

Abstract
Formation of 100S ribosome dimer is generally associated with translation suppression in bacteria. Trans-acting factors ribosome modulation factor (RMF) and hibernating promoting factor (HPF) were shown to directly mediate this process in E. coli. Gram-positive S. aureus lacks an RMF homolog and the structural basis for its 100S formation was not known. Here we report the cryo-electron microscopy structure of the native 100S ribosome from S. aureus, revealing the molecular mechanism of its formation. The structure is distinct from previously reported analogs and relies on the HPF C-terminal extension forming the binding platform for the interactions between both of the small ribosomal subunits. The 100S dimer is formed through interactions between rRNA h26, h40, and protein uS2, involving conformational changes of the head as well as surface regions that could potentially prevent RNA polymerase from docking to the ribosome.
Under conditions of nutrient limitation, bacterial ribosomes undergo dimerization, forming a 100S complex that is translationally inactive. Here the authors present the structural basis for formation of the 100S complexes in Gram-positive bacteria, shedding light on the mechanism of translation suppression by the ribosome-silencing factors.
Introduction
The ribosome is the universal cellular ribonucleoprotein assembly that translates the genetic code into proteins. Suppressing the translation process under stress conditions is an adaptation mechanism, as protein synthesis is a highly energy-consuming function1–4. Gammaproteobacteria such as Escherichia coli possess three ribosome-silencing factors that bind to the small ribosomal subunit and block the binding pocket of messenger RNA (mRNA) and the anticodon region of A-, P-, and E-transfer RNA (tRNA)5, 6. YfiA promotes the inactivation of the 70S, as it prevents ribosomal recycling for translation initiation7, 8, ribosome modulation factor (RMF), and hibernating promoting factor (HPF), which concertedly induce the dimerization of two 70S ribosomes into a translationally inactive, hibernating 100S complex9–11. E. coli 100S dimers are mostly found during the stationary growth phase when nutritional sources are scarce12–14. This process is reversible, as once fresh nutrient sources are introduced, the 100S dimers can dissociate within minutes back into active 70S ribosomes13, 14. In vitro studies and cryo-electron microscopy (cryo-EM) showed that the two 70S ribosomes are conjoined together through their small subunits in a ‘head-to-head’ configuration to form the 100S complex15–17.
Ribosomes of Gram-positive bacteria such as Staphylococcus aureus also form 100S dimers. However, they can be found throughout all growth phases, even when nutrients are ample15, 18–21. The significance of the existence of hibernating 100S during the exponential growth phase has not been completely understood. Recent studies suggested that the formation of 100S ribosomes in the logarithmic growth phase of S. aureus reduces the translation efficiency of a fraction of genes22. Similarly, it was demonstrated that loss of S. aureus HPF (HPFSA) causes massive ribosome breakdown upon entering the stationary phase that correlates with the onset of cell death22. In addition, deletion of HPF in Lactococcus lactis exhibited decreased viability after resuscitation from starvation conditions17 and HPF-depleted Bacillus subtilis cells ability to regrow from the stationary phase was decreased21. Finally, deletion of hpf in Listeria monocytogenes impaired the survival of the bacteria in a murine model of infection23 and compromised its tolerance to aminoglycosides24.
In contrast to E. coli, S. aureus does not carry RMF or YfiA homologs. Instead, it contains only an hpf gene that encodes another form of HPFSA, which is twice as long as HPFEC 9, 25. This long form of HPF is common among all Gram-positive bacteria and plant plastids (Supplementary Fig. 1)17, 20, 23, 26. To reveal the basis for ribosome dimerization in S. aureus, we have determined the structure of the native 100S complex (SA100S) using single particle cryo-EM. The structure revealed that the two 70S ribosomes intertwine intricately through their 30S subunits and these interactions involve ribosomal protein uS2, ribosomal RNA helices h26 and h40, and the C-terminal domain of HPFSA(C-HPFSA). A loss of dimerization sensitizes cells to heat stress. In addition, the N-terminal domain of HPFSA (N-HPFSA) blocks the mRNA and the anti-codon tRNA binding sites, demonstrating the dual functionality of the HPFSA. Finally, the structure of the S. aureus 100S dimer was found to be distinct from its analogous E. coli complex and therefore may be a potential species-specific therapeutic target.
Results
Structure determination of the SA100S complex
For cryo-EM analysis, 100S ribosome dimers were isolated and purified from a hpf-knockout S. aureus strain (USA300) harboring a high-copy plasmid encoding for the hpf allele. Tenuous density corresponding to one of the 70S monomers in the two-dimensional (2D) class averages suggested that the connection between the two 70S ribosomes is generally flexible. This was confirmed by computational sorting of the three-dimensional (3D) classes, where one ribosome was typically sharper than its interacting counterpart. To overcome the conformational flexibility of the 100S, we collected a large data set of 12,504 micrographs and used in silico classification to select a distinct homogeneous sub-population of a sufficient number of particles with RELION 2.027 (Supplementary Fig. 2). Consequently, a single 3D class of 12,570 particles, representing 5.86% of the total particles, with a well-defined orientational state for both 70S ribosomes, was obtained. These particles were further refined and reconstructed to a 8.2 Å resolution cryo-EM map. Inspection of the two 70S copies in the asymmetric reconstruction suggested that there was a clear axis of twofold symmetry within the 100S particle in agreement with the previous studies15, 16. Thus, a refinement with C2 symmetry was applied and improved the overall resolution to 6.8 Å, sufficient for molecular interpretation (Supplementary Figs. 2 and 3). Features of the ribosomes were readily identified in this map and coordinates of the 70S were fitted (Fig. 1).
Fig. 1.
