolution, which show a distinct rotational freedom of movement of 55° around the interface. We furthermore obtained a density-map of the L. lactis 70S ribosome as part of the dimeric complex at 5.6 Å resolution, and were able to confidently align two copies of the ribosome to reconstruct the 100S structure thus significantly improving the density at the interface. Using the recently published structure of the 70S ribosome of the Gram-positive bacterium Bacillus subtilis (PDB-code 3J9W21) as a starting model, as well as the highest resolution E. coli 70S structure (PDB-code 4YBB22), we were able to model the structure of the L. lactis 70S ribosome. Moreover, we located both the N- and C-terminal domains of HPFlong in the 100S ribosome as well as the interaction sites of the 70S particles within the dimer. We now present a second mechanism of ribosome dimerization that may be widely used in the bacterial kingdom and is distinctly different from the one proposed for E. coli.
Results
EM maps of the entire 100S dimer
The dimerization interface of 100S structures is formed via interactions between the two 30S subunits. After a classification of 100S dimers into 32 classes, 62,499 of 163,121 particles were present in nine classes with a resolution better than 30 Å. Three different conformations were found (Supplementary Fig. 1). After aligning the bottom halves of the 100S ribosomes, a distinct change in position of the L1 stalk can be observed. The three conformations indicate a rotation of up to 55° around the 30S–30S interface. Also, the distance between the two ribosome monomers with respect to the interface increases from panel a to c; the different states are termed closed, intermediate, and open. The position of the L1 stalk does not change between particles of the most dominant closed state (59% of particles), and, contrary to what has been reported for dimers from E. coli 9, those dimers show an unexpected lack of flexibility around the interface, despite the very narrow dimer connection. In contrast, in particles belonging to the open conformation (35%), the position of the L1 stalk is displaced perpendicular to the interface, demonstrating an increased mobility, with respect to the closed state.
EM map of a monomeric ribosome within the dimer
Due to the conformational heterogeneity of the dimeric ribosomes, we used masks to refine the 30 and 50S subunits separately. A mask around the 30S subunit of one of the monomers yielded a density map at 5.9 Å (data not shown), and masking the 50S subunit of one of the monomers (Supplementary Fig. 2), yielded a density map at 5.6 Å resolution (Supplementary Figs. 3–5). The 5.9 Å (30S-masked) density map confirms the 5.6 Å (50S-masked) map. Therefore only the latter was used for modeling. The resolution is the highest in the center of the 50S subunit and follows a gradient through the 30S and toward the interface (Supplementary Fig. 3). A lower resolution for the 30S subunit is common in EM maps of ribosomes and is attributed to movement and flexibility within this part of the ribosome.
Model of the 70S ribosome
The genome of L. lactis (subsp. cremoris, MG1363) (NCBI Reference Sequence: NC_009004.1) encodes 20 and 32 proteins of the 30 and 50S subunit, respectively. Compared to B. subtilis, L. lactis lacks proteins L7a and bL25, and proteins uS14 and bL31 are present only as a single variant. The amino-acid conservation in ribosomal proteins is high. Along with the rRNA, we successfully assigned most of the ribosomal proteins (Fig. 1; Supplementary Fig. 6; Table 1) except uL1, L7/ L12, bL9, uL10, and uL11. Ribosomal protein bL9 may have been lost, due to a loose association with the ribosome18. The other missing proteins are known to be very flexible and, similar to the Staphylococcus aureus ribosome, lack sufficient density for modeling20. Two genes of L. lactis, llmg 0899 and llmg 2078, have been annotated as encoding ribosomal protein uS15. However, a BLAST search with the sequence alignment of their products revealed that only llmg 0899 truly encodes protein uS15. Additionally, we assigned protein bS21 in our map, which is absent in the model of B. subtilis ribosome21. For most of the protein subunits, the quality of the density was sufficient to trace the main chain, except for a few flexible loops and termini. Although at this resolution it is not possible to unambiguously assign amino-acid side chains, the quality of the map allowed their tentative assignment. The high sequence similarity between ribosomal proteins from L. lactis (PDB-code 5MYJ) and B. subtilis (PDB-code 3J9W) is reflected in their structural conservation, with the overall rmsd values for their 50 and 30S subunits of 1.65 and 2.2 Å, respectively.
Fig. 1.
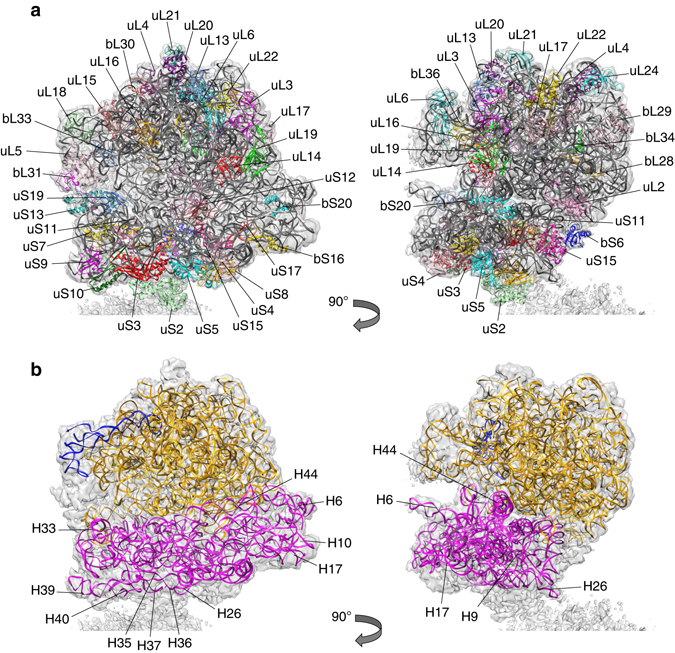
The complete 70S model of L. lactis. a Protein models fitted into the EM density and rotated over 90° with RNA in gray and proteins colored differently. Protein names are based on ref. 3. b rRNA models fitted into the EM density and rotated over 90°
Table 1.
Data collection, refinement, and validation statistics
| Data collection | |
| Microscope | Titan KRIOS with Cs-corrector |
| Camera | Falcon 2 |
| Voltage | 300 kV |
| Pixel size (Å) | 1.105 |
| Total dose (e−Å−2) | 25 |
| Micrographs collected (#) | 5275 |
| Refinement | |
| Total number of particles (#) | 163,121 |
| Particles in 50S masked map (#) | 43,530 |
| Resolution (Å; at FSCa = 0.143) | 5.6 |
| CCa (model to map fitb) | 0.800 |
| RMS a deviations | |
| Bonds (Å) | 0.008 |
| Angles (°) | 1.262 |
| Chirality (°) | 0.06 |
| Planarity (°) | 0.008 |
| Validation c | |
| Clash-scored | 18.69 |
| Proteins | |
| MolProbity score | 2.55 |
| Favored rotamers | 91.3% |
| Ramachandran favored | 82.31% |
| Ramachandran allowed | 17.54% |
| Ramachandran outliers | 0.15% |
| RNA | |
| Correct sugar puckers | 99.49% |
Model of HPFlong
In order to improve the density of the map at the dimer interface a 100S map was reconstructed (Supplementary Fig. 7). After localization of all known ribosomal protein subunits, two regions of undescribed density remained, which we assign to HPFlong (Fig. 2). One density is located at the interface between the 30 and 50S subunits, and the other at the interface between both 30S subunits. This assignment was confirmed by the high similarity between the map densities and the respective homologous structures: YfiA from E. coli bound to the 70S ribosome of T. thermophilus (PDB-code 4V8I) and the C-terminal domain of ribosome-associated protein Y (PSrp-1) from Clostridium acetobutylicum (PDB-code 3KA5; Supplementary Fig. 8).
