

Transcellular tunnels in endothelial cells can be formed by leukocytes and pathogens as a way of crossing the endothelial barrier. Using force microscopy and fluorescence microscopy, we find that the actin cytoskeleton provides the primary mechanical barrier to transcellular tunnel formation, which can be overcome by force or by toxins.
Abstract
The endothelium serves as a protective semipermeable barrier in blood vessels and lymphatic vessels. Leukocytes and pathogens can pass directly through the endothelium by opening holes in endothelial cells, known as transcellular tunnels, which are formed by contact and self-fusion of the apical and basal plasma membranes. Here we test the hypothesis that the actin cytoskeleton is the primary barrier to transcellular tunnel formation using a combination of atomic force microscopy and fluorescence microscopy of live cells. We find that localized mechanical forces are sufficient to induce the formation of transcellular tunnels in human umbilical vein endothelial cells (HUVECs). When HUVECs are exposed to the bacterial toxin called epidermal cell differentiation inhibitor (EDIN), which can induce spontaneous transcellular tunnels, less mechanical work is required to form tunnels due to the reduced cytoskeletal stiffness and thickness of these cells, similarly to the effects of a Rho-associated protein kinase (ROCK) inhibitor. We also observe actin enrichment in response to mechanical indentation that is reduced in cells exposed to the bacterial toxin. Our study shows that the actin cytoskeleton of endothelial cells provides both passive and active resistance against transcellular tunnel formation, serving as a mechanical barrier that can be overcome by mechanical force as well as disruption of the cytoskeleton.
INTRODUCTION
Endothelial cells adhere together to form a semipermeable monolayer that lines the inner surface of blood vessels and lymphatic vessels in animals. These barriers are essential to prevent pathogen dissemination in the underlying tissues during bacteremia and at the same time control the inward and outward passage of white blood cells through vessels in response to inflammatory signals (Carman and Springer, 2008). The diapedesis of leukocytes through endothelial cells can occur between two adjacent endothelial cells after dissociation of intercellular junctions or directly through a single endothelial cell via transcellular tunnels induced by leukocytes (Carman and Springer, 2008). Formation of these transcellular tunnels involves the self-fusion of the endothelial cell’s apical and basal plasma membrane. While migration of leukocytes through endothelial cells has been documented, the physical principles at play during the initiation of transcellular tunnels are not well characterized.
Transcellular migration of leukocytes is accompanied by reorganization of the endothelial cytoskeleton, recruitment of membrane regulating proteins, such as the fusion proteins SNAREs, and docking of intracellular vesicles at the fusion site (Carman and Springer, 2008; Carman, 2009), as well as accumulation of proteins including caveolin at the membrane after fusion (Boyer et al., 2006). Invadosome-like protrusions (ILPs) by leukocytes, which are used to exert forces on endothelial cells and thought to probe for mechanically soft regions, have also been implicated in successful tunnel formation (Carman et al., 2003, 2007; Carman and Springer, 2008). Depolymerization of actin by treatment of endothelial cells with cytochalasin D resulted in more successful transcellular migration of leukocytes (Martinelli et al., 2014), and measurements of endothelial cell elasticity underneath transmigrating cells show a lower elasticity than other areas of the cell (Isac et al., 2011). These observations suggest a crucial role for the endothelial cell actin cytoskeleton in resisting transcellular tunnel formation.
Interestingly, transcellular tunnels can form in the absence of a transmigrating cell. Several bacterial toxins have been found to induce spontaneous transcellular tunnels that are strikingly similar in morphology to tunnels formed during leukocyte diapedesis (Boyer et al., 2006; Carman and Springer, 2008; Maddugoda et al., 2011). Exposure of the endothelial cells to C3, epidermal cell differentiation inhibitor (EDIN), or edema toxin results in loss of central actin stress fibers through inhibition of RhoA or increase of intracellular cAMP, leading to increased spread area and spontaneous formation of large transcellular tunnels, giving rise to vascular leakiness in vivo (Boyer et al., 2006; Maddugoda et al., 2011).
EDIN is a mono-ADP-ribosyltransferase (mART) protein that catalyzes the ADP ribosylation of RhoA, resulting in its sequestration by Rho guanine nucleotide dissociation inhibitor (RhoGDI) in the cytosol (Chardin et al., 1989; Genth et al., 2003; Wilde et al., 2003). This form of RhoA cannot be activated by guanine nucleotide exchange factor. Hence, EDIN locks RhoA in its inactivated form and can essentially block the activation of the downstream effectors such as Rho-associated protein kinase (ROCK), diaphanous-related formins (DIA), focal adhesion kinase (FAK), and phosphoplipase D (PLD), which are important regulators of myosin, actin, focal adhesions, and membrane remodeling (Watanabe et al., 1999; Boyer et al., 2006; Bruntz et al., 2014). Through ROCK, RhoA mediates the phosphorylation/inactivation of the regulatory subunit of the myosin-II phosphatase myosin phosphatase target subunit 1 (MYPT1) (Kimura et al., 1996). Through the mDIA formin, RhoA drives the elongation of actin filaments (Watanabe et al., 1999).
EDIN was previously found to induce transcellular tunnels in nonendothelial cell types, including fibroblasts, osteosarcoma cells, and myoblasts (Boyer et al., 2006), as were cytoskeletal inhibitors, though at a lower frequency than the toxin (Boyer et al., 2006; Rolando et al., 2009; Cai et al., 2010). The closure of these tunnels involves recruitment of actin and missing in metastasis (MIM), which senses and generates membrane curvature via its I-BAR (inverse-BAR [BIN/amphiphysin and RVS167]) domain and drives actin polymerization via its interaction with actin-related protein 2/3 (Arp2/3) (Maddugoda et al., 2011). These findings indicate that bacterial pathogens may disrupt the actin cytoskeleton to promote transcellular tunnel formation.
Here we test the hypothesis that the actin cytoskeleton of endothelial cells is the dominant barrier against transcellular tunnel formation. We use atomic force microscopy (AFM) to apply a localized mechanical force on human umbilical vein endothelial cells (HUVECs), and we combine AFM with total internal reflection fluorescence (TIRF) microscopy to visualize the formation of transcellular tunnels and recruitment of associated proteins in live cells. This technique enables us to quantify the mechanical work needed to drive transcellular tunnel formation in the absence of leukocyte signaling or bacterial toxin activity. By adding toxins and cytoskeletal inhibitors and performing the same measurements, we are able to isolate their effect on the actin cytoskeleton and tunnel formation.
We find that mechanical force alone can induce transcellular tunnel formation in HUVECs in the absence of toxins, leukocytes, or cytoskeletal inhibitors. The transcellular tunnels formed mechanically are locally enriched in the I-BAR domain of MIM and actin, consistent with those formed by toxins. Using the AFM, we quantitatively compare the mechanical properties of control cells and cells exposed to the bacterial toxin EDIN and the ROCK inhibitor Y-27632, and we calculate the mechanical work (integrated force over thickness) necessary for transcellular tunnel formation in each. Cells exposed to EDIN were thinner and softer than control cells, leading to a reduction in work required for tunnel formation and an increase in the probability of spontaneous tunnel formation. Surprisingly, we also found that passive resistance to transcellular tunnel formation by the actin cytoskeleton of endothelial cells is complemented by an active response to mechanical force, marked by actin enrichment at the indentation site. The static and dynamic mechanical properties of the actin cytoskeleton thus provide crucial support for the barrier function of the endothelium, one that pathogens and leukocytes can overcome either by localized force or by biochemical destabilization of the cytoskeleton.
