

Abstract
We report a new strategy to regulate microRNAs (miRNAs) biogenesis by using bi-functional small molecules that consist of a pre-miRNA binding unit connected by a linker to a Dicer inhibiting unit. In this effort, fluorescence polarization-based screening was used to identify neomycin as a pre-miR-21 binding ligand. Although neomycin cannot inhibit miR-21 maturation, linking it to the RNase inhibitor 1 forms the bi-functional conjugate 7A, which inhibits the production of miR-21. We expect that this strategy will be applicable to design other molecules for miRNA regulation.
SYNOPSIS TOC
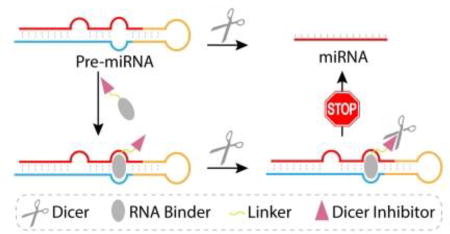
MicroRNAs (miRNAs) are small non-coding RNAs, which regulate gene expression by targeting mRNAs for translational repression or destabilization. Various biological processes are controlled by miRNAs,1 and dysregulation of miRNAs is associated with many human diseases.2 As a result, methods to regulate the expression or function of miRNAs should be useful in studies aimed at probing their function as well as the development of novel therapies for human diseases.
Chemically modified antisense oligonucleotides (ASO) have been used for blocking the function of miRNA.3 However, applications of ASO are limited by their poor cell permeability, unproductive accumulation in endosomes, inherent off-target effects, and poor pharmacokinetic and bio-distribution profiles.4 These limitations demonstrate that alternative methods are needed to regulate miRNAs. Consequently, the development of small molecules capable of modulating miRNA production has emerged as a promising approach to overcome these limitations.5
The biogenesis of miRNAs is a multistep process,6 that starts with transcription of the miRNA gene in the nucleus to generate the primary transcript (pri-miRNA). Pri-miRNA is processed by the RNase Drosha to form precursor miRNA (pre-miRNA), which is then exported to the cytoplasm and further processed by the RNase Dicer to produce the miRNA duplex. One strand of the duplex, the mature miRNA, is incorporated into the RNA-induced silencing complex (RISC) to guide the translational regulation of targeted mRNAs.
A small molecule that targets any step in this sequence can potentially serve as a miRNA regulator.5 For example, Deiters and co-workers showed that miRNAs can be regulated by using small molecules that repress their transcription.7 In addition, Disney and co-workers developed a computational approach to identify small molecules that bind to Drosha or Dicer processing sites within pri-miRNA or pre-miRNA, which thereby inhibit the biosynthesis of the mature miRNA.8 Other inhibitors interfering with the miRNA maturation process have been discovered through screening of chemical libraries.5,9
It is not uncommon that small molecules, which are selected using binding-based screening against miRNA precursors, are unable to block miRNA maturation.10 This phenomenon might be a consequence of promiscuous targeting and/or poor cell permeability of these small molecules. In addition, these small molecules might possess binding affinities that are insufficiently low to compete with the extensive macromolecule interactions occurring between RNAs and other binding partners. To enhance binding between small molecules and targeted miRNA precursors, rationally designed bi-functional molecules have been developed in which additional binding moieties are conjugated to original RNA-recognizing molecules. For example, Duca and co-workers designed neomycin-nucleobase conjugates that have additional base pairing driven affinities against pre-miRNAs to give new inhibitors that block maturation of miR-372 and -373.11 Maiti and co-workers developed dual binding neomycin-bisbenzimidazole conjugates, which contain an intercalating unit to enhance binding of neomycin, and showed that the conjugates serve as inhibitors for oncogenic miR-27a.12
In this work, we devised and tested a new strategy to regulate miRNA biogenesis that relies on the use of newly designed bi-functional small molecules (Figure 1). The purpose of previously developed bi-functional molecules is to target two closely located binding sites and utilize bi-valent recognition to enhance binding affinity. In contrast, our bi-functional molecules are designed to deliver a specific Dicer inhibitor to the Dicer-catalyzed cleavage site in the target pre-miRNA/Dicer complex. The new conjugates consist of a high affinity pre-miRNA binding unit linked to a low affinity Dicer inhibition unit. The Dicer inhibitor in the conjugate is selected so that it alone does not inhibit Dicer to a significant extent. As a result, the pan-cellular function of Dicer will not be disrupted by the bi-funtional molecule. However, recognition of a target pre-miRNA by the RNA binding unit in the conjugate brings the Dicer inhibitor unit into close proximity with the pre-miRNA cleavage site of bound Dicer, thus causing blockage of the activity of Dicer. Importantly, by functing in this manner, the inhibition unit will promote inhibition of the production of the target miRNA.