Fig. 2.
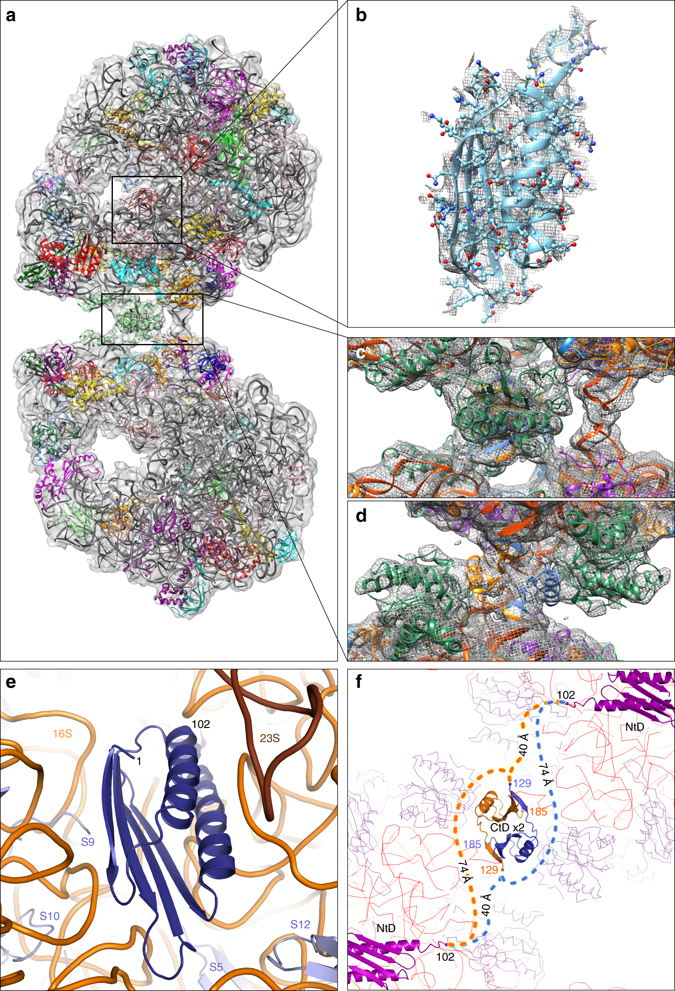
Location of the N- and C-terminal domain of HPFlong. The extra, unmodeled densities in the map, indicated by the boxes of a, correspond to HPFlong N-terminal domain (b and Supplementary Movie 1) and C-terminal domain (c, d and in Supplementary Movie 2; colored gold and blue). d has been rotated 90° with respect to a, c, showing the dimeric interaction of the C-terminal domains. e The surrounding proteins (steel blue) and RNA (orange) of the N-terminal domain. f The linking of the distant N- and C-terminal domains (NtD and CtD) is shown schematically. Their respective locations in the dimer are clarified by the surrounding ribosomal proteins (purple lines) and 16S rRNA (red lines)
Structure prediction of HPFlong by the Predict Protein Server23 (Supplementary Fig. 9) revealed a two-domain architecture connected through an unstructured, solvent-exposed linker. The HPFlong N-terminal domain (residues 1-102, Fig. 2b), located between the 30 and 50S subunits, is in close proximity to proteins uS7 and uS12 and interacts with the 16S RNA at several locations (Fig. 2e). The C-terminal domain is located at the center of the 30S–30S interface (Fig. 2c). In the PSrp-1 dimer, the interface is formed by two hydrophobic patches at the surface of a β-sheet of each monomer (three Val, one Phe, and one Ile residue on each monomer). In the L. lactis HPFlong homology model, these residues are conserved, except for a single Val to Thr substitution (Supplementary Fig. 10). Furthermore, the PSrp-1 dimer is stabilized by a parallel β-sheet formed via the interaction of the first β-strand of each monomer with the last β-strand of the other monomer. The same hydrophobic and β-strand dimer stabilization is predicted for L. lactis HPFlong (Supplementary Fig. 9). The C-terminal domain of HPFlong is thus a dimer stabilized by hydrophobic patches and intermolecular β-sheet formation.
The EM map shows almost no density for residues 103–128 that connect the N- and C-terminal domains of HPFlong, which is indicative of a high flexibility. This is in agreement with the secondary structure predictions and the lack of sequence conservation among the linker domains in HPFlong (Supplementary Figs. 9 and 10). The length of the linker is rather well conserved and typically between 24 and 34 residues in ~4600 sequences analyzed (vide infra). The first visible residue (Asp129) of the C-terminal domain of HPFlong is at a distance of either 40 or 74 Å from residue 102, depending on which of the two copies of the protein is considered (Fig. 2f). We cannot unambiguously determine which route the connecting loop follows, but the maximum reaching distance (99 Å for 26 residues, assuming an average Cα–Cα distance of 3.8 Å per residue) suggests that the long route for spanning two opposing ribosomes is well possible.
Dimer interface
Each ribosome in the 100S particle interacts at the dimer interface with both copies of the HPFlong C-terminal domain (Fig. 2c, d). From the perspective of one 70S ribosome in the dimer, the main interaction is between its uS2 protein and copy A (randomly assigned) of the HPFlong C-terminal domains; whereas RNA helix 40 and most likely the N terminus of bS18, interact with the copy B. Chain bS18 also interacts with RNA helix 26, which seems to be involved in the dimerization through interactions with helix 26 of the opposing ribosome (at least in L. lactis) (Fig. 2c).
Rotation in the 100S ribosomes
The model of the 70S ribosome can be used to compare three conformations of 100S ribosomes (Supplementary Fig. 1). Two copies of the 70S model without the C-terminal domains of HPFlong were rigid-body fitted in each map (Fig. 3). The 55° rotational difference manifests in a change of the orientation of the helices 26 of the 16S RNAs from facing each other in the closed state to interacting with protein uS2 of the opposing ribosome in the open state. The intermediate state has a lower resolution and a less defined interface, although its well-defined L1 Stalk points to a stable conformation. The fit of the two models in the intermediate map indicates that the two RNA helices 26 are interacting in this conformation; the density for HPFlong seems to be (partially) lost. Whether ribosome dimers assemble into these different conformations or are able to switch after assembly remains to be studied.
Fig. 3.