RESULTS
Mechanical force is sufficient to form transcellular tunnels in HUVECs
We utilized atomic force microscopy to apply controlled forces to cultured HUVECs with high temporal, force, and spatial precision. We combined this technique with TIRF microscopy to enable simultaneous imaging of protein localization dynamics near the ventral membrane and to track morphological changes of the cell, specifically tunnel opening and closing. The experimental setup is depicted in Figure 1A. An AFM cantilever with a pyramidal tip (∼100 nm tip diameter) is used to indent a spread cell near the cell edge. First, a force clamp of 5 nN is applied to establish contact with the cell. Then the force is linearly increased until a tunnel forms or until the AFM piezo has been extended by 5 µm. Then the position of the cantilever is held for 20–120 s before the tip is retracted.
FIGURE 1:

Local force application by the tip of an atomic force microscope cantilever is sufficient to induce transcellular tunnels in endothelial cells. (A) Schematic diagram of the experimental setup. Using the AFM, a tipped cantilever is extended toward a spread HUVEC until the tip is in contact with the cell at 5 nN force. Then the applied force is slowly increased at a rate of 1 nN/s until a tunnel is observed by TIRF imaging of the cell membrane or the tip has been extended by 3–5 µm. The cantilever is retracted while the progression of the opening and closing of the tunnel is tracked by time-lapse imaging. (B) Time-lapse TIRF images of a HUVEC forming a tunnel when indented by the tip of an AFM cantilever. The cell was expressing GFP-MIM-I-BAR and LifeAct-mCherry and labeled with the CellMask far red membrane dye. Opening of the tunnel was followed by localization of MIM around the aperture and actin-rich wave formation to close the aperture. The arrowheads indicate location of the AFM cantilever tip. The time points of each image relative to tip contact (t = 0 s) are displayed above the image. Scale bar: 10 µm. (C) Example force trace during an indentation experiment. (D) Kymograph of a region of the cell in A, outlined by the yellow box, showing tunnel opening at a higher temporal resolution of 2 s intervals. The yellow dotted line marks the location of the tip and the yellow solid lines trace the opening of the tunnel. (E) Trace of AFM tip distance from the glass substrate, which corresponds to the force trace in C. This trace also shows the change in cell height as the tip continued to deform the cell at higher forces. The arrows in C–E indicate the point when the tunnel opened. Inset shows tip distance trace when set occurred. (F) Example force vs. tip-substrate distance trace from a different cell, showing a gradual increase in force at the beginning of indentation, followed by a steeper increase in force as a function of deformation. A step displacement of the tip occurred at constant force due to full penetration of the cell when a tunnel opens.
Using HUVECs cultured in control conditions, we observed tunnel formation in many of the cells (∼82%, n = 49 cells) that were indented. These tunnels were very transient in nature, reaching an average size of 0.6 µm and typically closing before or as soon as the AFM cantilever tip was retracted. Figure 1B and Supplemental Video 1 show a sequence of images of the ventral plasma membrane, labeled with a membrane dye, where tunnel opening and closing was observed. A kymograph of the tunnel opening process is shown in Figure 1D. The MIM I-BAR domain and actin were also observed at the tunnel edges, which represent hallmarks of toxin-induced transcellular tunnel formation (Maddugoda et al., 2011) (Figure 1B).
By correlating timing of the force trace with the time lapse image, we could obtain the magnitude of the applied force at the time point when the tunnel opened, marked by the arrow (Figure 1C). Interestingly, plasma membrane fusion and opening of tunnels in the cells often coincided with a step displacement of the tip, with a median size of 11 nm, consistent with the thickness of two plasma membranes (Figure 1, C and F; Supplemental Figure S1). We termed the force at which the step displacement occurs the penetration force (Figure 1F). These results demonstrate that mechanical force alone is sufficient to induce transcellular tunnels in HUVECs.
Disruption of RhoA and ROCK signaling alters cell mechanics
We postulated that lowered myosin activity would compromise the integrity and reduce the stiffness of the actin cytoskeleton, thus reducing the barrier against tunnel formation in endothelial cells. To test this, we acquired images of the actin cytoskeleton in cells under the three different conditions—RhoA inhibition, ROCK inhibition, and control. For the inhibition conditions, HUVECs were transfected with plasmids encoding for EDIN, a RhoA inhibiting toxin from Staphylococcus aureus, or treated with the ROCK inhibitor Y-27632 at 50 µM to disrupt myosin activity. We found that the actin cytoskeletal organization was very different in EDIN-expressing and Y-27632–treated cells compared with control cells, with the actin bundles in control cells appearing thicker, straighter, and less peripheral than in EDIN-expressing and Y-27632–treated cells (Figure 2A; Supplemental Figure S1). As expected, indentation of the EDIN-expressing and Y-27632–treated cells with an AFM tip also resulted in tunnel formation (Figure 2, B and C; Supplemental Figure S1).
FIGURE 2:

Bacterial toxin, EDIN, and ROCK inhibitor drug, Y-27632, alter mechanical properties of the cell. (A) Confocal images of LifeAct-GFP in fixed control and EDIN-expressing HUVECs. Very few central actin fiber bundles were observed in the presence of EDIN. The image of LifeAct-GFP in a live Y-27632–treated cell was taken by TIRF microscopy. White arrowheads indicate location of some transcellular tunnels. Scale bar: 10 µm. Inhibition of ROCK activity also disrupted central stress fiber formation. Mechanical force also induces tunnel formation in EDIN-expressing (B) and Y-27632–treated (C) HUVECs. Kymographs show evolution of tunnel opening at 2 s intervals. The yellow dotted line marks the location of the tip after contact and the yellow solid lines trace the opening of the tunnel. The regions of the cell depicted in the kymographs are outlined in the left panel. (D) Thickness of the cell at the indentation site for the three conditions. Box plots show the median and the 25th and 75th percentiles of distribution. Open squares show mean thickness. Thickness was measured from initial cell contact point until tip-substrate contact. On average, cells expressing EDIN were thinner than control cells at the sites where the cells were indented. NCTRL = 51, NEDIN = 42, NY-27632 = 16. (E) Young’s modulus of cells measured by indenting the cell body with an AFM tip and fitting the cantilever deflection curve with a modified Hertz model. On average, cells expressing EDIN and treated with Y-27632 were much softer than control cells. NCTRL = 9, NEDIN = 7, NY-27632 = 5. (F) Cells expressing EDIN had lower membrane tension than control cells. Membrane tension was measured by pulling membrane tethers from the cell and looking at the tether rupture force. NCTRL = 15, NEDIN = 11, NY-27632 = 8. Significance of difference was tested using the nonparametric Wilcoxon rank sum test. Asterisks above each box indicate difference from control cells. Horizontal bar shows comparison between EDIN and ROCK inhibition conditions *p < 0.05, **p < 0.01, ***p < 0.001.
