

Abstract
Palladium catalyzed conjunctive cross-coupling is used for the synthesis of enantioenriched allylboron reagents. This reaction employs non-symmetric bis(alkenyl)borates as substrates and appears to occur by a mechanism that involves selective activation of the less substituted alkene, followed by migration of the more substituted alkene during the course of a Pd-induced metalate rearrangement.
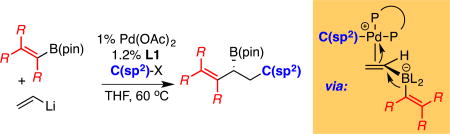
Because they engage in an array of stereospecific and stereoselective transformations1, chiral allylboron reagents hold a distinctive place in organic chemistry.2 Importantly, these reactive intermediates can be readily transformed into allylic alcohols3 and amines4, and they can be employed in carbon-carbon bond forming reactions such as carbonyl and imine allylation.5 Moreover, the ability to accomplish stereospecific α- and γ-selective cross-coupling6 reactions with high levels of chirality-transfer adds to the utility of these special reagents. Due to the utility of allyl boronates, a number of catalytic asymmetric synthesis routes have been studied for their production. Whereas construction of terminal allylic boronates (i.e. A, Scheme 1B) can be achieved by Cu-catalyzed enantioselective allylic borylation7 or allylic alkylation8, catalytic construction of internal allylic boronates (i.e. C and D) by substitution reactions generally requires use of non-racemic allylic alcohol derivatives.9 Other useful catalytic enantioselective routes to internal allyl boron reagents – for example, diene diboration,10 cross-coupling with geminal bis(boronates)11, and conjugate 1,4 or 1,6-borylation of activated dienes12–generally result in products bearing extraneous functionality. While the above-described examples are powerful catalytic methods, none of them provide access to chiral allylboron reagents with a broad array of product substitution patterns. Only Aggarwal’s stoichiometric sparteine-mediated homologation reaction is able to access the full complement of both terminal and internal nonracemic allyl boronates structures.13 In this report, we describe the application of catalytic enantioselective conjunctive cross-coupling to a broad array of alkenyl(vinyl)borate substrates (Scheme 1B) and show that this method provides an inroad to diverse allyl boronate structures in an efficient asymmetric fashion.
Scheme 1.