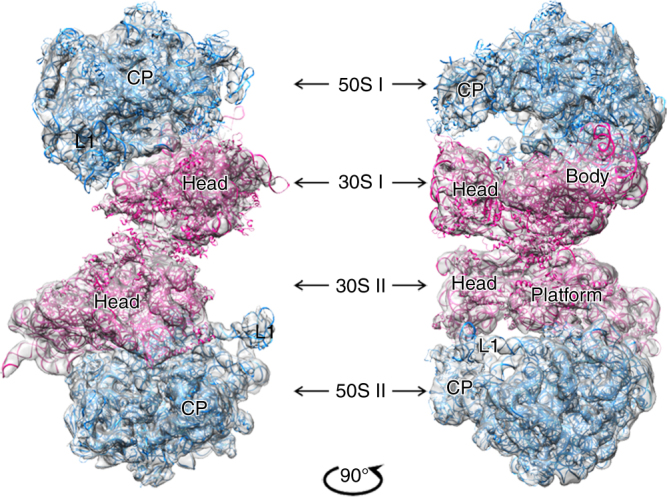
Overall structure of the SA100S ribosome dimer. The paired ribosomes within the 6.8 Å cryo-EM map of the 100S are connected to each other through the 30S subunits (colored in pink) and are oriented in a twofold symmetry relative to each other. The 50S subunits are colored in blue. Domain labels are: CP, central protuberance; L1, uL1 arm; Head, 30S subunit head; platform, 30S subunit platform; Body, 30S subunit body
In order to examine the detailed structure of HPFSA and its interactions with the ribosome, the data were re-processed as monomers, i.e., the 70S ribosomes composing the dimeric particles were treated individually, which yielded a 3.0 Å cryo-EM density map for the 70S particle (Supplementary Fig. 4). To further improve the quality of the maps and to permit accurate model building, the small and large subunits were refined separately by applying a mask on the specific subunit during its refinement. This resulted in a 3.2 and 2.9 Å resolution maps of the 30S and 50S subunits, respectively, which allowed accurate building of an atomic model of both subunits into their corresponding maps.
C-HPF domain provides binding platform for 30S dimerization
The first 100 residues that form the N-HPFSA were clearly identified and modeled using the masked 30S density map (Supplementary Fig. 5). This structure revealed that the N-HPFSA is involved in several interactions with the 16S rRNA (Fig. 2a) and it is bound between the head and the body of the small ribosomal subunit in a pocket that is composed of rRNA helices h18, h23, h25, h28-h31, h34, h44, and ribosomal proteins uS7, uS9, and uS11. Superposition of Thermus thermophilus 70S (Tth70S) complex with mRNA and three tRNAs at their binding sites (PDB code 4W2F28) on one of the 70S ribosomes composing the SA100S dimer presented here demonstrates that N-HPFSA physically blocking the binding pocket of mRNA and tRNA (Fig. 2b).The first step of prokaryotic protein synthesis involves the binding of a specific initiator methionyl tRNA and mRNA to the small ribosomal subunit to form an initiation complex. The large ribosomal subunit then joins the complex to form an elongation-competent ribosome. The adaptor function of the tRNAs involves two distinct regions of the molecule. All tRNAs possess CCA at their 3′-terminus and amino acids are covalently attached to the ribose of the terminal adenosine, whereas the mRNA template is recognized by the anticodon loop, located at the other end of the tRNA through complementary base pairing. Hence, blocking the binding pockets of the mRNA and tRNA molecules inhibits the translation process. The sequences of the N-terminal 100 residues of HPFSA and the short HPFEC have 29% identity, with high structural conservation (Supplementary Fig. 1). RMFEC also shares a similar degree of identity with the middle part of HPFSA; therefore, it was presumed that S. aureus long HPF might be a hybrid of the short HPF and RMF. However, superposition of the crystal structure of Tth70S in complex with RMFEC 5 to this HPFSA-70S map, revealed no extra density at RMFEC-binding pocket or at its vicinity (Supplementary Fig. 6).
Fig. 2.
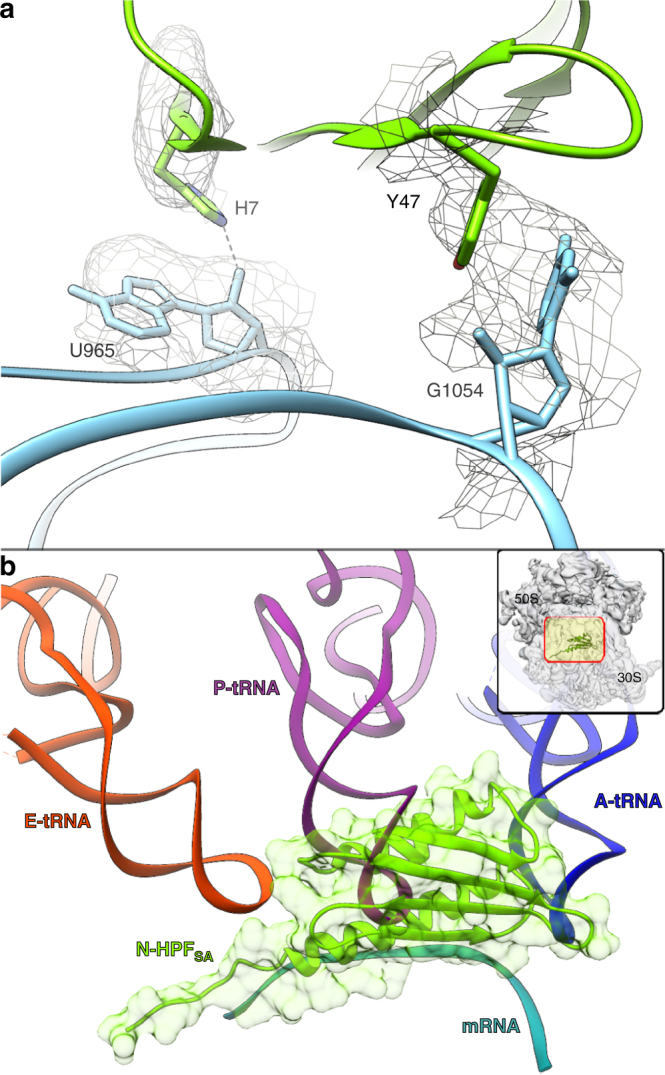
Structure of the N-terminal domain of HPF. a The N-HPFSA (chartreuse) is interacting with the rRNA (light blue) in several locations. For example, H7 form hydrogen bonds with U965 (E. coli numbering) and Y47 is stacked with the base of G1054. b Superposition of Tth70S complex with mRNA (cyan) and three tRNAs (A-site in blue, P-site in magenta and E-site in orange, PDB code 4W2F) on one of the 70S ribosome composing the SA100S dimer presented here, demonstrating that the N-HPFSA blocks the binding sites of mRNA and the anti-codon region of all three tRNAs. The N-HPFSA location on a single 70S monomer is shown in the top right inset