We found that EDIN-expressing cells (median: 332 nm) were significantly thinner than control cells (median: 462 nm) near the edges, where the tip indentation experiments were conducted, whereas cells treated with Y-27632 (median: 365 nm) were not (Figure 2D). Using the AFM, we measured the Young’s moduli of elasticity of the cells and found that the EDIN-expressing (median: 0.3 kPa) and Y-27632–treated cells (median: 3 kPa) had a much softer actomyosin cortex than control cells (median: 9 kPa), with EDIN-expressing cells having the lowest elasticity (Figure 2E). These results show that the mechanical properties of cells are altered in the presence of EDIN or Y-27632, with a more pronounced effect induced by EDIN.
Reduction in myosin activity has been shown to lead to larger spread area and potentially higher membrane tension (Cai et al., 2010; Gonzalez-Rodriguez et al., 2012). Using the AFM (Diz-Muñoz et al., 2010, 2013), we were able to directly quantify and compare the plasma membrane effective tension (a combination of in-plane tension and membrane-to-cortex attachment) of EDIN-expressing and ROCK-inhibited cells to that of control cells. Surprisingly, we observed a decrease in plasma membrane tension in the presence of EDIN (Figure 2F). We did not observe a change in plasma membrane tension after ROCK inhibition (Figure 2F; medians: control = 0.44, EDIN = 0.25, ROCK-inhibited = 0.38 mN/m).
These mechanical measurements show that EDIN causes HUVECs to thin, soften, and reduce plasma membrane effective tension, perhaps through reduced membrane-to-cortex attachment, all of which could contribute to reducing the mechanical barrier against transcellular tunnel formation.
EDIN toxin reduces cells stiffness and lowers mechanical work to form transcellular tunnel
Considering the effects of EDIN on the thickness, elasticity, and plasma membrane tension of endothelial cells, we sought to quantify the mechanical barrier against tunnel formation in the presence of this toxin. From the force-versus-height curves obtained during cell indentation, we could determine the penetration step size as well as the applied force on tunnel opening (Figure 3A). We observed no difference in penetration step size among control, EDIN-expressing, and ROCK-inhibited cells, suggesting a common mechanism during the final step of tunnel opening, presumably fusion of the two plasma membranes (Figure 3B). We found that the average penetration force of cells expressing EDIN was lower than that of control cells (Figure 3C; Supplemental Figure S2; means: control = 39, EDIN = 19, ROCK-inhibited = 40 nN). Consistent with the lower average penetration force, the number of tunnels that formed at low forces (F ≤ 20 nN) was higher in EDIN-treated cells than in control cells (Figure 3D). Surprisingly, in ROCK-inhibited cells we observed fewer tunnels forming under a 20 nN force and a higher average penetration force (Figure 3, C and D).
FIGURE 3:

Altered cell mechanics by EDIN decreases the barrier against tunnel opening. (A) Cartoon plot of force-vs.-height curve. The point of tip-cell contact is indicated by an arrow. The penetration step size during tunnel opening is measured as shown on the cartoon. Penetration work is measured by computing the shaded area under the curve. Area is computed from the cell contact point until right before cell penetration occurs. (B) Penetration step sizes for control, EDIN-expressing and Y-27632–treated cells. Box plots show the median and the 25th and 75th percentiles of distribution. Open squares show mean thickness. No difference observed among the three conditions. NCTRL = 32, NEDIN = 27, NY-27632 = 12. (C) Force required to induce tunnels for the three conditions. On average, the penetration force for cells expressing EDIN was lower than for control cells. NCTRL = 38, NEDIN = 27, NY-27632 = 16. (D) Percentage of indentation experiments that induced tunnels at forces equal to or below 20 nN. A larger fraction of EDIN-expressing, but not ROCK-inhibited, cells formed tunnels at lower forces compared with control cells. (E) Work done to fully indent the cell and form a tunnel. Cells expressing EDIN required less work than control and Y-27632–treated cells to induce tunnels. NCTRL = 34, NEDIN = 27, NY-27632 = 16. (F) To evaluate the contribution of actomyosin network density on the barrier against tunnel formation, the indentation experiments were repeated using a 5.66 µm bead glued to the end of a tipless cantilever, which increases the surface area of indentation. The tunnel barrier was increased with the use of a bead to indent cells for both control and EDIN conditions. Control cells showed a larger fold increase in penetration work compared with EDIN-expressing cells. NCTRL = 7, NEDIN = 9. Statistical analyses were performed using the nonparametric Wilcoxon rank sum test. Statistical significance of distribution differences between EDIN or ROCK inhibition data and control data are noted above the box. The horizontal bars show comparison between EDIN and ROCK inhibition conditions. *p < 0.05, **p < 0.01, ***p < 0.001.
To better capture the effects of EDIN and Y-27632 on the mechanical barrier presented by the actin cytoskeleton, we computed the total work done to penetrate a cell by integrating force versus height for each indentation, as depicted by the shaded area in Figure 3A. We found that the distribution of cell penetration work was shifted to lower values in EDIN-expressing cells (Figure 3E). There were no significant differences in the average penetration forces or work required to form tunnels in ROCK-inhibited and control cells.
If the actin cytoskeleton compressed by the AFM tip is the primary barrier to tunnel formation, then a larger AFM tip should encounter an increased barrier. We replaced the pyramidal tipped cantilever with a 5.66 µm silica bead and repeated the force ramp indentations on control and EDIN-expressing cells. The work required to indent a cell using a bead and a cantilever tip can be estimated by integration of the modified Hertz’s model for the respective indentation geometries (Azeloglu and Costa, 2011). Consistent with the key role of the cytoskeleton in resisting tunnel formation, we measured 5.5-fold greater penetration work in control cells compared with EDIN-expressing cells when a bead indenter was used. In theory, the work required to indent a cell using a bead should be ∼9-fold higher than using a cantilever tip on the same cell if the assumptions of the Herz model hold. We found that the penetration work with the bead was 7.5-fold greater in control cells than with the pyramidal tip but only 4-fold greater when measured on EDIN-expressing cells (Figure 3F). The ∼50% lower penetration work measured in the EDIN-expressing cells suggests that it is not well described by the linear elastic and homogenous material assumptions of the Hertz model. These results are consistent with the barrier function of the actin cytoskeleton against transcellular tunnel formation.