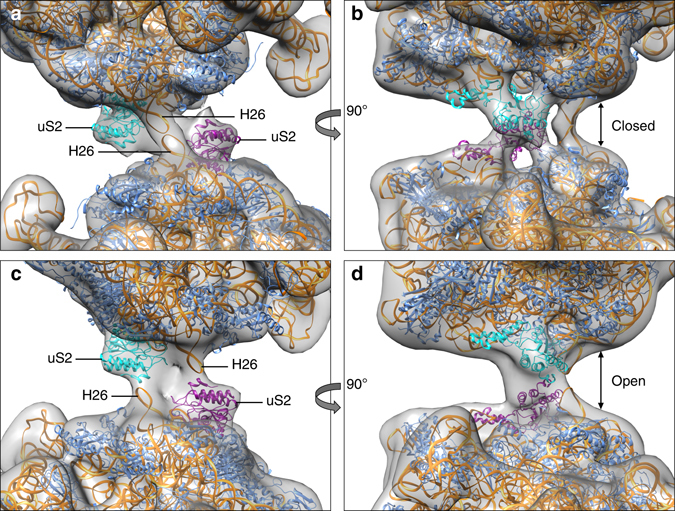
Different conformational states of 100S ribosomes. Rigid-body fits of two 70S models without the C-terminal domains of HPFlong into the low-resolution maps of the two most extreme conformations, closed state (a, b) and open state (c, d). The difference between the two classes depicts a 55° rotation around the interface. The ‘closed’ conformation closely resembles our 5.6 Å map. Colors indicate 16S rRNA (orange), protein uS2 (cyan and purple) and other proteins (blue). The rotation causes H26 to change its interaction from H26 to protein uS2 from the other ribosome, effectively widening the space between the two 70S ribosomes within the dimer (arrows in b, d)
When assigning the two C-terminal domains of the HPFlong dimer each to a specific ribosome, it was not possible to account for the two most rotated dimer conformations (Supplementary Fig. 1a, c); the C-terminal dimer was torn apart or crashed into itself. Both 100S conformations can only be accounted for when the C-terminal domains are fitted together as an intact dimer. The best fit is obtained by moving HPFlong such that the two-fold symmetry axes of the 100S and HPFlong coincide again. From the closed (Supplementary Fig. 1a) to the open state (Supplementary Fig. 1c) the dimer shifts ~6 Å and rotates 27° over the long axis. This compensates the increased distance between the two 70S ribosomes and the rotation. Most likely, the HPFlong C-terminal domains are not flexible and maintain their dimer interface, while the flexibility in protein uS2 allows the rotation.
Oligomeric state of HPFlong in solution
We performed size-exclusion chromatography coupled to multi-angle laser light scattering (SEC-MALLS) measurements to determine the oligomeric state of HPFlong in solution. Purified HPFlong in the native state has a molecular mass of ~40.5 kDa, corresponding to about twice the calculated mass on the basis of the amino-acid sequence (21.3 kDa). Thus, HPFlong forms a stable dimer in solution (Fig. 4).
Fig. 4.
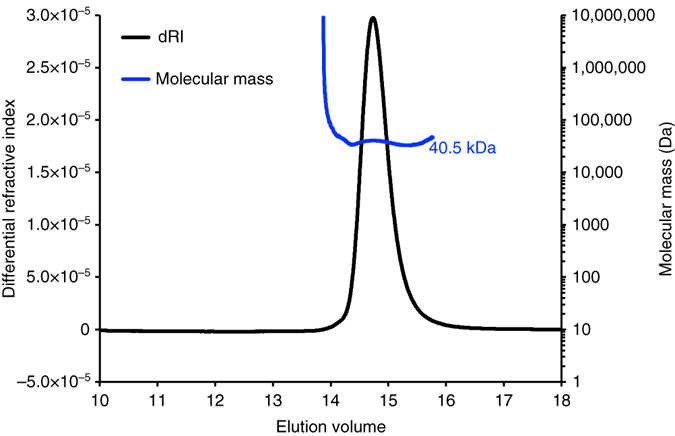
Oligomeric state analysis of HPFlong by SEC-MALLS. The Superdex 200 10/300 column (GE Healthcare) was equilibrated with 100 mM Tris-HCl, 150 mM NaCl (pH 8.0), and the protein was injected in the same buffer. The chromatogram (elution volume is indicated on the x axis) shows the readings of refractive index (RI) detector in black (the scale for the RI detector is shown in the left-hand axis). The thick blue line indicates the calculated molecular mass of the eluting protein throughout the chromatogram (scale on the right-hand axis). The calculated molecular weight is 40.5 kDa (molecular weight of a monomer is 21.3 kDa)
Distribution and phylogenetics of HPFlong
A BLASTp search for HPFlong homologs yielded about 7000 plausible hits with at least 30% sequence identity. Sorting by sequence coverage showed a steep drop from full-length (~185 residues) to half-size proteins after about 4600 hits. HPFlong homologs were present in almost all bacterial phyla, but not in Gram-positive Acholeplasma sp., Gram-negative Borrelia sp., Euryachaeota, and Crenarchaeota.
Alignment of a selected set of 110 homologs (Supplementary Fig. 10) revealed that 95 sequences cover all three regions of HPFlong, and they align well with both N- and C-terminal domains; 15 sequences covered only the N-terminal domain. In the ‘full-length’ homologs, the linker between the two domains showed very little conservation in sequence and composition, and its length varied between 16 and 62 residues; the majority being between 24 and 34 residues. In the C-terminal domain, the five hydrophobic residues at the dimer interface are highly conserved: three out of five are (almost) invariantly present, while for the remaining two a hydrophobic substitution is typically present (e.g., valine to isoleucine) (Supplementary Fig. 10).
A phylogenetic tree constructed for the 110 sequences (Fig. 5) revealed that the distribution and evolutionary relationship of HPFlong homologs is reflected in the distribution of bacterial phyla. L. lactis HPFlong appears in a clade representing (almost) all Gram-positive, low G+C phyla at one ‘end’ of the phylogenetic tree. This clade (with the exception of one S. aureus species) contains full-length HPFlong homologs with the inter-domain linker length comparable to that of L. lactis (31 residues). In the Gram-positive, high G+C actinobacteria at the other ‘end’ of the phylogenetic distribution, the linker is significantly longer. The shortest linkers are observed in the sequences from Thermodesulfobacteria. Gram-negative bacterial phyla are situated in the ‘middle’ of the phylogenetic tree. Notably, the β- and γ-proteobacteria (Neisseria and Escherichia species) only contain short YfiA homologs (originally described as HPF and YfiA). Interestingly, RMF homologs were only found (by BLASTp) in γ-proteobacteria.
Fig. 5.