To further improve the density at the 100S ribosome dimer interface, we performed a focused refinement on this region with both 50S subunits masked out and applying C2 symmetry. This yielded a 6.7 Å resolution map of the dimer interface (Supplementary Fig. 2). The improved density allowed interpretation of the interaction network between the two 30S subunits. In our 100S model, protein uS2 and helix h26 of the 16S rRNA are intertwined so that h26 of one ribosome is interacting with uS2 of the other ribosome and vice versa (Fig. 3a, b). At the interface of the two subunits an additional density was observed (Supplementary Fig. 7). This density was also characterized by a twofold symmetry, similar to the twofold between the two 70S ribosomes that form the 100S dimer, hence improved when C2 symmetry was applied (Supplementary Fig. 2). Given that all the ribosomal proteins were built, as well as the N-HPFSA, we assigned this density to the C-HPFSA. Homology modeling based on the available crystal structure of a ribosome-associated factor Y from the Gram-positive bacteria Streptococcus pyogenes (PDB ID: 3LYV) and on biochemical assays (Supplementary Fig. 7D–H), we suggest dimerization of the C-HPFSA (Supplementary Fig. 7C). The local resolution of ~ 6 Å in this region allowed fitting the C-HPFSA dimer at the 100S interface, showing that it further interacts with helix h40 of the adjacent 30S (Figs. 3a, b). Owing to the limited resolution of the cryo-EM maps in this region, the specific interactions between the two C-HPFSA domains that allows self-dimerization could not be characterized. However, the homology model suggests a 3D domain-swapping mechanism in which the β-strand of one C-HPFSA is packed in parallel against the antiparallel β-sheet of its dimer counterpart (Fig. 3c). The fact that the intra β-strand interaction is parallel to the β-sheet makes the dimer less stable.
Fig. 3.

Close-up view of interface between the two 30S subunits. a h26 of the top ribosome (pink) is interacting with uS2 of the bottom ribosome (blue) and vice versa (h26 in magenta and uS2 in light blue). b h40 of the top ribosome (yellow) is interacting with C terminal HPFSA (C-HPFSA) of the bottom ribosome (dark green) and vice versa (h40 in orange and the C-HPFSA in light green). View of the whole 100S and the interface region within the 100S structure of both a and b is shown in the upper inset. c Homology model of the C-HPFSA suggests the two domains self-dimerized via β-sheet interactions. d Superposition of SA100S and S. aureus 70S (SA70S PDB code 5LI0). Helices 26 and 40 are in blue and hot pink, respectively. Ribosomal protein uS2 of SA100S is in light blue, uS2 of SA70S is in light pink, and the rRNA of both structures is in gray
To confirm the cryo-EM structure observation, we constructed S. aureus strains harboring the N-terminal domain (NTD, residues 1–100) and C-terminal domain (CTD, residues 121-190). Although ribosomes purified from the NTD strain lose the ability of forming the 100S dimer, the CTD strain ribosomes behave similarly to the strain harboring the full-length HPFSA (Supplementary Fig. 7D, E). In addition, in vitro assays demonstrated that the full-length HPFSA protein as well as the C-HPFSA construct are able to self-dimerize (Supplementary Fig. 7F–H). The 70S dimerization is critical for stationary phase cell survival under acute heat stress. At non-permissive temperature (62 °C), the empty vector control and NTD strains were > 100-fold more susceptible to heat killing than the full-length HPF and CTD strains (Supplementary Fig. 8). Taken together, our data suggest that C-HPFSA forms the binding platform for the adjacent 30S, which is essential for S. aureus 100S dimerization. Furthermore, the superposition of our structure and the native SA70S19 revealed that the presence of HPFSA induces a conformational change of helices h26 and h40 (Fig. 3d). Previous studies demonstrated both in S. aureus and L. lactis that the 100S dimers cannot be formed without the presence of HPF17, 22. Therefore, we suggest that upon HPFSA binding a conformational change in h26 and h40 occur, which allow them to interact with uS2 and C-HPFSA to further stabilize the dimer.
30S conformational changes is induced by 100S stabilization
Ribosomal 30S head is a highly dynamic domain able to adopt major swiveling rearrangements. To reveal the degree of relation between the 100S dimerization and the head conformational changes, we used focused masked classification over HPFSA and refined ribosomes as monomers (Supplementary Fig. 3). Comparison of the distinct 3D classes showed that upon HPFSA binding a concerted movement of the head and the body occurs, similar to the ratcheting motion of the head during translocation. The head of the 30S subunit rotates with respect to the body by an angle of about 5°, resulting in a maximum ~ 3 Å displacement of the shoulder helix h16 and ~ 8 Å displacement of the beak helix h33 (Fig. 4a and Supplementary Movie 1). The rotation is counterclockwise when seen from the solvent side of the 30S subunit and results in decreased contact surface between the head domain and the body.