Active actin cytoskeletal response resists transcellular tunnel formation in wild-type endothelial cells
In addition to providing passive resistance to deformation, the actin cytoskeleton is known to actively respond to external mechanical perturbations (Uyeda et al., 2011; Luo et al., 2012, 2013). We sought to determine if control endothelial cells have a different active response compared with EDIN-expressing and ROCK-inhibited cells. On characterization of the force-induced transcellular tunnels, we found that more than 60% of the tunnels formed in EDIN-expressing and Y-27632–treated cells were larger than 0.6 µm in diameter and/or persisted at least more than 4 s after retraction of the cantilever tip (Figure 4A). We observed a gradual accumulation of actin at the site of tip indentation, with some tunnel formation events showing stronger actin enrichment than others (Figure 4B). While actin cable formation around transcellular tunnels has recently been observed (Stefani et al., 2017), the degree of actin accumulation in control cells compared with toxin expressing cells is not known. We quantified the level of actin enrichment by measuring the fold change in mean intensity around the site of tip indentation with respect to the mean intensity on initial tip contact with the cell. We found that 61% of the tunnels formed in control cells showed strong actin enrichment, defined as more than 1.25-fold increase in normalized actin intensity, whereas less than 33.3% of the tunnels showed the same level of enrichment in EDIN-expressing and Y-27632–treated cells (Figure 4C). The large fraction of strong actin enrichment in control cells could be due to an active actin response machinery, which may be disrupted by the EDIN toxin or inhibition of ROCK.
FIGURE 4:

Active response of cells to indentation by AFM resists tunnel formation and limits tunnel size and persistence. (A) Percentage tunnels that were large (>0.6 µm) or persistent (remained open for >4 s after tip retraction). EDIN expression and Y-27632 treatment resulted in larger and more persistent AFM-induced tunnels. (B) Time-lapse images showing examples of the strong (>1.25-fold increase) or weak (<1.25-fold increase) actin enrichment around the AFM-induced tunnel. Scale bar: 2 µm. (C) Summary of the fraction of indentation experiments that induced strong or weak actin responses in control, EDIN-expressing, and ROCK-inhibited cells. A large fraction of the indentations of control cells induced strong accumulation of actin, whereas more EDIN-expressing and ROCK-inhibited cells had larger tunnels with little actin accumulation. (D) To determine whether membrane deformation could induce actin enrichment, cells were indented with an AFM tip at a low force of 2–5 nN, and the applied force was held for 2–3 min while imaging LifeAct-GFP with TIRF microscopy. (E) Image sequence of actin enrichment at the tip indentation site of a control cell. Arrowheads indicate location of AFM cantilever tip. Scale bar: 5 µm. (F) Example traces of normalized mean actin intensity change in a control (black) and EDIN-expressing (purple) cell. Note that the actin accumulation occurs more than 60 s after the cantilever indentation. (G) The corresponding height change traces for the same cells in F. The force generated by actin polymerization appeared to be sufficient to push on the AFM tip clamped at 5 nN. (H) Percentage of cells that showed actin enrichment based on a 1.25× cutoff threshold for the three conditions. (I) Distribution of the maximum normalized actin intensity change for the three conditions. Box plots show the median and 25th and 75th percentiles of distribution. Open squares show mean thickness. Statistical analyses were performed using the nonparametric Wilcoxon rank sum test. The difference between EDIN-expressing and control cells was only weakly significant. NCTRL = 16, NEDIN = 8, NY-27632 = 5. (J) Changes in cell height after indenting the cells. NCTRL = 16, NEDIN = 8, NY-27632 = 5. No significant difference among the three conditions.
The forces applied to the cells in our indentation experiments involved large-scale deformations of the actomyosin network. To evaluate whether actin accumulation occurs in the absence of tunnel formation, we applied a low force of 2–5 nN and clamped the force for 2–3 min to impose a small deformation on control cells without fully penetrating them (Figure 4D). Consistent with our observations during transcellular tunnel formation, we saw actin accumulation under the tip within 1 min after contact (Figure 4E). An example trace of mean actin intensity at the site of tip indentation is shown in Figure 4F. In the same cell, we also measured an increase in cell height of >100 nm, indicating a pushing back of the cantilever tip against the small force clamp (Figure 4G). We observed strong accumulation of actin (>1.25-fold mean intensity increase) in >80% of the weakly indented control cells. A smaller fraction of cells expressing EDIN (25%) also showed strong actin enrichment. Example traces of mean intensity and cell height of an EDIN-expressing cell showing weak actin enrichment are shown in Figure 4, F and G. The fraction of cells that showed actin enrichment was lowest in the EDIN-expressing cells (Figure 4H). Owing to the binary nature of the actin response observed in EDIN-expressing cells (Supplemental Figure S3), whereby enrichment was either very strong or extremely weak, possibly due to different expression levels, the difference in average actin enrichment between control and EDIN conditions was only weakly significant (Figure 4I). The average increase in cell height at the tip indentation site was not statistically significantly different among the three conditions, though the control cells showed the largest spread in height with increase up to 500 nm, whereas the Y-27632–treated cells showed little change in cell height (Figure 4J). These results demonstrate the ability of control cells to actively resist small membrane deformations by actin accumulation, a response that is diminished in cells exposed to EDIN.
DISCUSSION
Force application by AFM is sufficient to form transcellular tunnels
In this study, we established an AFM-based technique to isolate the role of the actin cytoskeleton and mechanical force in formation of transcellular tunnels in endothelial cells. By using an AFM cantilever tip to apply a localized force on a cell, we eliminate contributions from other biophysical and biochemical processes that often take place during leukocyte transcellular migration. In addition, the length scale of the pyramidal cantilever tip is on the same order of magnitude as that of protrusions, or podosomes, of leukocytes. The width of leukocyte podosomes is ∼500 nm while the length ranges from 100 to 2000 nm, with some spanning nearly the entire endothelial cell thickness (Carman et al., 2007). By combining the use of AFM with TIRF imaging of the cell membrane, we can probe the resistance of localized regions in the actomyosin network to indentation and explore the spatial heterogeneity of the actomyosin network of single cells. We focused on HUVECs because this cell type was already widely used in other studies related to transcellular tunnels. As has been previously shown, EDIN triggers the formation of transcellular tunnels in endothelial cells more efficiently than in other cell types tested (Boyer et al., 2006).
The distribution of measured cell penetration forces was very broad, ranging from 5 to 100 nN. The large variation in forces likely results from the spatial heterogeneity of cytoskeletal structures in cells and, thus, the microscale mechanics may determine precisely where a tunnel is most likely to open under a specific load (Solon et al., 2007). Interestingly, the average protrusive forces by podosomes have been measured to be around 90 nN (Labernadie et al., 2014), which are consistent with what we estimate to be required for tunnel formation. The distribution of cell penetration forces for the EDIN-expressing cells were equally broad, with a slight skew to the lower forces, while the peak of the distribution for Y-27632–treated cells was shifted to a higher force. Therefore the spatial heterogeneity of cytoskeletal structure was still evident even when actomyosin contractility was globally challenged.