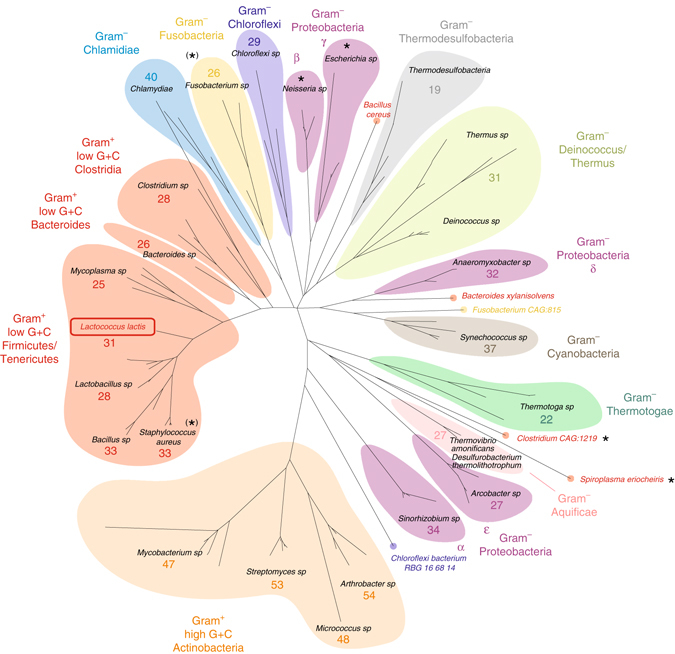
Phylogenetic tree of HPF homologs. The tree is based on 110 proteins homologous to HPFlong from L. lactis. The average linker length of HPFlong is given by the number of residues in each of the lineages
Discussion
The here-reported structure of the L. lactis 70S ribosome is that of the fifth bacterial species. Structural variations in ribosomes have been reviewed by Khusainov et al.20; and some of them (e.g., variations in the lengths of rRNA loops) could point to the involvement of rRNA extensions in translation control in a species-specific manner. Most knowledge on initiation, translation, hibernation, mRNA recognition, and ribosome management is based on studies in E. coli. The lack of (structural) studies on ribosomes across the bacterial kingdom is one of the most important shortcomings in understanding vital and clinically relevant species-specific regulation of translation and phenomena such as antibiotic resistance20.
We report a novel mechanism of ribosome dimerization via a long-HPF-type two-domain protein. The N-terminal domain of HPFlong is structurally conserved and has a similar function and binding site as YfiA and HPFshort from E. coli (Supplementary Fig. 11). It blocks ribosome function by binding to the sites for tRNA, initiation factors IF1 and IF3, and elongation factor G11, thereby inhibiting ribosome activity and preventing the dissociation of the 70S particles into the 30 and 50S subunits24–26. Analogous to HPFshort from E. coli, the N-terminal domain of HPFlong strengthens the association of the 30 and 50S into 70S ribosomes, which renders them less susceptible to endoribonuclease action and degradation27.
In contrast to RMF from E. coli, the HPFlong C-terminal domain does not interact with the SD-aSD sequence but with protein uS2 at the dimer interface. Protein uS2 is essential for ribosome function28, 29 and is also part of the mRNA ‘platform binding center’ (PBC) along with proteins uS7 and uS11 and 16S rRNA helices 26 and 4030, 31. This platform has been proposed to be a common site dedicated to binding mRNAs prior to the actual translation initiation to increase control over translation30. Supplementary Fig. 12 clearly demonstrates the overlap between the L. lactis dimer interface and the locked mRNA structure. We therefore propose that the HPFlong C-terminal domain blocks the PBC by acting as a general, rather than a specific, inhibitor of mRNA translation initiation.
We could not unambiguously connect the N- and C-terminal domains of HPFlong in 100S ribosomes. The linker is sufficiently long to link each N-terminal domain to each C-terminal domain and allow the rotation over the dimer interface (Fig. 3). The shorter (~40 Å) and longer route (~74 Å) can be bridged by the linker in the majority of HPFlong homologs. It is even possible that both routes are used, because HPFlong is a natural dimer, strongly interacting via the two C-terminal domains. This is supported by (i) the finding that a dimeric HPFlong fits both main conformations of the 100S ribosome (Fig. 3); (ii) the hydrophobic nature of the dimer interface; (iii) the fact that these hydrophobic residues are conserved in all proteins that carry the C-terminal domain (Supplementary Fig. 10); (iv) the X-ray crystal structures of two homologs (PDB-codes 3K2T and 3KA5, see Methods section); and (v) the SEC-MALLS experiments (Fig. 4).
We compared the ribosome dimerization mechanism by HPFlong in L. lactis with that of HPFshort and RMF in E.coli 5, 9 (Fig. 6). We used the map of Kato et al.9, because it is of higher resolution (~18 Å) and in better agreement with the structure compared to the map from Ortiz et al.5 (Supplementary Fig. 13). Superposition of the maps from E. coli and L. lactis based on the bottom ribosomes reveals the enormous difference in overall orientation of the top ribosome. The first dimer interaction site between the two 30S head-domains5, 11 is not present in L. lactis, and we propose that also in E. coli only one interaction site exists, in agreement with the map from Kato et al.9, and the very weak and loose nature of the E. coli dimer interface5, 9.
Fig. 6.
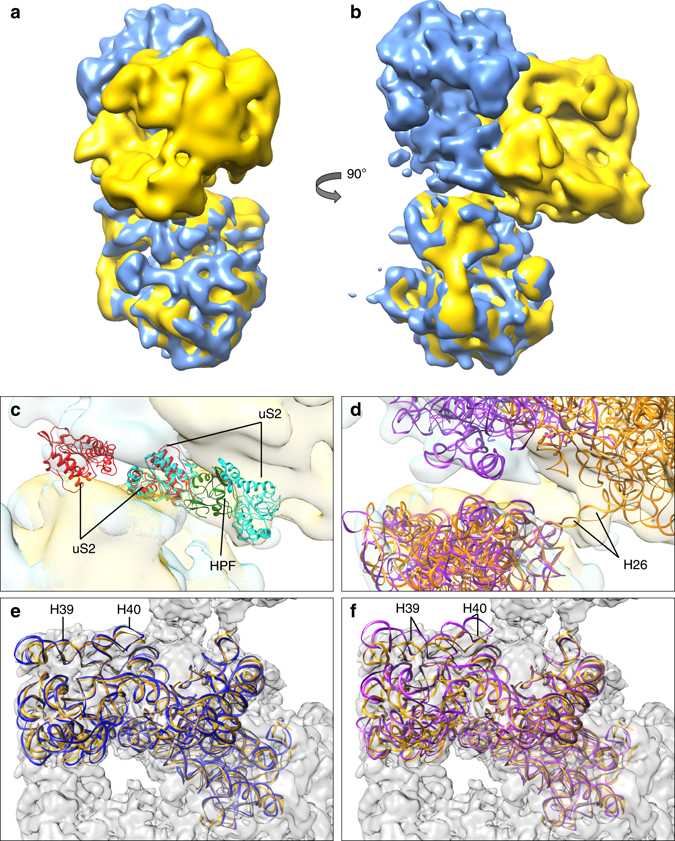
Ribosome dimerization: RMF vs. HPFlong. a–d Comparison of 3D maps of E. coli (blue 9) and L. lactis (yellow) 100S ribosomes. The density for the E. coli ribosome was obtained by taking the maximum voxel value of two copies of the EM-density map from Kato et al.9. Two copies of the L. lactis model and two copies of the T. thermophilus structure with RMF (PDB-code 4V8G), thus displaying the mechanism of E. coli, not T. thermophilus, were fitted into the corresponding density (c, d). c Zoom in of dimer interface showing proteins uS2 (marked red for T. thermophilus and cyan for L. lactis). In L. lactis the dimerization process is mediated by the C-terminal domain of HPFlong (green), whereas in T. thermophilus uS2 interacts with proteins uS3, uS4, and uS5 from the opposing ribosome (previously described as second contact site11). d The RNA chains of the L. lactis 100S (orange) and T. thermophilus 100S structures (purple). Helix 26 plays a key role in dimerization of L. lactis ribosomes but not in T. thermophilus. e, f Comparison of the 16S rRNA of ribosomes from L. lactis (orange) to T. thermophilus in apo-state (e, blue) and RMF-bound state (f, purple). The 100S map from L. lactis is depicted in gray in the background. By looking at the helices 39 and 40 of the head domain, it is clear that the ribosome from L. lactis is in the apo-state and does not undergo a conformational change upon binding of HPFlong
Despite the large difference in the overall 100S structures, the main protein involved in the interface of E. coli and L. lactis dimers is uS2. The actual binding site on uS2 is not conserved (Fig. 6c). In E. coli, this dimer interaction site comprises the binding of uS2 to uS3, uS4, and uS5 from two opposing ribosomes5, 9, 11 (Supplementary Fig. 13). The uS3–uS5 proteins form the mRNA entrance tunnel, which is blocked by the dimerization. In L. lactis, uS2 binds to the C-terminal domain of HPFlong, which causes the actual dimerization, and proteins uS3–uS5 are not part of the interface.