Catalytic conjunctive cross-coupling (A) and its application to bis(alkenyl) "ate" complexes (B).
Recently, we reported the catalytic enantioselective conjunctive cross-coupling reaction to furnish chiral secondary boronic esters (Scheme 1, eq. 1).14 This three-component reaction employs a Pd complex in conjunction with the diphosphine ligand, Mandyphos (L1), to couple a boronic ester, an organolithium reagent, and an organotriflate electrophile. During the course of the reaction, a palladium induced 1,2-metalate shift (Scheme 1A) establishes the first C-C bond and installs the borylated stereogenic center; subsequent reductive elimination from a dialkyl Pd complex forges the second C-C bond. Whereas previously described conjunctive coupling reactions employed aryl and alkyl migrating groups (RM in Scheme 1A), we considered that with a migrating alkene, the reaction might provide a general route to allyl boronate reagents (Scheme 1B). In terms of reaction development, the demonstrated ability of conjunctive cross-coupling reactions to accommodate aromatic C(sp2) migrating groups was encouraging; however, for alkene migration several critical issues remained uncertain. First, it was not clear whether the Pd-complex could selectively bind and activate one alkene in the bis(alkenyl)borate substrate, or whether indiscriminant activation of both alkenes would lead to product mixtures. Second, it wasn't clear whether the allyl boronate functional group in the product – a motif that can engage in rapid transmetalation15 – would be consumed by direct Suzuki reactions with the organic electrophile. Lastly, both the chemical and configurational stability of the "ate" complex and reaction products in the conjunctive coupling process was uncertain.
We began our investigation into the conjunctive cross-coupling of bis(alkenyl) borates by treating 1-hexenylB(pin) (1), with halide-free vinyllithium and phenyl triflate in the presence of 1% Pd(OAc)2 and 1.2% MandyPhos ligand L1 (Table 1, entry 1). This experiment afforded the allyl boronate product (2) in good isolated yield and high enantiomeric purity. In addition to the conjunctive coupling product 2, the reaction mixture contained detectable amounts of direct Suzuki-Miyaura16 cross-coupling products (3 and 4). Of note, only a single regioisomer of conjunctive coupling product 2 was formed and its identity was consistent with activation of the unsubstituted vinyl ligand as opposed to the 1-hexenyl ligand. Importantly, no evidence for alkene isomerization could be detected. As depicted in Table 1, other ligand frameworks were also capable of delivering the conjunctive cross-coupling product and, while these provided uniformly regioselective reactions, the chemoselectivity (2:3+4) and enantioselectivity with other ligand classes generally doesn't measure up to that obtained with the MandyPhos class of ligands (L1–L6). Further optimization of reaction conditions and boronic ester structure did not lead to additional improvements and, accordingly, these conditions with ligand L1 were employed in subsequent studies.
Table 1.
Conjunctive Cross-Coupling with Bis(alkenyl)borates.
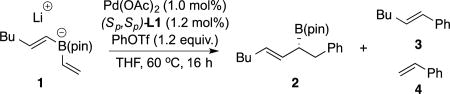
| ||||
|---|---|---|---|---|
|
| ||||
| entry | ligand | yield (%) a | 2:(3+4)a | erb |
| 1 | L1 | 85 | 8:1 | 92:8 |
| 2 | L2 | 77 | 8:1 | 89:11 |
| 3 | L3 | 70 | 5:1 | 89:11 |
| 4 | L4 | <2 | na | na |
| 5 | L5 | 85 | 6:1 | 92:8 |
| 6 | L6 | 78 | 7:1 | 72:28 |
| 7 | L7 | 40 | 1:1 | 77:23 |
| 8 | L8 | 30 | >10:1 | 59:41 |
| 9 | L9 | 58 | >20:1 | 70:30 |
| 10 | L10 | 29 | 1:1 | 69:31 |
| 11 | L11 | 40 | >20:1 | 53:47 |
Determined by NMR versus an internal standard.
Determined by chiral SFC analysis.
With the prospect for regio- and stereoselective alkenyl migration in conjunctive coupling established, other "ate" complexes were examined with phenyl tri-flate as the electrophile (Table 2). Across a range of substrates, conjunctive couplings were found to reliably furnish stable allylboronate products in good yield and with enantioselectivities from 77:23 to 96:4er. Simple E-alkyl substitution appears generally tolerated delivering substituted allylboronate products (2, 5 – 8) in good yield and generally high enantiomeric purity. In addition, aryl E-disubstituted allyl boronates (10 – 14) as well as a dienyl boronate (9) could be accessed with this strategy. Of note, a terminal allylboronate (15) could also be accessed efficiently and in good selectivity by conjunctive coupling involving a divinylB(pin) "ate" complex. Importantly, with Z-disubstituted alkenyl derivatives (16–17), the conjunctive coupling occurred without isomerization of the migrating alkene suggesting that product olefin configuration can be controlled by appropriate substrate design. In addition, β,β-disubstituted (18), α-substituted (21–23), and cyclic alkenyl boronic esters (19–20) are also competent substrates. In terms of functional group compatibility, substrates containing a silyl ether (5), furyl (11), and benzodioxole (14) were compatible in the reaction as was a Boc-protected amine (20), aryl fluoride (12), and a vinyl silane (23). Lastly, it was demonstrated that the reaction can be accomplished on larger scale: reaction to form 2 on 5 mmol scale gave similar yield and selectivity as smaller scale reactions.
Table 2.
Conjunctive Cross-Coupling of Bis(alkenyl)borate Complexes.a

|
Reaction times vary from 1.5–16 h, See SI for conditions. Isolated yield and er values represent an average of two experiments.
With 2% Pd(OAc)2 and 2.2% L1.
With 3% Pd(OAc)2 and 3.2% L1.
Isolated as the derived alcohol.
Reaction solvent is 1:1 THF:DMSO.
Reaction conducted on 5 mmol scale.
To learn about the ability of the reaction to accommodate different electrophilic reagents, the examples in Table 3 were examined. It was found that various aryl and alkenyl trifluoromethanesulfonates were processed efficiently. In the case of electron deficient electrophiles, a modest reduction in enantioselectivity was observed (24) while electron rich electrophiles reacted with uniformly high selectivity (25–27). The reaction employing organohalide electrophiles in place of organotriflates was unsuccessful in the absence of additives. Attributing this outcome to the inhibiting effect of halide byproducts of the reaction the use of alkali metal triflate additives was examined.17 Of note, the reaction in the presence of KOTf enabled the reaction of bromide electrophiles; however, in some cases the use of a bromide electrophile gave diminished yield and, more surprisingly, lowered enantioselectivity when compared to the triflate (for example, compound 29 was formed in 58% yield and 78:22 er when the aryl bromide electrophile was employed). As depicted in Table 3, a quinoline was also accommodated in the reaction (28, 38).
Table 3.
Conjunctive Cross-Coupling of Bis(alkenyl)borate Complexes with Various Electrophiles.a