Fusion of inner leaflets during transcellular tunnel formation differs from fusion of outer leaflets of the plasma membrane during cell-cell fusion (Shilagardi et al., 2013; Kim et al., 2015) or self-fusion (Sumida and Yamada, 2013) in important ways. First, tunnel formation requires that two inner leaflets must find each other through cytoplasm filled with large physical obstructions, such as the cortical actin cytoskeleton and various organelles, that normally prevent tunnel formation in wild-type cells. Close apposition of extracellular leaflets during cell-cell fusion may also be prevented by physical obstructions, but through different proteins, such as the extracellular matrix, regulated in different ways. Second, the properties of the inner leaflet of the plasma membrane differ in lipid and protein composition compared with the outer leaflet, and thus the requirements for fusion may also differ (van Meer, 2011; Bucci, 2013). Proposed models of inner leaflet fusion involve either divalent cations and negatively charged lipids (Mondal Roy and Sarkar, 2011) or fusogenic proteins such as SNARE (Carman, 2009). For transcellular tunnel formation, clearance of the actin cytoskeleton appears to be the main barrier that keeps the dorsal and ventral membranes apart and prevents fusion under most circumstances. Our results show that mechanical force is sufficient to overcome that cytoskeletal barrier and induce inner leaflet fusion.
EDIN toxin reduces the endothelial mechanical barrier against tunnel formation
We found a significant decrease in thickness and elasticity in cells expressing EDIN compared with control cells. We also observed a decrease in elasticity of cells treated with Y-27632 but to a much lesser degree. The decreased thickness and elasticity could increase the likelihood that the dorsal and ventral membranes of the cell transiently come into contact by membrane fluctuations. Moreover, cells with a thinner and softer actomyosin network would be much easier to fully indent with the AFM cantilever tip, thus requiring less work. As predicted, we observed a significantly lower average penetration work in EDIN-expressing cells. However, disruption of ROCK activity by Y-27632 drug treatment did not result in a decrease in average penetration work. Previous findings have shown the much lower efficiency of direct ROCK inhibition on tunnel formation in endothelial cells (Boyer et al., 2006), demonstrating the importance of other effectors of RhoA, in addition to ROCK, in mediating spontaneous tunnel formation in cells. Dispersal of thick actin cables into thin filaments by ROCK inhibition may alter the organization of actin filaments but not their resistance to compression, thus rendering the average penetration work unchanged compared with control cells, whereas reduction of the total F-actin content in the EDIN-expressing cells may have a more significant impact on the barrier function of the cell (Boyer et al., 2006). Other studies have also proposed that DIA1, which is another effector of RhoA, acts in parallel with ROCK to regulate actin cytoskeletal architecture and mediates force-induced focal contact growth in a ROCK-independent manner (Watanabe et al., 1999; Riveline et al., 2001; Vicente-Manzanares et al., 2003). Therefore actin polymerization by DIA1 may be equally important in keeping the membranes apart to prevent tunnel formation.
The average size of force-induced tunnels in EDIN-expressing cells (∼0.7 µm) was generally smaller than the spontaneous tunnels observed in cells after EDIN exposure (Boyer et al., 2006; Maddugoda et al., 2011; Gonzalez-Rodriguez et al., 2012). This may be due to differences in the level of RhoA inhibition between cells directly exposed to purified recombinant toxin in previous literature and cells expressing EDIN via plasmid transfection. The size of tunnels induced has been shown to be proportional to the efficiency of RhoA inhibition (Rolando et al., 2009). Furthermore, for a tunnel to form spontaneously, a larger area of the actin network may need to be disrupted so the upper and lower membranes can come into contact and fuse. Tunnels formed by the AFM cantilever tip, on the other hand, could be induced in an area of the cell that has a thicker actin meshwork, and thus enlargement of the tunnel could be more restricted.
Interestingly, the plasma membrane effective tension of EDIN-expressing cells was measured to be lower than in control cells. Interpretation of tether pulling experiments in terms of effective tension is not straightforward since the tether force can be influenced by interactions between cortex and lipid membrane, in addition to the tension of the lipid membrane itself (Hochmuth et al., 1996). The observed reduction in plasma membrane effective tension in EDIN-expressing cells could potentially be due to a decrease in membrane-to-cortex attachment to a sparser actin cytoskeleton, as suggested by the much-lower Young modulus than in control cells, but it could also result from a reduced in-plane bilayer tension. Reduced membrane tension could in principle affect both the likelihood of a fusion event, as well as the ability to nucleate a tunnel. A cellular dewetting model has been previously proposed to describe regulation of tunnel opening and stabilization by the balance between membrane tension and line tension (Gonzalez-Rodriguez et al., 2012; Lemichez et al., 2013). In this model, tunnel nucleation was modeled in analogy with hole nucleation in a fluid layer and is expected to directly depend on membrane tension. As discussed above, our measurements show that the tunnel nucleation barrier is lower in the EDIN-expressing cells than in control cells despite the lower tension, suggesting that a more complete model of transcellular tunnel formation will need to account for the cytoskeletal changes induced by the EDIN toxin. A lower surface tension would also in principle lead to more narrow tunnels (Gonzalez-Rodriguez et al., 2012). However, recent results (Stefani et al., 2017) suggest that line tension, which limits tunnel opening, varies with time and might reach its plateau value over a longer period when the cytoskeleton is affected by EDIN, thus leading to larger tunnels.
Active cytoskeletal response to force-induced transcellular tunnels
In addition to changes in average transcellular tunnel formation forces in EDIN-expressing cells compared with control cells, we observed different distributions in the types of actin response to indentation by AFM. A large fraction of the indentations of control cells led to bright punctate or ring-shaped actin accumulation at the indentation site. In contrast, most of the EDIN-expressing and Y-27632–treated cells that were indented showed very weak actin enrichment around the tunnels throughout the duration of the experiments, which often resulted in larger tunnel sizes and longer persistence of the tunnels. We found even with low forces not resulting in membrane fusion that actin accumulates at the contact point and pushes back on the AFM tip, increasing tip-substrate separation for a constant force. Previous work has shown that myosin II can sense and localize to dilated and stretched regions of the actin cytoskeleton, such as the tip of an aspirated cell in a micropipette (Luo et al., 2013). Actin rings have also been observed around transmigrating leukocytes during diapedesis (Heemskerk et al., 2016). ROCK inhibition did not alter the fraction of cells that showed strong actin enrichment after tip indentation compared with control cells but shifted the distribution of change in cell height in response to light indentation with the AFM tip to lower values. These observations suggest that ROCK-mediated myosin activity could play an important role in the active pushback against external forces that indent the cell. Since membrane curvature during partial indentation differs from membrane curvature within a tunnel, MIM localization is not expected, so local nucleation of actin must be driven by other nucleation promoting factors (Maddugoda et al., 2011; Lemichez et al., 2013). The active response of actin to deformations by the AFM tip suggests an additional role of the actomyosin network in regulating cell shape in the presence of dynamic mechanical perturbations through force-dependent actin reinforcement.