In E. coli, HPF and YfiA are not completely able to prevent the separation of 30 and 50S for initiation32. Instead, they specifically hinder translation of leaderless mRNA14, slowing down their translation, but also reducing translation errors33. RMF is essential to further block translation. It induces dimerization by a conformational change in the ribosome 30S head-domains. While the N-terminal domain of HPFlong is analogous to YfiA and HPFshort, the binding-site of the C-terminal domain from L. lactis HPFlong is not the same as that of RMF. Still, the HPFlong C-terminal domain likely also completes the ribosome inactivation. The direct involvement of HPFlong C-terminal domain at the dimer interface leaves the SD-aSD sequence accessible in the L. lactis ribosome dimers. Also, contrasting our previous analysis6, there is no conformational change in L. lactis ribosomes upon binding of HPFlong (Fig. 6e, f).
The existence of two different mechanisms of ribosome dimerization, and the conservation of protein uS2 as main interaction partner, supports the idea that dimerization has been rescued in γ-proteobacteria by the rmf gene product upon loss of the C-terminal domain of HPFlong. Given the fact that RMF only exists in γ-proteobacteria, our homology search confirms and extends the conclusion that the E. coli dimerization mechanism, albeit best studied, is in fact not the most widely spread strategy14. Full-length homologs of L. lactis HPFlong were found in almost all bacterial phyla. Few lacked the N-terminal domain (e.g., some M. tuberculosis, S. aureus sp.) or even an HPF homolog at all (e.g., Acholaplasma and Borrelia sp. as well as Archaea); all Neisseria sp. lacked both a C-terminal domain as well as RMF in their genome. These organisms may lack or have alternative mechanisms of translation regulation and ribosome stabilization.
One of the key differences between ribosome hibernation in L. lactis and E. coli relates to the states of HPFlong and RMF. While in E. coli two unoccupied ribosomes each collect one copy of RMF and then dimerize, HPFlong is a dimer in solution and therefore it can in principle initially gather one, and then the other ribosome (Fig. 7). Our findings indicate that the 70S–70S interactions in L. lactis are due to HPFlong exclusively. With increasing levels of HPFlong, more 100S ribosomes are formed but there will be an optimum. If HPFlong is produced in excess of ribosomes, the equilibrium will shift from 100S to 70S-HPFlong, i.e., monomeric ribosomes with dimeric HPFlong bound (Fig. 7). Both 100S and 70S-HPFlong are inactive, but the dimeric state may protect against degradation by shielding protein uS2, which forms the main dimer interaction site in both L. lactis and E. coli. Next to the HPFlong/ribosome ratio, the presence of mRNA and the availability of amino acyl-tRNA (more abundant in the exponential than in the stationary phase) determine whether the ribosomes are monomeric or dimeric.
Fig. 7.
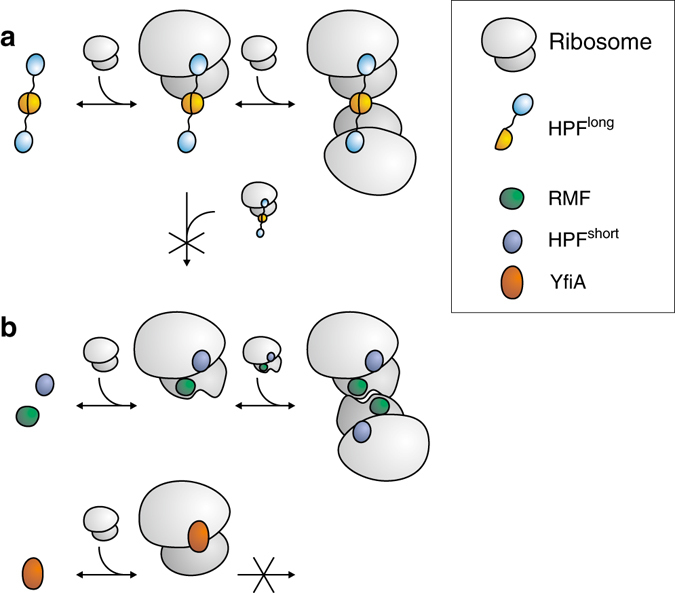
Schematic of dimerization of ribosomes in L. lactis and E. coli. Because HPFlong is a dimer in solution it will collect two ribosomes consecutively (a). Prolonged exposure to an excess of HPFlong shifts the equilibrium toward monomeric inactive ribosomes. In E. coli, (b) each ribosome first collects a copy of HPFshort and RMF. The conformational change upon binding of RMF allows the two ribosomes to dimerize. When YfiA binds at the location of HPFshort, RMF can no longer bind and the ribosomes do not dimerize
Ueta et al.18 have proposed that in the exponential phase of growth HPFlong associates with 100S but not with the 70S particles. The expression of HPFlong increases at the onset of the stationary phase of growth and thus more dimers are formed. During prolonged stationary growth, the number of 70S ribosomes associated with HPFlong increases and the number of dimers decreases. We now explain these observations by the fact that HPFlong is a dimer. As occasionally a 100S particle gets reactivated, the balance shifts from 100 to 70S. Notably, in E. coli, HPFshort and YfiA compete for the same binding site where YfiA, but not HPFshort, prevents binding of RMF. This can also lead to two populations of inactivated ribosomes, but in this case the 70S particles have YfiA bound and the 100S particles have HPF and RMF bound.
In this study, we present a new HPFlong-mediated mechanism of ribosome hibernation, which may be present in nearly all bacterial species and shows stark differences to the RMF-induced dimerization in γ-proteobacteria. HPFlong is a dual domain protein that forms a dimer in solution. Although the N-terminal domain of HPFlong binds in the same location as YfiA and HPFshort from E. coli, the location of the C-terminal domain does not overlap with that of RMF. While RMF induces a conformational change to allow dimerization, the dimeric C-terminal domains of HPFlong directly form the dimer, to which the two ribosomes, remaining in apo-state, are bound. In E. coli, ribosomal activity is inhibited by dimer formation, which blocks the mRNA entrance tunnel, and by RMF itself, which interacts with the SD-aSD sequence. In contrast, in L. lactis the SD-aSD and mRNA tunnel are still accessable, and ribosome activity is hindered by the C-terminal domain of HPFlong, which blocks the PBC. Interestingly, both species are able to shift ribosome hibernation from a dimeric inactive to a monomeric inactive state. In E. coli, this can be realised by different expression levels for YfiA and HPFshort, while in L. lactis HPFlong alone is responsible. Thus, our study gives an example of how vital functions in bacterial physiology can evolve through the acquisition of different molecular mechanisms in different species.