Figure 1.

A schematic illustration of the new approach to regulate miRNA biogenesis by using bi-functional small molecule that target pre-miRNA.
Oncogenic miR-21 is upregulated in many cancers.13 As a result, small molecules that inhibit the production of miR-21 can serve as useful tools to study the roles played by miR-21 in cancers and as potential cancer therapeutic agents. To demostrate the feasibility of the new strategy described above, we designed, prepared and tested bi-functional inhibitors that block the biogenesis of human miR-21.
The first stage of assembling the newly designed bi-functional miR-21 inhibitors involves identification of a pre-miR-21 targeting small molecule. Fluorescence polarization (FP), a powerful tool for studying molecular interactions, was utilized for this purpose.14 In this assay, the degree of FP from a fluorophore-labeled small molecule increases when it forms a complex with a macromolecule owing to a reduction in the rate of fluorophore tumbling. To identify small molecules that bind pre-miR-21, we used the FP-based competitive binding assay to screen ligands that can compete with a moderate affinity ligand for binding to pre-miR-21. Because kanamycin binds to several RNAs,15 we expected that it would bind pre-miR-21 and, as a result, serve as the standard ligand in the FP-based screening protocol. To verify this assumption, kanamycin derivatives, containing a 1,8-naphthalimide fluorophore at either the primary hydroxyl (KOF) or the primary amine group (KNF) (Scheme S1, ESI) were prepared and their binding to pre-miR-21 was determined using the FP assay (Figure S1, ESI). Based on the saturation binding curve of KOF versus pre-miR-21 from the FP assay, a binding affinity (Kd) of 419 ± 83 nM was obtained (Figure S1b). On the other hand, the binding affinity of KNF to pre-miR-21 was found to be too small to be determined (Figure S1c).
Based on these results, KOF was employed as the standard pre-miR-21 binding ligand in the FP-based screening protocol. Screening was carried out by incubating the pre-formed KOF/pre- miR-21 complex with 13 commercially available aminoglycosides, which are one of the best studied classes of molecules that target RNAs.14–16 Displacement of KOF from pre-miR-21 by an aminoglycoside with higher affinity against pre-miR-21 is reflected in a decrease of the FP value. Among the tested substances, neomycin was found to reduce the FP of KOF/pre-miR-21 to a value that matches that of free KOF, indicating a complete displacement (Figure 2). To determine the binding affinity of neomycin to pre-miR-21, the hydroxyl fluorophore-tagged derivative of neomycin, NF, was prepared (Scheme S2). Using the FP assay, we determined that the Kd of NF to pre-miR-21 is 38 ± 2 nM (Figure S2), which is 10 fold smaller than that of KOF. This result confirms that neomycin strongly binds to pre-miR-21 and led to the selection of neomycin as the pre-miR-21 binding unit in the new bi-functional molecules. Importantly, the above result indicates that modification of the primary hydroxyl group on neomycin does not disrupt binding, therefore, this hydroxyl group can be a suitable position to conjugate the Dicer inhibition unit.
Figure 2.

Fluorescence polarization of KOF in the absence or presence of pre-miR-21 and different aminoglycosides. The error bars represent the standard error of mean (N = 3).
The active site of Dicer is located within the RNase III domain, which cleaves pre-miRNAs through a mechanism in which two metal ions (Mg2+) catalyze the phosphoryl transfer reaction.17 The distance between the two Mg2+ is around 4 Å, similar to those in active sites of other mechanistically similar RNases. Many RNase inhibitors with metal chelating properties have been developed,18 including the N-hydroxyimide 1 (Scheme 1a) that inhibits influenza endo-nuclease at micomolar concentrations.18b As a result, 1 was used as the Dicer inhibiting unit in the new bi-functional molecules.
Scheme 1.

(a) Structure of an influenza endo-nuclease inhibitor. (b) Synthesis of inhibitor building blocks. (c) Synthesis of bi-functional small molecules.