Maintaining a selective barrier to transcellular tunnel formation
The endothelial monolayer provides a physical barrier against large living and nonliving objects in the bloodstream or lymph from entering the underlying tissue. Controlled passage through this barrier can occur via transcellular or paracellular routes. Several studies have shown that the transcellular route involves the actin cytoskeleton. In this study, we present quantitative evidence that the actin cytoskeleton physically keeps the dorsal and ventral membranes apart and actively resists external mechanical stress that compress the network. Localized mechanical indentation is sufficient to overcome the actin cytoskeleton and bring the two inner leaflets in close enough proximity for fusion to occur. Some pathogens indiscriminately secrete actin depolymerizing toxins, such as EDIN, to globally weaken the endothelial barrier and induce spontaneous transcellular tunnels. These tunnels enable the pathogens to cross the barrier without generating force themselves, but such tunnels also lead to disruption of tissue homeostasis, giving rise to symptoms such as edema. Leukocytes, however, are able to undergo successful transcellular diapedesis without detrimental side effects. This is likely possible due to the fine balance between active protrusions and ligand-receptor engagement to spatially and temporally regulate the actin cytoskeletal state inside the endothelial cells such that the tunnel width is adjusted to the size of leukocytes (see Heemskerk et al., 2016) and immediately closed after the leukocyte has completed its migration.
MATERIALS AND METHODS
Cell culture, transfection, and sample preparation
Human umbilical vein endothelial cells, obtained from the UCB Cell Culture Facility, were cultured in RPMI-1640 (Life Technologies, Carlsbad, CA) supplemented with 20% fetal bovine serum (FBS; Life Technologies), 1% penicillin/streptomycin (Sigma, St. Louis, MO), 0.0275 mg/ml heparin (Sigma), and 0.05 mg/ml endothelial cell growth supplement (Alfa Aesar, Ward Hill, MA). Testing for mycoplasma contamination was conducted every 6 mo. Cells were collected by incubation with 0.25% trypsin (Sigma), neutralized with regular medium, and plated on glass-bottom chambers that were precoated with 0.2% gelatin wt/vol (BD Biosciences) for 10 min. Cells were allowed to adhere and spread for 2–4 h before experiments were conducted.
Stable expression of LifeAct-GFP and LifeAct-mCherry in HUVECs was performed by lentiviral transduction of the genes of interest cloned into pHR vectors (kind gift from R. Vale, University of California, San Francisco [UCSF]). HUVECs with stable expression of tagRFP-EDIN were also created by lentiviral transduction of the fusion gene cloned into a pLVX vector that contains the tetracycline-inducible promoter (kind gift from O. Weiner, UCSF). These cells were cultured in media supplemented with tetracycline-free FBS (Clontech, San Francisco, CA) to eliminate baseline expression of EDIN. Expression of tagRFP-EDIN in these cells was induced by incubating them in 1 µg/ml tetracycline for 6–16 h prior to experiments. A majority of the experiments testing the effects of EDIN were conducted using HUVECs transiently transfected with pEDIN and MIM-I-BAR-GFP (Maddugoda et al., 2011), or tagRFP-EDIN. The tagRFP-EDIN construct was made by cloning PCR amplified tagRFP into the pEDIN construct. Cells were transfected by electroporation (Life Technologies) according to manufacturer’s protocol and immediately plated on gelatin-coated glass bottom chambers for experiments 6–18 h after transfection. Cells that were transiently transfected with EDIN were used in all the experiments, whereas some of the force, work, height, step size, and actin intensity measurements included cells that were stably expressing EDIN. DNA sequences of the inserted gene portions into the pLVX and pEDIN constructs are provided in the Supplemental Information. Proper insertion of the genes were verified by DNA sequencing.
HUVECs were incubated in media containing 50 μM of Y-27632 (Sigma) for at least 1 h before experiments to inhibit ROCK activity. All cells were incubated with CellMask Far red (Invitrogen) during the experiments to visualize the plasma membrane and to track tunnel formation.
Cell indentation by atomic force microscopy
To isolate the role of physical forces on transcellular tunnel formation, we used atomic force microscopy to apply localized forces with tipped cantilevers, which have a geometry that is of the same length scale as protrusions extended by leukocytes into endothelial cells. Experiments were conducted using a BioScope Catalyst Atomic Force Microscope (Bruker AXS, Santa Barbara, CA) with a temperature-controlled stage set to 37°C, mounted atop an inverted optical microscope (Axio Observer.Z1; Carl Zeiss, Thornwood, NY). Data acquisition and AFM control were done using a signal access module and custom-designed software (LabVIEW, National Instruments, Austin, TX). DNP cantilevers (Bruker) with nominal spring constants of 0.35 N/m were used in all indentation experiment. The actual spring constant of each cantilever was determined by fitting thermal fluctuations in air.
During an indentation experiment, a region of interest was first identified near the cell edge, and the stage was translated to position the region of interest under the mounted cantilever tip. The regions of interest were chosen based on the actin cytoskeleton architecture such that areas rich in thick actin bundles were avoided. This was done to minimize variations in force measurements due to indentation of stress fiber bundles, in addition to the actin cortex. A 5 nN force clamp was applied to establish contact with the cell. Then, the magnitude of the clamped force was linearly increased at a rate of 1 nN/s until a tunnel was observed by TIRF imaging. Extension of the piezo was halted a few seconds after tunnel opening. If no tunnel was observed, then ramping of the piezo was halted after an additional 5 µm extension beyond the cell contact point. In cases where the tunnel size was very small, the cantilever may not be retracted immediately after the tunnel was formed because confirmation of tunnel opening was determined during analysis post experiment. For tunnels that could be seen to open during the experiments, we typically halted the further indentation of the cantilever (further increase in force applied) and maintained the position of the cantilever for an additional few seconds to avoid tether pulling effects, which occasionally happened due to nonspecific binding of the plasma membrane to the cantilever tip. The tip was retracted at a rate of 100 nm/s 20–120 s after stopping piezo extension.
To investigate the active response of cells to small indentations, a 3- to 5 nN force clamp was applied to establish contact, and the force was held for 2–5 min while time-lapse imaging was acquired at 2 s intervals. The cantilevers were passivated by plasma cleaning for 30 s, followed by incubation with 3 mg/ml poly(l-lysine)-poly(ethylene glycol) (PLL-PEG) for 1 h. Surface passivation of the cantilever tip prevents nonspecific binding of the plasma membrane, which sometimes result in tether pulling and tunnel formation during retraction.
Elasticity measurements
MLCT cantilevers (Bruker) with nominal spring constants of 0.01 N/m were used to measure the Young’s modulus of elasticity of control, EDIN-expressing and Y-27632–treated cells. Cells were indented at a rate of 1 µm/s and the extension-deflection curves were fitted to a modified Hertz equation for a pyramidal tip, as previously described (Rosenbluth et al., 2006), using a code written in Matlab (Mathworks, Natick, MA). Experiments were conducted at 37°C.