Methods
Cell growth and purification
Cells were grown and ribosomes collected as described previously6. In short: L. lactis MG1363 strain were grown as standing cultures in M17 medium (Difco Laboratories, Detroit, MA, USA), supplemented with 0.5% (w/v) glucose (GM17); 5 µg ml−1 chloramphenicol and/or 2.5 µg ml−1 erythromycin were added when cells were transformed with the required plasmid(s).
L. lactis cells were collected by centrifugation (7000×g, 10 min, 4 °C) after 3 h (logarithmic growth phase, 70S fraction) or 7 h (stationary growth phase, 100S fraction). The cell pellet was resuspended in buffer I (20 mM Tris-HCl, pH 7.6), 15 mM magnesium acetate, 100 mM ammonium acetate, and 6 mM 2-mercaptoethanol containing 1 mM phenylmethylsulphonyl fluoride (PMSF), and lysed by vortexing with 0.2 mg glass beads in an ice-cold Tissue lyser (Qiagen, Venlo, The Netherlands). The homogenate was centrifuged (9000×g, 15 min, 4 °C), the supernatant was collected and the pellet resuspended in buffer I supplemented with 1 mM PMSF. The suspension was centrifuged again and the combined supernatants were layered onto a 30% sucrose cushion in buffer I and centrifuged (206,000×g for 3 h at 4 °C). Resuspension of the pellet in buffer I gave a crude ribosome preparation.
To further purify ribosome monomers and dimers, 400 µl of ribosome preparation was loaded onto a linear 10–40% 12 ml sucrose density gradient column in buffer I and centrifuged in a SW 32.1 Ti rotor (Beckman) at 125,000×g for 80 min at 4 °C. The gradient was divided into 400 µl fractions after which their 260 nm absorbance was measured using a UV-1700 CARY bio UV-Visible Spectrophotometer (Agilent Technologies, Palo Alto, CA, USA). The fractions containing the ribosome monomers (70S) or dimers (100S) were dialyzed at 4 °C against buffer I and prepared for electron microscopy.
Electron microscopy
A volume of 3 µl of ribosome sample of either 100- or 70S-purified ribosomes was pipetted onto glow-discharged 400 mesh holey carbon grids (Quantifoil, 2/2) and plunge-frozen in liquid ethane with a Vitrobot (FEI, Eindhoven, The Netherlands). All images were recorded on a Titan KRIOS (FEI) equipped with a high brightness field-emission gun operated at 300 kV, a Cs-corrector (CEOS, Heidelberg, Germany) and a Falcon 2 DED camera (FEI) at the NeCEN (Leiden, The Netherlands). Images were taken automatically with EPU software (FEI) using a dose of 25 e− per Å2 and focus settings of 1000, 1600, and 2500 nm defocus with a final pixel size of 1.105 Å. No movie-frames were recorded.
Data processing
From the raw images, the purified 70S peak-fraction micrographs seemed to contain as many dimers as the 100S peak. Likely, HPFlong dimers were co-purified with monomeric ribosomes in the 70S fraction18, 34. During 2D and 3D processing, 100S particles from cells in the exponential and stationary phase of growth did not separate into different classes, but mixed in equal amounts. Therefore both data sets were combined to increase the amount of data.
CTF parameters were estimated using CTFFIND435. The resulting _avrot.txt files were used to select micrographs with a CTF fit better than 7 Å, by applying a 0.1 threshold for the drop in the cross-correlation between calculated fit and data. This yielded a total of 2713 micrographs of the original 100S fraction and an additional 2562 micrographs of the 70S fraction.
RELION 1.3 and 1.436 were used for all processing apart from the initial model. Due to the lack of mass in the center of the dimers, automated particle picking resulted in uncentered picks. On top of that, the high similarity between 70 and 100S particles, which were both present in all micrographs, rendered 2D and 3D cleaning insufficient. To avoid the presence of monomeric particles in the dimeric structure, particle picking was done manually. This was done in RELION 1.3 for the 2500 nm defocus images and in Xmipp 3.037 for the micrographs that were taken with less defocus, making use of contrast enhancement and band-pass filtering.
Initially, particles were down-sized four times and the dataset was cleaned using reference-free 2D classification in RELION. To obtain an initial model, the last 2D class-sums were exported into Xmipp 3.0 to run two rounds of RANSAC38 feeding the best result of the first as a model for the second round after which ten very similar models were obtained, one of which served then as initial model in RELION 3.1 for the first 3D classifications. All references were low-pass filtered to 60 Å to avoid a strong influence of the reference on the results.
Visualization and comparison of 3D models were done using Chimera39. Volume alignments based on one half of the dimer were obtained by applying the segmentation tool40 and grouping the segments until two monomeric maps remained. The freedom of rotation within the dimer was measured taking the two most extreme conformations and first fitting one class on the bottom segment of the other using the fit in volume tool. Secondly, that same class was fitted onto the top segment. The tool gives the applied rotation.
The clean set contained 62,499 particles and was further processed with unbinned pixels. To compensate the movement, the next classifications and refinement used several soft-edged masks (Supplementary Fig. 2). Simple circular masks were created in Xmipp 3.0 and tighter masks were created in RELION 1.3 based on sub-volumes that were created using the segmentation tool in Chimera. The final masked map totaled 43,530 particles (28,747 originating from the 100S and 14,782 from the 70S fraction) and was refined in RELION 1.4 based on a 50S mask, while the auto-mask function of RELION 1.4 was used to create the final masks for post-processing to allow for a tighter mask and obtain information beyond the 50S mask. Post-processing was done in RELION 1.4, correcting the map for the modulation transfer function of the detector. For sharpening, two ad-hoc b-factors were used: −150 and −100. To limit the sharpening of noise in the 100S interface, the second, more conservative value was used prior to reconstructing the 100S map from two copies of the map (vide infra). Reported resolutions are based on the gold-standard FSC = 0.143 criterion41 and local resolutions were determined in Resmap42. Since the final map from the 50S mask (5.6 Å) and 30S mask (5.9 Å) were in agreement, all modeling was based on the 50S map.
The 100S density map was reconstructed from two copies of the 5.6 Å resolution map (Supplementary Figs. 3 and 7), which were first fitted into the best 100S class (19 Å resolution, Supplementary Fig. 1a) and then into each other using the Fit Volume tool in Chimera. The single 100S was created by taking the maximum voxel value between the two aligned maps, getting for each voxel the largest value. This two-fold symmetrical 100S map with improved density at the interface was only used to model the interface.
Modeling
The structural modeling was started by rigid-body fitting the B. subtilis structure (PDB-code 3J9W21) in the density map in COOT43. RNA sequences for L. lactis were taken from the RNAcentral database (http://rnacentral.org) and models were built using the B. subtilis ribosome as a template. The RNA was adjusted manually and subsequently refined using ERRASER44. The protein models were either generated in Phenix45 or with Phyre2 server46. The obtained model was refined using Phenix real-space refinement47 at the resolution of 5.6 Å including the simulated annealing protocol. In addition the E. coli 70S ribosome model (4YBB) was used as a reference for modeling the conserved part of the model, because it is currently resolved to the highest resolution (2.1 Å).