|
Isolated yield and er values represent an average of two experiments.
When X=Br, 1.2 equiv. of KOTf added; see SI for conditions.
This experiment conducted with 2% Pd(OAc)2 and 2.2% L1.
Isolated as the derived alcohol.
This experiment conducted with 3% Pd(OAc)2 and 3.2% L1, reaction time of 5 h.
The data in Tables 1–3 was obtained from reactions that employed halide-free vinyllithium prepared from tetravinylstannane by lithium-tin exchange. Recently, we established that halide-containing vinyllithium (prepared by Li-halogen exchange) and vinyl Grignard reagents can be employed in conjunctive cross-couplings as long as appropriate metal triflates are added to the reaction.17 To determine whether these strategies could also be employed in the reactions of bis(alkenyl)borate complexes, the experiments in Scheme 2 were undertaken. With vinylmagnesium chloride as the nucleophile (eq. 1), it was necessary to add two equivalents of sodium triflate and to use 3:1 THF:DMSO as the reaction solvent. While the reaction is markedly slower than with halide-free vinyllithium, with these conditions, the process occurs with the same level of enantioselectivity (92:8 er), and with only a modestly diminished yield (69% versus 83%). To determine whether halide-containing vinyllithium might be employed in the reaction, vinyl bromide was treated with t-butyllithium at −78 °C in ether/pentane and then the alkenyl boronic ester added to form the "ate" complex. After warming to room temperature the solvent was removed and a conjunctive cross-coupling conducted in THF solvent. While it was found that KOTf (2 equiv) was indeed required, the reaction nonetheless proceeded in similar levels of enantioselectivity as the halide-free reaction.
Scheme 2.

Conjunctive coupling of Grignard-derived and halide-containing borates.
During the course of the experiments above, it was found that the intermediate "ate" complexes are often free-flowing crystalline solids. Thus, it was of interest to determine the long-term stability of these materials as potential "off-the-shelf" coupling partners. To probe this feature, the bis(alkenyl) ate complex 39 was prepared on gram scale and stored under argon atmosphere at room temperature. As depicted in the 11B NMR spectrum in Scheme 3A, even after three months of storage, the "ate" complex exhibits very little decomposition to other boron-containing species. Importantly, this material engages in efficient and selective conjunctive cross-coupling with an outcome similar to that obtained with freshly prepared reactants (eq. 3, Scheme 3B).
Scheme 3.

Utility of a storable, stable alkenyl(vinyl)borate complex.
To highlight the synthetic utility of allylboron reagents prepared by the conjunctive cross-coupling reaction, the experiments in Scheme 4 were conducted. In the first (eq 4), it was shown that peroxide oxidation proceeds uneventfully and furnishes the allylic alcohol in outstanding yield and with preservation of enantiomeric purity. Next it was shown that direct stereospecific amination of the boronic ester can be accomplished with MeONH2/BuLi to furnish the derived allylic amine derivative41(eq. 5); to our knowledge, this is the first time that that stereospecificity in the amination of non-racemic allyl boronates has been demonstrated. Lastly, we examined carbonyl allylation reactions: whereas direct allylation of benzaldehyde with allyl boronate 2 provides stereoisomeric mixtures (data not shown), when the reaction was conducted through the intermediacy of the borinic ester (eq. 6) as prescribed by Aggarwal18, the allylation occurs with excellent selectivity.
Scheme 4.