Membrane tension measurements
Plasma membrane effective tension, which includes in-plane tension and membrane-to-cortex attachment, was measured with the atomic force microscope. We measured membrane tension by quantifying the static retraction force of membrane tubes pulled from the plasma membrane (Hochmuith et al., 1996). Very soft BioLever (Asylum Research, Santa Barbara, CA) or OBL (Bruker) probes with nominal spring constants of 0.006 N/m were used due to their high force resolution and low noise characteristics. Experiments were conducted at 37°C. The cantilevers were incubated with 2.5 mg/ml concanavalin A (Sigma) for 1 h to enable membrane adhesion to the tip when placed in contact with the cell. The mounted cantilever was first extended at a rate of 1 µm/s until the tip was in contact with the cell at 200 pN force. The tip was held at a constant force for 5–25 s to enable sufficient binding of the membrane to the cantilever tip before it was retracted by 5 µm at a rate of 5 µm/s to pull a membrane tether. The tip was then held at constant height to allow for the tether force to equilibrate as viscous effects from the membrane-to-cortex attachment during tether pulling dissipated. The resulting force, which was constant over time, reflects the static force (zero velocity force) required to stably hold the membrane tether away from the cell (Dai and Sheetz, 1995). To account for any cantilever or sample drift that could introduce errors into the force measurement, we waited for the static tether to detach from the cantilever tip and recorded the zero-point deflection of the cantilever, which was subtracted from the cantilever deflection required to hold the static membrane tether to obtain the static tether force. To ensure that tether detachment occurred within several minutes of tether pulling, we titrated the concentration of concanavalin A on the tip to a level where the tether was sufficiently attached to allow tether pulling but not so strongly attached that the tether did not retract. Only cells that exhibited stable tether forces were included in the data set. By measuring this drift-corrected tether force, we could compute the plasma membrane effective tension using the following equation (Hochmuth et al., 1996):
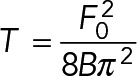 |
where T is the membrane tension, F0 is the tether rupture force, and B is the membrane bending stiffness. B was estimated to be 85.6 pN·nm (20 KBT) when computing membrane tension.
Microscopy and image analysis
Images of the cell membrane, actin, and other fluorescently tagged proteins were acquired by total internal reflection fluorescence microscopy (TIRFM) on an inverted microscope (Axio Observer.Z1; Zeiss), equipped with a 100×/1.46 NA oil immersion objective (Zeiss Plan-Apochromat) and an electron multiplying charge-coupled device (EMCCD) camera (iXon; Andor, South Windsor, CT). Images of fixed cells were acquired on a spinning disk confocal microscope (Axio Observer.Z1, Zeiss) with a 63×/1.4 NA oil immersion objective (Zeiss Plan-Apochromat). Time-lapse images were taken at 2 s intervals to track tunnel opening during indentation. Analysis of actin accumulation at the indentation site was performed using ImageJ (National Institutes of Health). The intensity of actin was measured by computing the mean pixel intensity within a manually drawn region of interest around the indentation site of time lapse images of LifeAct. Actin intensity at the indentation site was normalized to actin intensity in an adjacent region of the cell to account for fluctuations in intensity within the cell. Actin accumulation was quantified by dividing the normalized mean intensity measurements to the initial normalized mean intensity on indentation of cell.
Bead indentation of cells
To test the effects of increasing surface area on the work required to induce tunnels in cells, we repeated the indentation experiments with a 5.6-µm-diameter silica bead attached to a tipless cantilever (DNP, Bruker) by UV glue (NOA63; Norland Products, Cranbury, NJ). The nominal radius of curvature of the cantilever tip is 50 nm.
Data analysis
Data analyses were performed using Microsoft Excel and Igor Pro (WaveMetrics, Portland, OR). Force and tip distance traces that are presented in the figures were smoothed by median filtering to remove high-frequency thermal noise. Twenty- to forty-millisecond segments of the traces where vibrations of the filter wheel instrument were picked up by the AFM were also removed to avoid artifacts in the final traces. Penetration forces were measured by identifying the force value at which a step in cell height occurred or when a tunnel was observed in the time-lapse movie. Penetration work is defined as the total area under the force-versus-cell height curve, excluding the penetration step when tunnel opening occurs. Significant actin enrichment was determined using a 1.25× increase threshold.
Statistical analysis
Tests for significance in distribution differences between two conditions were conducted using the Wilcoxon rank sum test, with a two-tailed, p ≤ 0.05 threshold for significance, unless indicated otherwise. Average values were presented as means and medians, and the variance in distribution was represented by the 25th and 75th percentiles in a box plot. Statistical tests were conducted using Igor Pro.
Supplementary Material
Acknowledgments
We thank C. Chan and S. Son for help with initial test experiments, A. Diz-Muñoz for help with tether pulling experiments and cloning design, M. Taylor and R. Vale for the LifeAct lentiviral constructs, and the rest of the Fletcher lab for helpful feedback and technical consultation. This work was supported by the France-Berkeley Fund (P.B., E.L., D.A.F.), National Institutes of Health R01 GM114671 (D.A.F.), a Siebel Fellowship (W.P.N.), and Agence Nationale de la Recherche Grant ANR-15-CE18-0016-01 to E.L. and P.B.
Abbreviations used:
- AFM
atomic force microscopy
- HUVEC
human umbilical vein endothelial cells.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E17-01-0080) on August 9, 2017.
REFERENCES
- Azeloglu EU, Costa KD. Atomic force microscopy in mechanobiology: measuring microelastic heterogeneity of living cells. Methods Mol Biol. 2011;736:303–329. doi: 10.1007/978-1-61779-105-5_19. [DOI] [PubMed] [Google Scholar]
- Boyer L, Doye A, Rolando M, Flatau G, Munro P, Gounon P, Clement R, Pulcini C, Popoff MR, Mettouchi A, et al. Induction of transient macroapertures in endothelial cells through RhoA inhibition by Staphylococcus aureus factors. J Cell Biol. 2006;173:809–819. doi: 10.1083/jcb.200509009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruntz RC, Lindsley CW, Brown HA. Phospholipase D signaling pathways and phosphatidic acid as therapeutic targets in cancer. Pharmacol Rev. 2014;66:1033–1079. doi: 10.1124/pr.114.009217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci M. Lipids: leaflets out of order. Nat Chem Biol. 2013;9:67–67. [Google Scholar]
- Cai Y, Rossier O, Gauthier NC, Biais N, Fardin M-A, Zhang X, Miller LW, Ladoux B, Cornish VW, Sheetz MP. Cytoskeletal coherence requires myosin-IIA contractility. J Cell Sci. 2010;123:413–423. doi: 10.1242/jcs.058297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV. Mechanisms for transcellular diapedesis: probing and pathfinding by “invadosome-like protrusions.”. J Cell Sci. 2009;122:3025–3035. doi: 10.1242/jcs.047522. [DOI] [PubMed] [Google Scholar]