Fig. 4.

Structural differences of C-HPFSA bound/unbound 30S subunits and S. aureus vs. E. coli 100S structures. a Superposition of two small ribosomal subunits; bound and unbound to HPF from two views demonstrates conformation change upon C-HPFSA binding. The bound 30S is in blue and the unbound is in gray. The density of C-HPFSA is shown on the top panel. Arrows demonstrate the movement directions of the 30S head and body upon binding. b, c Superposition of SA100S (in blue) on b. Single-particle cryo-EM map of part of EC100S (in yellow, EMDB code EMD-1750) and c cryo-EM tomogram of EC100S (in green, EMDB code EMD-5174)
According to the structures of Tth70S in complex with RMFEC and HPFEC (PDB code 4V8G and 4V8H, respectively), upon binding of RMFEC the head of the 30S subunit undergoes a conformational change, which facilitates the formation of the 100S dimer5. A similar conformational change was observed upon HPFEC binding to Tth70S. However, as shown above, the N-HPFSA that is structurally similar to HPFEC, is not sufficient by itself to form 100S dimers in S. aureus. Thus, it seems that the conformational change induced by HPFSA binding stabilizes the conformation of uS2 and enables participation in dimer formation. Indeed, protein uS2 is better resolved in our maps where HPFSA is bound compared with apo-ribosomes.
Distinct structural differences between SA100S and EC100S
S. aureus 100S complex formation involves interactions between protein uS2 and helices h26 and h40 of the 16S rRNA. Two modes of E. coli 100S (EC100S) complex formation have been suggested according to two independent cryo-EM studies; In the first, the ribosomal proteins uS2, uS3, and uS5 of the 30S are involved in dimer formation15, whereas in the second, uS2, uS9, uS10, and helix h39 are involved in this process16. In the SA100S, only the head (uS2 and h40) and platform (h26) of the 30S subunits take part in the dimerization process, whereas in the EC100S, the body (uS2, uS9, uS10, and h39) participates too in 100S dimerization. Alternatively, Kato et al.15 have proposed that the body (uS5) domain of the 30S is also part of the EC100S formation. Superposition of our SA100S with EC100S maps (EMDB 1750, 5174) through alignment of one of the paired 70S, revealed significantly different conformations of the dimer formed between the two 30S subunits (Fig. 4b, c). In either EC100S reconstruction, one 70S ribosome is rotated with respect to its counterpart in the SA100S by an angle of ~ 110° such that the interface between the two 30S in EC100S spans over a much larger surface area. Therefore, the essence of interactions forming the 100S and the structural elements involved is not universal, but species dependent. In S. aureus, rRNA helices h26, h40, and ribosomal protein uS2 form the dimerization interface, whereas in E. coli, rRNA helix h39 and proteins uS9, uS10 or uS2, uS3, and uS5 are involved in a more expanded interaction surface.
Discussion
The 100S complex was first identified in bacteria over 50 years ago29, 30. Extensive biochemical studies were performed since then, in order to identify which factors take part in the dimer formation, as well as understanding the underlying molecular mechanism and significance of this phenomenon. Two distinct types of bacterial 100S ribosomes have been identified; the first in Gammaproteobacteria and Betaproteobacteria during the stationary growth phase that is induced by two proteins, short HPF, and RMF. The other type of 100S dimer is typical for Gram-positive bacteria and is present throughout all growth phases. Structural studies performed using T. thermophilus 100S, revealed how E. coli HPF and RMF interactions with the heterologous ribosome enable its hibernating form. Cryo-EM density maps reconstructed from E. coli 100S particles revealed that the two ribosomes composing the dimer are connected through their small subunit. However, due to their low resolution, the nature of these interactions has remained ambiguous. Although parts of the 30S dimerization machinery in E. coli have been described; the formation of 100S dimers mediated by long HPFs found in Gram-positive bacteria is even less understood.
In this study, the initial 6.8 Å density map reconstructed from single particles of 100S ribosomes of S. aureus allowed a more accurate fitting of an atomic model, which revealed that similar to EC100S dimers, the ribosomal pair of the SA100S is bound through their small subunits. The 30S–30S interactions are mediated by a homo-dimer of CTDs from two HPFSA, which allows for rRNA and ribosomal proteins to interlock and further secure the connection between the two 70S ribosomes. Such dimerization presents two recognition sites for rRNA binding, in this case h26 form both 30S subunits and can therefore provide a cooperative interaction that strengthens the affinity of the CTD for the rRNA31. Notably, although the interactions between the two 70S should be strong, they certainly are not rigid, as the 3D classification shows that the large majority of the dimers exhibit flexibility between to two ribosomes. Indeed, the dimer stabilizing parallel β-sheet interactions facilitate weaker dimer interaction (compared with an antiparallel arrangement).
Re-processing the cryo-EM data and reconstructing density maps from individual 70S ribosomes that composed the dimer yielded density maps at a resolution of 2.9–3.2 Å that allowed us to trace the N-HPFSA. The binding site of N-HPFSA overlaps with that of the mRNA and the tRNA anti-codon loop, thus explaining how HPF halts protein synthesis by steric block of the decoding site. Although the NTD is well resolved within the high-resolution maps, no density was found to be connecting between this domain and the CTD. This may indicate that the two domains are linked via an unstructured, highly flexible loop. These observations alongside sucrose density analysis and negative stain of HPF-truncated strains led to the conclusion that HPFSA has dual functionality. The NTD is responsible for silencing the ribosomes, whereas the CTD is directly involved in the dimer formation. As the NTD by itself is sufficient for silencing the ribosome, much similar to the protein YfiA, the biological significance of the ribosome dimerization remains unclear. It is possible that the 30S–30S dimerization interface precludes the accessibility of RNases, because the abundance of SA100S is inversely correlated with the degree of ribosome degradation when nutrient is depleted22. Under heat stress, dimerization appears to have a protective role in mitigating ribosome damage that is manifested by cell death (Supplementary Fig. 8). Furthermore, bacterial translation and transcription are coupled and the binding sites (e.g., uS2) of RNA polymerase (RNAP) on the 70S ribosome32 coincidently overlap with the 70S dimerizing interface. Therefore, one might speculate that homo-dimerizing 70S prevent spurious interactions between the RNAP and ribosome in the crowded cytoplasm when translation should not be occurring.