Functionalization of allylboron reagents.
In conclusion, we have described a general catalytic enantioselective strategy for construction of both terminal and internal α-chiral allyl boronate reagents by catalytic conjunctive cross-coupling reactions.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM-118641). We thank Solvias for providing MandyPhos ligand.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Review of stereospecific transformations of boronic esters, see: Thomas SP, French RM, Jheengut V, Aggarwal VK. Chem. Rec. 2009;9:24. doi: 10.1002/tcr.20168.
- 2.For an excellent recent review: Diner C, Szabó KJ. J. Am. Chem. Soc. 2017;139:2. doi: 10.1021/jacs.6b10017.
- 3.Review of borane oxidation: Brown HC, Snyder C, Rao BCS, Zweifel G. Tetrahedron. 1986;42:5505.Oxidation with PhNO occurs with allyl migration: Kyne RE, Ryan MC, Kliman LT, Morken JP. Org. Lett. 2010;12:3796. doi: 10.1021/ol101472k.
- 4.Direct amination of allylB(pin) derivatives: Mlynarski SN, Karns AS, Morken JP. J. Am. Chem. Soc. 2012;134:16449. doi: 10.1021/ja305448w.
- 5.Hall DG, Lachance H. Allylboration of Carbonyl Com-pounds. Wiley; Hoboken, NJ: 2012. [Google Scholar]
- 6.(a) Glasspoole BW, Ghozati K, Moir JW, Crudden CM. Chem. Commun. 2012;48:1230. doi: 10.1039/c2cc16076e. [DOI] [PubMed] [Google Scholar]; (b) Chausset-Boissarie L, Ghozati K, LaBine E, Chen JL-Y, Aggarwal VK, Crudden CM. Chem. - Eur. J. 2013;19:17698. doi: 10.1002/chem.201303683. [DOI] [PubMed] [Google Scholar]; (c) Yang Y, Buchwald SL. J. Am. Chem. Soc. 2013;135:10642. doi: 10.1021/ja405950c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Farmer JL, Hunter HN, Organ MG. J. Am. Chem. Soc. 2012;134:17470. doi: 10.1021/ja308613b. [DOI] [PubMed] [Google Scholar]; (e) Rybak T, Hall DG. Org. Lett. 2015;17:4156. doi: 10.1021/acs.orglett.5b01906. [DOI] [PubMed] [Google Scholar]; (f) Potter B, Edelstein EK, Morken JP. Org. Lett. 2016;18:3286. doi: 10.1021/acs.orglett.6b01580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Ito H, Ito S, Sasaki Y, Matsuura K, Sawamura M. J. Am. Chem. Soc. 2007;129:14856. doi: 10.1021/ja076634o. [DOI] [PubMed] [Google Scholar]; (b) Guzman-Martinez A, Hov-eyda AH. J. Am. Chem. Soc. 2010;132:10634. doi: 10.1021/ja104254d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Park JK, Lackey HH, Ondrusek BA, McQuade DT. J. Am. Chem. Soc. 2011;133:2410. doi: 10.1021/ja1112518. [DOI] [PubMed] [Google Scholar]; (d) Hojoh K, Shido Y, Ohmiya H, Sawamura M. Angew. Chem. Int. Ed. 2014;53:4954. doi: 10.1002/anie.201402386. [DOI] [PubMed] [Google Scholar]
- 8.Carosi L, Hall DG. Angew. Chem. Int. Ed. 2007;46:5913. doi: 10.1002/anie.200700975. [DOI] [PubMed] [Google Scholar]
- 9.(a) Ito H, Kawakami C, Sawamura M. J. Am. Chem. Soc. 2005;127:16034. doi: 10.1021/ja056099x. [DOI] [PubMed] [Google Scholar]; (b) Zhou Q, Srinivas HD, Zhang S, Watson MP. J. Am. Chem. Soc. 2016;138:11989. doi: 10.1021/jacs.6b07396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Pelz NF, Woodward AR, Burks HE, Siebr JD, Morken JP. J. Am. Chem. Soc. 2004;126:16328. doi: 10.1021/ja044167u. [DOI] [PubMed] [Google Scholar]; (b) Kliman LT, Mlynarski SN, Morken JP. J. Am. Chem. Soc. 2012;51:521. [Google Scholar]
- 11.Potter B, Szymaniak AA, Edelstein EK, Morken JP. J. Am. Chem. Soc. 2014;136:17918. doi: 10.1021/ja510266x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Luo Y, Roy ID, Madec AGE, Lam HW. Angew. Chem. Int. Ed. 2014;53:4186. doi: 10.1002/anie.201310380. [DOI] [PubMed] [Google Scholar]; (b) Sim H-S, Feng X, Yun J. Chem. Eur. J. 2009;15:1939. doi: 10.1002/chem.200802150. [DOI] [PubMed] [Google Scholar]
- 13.Chen JL-Y, Aggarwal VK. Angew. Chem. Int. Ed. 2014;53:10992–10996. doi: 10.1002/anie.201407127. [DOI] [PubMed] [Google Scholar]
- 14.Zhang L, Lovinger GJ, Edelstein EK, Szymaniak AA, Chierchia MP, Morken JP. Science. 2016;351:70. doi: 10.1126/science.aad6080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Yamamoto Y, Takada S, Miyaura N. Organometallics. 2009;28:152. [Google Scholar]; (b) Ardolino MJ, Morken JP. J. Am. Chem. Soc. 2014;136:7092. doi: 10.1021/ja502280w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Review: Suzuki A. J. Organomet. Chem. 1999;576:147.
- 17.Lovinger GJ, Aparece MD, Morken JP. J. Am. Chem. Soc. 2017;139:3153. doi: 10.1021/jacs.6b12663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen JLY, Scott HK, Hesse MJ, Willis CL, Ag-garwal VK. J. Am. Chem. Soc. 2013;135:5316. doi: 10.1021/ja401564z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.