Reagents and conditions: (a) para-methoxybenzyloxyamine, toluene, reflux; (b) trifloroacetic acid, DCM; (c) i) (Boc)2O, Et3N, DMF, H2O, 60 °C; ii) TPS-Cl, pyridine, r.t.; iii) Cysteamine hyrochloride, Cs2CO3, DMF; (d) 6-azido-hexanoic acid, EDCI, TEA, DCM; (e) i) CuSO4, sodium ascorbate, DMSO/H2O; ii) trifloroacetic acid, DCM.
The route to synthesize bi-functional inhibitors began with preparation of a series of homophthalic acids (2A–D) possessing alkynyl polyethylene glycol tails of different lengths (Scheme 1b & S3). Reactions of 2A–D with para-methoxybenzyl (PMB) oxyamine produced O-PMB-oxyimides 3A–D that were later coupled to neomycin. The free imide 4, used as a control in activity assays, was produced by removing the PMB group in 3A with trifloroacetic acid. Employing the reported method,14 the amino groups in neomycin were protected and the primary hydroxyl group was tranformed to an amino group to give 5 (Scheme 1c). Reaction of 5 with 6-azido-hexanoic acid gave the azide 6, which was then coupled with 3A–D using click chemistry. Subsequent removal of the Boc and PMB protecting groups from the click products by using trifloroacetic acid generated bi-functional molecules 7A–D containing tethers of different length linking the two functional units.
The ability of 7A–D to inhibit Dicer-mediated pre-miR-21 cleavage in vitro was determined. For this purpose, 32P-labeled pre-miR-21 was prepared by in vitro transcription,19 and then incubated (37 °C, 2.5 h) with recombinant human Dicer in the absence or presence of 7A–D, or either the Dicer inhibition unit alone (i.e. compound 4), the pre-miR-21 binding unit alone (i.e., neomycin), or a commercial morpholino-based anti-miR-21 ASO designed to block miR-21 maturation (anti-miR-21 A). The extent of cleavage of pre-miR-21 (72 nt) into mature miR-21 (22 nt) was then determined utilizing a 15% denaturing polyacrylamide gel and phosphor-imaging (Figure S3). As shown in Figure 3, Dicer cleaves pre-miR-21 completely to form mature miR-21. In addition, anti-miR-21 A, as a positive control, inhibits cleavage by 70% at 5 µM, indicating that the assay performs correctly. Importantly, bi-functional molecule 7A inhibits Dicer promoted cleavage in a dose dependent manner, reducing the level of miR-21 formation by 90% at 5 µM (Figure 3 & S4). A comparison of the results coming from studies with 7A–D shows that Dicer catalyzed miR-21 formation increases as the length of the linker between the two functional units increases, demonstrating that inhibitory activity is critically altered by the distance between the two functional units. In contrast to the bi-functional molecules, unmodified neomycin, at concentrations up to 1 mM, does not inhibit Dicer activity (Figure 3 & S5). Furthermore, the inhibitor unit alone (i.e., 4) displays no inhibition at concentrations up to 50 µM (Figure 3 & S6). The combined results clearly show that conjugation of neomycin, the pre-miR-21 recognition unit, to the Dicer inhibitory unit 1 significantly enhances the Dicer inhibition potency (>200 fold comparing 7A to neomycin) and that the presence of a linker of proper length between the two units is essential for the activity.
Figure 3.

(a) The representative image of the electrophoresis analysis of Dicer-mediated pre-miR-21 cleavage in the presence of tested compounds. (b) Densitometric quantitative analysis of miR-21 levels from three independent assays as in (a). The error bars represent the standard error of mean (N = 3).
Because 7A is the most potent Dicer inhibitor in the in vitro assay, it was used for in cell investigations. To study the in cell inhibitory activity of 7A, mature miR-21 expression levels were determined by utilizing the RT-qPCR assay. HEK293T cells were transfected with pCMV-miR21 plasmid expressing pre-miR-21 and treated at the time of transfection with neomycin, a commericial phosphorothioate-based anti-miR-21 ASO (anti-miR-21 B), or various concentrations of 7A. As shown in Figure 4a, 7A inhibits the expression of miR-21 in a dose-dependent manner (76% reduction at 5 µM when compared to non-treated cells). The activity of 7A is similar to that of anti-miR-21 B. In contrast, neomycin does not cause a significant inhibition of miR-21 expression at 100 µM. When 7A was added 4 h after transfection, no inhibititory activtiy was observed, which suggests the transfection procedure may assist cellular delivery of 7A.