Even though the 100S complexes of gammaproteobacteria and Gram-positive bacteria serve the same purpose, the structural basis for dimerization is different. It is quite likely to be that all bacteria carrying a long HPF protein form 100S complexes in the same manner presented here. Hence, hampering the dimer formation by targeting their signature interface between the two ribosomes may offer a unique Gram-positive specific anti-bacterial treatment.
Methods
Bacterial strains and growth conditions
S. aureus strain JE2 and its hpf-null mutant22 are derivatives of the community-associated methicillin-resistant USA300 isolate (NR-46543, BEI Resources)33. Restoration of the 100S ribosome formation in the hpf-null background was tested with the complementing plasmids harboring either the full-length hpf (pLI50hpf), CTD deletion (pLI50hpf (1-100)), or NTD deletion (pLI50hpf (121-190)). The full-length hpf was PCR amplified with primers P651 (5′-CGG GAT CCA TAC AAC TGG ATT AAC AAT TCA TCG TGC AGG GTG-3′) and P627 (5′-TGA AGC TTT AAA CTT AAT TTA TTG TTC ACT AGT TTG AAT CAA GCC-3′) and the genomic DNA of JE2 as a template. A PCR fragment corresponding to the 1-100 amino acids of hpf was amplified with primers P651 and P722 (5′-TGA AGC TTT TAC TTA CGA TTA ATA CGT GTT TTA TAT TTT CG-3′). To obtain hpf(121–190) region, the NTD deletion was introduced by two-step crossover PCR34 using four primers (P651; P1080 (5′-ATC ATC GTA AGC GTC ATT ATC CAT AGT AAT CTC TCC TTA AAC CTC TTT AT-3′); P1079 (5′-ATA AAG AGG TTT AAG GAG AGA TTA CTA TGG ATA ATG ACG CTT ACG ATG AT-3′);P627). The primer pairs P651/P1080 and P1079/P627 were used in the first PCR reaction with the JE2 genomic DNA as a template. The P651/P627 pair was used in the second reaction using PCR products of the first reaction as the templates. The hpf PCR fragments were then cloned into the BamHI and HindIII sites of pLI5035, in which the expression of hpf variants is under the control of its native promoter and Shine-Dalgarno sequence. Plasmids were passaged through the restriction-deficient S. aureus strain RN422036. The plasmids were isolated from strain RN4220 and transformed into the S. aureus JE2Δhpf mutant. S. aureus strains carrying pLI50 derivatives were routinely grown in tryptic soy broth (TSB, BD Difco) supplemented with chloramphenicol (10 μg ml−1).
The overexpression and purification of recombinant His-tagged full-length HPF from pET28a (Novagen) have been previously described22, 37. To overexpress the HPF-CTD, heptahistidine tag was incorporated to the N terminus of CTD by PCR amplification using primers P1076 (5′-TAC CAT GGG CCA TCA TCA CCA TCA CCA TCA TAG CAG CGG CAT AGA AAT TAT TCG TTC AAA-3′) and P1078 (5′-CGC TCG AGT TAT TGT TCA CTA GTT TGA ATC-3′). The DNA fragment was cloned into the NcoI and XhoI sites of pET28a, yielding plasmid pEThpf(121–190). A mini Ser-Ser-Gly linker was introduced downstream of the heptahistidine preceding the 121 residue of hpf. E. coli BL21(DE3) bearing pET28a derivatives was grown in LB supplemented with 50 μg ml−1 kanamycin and induced with a final 0.5 mM isopropyl β-d-1-thiogalactopyranoside at cell density OD600 ~ 0.45. All strains were cultured at 37 °C unless otherwise noted.
Ribosome purification
Cells were grown in TSB medium to an OD600 of 0.7–0.8 and collected by centrifugation at 11,540 g for 30 min at 4 °C. The collected cells were immediately suspended in ice-cold buffer A (20 mM Hepes-KOH (pH 7.5), 14 mM Mg2+ acetate, 100 mM KCl, 1 mM dithiothreitol and 0.5 mM phenylmethylsufonyl fluoride). The suspended cells were flash frozen as bacterial spheres by slowly pipetting the slurry into liquid nitrogen and stored at −80 °C. To disrupt the cells membrane, the frozen bacterial spheres were cryo-milled into a fine powder, which was then suspended in buffer A. The cells debris was discarded by centrifuging the lysate at 30,000 g for 20 min at 4 °C. The cell extract was collected and layered onto a 1.1 M sucrose cushion in buffer A and centrifuged at 310,000 g for 17 h at 4 °C. The ribosomal pellet was re-suspended in buffer A and layered onto a 10–40% sucrose gradient and centrifuged using a SW-28 rotor, at 18,000 r.p.m. for 17 h at 4 °C. The desired fractions were collected, combined, and centrifuged overnight at 320,000 g, 4 °C. The ribosomal pellet was re-suspended in buffer B (20 mM Hepes-KOH, 10 mM Mg2+ acetate, 100 mM KCl) and centrifuged again, using a TLA-100.2 rotor at 75,000 r.p.m. for 1.5 h at 4 °C. The ribosomal pellet was re-suspended in buffer B, diluted to a concentration of ~ 1mg ml−1, aliquoted, and flash frozen in liquid nitrogen.