In order to model the densities corresponding to HPFlong (185 residues), first the crystal structure of the T. thermophilus 70S ribosome bound to protein Y (YfiA) (PDB-code 4Y4O) was superposed on top of our 70S map, showing that YfiA coincides well with one patch of the density. The N-terminal domain of HPFlong (residues 1–102) was placed there and subsequently refined using the original model as a guide. To model the C-terminal domain (residues 129–185) of HPFlong, we threaded the L. lactis HPFlong sequence based on the best fitting template from a symmetry search in COTH48 and performed a Phyre2 search46. The best Phyre2 result was the model of residues 119–174 of ribosome-associated protein Y (PSrp-1) from C. acetobutylicum (PDB-code 3KA5). The most homologous structure was of Lmo2511 protein from L. monocytogenes (3K2T). Both homologs and the symmetry prediction template had the same symmetry and a highly comparable secondary structure and conserved dimer interface. The structure of PSrp-1 was used as a template for one-to-one threading, because of its annotation to protein Y (as opposed to unknown), higher resolution (1.8 vs. 2.4 Å) and the slightly better model quality. The PSrp-1 crystal structure was modeled as a dimer in the asymmetric unit.
All chains, except the HPFlong C-terminal domain, were modeled to the single 5.6 Å map. The C-terminal domain of HPFlong and dimer interaction of H26–H26 were modeled to the 100S map that was created from two copies of the map and started with a rigid body fit of two copies of the ribosome 70S model and the HPFlong C-terminal dimer. Refinement in Phenix at 6 Å resolution yielded spontaneous base-pairing in H26 without further manual adjustments.
Homology searches and phylogenetic tree
To search for L. lactis HPFlong homologs, its sequence (NCBI accession number WP_011834629.1) was used as a query sequence in BLASTp (http://www.ncbi.nlm.nih.gov/BLAST/), setting a limit of 10,000 (number of homologs to identify). We manually removed false hits and set additional thresholds at 30% sequence identity and 40% coverage; the latter to draw out HPFshort homologs as well.
We then selected 110 homologs from as many different bacterial phyla as possible49, choosing (if present) four sequences with full coverage and two sequences with ‘half’ coverage (Supplementary Fig. 10). Sequence alignment was performed by MUSCLE in MEGA750, using default parameters. Phylogenetic analysis was also done in MEGA7 (using the Maximum Likelihood method and the JTT matrix based model, with partial deletion of positions containing gaps and missing data). In addition, a BLASTp search with the E. coli RMF sequence was performed, yielding 711 homologs.
Oligomeric state of HPFlong
We performed SEC-MALLS to study the oligomeric state of HPFlong in vitro. MALLS is a spectroscopic technique, which has proven to be one of the best for the determination of oligomeric states of polymers, including proteins in solution51. For this purpose, HPFlong from L. lactis encoded with N-terminal Strep-tag was overexpressed in L. lactis NZ9000 and purified on a Strep-tactin Sepharose column (IBA-GmbH, Göttingen, Germany) as described previously6. Purified protein was concentrated (to about 4 mg ml−1) in buffer S (100 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM EDTA) and immediately subjected to SEC-MALLS using Minidawn TREOS detector (Wyatt Technologies, USA) and, based on the obtained profile, the molecular mass was calculated using ASTRA software (Wyatt Technologies, USA). In total, three batches of HPFlong were purified and subjected to SEC-MALLS. Two included 10% glycerol in the buffer and the final measurement was done in the absence of glycerol to flatten the baseline for the refractive index measurements. All three data sets were in agreement and showed that HPFlong is a dimer.
Data availability
The raw data that support the findings of this study are available from the corresponding author on request. Final EM maps and model are available online under PDB accession code 5MYJ and EMDB accession code EMD-3581.
Electronic supplementary material
Acknowledgements
We are grateful to Dr Rishi Matadeen for support at the NeCEN, Dr Laura van Bezouwen for valuable discussions regarding particle processing, Michiel Punter for his computer support and the Center for Information Technology of the University of Groningen, in particular Dr Bob Dröge, for all their computer support and for providing access to the Peregrine high-performance computing cluster. This research is financed in part by the BioSolar Cells open innovation consortium, supported by the Dutch Ministry of Economic Affairs, Agriculture and Innovation (LEF). The work was further funded by a NWO TOPGO (L.10.060), ERC Advanced grant (ABCVolume) to B.P. and NWO Vidi grant (723.014.002) to A.G.
Author contributions
Concept supervision and design: A.G., E.J.B., and B.P.; SEC-MALLS experiments: V.A.; sample preparation: P.P.; data acquisition: G.T.O., data processing: L.E.F.; modeling and interpretation: L.E.F., T.P., and A.G.; manuscript writing: L.E.F., G.T.O., T.P., B.P., E.J.B., and A.G.
Competing interests
The authors declare no competing financial interests.
Footnotes
Electronic supplementary material
Supplementary Information accompanies this paper at doi:10.1038/s41467-017-00718-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Poehlsgaard J, Douthwaite S. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 2005;3:870–881. doi: 10.1038/nrmicro1265. [DOI] [PubMed] [Google Scholar]
- 2.Schellhorn HE, Audia JP, Wei LI, Chang L. Identification of conserved, RpoS-dependent stationary-phase genes of Escherichia coli. J. Bacteriol. 1998;180:6283–6291. doi: 10.1128/jb.180.23.6283-6291.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutiérrez-Ríos RM, et al. Regulatory network of Escherichia coli: consistency between literature knowledge and microarray profiles. Genome Res. 2003;13:2435–2443. doi: 10.1101/gr.1387003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Starosta AL, Lassak J, Jung K, Wilson DN. The bacterial translation stress response. FEMS. Microbiol. Rev. 2014;38:1172–1201. doi: 10.1111/1574-6976.12083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ortiz JO, et al. Structure of hibernating ribosomes studied by cryoelectron tomography in vitro and in situ. J. Cell. Biol. 2010;190:613–621. doi: 10.1083/jcb.201005007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puri P, et al. Lactococcus lactis YfiA is necessary and sufficient for ribosome dimerization. Mol. Microbiol. 2014;91:394–407. doi: 10.1111/mmi.12468. [DOI] [PubMed] [Google Scholar]
- 7.Yoshida H, Wada A. The 100S ribosome: ribosomal hibernation induced by stress. Wiley Interdiscip. Rev. RNA. 2014;5:723–732. doi: 10.1002/wrna.1222. [DOI] [PubMed] [Google Scholar]
- 8.Krokowski D, et al. Characterization of hibernating ribosomes in mammalian cells. Cell Cycle. 2011;10:2691–2702. doi: 10.4161/cc.10.16.16844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato T, et al. Structure of the 100S ribosome in the hibernation stage revealed by electron cryomicroscopy. Structure. 2010;18:719–724. doi: 10.1016/j.str.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida H, et al. The ribosome modulation factor (RMF) binding site on the 100S ribosome of Escherichia coli. J. Biochem. 2002;132:983–989. doi: 10.1093/oxfordjournals.jbchem.a003313. [DOI] [PubMed] [Google Scholar]