Figure 4.

(a) RT-qPCR analysis of mature miR-21 expression levels. (b) The representative image of western blotting analysis of PDCD4 levels in pre-miR-21 expressing HEK293T cells with or without 7A treatment. (c) Densitometric quantitative analysis of PDCD4 levels from three independent assays as in (b). The error bars represent the standard error of mean (N = 3).
Programmed cell death protein 4 (PDCD4) is a tumor suppressor protein, whose mRNA is a direct target of miR-21.20 Expression of miR-21 represses PDCD4 expression in HEK293T cells and repression of miR-21 should de-repress PDCD4 production. As expected, the expression level of PDCD4 protein, determined by using western blotting and compared to non-treated cells, increases by 90% when cells are treated with 7A (Figure 4b & 4c). The cellular assay results demonstrate that 7A is highly active in repressing miR-21 formation in cells.
In the design of bi-functional molecules employed in this study, we utilized aminoglycosides as the pre-miRNA binding unit because they are well-known RNA-binders. However, aminoglycosides, including neomycin, are known to target a number of RNA sequences and, as a result, the new bifunctional substances could suffer from selectivity issues.11–12,14–16
To investigate the miRNA regulation selectivity of the neomycin-based bi-functional molecule 7A, its ability to inhibit another randomly chosen miRNA, miR-34a, in HEK293T cells was determined using RT-qPCR. The results show that 7A indeed reduces the expression of miR-34a (40% reduction at 5 µM when compared to non-treated cells), but not at the same level of potency as it does against miR-21 at the same concentration (Figure S7). Thus, although 7A displays some degree of selectivity, the incorporation of more selective pre-miRNA recognition moieties other than aminoglycosides in the bi-functional design may lead to improved RNA selectivity.
Because neomycin is known to target ribosomal RNA and interfere translation, effects of 7A on translation were tested by transfecting HEK293T and CHO cells with an EGFP reporter followed by 7A treatment.8b No significant change on EGFP expression was observed at the concentration (5 µM) that inhibits miR-21 maturation (Figure S8). Furthermore, no cytotoxicity was observed (analyzed by the MTT assay) when cells were treated with 7A at the same concentration (Figure S9). These results show that under the condition of miR-21 inhibition, 7A does not affect translation or cause toxicity.
In summary, a new approach to regulate miRNA maturation was developed that relies on the use of a Dicer-inhibiting bi-functional small molecule. The study showed that 7A, which contains neomycin as a pre-miR-21 binding unit and the N-hydroxyimide 1 as the Dicer inhibitory unit, inhibits miR-21 maturation in vitro and in cells. Together, these observations demonstrate that the new bi-functional strategy is applicable to the design of active miR-21 inhibitors based on an otherwise non-active pre-miR-21 binder. We expect that the strategy will be affective in generating new bi-functional regulators for miRNAs.
Supplementary Material
Acknowledgments
This research was supported by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (P20GM103451).
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website. Figure S1–S9, Scheme S1–S3 and experimental details (PDF)
The authors declare no competing financial interests.
References
- 1.Ameres SL, Zamore PD. Nat. Rev. Mol. Cell Biol. 2013;14:475. doi: 10.1038/nrm3611. [DOI] [PubMed] [Google Scholar]