Cryo-EM data collection and refinement
For the data set used to obtain the high-resolution structure of the 70S ribosome, 3 µl of purified ribosomes (OD260 nm = 3.0) were applied on to freshly glow discharged holey carbon grids (Quantifoil R2/2 Cu) pre-coated with a home-made continuous carbon film ( ~ 30 Å thick) and incubated for 30 s at 4 °C, 100 % humidity in a FEI Vitrobot Mk IV system. The grids were blotted for 3 s before plunge cooling in liquid ethane. In the case of the 100S ribosome dimer, three data sets were collected over several non-consecutive days and combined in attempts to minimize orientation bias. To pick dimers confidently, a more diluted sample (OD260 nm = ~ 2.2) was used, and blotting and freezing parameters were kept the same as above.
Micrographs were acquired using an FEI Titan Krios electron microscope operated at 300 kV. Images were acquired using FEI’s automated acquisition software (EPU) using a FEI Falcon II detector at a pixel size of (1.07 Å per pixel). All data sets were acquired at 17 frames per second and the total dose used was 25 electrons per Å2. Movies were processed using Motioncor238 for patched frame motion correction and dose weighting, Gctf39 was used for estimation of the contrast transfer function parameters, and RELION-2.027 for all other image processing steps. Templates for reference-based particle picking were obtained from 2D class averages that were calculated from a manually picked subset of the micrographs. Template-based particle picking was employed to pick particles for both the 100S dimer and 70S monomers. In either case, one round of reference-free 2D classification was used to prune falsely picked particles. For the 100S particle reconstruction, one round of 3D classification was used where initially a synthetically generated ribosome dimer used as an initial model to identify one class consisting of 12,570 particles that displayed two 70S particles well ordered with respect to each other (Supplementary Table 1).
In the case for the high-resolution reconstruction of the 70S particle, one round of 3D classification was used to remove particles clearly lacking the small subunit and the remaining particles were refined into a single reconstruction initially. As it was apparent that there was further unresolved heterogeneity present in the resulting reconstruction, particularly in the small subunit, fine angular 3D classification was employed to further classify the data. Among classes that clearly lacked the small subunit, we identified one major class that showed good density for both the large and small subunit. This subset of data (224,554 particles) was used for the final reconstruction of the 70S particle.
Reported resolutions are based on the gold-standard Fourier shell correlation (FSC) = 0.143 criterion and FSC curves were corrected for the effects of a soft mask on the FSC curve using high-resolution noise substitution40. All 3D refinements were started from a 60 Å low-pass-filtered initial model. Before visualization, all density maps were corrected for the modulation transfer function of the detector and then sharpened by applying a negative B-factor that was estimated using automated procedures41.
Model building and refinement
Model building of 100S of S. aureus rRNA and proteins was executed by combining template guided and de novo model building. The previously published structure of S. aureus 50S42 was used as an initial template for the large ribosomal subunit. The structures of T. thermophilus 70S43 were used as an initial template for the small ribosomal subunit. Each model was docked into the proper masked and sharpened maps using UCSF chimera44, mutated and manually adjusted using COOT45. Model refinement was performed using an iterative approach including real space refinement and geometry regularization in COOT, followed by real space refinement using PHENIX46. Once the refinement of each ribosomal subunit was completed, two copies of each particle was fitted into the 100S map and further refined.
Negative stain
SA70S complex mixture with tRNA and mRNA (3.5 µl of a 0.5 mg ml−1 solution) was applied on pre-coated carbon grids for 30 s and then blotted with filter paper. Afterward the grid was wiped on water drop and three times on 2% uranyl acetate drop, blotted with filter paper in between.
Size exclusion chromatography
Size exclusion chromatography (SEC) was performed on a Superdex-200 Increase 10/300 GL column (GE Healthcare) equilibrated with SEC buffer (20 mM Tris (pH 7.9), 20 mM MgCl2, 250 mM NH4Cl, 10 mM KCl) at a flow rate of 0.5 ml min−1. Fifty micrograms of purified HPFSA proteins were used for analysis. Absorbance at 280 nm was used to detect proteins eluted from the column. Apparent molecular weights were estimated using a standard curve of the molecular masses of protein markers (BioRad): myoglobin (17 kDa), ovalbumin (44 kDa), IgG bovine (158 kDa), and thyroglobulin (670 kDa).
Acute heat-killing assay
Stationary phase TSB cultures (supplemented with 5 μg ml−1 chloramphenicol) of S. aureus hpf-null derivatives were diluted with TSB to an OD600 of 0.37–0.38, split into three portions and incubated at 37, 58, and 62 °C in a thermomixer. After 30 min incubation, cells were subject to 10-fold serial dilutions with the sterile 1 × PBS buffer. Five microliters of each dilution was spotted on TSB agar plates and incubated for 18 h at 37 °C. To enumerate CFU ml−1, aliquots (75 μl) of serially diluted cells were spread onto TSB agar plates and values of CFU ml−1 were determined after 1 day incubation at 37 °C. Two to three independent experiments were performed on separate day.
Data availability
Cryo-EM maps have been deposited in the Electron Microscopy Databank with accession code EMDB-3637 with PDB ID code 5NG8 and EMDB-3640 with PDB ID code 5NGM. The additional data that support the findings of this study are available from the corresponding authors upon request.