- 11.Polikanov YS, Blaha GM, Steitz TA. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science. 2012;336:915–918. doi: 10.1126/science.1218538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huxley HE, Zubay G. Electron microscope observations on the structure of microsomal particles from Escherichia coli. J. Mol. Biol. 1960;2:10–18. doi: 10.1016/S0022-2836(60)80003-4. [DOI] [Google Scholar]
- 13.McCarthy BJ. Variations in bacterial ribosomes. Biochim. Biophys. Acta. 1960;39:563–564. doi: 10.1016/0006-3002(60)90221-3. [DOI] [Google Scholar]
- 14.Ueta M, et al. Role of HPF (hibernation promoting factor) in translational activity in Escherichia coli. J. Biochem. 2008;143:425–433. doi: 10.1093/jb/mvm243. [DOI] [PubMed] [Google Scholar]
- 15.Maki Y, Yoshida H, Wada A. Two proteins, YfiA and YhbH, associated with resting ribosomes in stationary phase Escherichia coli. Genes Cells. 2000;5:965–974. doi: 10.1046/j.1365-2443.2000.00389.x. [DOI] [PubMed] [Google Scholar]
- 16.Sharma MR, et al. PSRP1 is not a ribosomal protein, but a ribosome-binding factor that is recycled by the ribosome-recycling factor (RRF) and elongation factor G (EF-G) J. Biol. Chem. 2010;285:4006–4014. doi: 10.1074/jbc.M109.062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ueta M, et al. Conservation of two distinct types of 100S ribosome in bacteria. Genes Cells. 2013;18:554–574. doi: 10.1111/gtc.12057. [DOI] [PubMed] [Google Scholar]
- 18.Ueta M, Wada C, Wada A. Formation of 100S ribosomes in Staphylococcus aureus by the hibernation promoting factor homolog SaHPF. Genes Cells. 2010;15:43–58. doi: 10.1111/j.1365-2443.2009.01364.x. [DOI] [PubMed] [Google Scholar]
- 19.Tagami K, et al. Expression of a small (p)ppGpp synthetase, YwaC, in the (p)ppGpp 0 mutant of Bacillus subtilistriggers YvyD-dependent dimerization of ribosome. MicrobiologyOpen. 2012;1:115–134. doi: 10.1002/mbo3.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khusainov I, et al. [A glimpse on Staphylococcus aureus translation machinery and its control] Mol. Biol. 2016;50:549–557. doi: 10.1134/S002689331604004X. [DOI] [PubMed] [Google Scholar]
- 21.Sohmen D, et al. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat. Commun. 2015;6:6941. doi: 10.1038/ncomms7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noeske J, et al. High-resolution structure of the Escherichia coli ribosome. Nat. Struct. Mol. Biol. 2015;22:336–341. doi: 10.1038/nsmb.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rost B, Liu J. The PredictProtein server. Nucleic Acids Res. 2003;31:3300–3304. doi: 10.1093/nar/gkg508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Agafonov DE, Kolb VA, Nazimov IV, Spirin AS. A protein residing at the subunit interface of the bacterial ribosome. Proc. Natl Acad. Sci. USA. 1999;96:12345–12349. doi: 10.1073/pnas.96.22.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavlov MY, Antoun A, Lovmar M, Ehrenberg M. Complementary roles of initiation factor 1 and ribosome recycling factor in 70S ribosome splitting. EMBO J. 2008;27:1706–1717. doi: 10.1038/emboj.2008.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janosi L, Hara H, Zhang S, Kaji A. Ribosome recycling by ribosome recycling factor (RRF)--an important but overlooked step of protein biosynthesis. Adv. Biophys. 1996;32:121–201. doi: 10.1016/0065-227X(96)84743-5. [DOI] [PubMed] [Google Scholar]
- 27.Zundel MA, Basturea GN, Deutscher MP. Initiation of ribosome degradation during starvation in Escherichia coli. RNA. 2009;15:977–983. doi: 10.1261/rna.1381309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuroda A, et al. Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science. 2001;293:705–708. doi: 10.1126/science.1061315. [DOI] [PubMed] [Google Scholar]
- 29.Bollen A, et al. A conditionally lethal mutation of Escherichia coli affecting the gene coding for ribosomal protein S2 (rpsB) J. Mol. Biol. 1979;132:219–233. doi: 10.1016/0022-2836(79)90392-9. [DOI] [PubMed] [Google Scholar]
- 30.Marzi S, et al. Structured mRNAs regulate translation initiation by binding to the platform of the ribosome. Cell. 2007;130:1019–1031. doi: 10.1016/j.cell.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 31.Boehringer D, Ban N. Trapping the ribosome to control gene expression. Cell. 2007;130:983–985. doi: 10.1016/j.cell.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Moll I, Hirokawa G, Kiel MC, Kaji A, Bläsi U. Translation initiation with 70S ribosomes: an alternative pathway for leaderless mRNAs. Nucleic Acids Res. 2004;32:3354–3363. doi: 10.1093/nar/gkh663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agafonov DE, Spirin AS. The ribosome-associated inhibitor A reduces translation errors. Biochem. Biophys. Res. Commun. 2004;320:354–358. doi: 10.1016/j.bbrc.2004.05.171. [DOI] [PubMed] [Google Scholar]
- 34.Kline BC, McKay SL, Tang WW, Portnoy DA. The Listeria monocytogenes hibernation-promoting factor is required for the formation of 100S ribosomes, optimal fitness, and pathogenesis. J. Bacteriol. 2015;197:581–591. doi: 10.1128/JB.02223-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rohou A, Grigorieff N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015;192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scheres SHW. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.de la Rosa-Trevín JM, et al. Xmipp 3.0: an improved software suite for image processing in electron microscopy. J. Struct. Biol. 2013;184:321–328. doi: 10.1016/j.jsb.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 38.Vargas J, Álvarez-Cabrera A-L, Marabini R, Carazo JM, Sorzano COS. Efficient initial volume determination from electron microscopy images of single particles. Bioinformatics. 2014;30:2891–2898. doi: 10.1093/bioinformatics/btu404. [DOI] [PubMed] [Google Scholar]
- 39.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 40.Pintilie GD, Zhang J, Goddard TD, Chiu W, Gossard DC. Quantitative analysis of cryo-EM density map segmentation by watershed and scale-space filtering, and fitting of structures by alignment to regions. J. Struct. Biol. 2010;170:427–438. doi: 10.1016/j.jsb.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scheres SHW, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kucukelbir A, Sigworth FJ, Tagare HD. Quantifying the local resolution of cryo-EM density maps. Nat. Methods. 2013;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chou F-C, Sripakdeevong P, Dibrov SM, Hermann T, Das R. Correcting pervasive errors in RNA crystallography through enumerative structure prediction. Nat. Methods. 2013;10:74–76. doi: 10.1038/nmeth.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Afonine PV, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mukherjee S, Zhang Y. Protein-protein complex structure predictions by multimeric threading and template recombination. Structure. 2011;19:955–966. doi: 10.1016/j.str.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salah P, et al. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 2009;37:2278–5588. doi: 10.1093/nar/gkp547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guskov, A. & Slotboom, D. J. in From Molecules to Living Organisms: an Interplay Between Biology and Physics (eds Pebay-Peyroula, E. et al.) 169–183 (Oxford University Press, 2016).
- 52.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding author on request. Final EM maps and model are available online under PDB accession code 5MYJ and EMDB accession code EMD-3581.