- 2.Li Y, Kowdley KV. Genomics, Proteomics Bioinf. 2012;10:246. doi: 10.1016/j.gpb.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stenvang J, Petri A, Lindow M, Obad S, Kauppinen S. Silence. 2012;3:1. doi: 10.1186/1758-907X-3-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juliano RL, Ming X, Nakagawa O. Acc. Chem. Res. 2012;45:1067. doi: 10.1021/ar2002123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Jayaraj GG, Nahar S, Maiti S. Chem. Commun. 2015;51:820. doi: 10.1039/c4cc04514a. [DOI] [PubMed] [Google Scholar]; (b) Velagapudi SP, Vummidi BR, Disney MD. Curr. Opin. Chem. Biol. 2015;24:97. doi: 10.1016/j.cbpa.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Nat. Cell Biol. 2009;11:228. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 7.Gumireddy K, Young DD, Xiong X, Hogenesch JB, Huang QH, Deiters A. Angew. Chem. Int. Ed. 2008;47:7482. doi: 10.1002/anie.200801555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Velagapudi SP, Gallo SM, Disney MD. Nat. Chem. Biol. 2014;10:291. doi: 10.1038/nchembio.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Childs-Disney JL, Disney MD. ACS Chem. Biol. 2016;11:375. doi: 10.1021/acschembio.5b00615. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Disney MD, Angelbello AJ. Acc. Chem. Res. 2016;49:2698. doi: 10.1021/acs.accounts.6b00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pai J, Hyun S, Hyun JY, Park SH, Kim WJ, Bae SH, Kim NK, Yu J, Shin I. J. Am. Chem. Soc. 2016;138:857. doi: 10.1021/jacs.5b09216. [DOI] [PubMed] [Google Scholar]
- 10.(a) Maiti M, Nauwelaerts K, Herdewijn P. Bioorg. Med. Chem. Lett. 2012;22:1709. doi: 10.1016/j.bmcl.2011.12.103. [DOI] [PubMed] [Google Scholar]; (b) Thi Phuong Anh T, Duc Duy V, Di Giorgio A, Duca M. Bioorg. Med. Chem. 2015;23:5334. doi: 10.1016/j.bmc.2015.07.062. [DOI] [PubMed] [Google Scholar]
- 11.(a) Vo DD, Staedel C, Zehnacker L, Benhida R, Darfeuille F, Duca M. ACS Chem. Biol. 2014;9:711. doi: 10.1021/cb400668h. [DOI] [PubMed] [Google Scholar]; (b) Vo DD, Tran TPA, Staedel C, Benhida R, Darfeuille F, Di Giorgio A, Duca M. Chem. Eur. J. 2016;22:5350. doi: 10.1002/chem.201505094. [DOI] [PubMed] [Google Scholar]
- 12.Nahar S, Ranjan N, Ray A, Arya DP, Maiti S. Chem. Sci. 2015;6:5837. doi: 10.1039/c5sc01969a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jazbutyte V, Thum T. Curr. Drug Targets. 2010;11:926. doi: 10.2174/138945010791591403. [DOI] [PubMed] [Google Scholar]
- 14.(a) Kirk SR, Luedtke NW, Tor Y. J. Am. Chem. Soc. 2000;122:980. [Google Scholar]; (b) Michael K, Wang H, Tor Y. Bioorg. Med. Chem. 1999;7:1361. doi: 10.1016/s0968-0896(99)00071-1. [DOI] [PubMed] [Google Scholar]
- 15.(a) Tok JBH, Cho JH, Rando RR. Biochemistry. 1999;38:199. doi: 10.1021/bi9819428. [DOI] [PubMed] [Google Scholar]; (b) Sucheck SJ, Greenberg WA, Tolbert TJ, Wong CH. Angew. Chem. Int. Ed. 2000;39:1080. doi: 10.1002/(sici)1521-3773(20000317)39:6<1080::aid-anie1080>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (c) Tor Y. Chembiochem. 2003;4:998. doi: 10.1002/cbic.200300680. [DOI] [PubMed] [Google Scholar]; (d) Thomas JR, Hergenrother PJ. Chem. Rev. 2008;108:1171. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 16.Zhao F, Zhao Q, Blount KF, Han Q, Tor Y, Hermann T. Angew. Chem. Int. Ed. 2005;44:5329. doi: 10.1002/anie.200500903. [DOI] [PubMed] [Google Scholar]
- 17.MacRae IJ, Zhou KH, Li F, Repic A, Brooks AN, Cande WZ, Adams PD, Doudna JA. Science. 2006;311:195. doi: 10.1126/science.1121638. [DOI] [PubMed] [Google Scholar]
- 18.(a) Parkes KEB, Ermert P, Fassler J, Ives J, Martin JA, Merrett JH, Obrecht D, Williams G, Klumpp K. J. Med. Chem. 2003;46:1153. doi: 10.1021/jm020334u. [DOI] [PubMed] [Google Scholar]; (b) Billamboz M, Bailly F, Barreca ML, De Luca L, Mouscadet JF, Calmels C, Andreola ML, Witvrouw M, Christ F, Debyser Z, Cotelle P. J. Med. Chem. 2008;51:7717. doi: 10.1021/jm8007085. [DOI] [PubMed] [Google Scholar]
- 19.Huang C, Yu Y-T. Curr. Protoc. Mol. Biol. John Wiley & Sons, Inc.; Hoboken, New Jersey: 2013. [Google Scholar]
- 20.Asangani IA, Rasheed SAK, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. Oncogene. 2008;27:2128. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.