Electronic supplementary material
Description of Additional Supplementary Files
Acknowledgements
The data were collected at the Cryo-EM Swedish National Facility. We thank Dr David Wood at the SLU Protein Core for assistance with SEC analysis, M. Carroni and J. Conrad for help with data collection, S. Fleischmann for computing support, Dr H. Rozenberg for fruitful discussions, and Y. Halfon and Z. Eyal S. Tel-Or for experimental support. Funding was provided by the FEBS Long-Term Fellowship (SA), Swedish Research Council (NT_2015-04107), Swedish Foundation for Strategic Research (Future Leaders Grant FFL15-0325), Ragnar Soderbergs foundation (Fellowship in Medicine M44/16), Raymond and Beverly Sackler Churchill college fellowship (A.A.); European Research Council Grant 322581; the Kimmelman Center for Macromolecular Assemblies; A.E.Y. holds the Martin S. and Helen Kimmel Professorial Chair at the Weizmann Institute of Science. M.-N.F.Y. is supported by the PEW Charitable Trusts, the Edward Mallinckrodt Jr. Foundation, and NIH GM121359.
Author contributions
M.-N.F.Y. and A.B. constructed the S. aureus strains and performed SEC. D.M. cultured the cells and purified the 100S ribosomes. S.A. collected and processed the cryo-EM data. D.M. performed the coordinate refinement of the atomic model. D.M. and S.A. interpreted the structure. A.A. supervised sample preparation, the EM analysis and overall project design and execution. A.B., E.Z., and A.E.Y. supervised model building and refinement and overall project design and execution. D.M., S.A., B.A., and A.A. wrote the manuscript and all authors have commented on its final version.
Competing interests
The authors declare no competing financial interests.
Footnotes
Donna Matzov and Shintaro Aibara contributed equally to this work.
Electronic supplementary material
Supplementary Information accompanies this paper at doi:10.1038/s41467-017-00753-8.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mee-Ngan F. Yap, Email: myap1@slu.edu
Alexey Amunts, Email: amunts@scilifelab.se.
Ada E. Yonath, Email: ada.yonath@weizmann.ac.il
References
- 1.Buttgereit F, Brand MD. A hierarchy of ATP-consuming processes in mammalian cells. Biochem. J. 1995;312:163–167. doi: 10.1042/bj3120163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Russell JB, Cook GM. Energetics of bacterial growth: balance of anabolic and catabolic reactions. Microbiol. Rev. 1995;59:48–62. doi: 10.1128/mr.59.1.48-62.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szaflarski W, Nierhaus KH. Question 7: optimized energy consumption for protein synthesis. Orig. Life. Evol. Biosphys. 2007;37:423–428. doi: 10.1007/s11084-007-9091-4. [DOI] [PubMed] [Google Scholar]
- 4.Yoshida H, Wada A. The 100S ribosome: ribosomal hibernation induced by stress. Wiley Interdisc. Rev. RNA. 2014;5:723–732. doi: 10.1002/wrna.1222. [DOI] [PubMed] [Google Scholar]
- 5.Polikanov YS, Blaha GM, Steitz TA. How hibernation factors RMF, HPF, and YfiA turn off protein synthesis. Science. 2012;336:915–918. doi: 10.1126/science.1218538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vila-Sanjurjo A, Schuwirth BS, Hau CW, Cate JH. Structural basis for the control of translation initiation during stress. Nat. Struct. Mol. Biol. 2004;11:1054–1059. doi: 10.1038/nsmb850. [DOI] [PubMed] [Google Scholar]
- 7.Agafonov DE, Kolb VA, Nazimov IV, Spirin AS. A protein residing at the subunit interface of the bacterial ribosome. Proc. Natl Acad. Sci. USA. 1999;96:12345–12349. doi: 10.1073/pnas.96.22.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ueta M, et al. Ribosome binding proteins YhbH and YfiA have opposite functions during 100S formation in the stationary phase of Escherichia coli. Genes Cells. 2005;10:1103–1112. doi: 10.1111/j.1365-2443.2005.00903.x. [DOI] [PubMed] [Google Scholar]
- 9.Ueta M, et al. Role of HPF (hibernation promoting factor) in translational activity in Escherichia coli. J. Biochem. 2008;143:425–433. doi: 10.1093/jb/mvm243. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida H, et al. The ribosome modulation factor (RMF) binding site on the 100S ribosome of Escherichia coli. J. Biochem. 2002;132:983–989. doi: 10.1093/oxfordjournals.jbchem.a003313. [DOI] [PubMed] [Google Scholar]
- 11.Wada A, Yamazaki Y, Fujita N, Ishihama A. Structure and probable genetic location of a “ribosome modulation factor” associated with 100S ribosomes in stationary-phase Escherichia coli cells. Proc. Natl Acad. Sci. USA. 1990;87:2657–2661. doi: 10.1073/pnas.87.7.2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maki Y, Yoshida H, Wada A. Two proteins, YfiA and YhbH, associated with resting ribosomes in stationary phase Escherichia coli. Genes Cells. 2000;5:965–974. doi: 10.1046/j.1365-2443.2000.00389.x. [DOI] [PubMed] [Google Scholar]
- 13.Wada A, Igarashi K, Yoshimura S, Aimoto S, Ishihama A. Ribosome modulation factor: stationary growth phase-specific inhibitor of ribosome functions from Escherichia coli. Biochem. Biophys. Res. Commun. 1995;214:410–417. doi: 10.1006/bbrc.1995.2302. [DOI] [PubMed] [Google Scholar]
- 14.Yamagishi M, et al. Regulation of the Escherichia coli rmf gene encoding the ribosome modulation factor: growth phase- and growth rate-dependent control. EMBO J. 1993;12:625–630. doi: 10.1002/j.1460-2075.1993.tb05695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato T, et al. Structure of the 100S ribosome in the hibernation stage revealed by electron cryomicroscopy. Structure. 2010;18:719–724. doi: 10.1016/j.str.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 16.Ortiz JO, et al. Structure of hibernating ribosomes studied by cryoelectron tomography in vitro and in situ. J. Cell Biol. 2010;190:613–621. doi: 10.1083/jcb.201005007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Puri P, et al. Lactococcus lactis YfiA is necessary and sufficient for ribosome dimerization. Mol. Microbiol. 2014;91:394–407. doi: 10.1111/mmi.12468. [DOI] [PubMed] [Google Scholar]
- 18.Izutsu K, Wada A, Wada C. Expression of ribosome modulation factor (RMF) in Escherichia coli requires ppGpp. Genes Cells. 2001;6:665–676. doi: 10.1046/j.1365-2443.2001.00457.x. [DOI] [PubMed] [Google Scholar]
- 19.Khusainov I, et al. Structure of the 70S ribosome from human pathogen Staphylococcus aureus. Nucleic Acids Res. 2016;44:10491–10504. doi: 10.1093/nar/gkw933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ueta M, et al. Conservation of two distinct types of 100S ribosome in bacteria. Genes Cells. 2013;18:554–574. doi: 10.1111/gtc.12057. [DOI] [PubMed] [Google Scholar]
- 21.Akanuma G, et al. Ribosome dimerization is essential for the efficient regrowth of Bacillus subtilis. Microbiology. 2016;162:448–458. doi: 10.1099/mic.0.000234. [DOI] [PubMed] [Google Scholar]
- 22.Basu A, Yap MN. Ribosome hibernation factor promotes Staphylococcal survival and differentially represses translation. Nucleic Acids Res. 2016;44:4881–4893. doi: 10.1093/nar/gkw180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kline BC, McKay SL, Tang WW, Portnoy DA. The Listeria monocytogenes hibernation-promoting factor is required for the formation of 100S ribosomes, optimal fitness, and pathogenesis. J. Bacteriol. 2015;197:581–591. doi: 10.1128/JB.02223-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McKay SL, Portnoy DA. Ribosome hibernation facilitates tolerance of stationary-phase bacteria to aminoglycosides. Antimicrob. Agents Chemother. 2015;59:6992–6999. doi: 10.1128/AAC.01532-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ueta M, Wada C, Wada A. Formation of 100S ribosomes in Staphylococcus aureus by the hibernation promoting factor homolog SaHPF. Genes Cells. 2010;15:43–58. doi: 10.1111/j.1365-2443.2009.01364.x. [DOI] [PubMed] [Google Scholar]
- 26.Tagami K, et al. Expression of a small (p)ppGpp synthetase, YwaC, in the (p)ppGpp(0) mutant of Bacillus subtilis triggers YvyD-dependent dimerization of ribosome. Microbiologyopen. 2012;1:115–134. doi: 10.1002/mbo3.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimanius D, Forsberg BO, Scheres SH, Lindahl E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife. 2016;15:18722. doi: 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Polikanov YS, et al. Amicoumacin a inhibits translation by stabilizing mRNA interaction with the ribosome. Mol. Cell. 2014;56:531–540. doi: 10.1016/j.molcel.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McCarthy B. Variations in bacterial ribosomes. Biochim. Biophys. Acta. 1960;39:563–564. doi: 10.1016/0006-3002(60)90221-3. [DOI] [Google Scholar]
- 30.Tissieres A, Watson JD, Schlessinger D, Hollingworth B. Ribonucleoprotein particles from Escherichia coli. J. Mol. Biol. 1959;1:221–233. doi: 10.1016/S0022-2836(59)80029-2. [DOI] [Google Scholar]
- 31.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007;8:479–490. doi: 10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohler R, Mooney R, Mills D, Landick R, Cramer P. Architecture of a transcribing-translating expressome. Science. 2017;356:194–197. doi: 10.1126/science.aal3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fey PD, et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 2013;4:e00537–00612. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Link AJ, Phillips D, Church GM. Methods for generating precise deletions and insertions in the genome of wild-type Escherichia coli: application to open reading frame characterization. J. Bacteriol. 1997;179:6228–6237. doi: 10.1128/jb.179.20.6228-6237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee CY, Buranen SL, Ye ZH. Construction of single-copy integration vectors for Staphylococcus aureus. Gene. 1991;103:101–105. doi: 10.1016/0378-1119(91)90399-V. [DOI] [PubMed] [Google Scholar]
- 36.Nair D, et al. Whole-genome sequencing of Staphylococcus aureus strain RN4220, a key laboratory strain used in virulence research, identifies mutations that affect not only virulence factors but also the fitness of the strain. J. Bacteriol. 2011;193:2332–2335. doi: 10.1128/JB.00027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davis AR, Gohara DW, Yap MN. Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl Acad. Sci. USA. 2014;111:15379–15384. doi: 10.1073/pnas.1410356111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng SQ, et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017;14:331–332. doi: 10.1038/nmeth.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S, et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 42.Eyal Z, et al. Structural insights into species-specific features of the ribosome from the pathogen Staphylococcus aureus. Proc. Natl Acad. Sci. USA. 2015;112:13. doi: 10.1073/pnas.1517952112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voorhees RM, Weixlbaumer A, Loakes D, Kelley AC, Ramakrishnan V. Insights into substrate stabilization from snapshots of the peptidyl transferase center of the intact 70S ribosome. Nat. Struct. Mol. Biol. 2009;16:528–533. doi: 10.1038/nsmb.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Afonine P, Headd J, Terwilliger T, Adams P. New tool: phenix. real_space_refine. Comput. Crystallogr. Newslett. 2013;4:43–44. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Cryo-EM maps have been deposited in the Electron Microscopy Databank with accession code EMDB-3637 with PDB ID code 5NG8 and EMDB-3640 with PDB ID code 5NGM. The additional data that support the findings of this study are available from the corresponding authors upon request.