- Carman CV, Jun C-D, Salas A, Springer TA. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol. 2003;171:6135–6144. doi: 10.4049/jimmunol.171.11.6135. [DOI] [PubMed] [Google Scholar]
- Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, Dvorak HF, Dvorak AM, Springer TA. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–797. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman CV, Springer TA. Trans-cellular migration: cell-cell contacts get intimate. Curr Opin Cell Biol. 2008;20:533–540. doi: 10.1016/j.ceb.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chardin P, Boquet P, Madaule P, Popoff MR, Rubin EJ, Gill DM. The mammalian G protein rhoC is ADP-ribosylated by Clostridium botulinum exoenzyme C3 and affects actin microfilaments in Vero cells. EMBO J. 1989;8:1087–1092. doi: 10.1002/j.1460-2075.1989.tb03477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Sheetz MP. Mechanical properties of neuronal growth cone membranes studied by tether formation with laser optical tweezers. Biophys J. 1995;68:988–996. doi: 10.1016/S0006-3495(95)80274-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diz-Muñoz A, Fletcher DA, Weiner OD. Use the force: membrane tension as an organizer of cell shape and motility. Trends Cell Biol. 2013;23:47–53. doi: 10.1016/j.tcb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diz-Muñoz A, Krieg M, Bergert M, Ibarlucea-Benitez I, Muller DJ, Paluch E, Heisenberg C-P. Control of directed cell migration in vivo by membrane-to-cortex attachment. PLoS Biol. 2010;8:e1000544. doi: 10.1371/journal.pbio.1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genth H, Gerhard R, Maeda A, Amano M, Kaibuchi K, Aktories K, Just I. Entrapment of Rho ADP-ribosylated by Clostridium botulinum C3 exoenzyme in the rho-guanine nucleotide dissociation inhibitor-1 complex. J Biol Chem. 2003;278:28523–28527. doi: 10.1074/jbc.M301915200. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez D, Maddugoda MP, Stefani C, Janel S, Lafont F, Cuvelier D, Lemichez E, Brochard-Wyart F. Cellular dewetting: opening of macroapertures in endothelial cells. Phys Rev Lett. 2012;108:218105. doi: 10.1103/PhysRevLett.108.218105. [DOI] [PubMed] [Google Scholar]
- Heemskerk N, Schimmel L, Oort C, Rijssel J van, Yin T, Ma B, Unen J van, Pitter B, Huveneers S, Goedhart J, et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun. 2016;7:10493. doi: 10.1038/ncomms10493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochmuth FM, Shao JY, Dai J, Sheetz MP. Deformation and flow of membrane into tethers extracted from neuronal growth cones. Biophys J. 1996;70:358–369. doi: 10.1016/S0006-3495(96)79577-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isac L, Thoelking G, Schwab A, Oberleithner H, Riethmuller C. Endothelial f-actin depolymerization enables leukocyte transmigration. Anal Bioanal Chem. 2011;399:2351–2358. doi: 10.1007/s00216-010-3978-z. [DOI] [PubMed] [Google Scholar]
- Kim JH, Ren Y, Ng WP, Li S, Son S, Kee Y-S, Zhang S, Zhang G, Fletcher DA, Robinson DN, et al. Mechanical tension drives cell membrane fusion. Dev Cell. 2015;32:561–573. doi: 10.1016/j.devcel.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Labernadie A, Bouissou A, Delobelle P, Balor S, Voituriez R, Proag A, Fourquaux I, Thibault C, Vieu C, Poincloux R, et al. Protrusion force microscopy reveals oscillatory force generation and mechanosensing activity of human macrophage podosomes. Nat Commun. 2014;5:5343. doi: 10.1038/ncomms6343. [DOI] [PubMed] [Google Scholar]
- Lemichez E, Gonzalez-Rodriguez D, Bassereau P, Brochard-Wyart F. Transcellular tunnel dynamics: Control of cellular dewetting by actomyosin contractility and I-BAR proteins. Biol Cell. 2013;105:109–117. doi: 10.1111/boc.201200063. [DOI] [PubMed] [Google Scholar]
- Luo T, Mohan K, Iglesias PA, Robinson DN. Molecular mechanisms of cellular mechanosensing. Nat Mater. 2013;12:1064–1071. doi: 10.1038/nmat3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo T, Mohan K, Srivastava V, Ren Y, Iglesias PA, Robinson DN. Understanding the cooperative interaction between myosin II and actin cross-linkers mediated by actin filaments during mechanosensation. Biophys J. 2012;102:238–247. doi: 10.1016/j.bpj.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddugoda MP, Stefani C, Gonzalez-Rodriguez D, Saarikangas J, Torrino S, Janel S, Munro P, Doye A, Prodon F, Aurrand-Lions M, et al. cAMP signaling by anthrax edema toxin induces transendothelial cell tunnels, which are resealed by MIM via Arp2/3-driven actin polymerization. Cell Host Microbe. 2011;10:464–474. doi: 10.1016/j.chom.2011.09.014. [DOI] [PubMed] [Google Scholar]
- Martinelli R, Zeiger AS, Whitfield M, Sciuto TE, Dvorak A, Van Vliet KJ, Greenwood J, Carman CV. Probing the biomechanical contribution of the endothelium to lymphocyte migration: diapedesis by the path of least resistance. J Cell Sci. 2014;127:3720–3734. doi: 10.1242/jcs.148619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondal Roy S, Sarkar M. Membrane fusion induced by small molecules and ions. J Lipids. 2011;2011:e528784. doi: 10.1155/2011/528784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riveline D, Zamir E, Balaban NQ, Schwarz US, Ishizaki T, Narumiya S, Kam Z, Geiger B, Bershadsky AD. Focal contacts as mechanosensors: externally applied local mechanical force induces growth of focal contacts by an mDia1-dependent and ROCK-independent mechanism. J Cell Biol. 2001;153:1175–1186. doi: 10.1083/jcb.153.6.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolando M, Munro P, Stefani C, Auberger P, Flatau G, Lemichez E. Injection of staphylococcus aureus EDIN by the Bacillus anthracis protective antigen machinery induces vascular permeability. Infect Immun. 2009;77:3596–3601. doi: 10.1128/IAI.00186-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluth MJ, Lam WA, Fletcher DA. Force microscopy of nonadherent cells: a comparison of leukemia cell deformability. Biophys J. 2006;90:2994–3003. doi: 10.1529/biophysj.105.067496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilagardi K, Li S, Luo F, Marikar F, Duan R, Jin P, Kim JH, Murnen K, Chen EH. Actin-propelled invasive membrane protrusions promote fusogenic protein engagement during cell-cell fusion. Science. 2013;340:359–363. doi: 10.1126/science.1234781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J. 2007;93:4453–4461. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani C, Gonzalez-Rodriguez D, Senju Y, Doye A, Efimova N, Janel S, Lipuma J, Tsai MC, Hamaoui D, Maddugoda MP, et al. Ezrin enhances line tension along transcellular tunnel edges via NMIIa driven actomyosin cable formation. Nat Commun. 2017;8:15839. doi: 10.1038/ncomms15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida GM, Yamada S. Self-contact elimination by membrane fusion. Proc Natl Acad Sci USA. 2013;110:18958–18963. doi: 10.1073/pnas.1311135110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyeda TQP, Iwadate Y, Umeki N, Nagasaki A, Yumura S. Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS ONE. 2011;6:e26200. doi: 10.1371/journal.pone.0026200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Meer G. Dynamic transbilayer lipid asymmetry. Cold Spring Harb Perspect Biol. 2011;3:a004671. doi: 10.1101/cshperspect.a004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Rey M, Pérez-Martínez M, Yáñez-Mó M, Sancho D, Cabrero JR, Barreiro O, de la Fuente H, Itoh K, Sánchez-Madrid F. The RhoA effector mDia is induced during T cell activation and regulates actin polymerization and cell migration in T lymphocytes. J Immunol. 2003;171:1023–1034. doi: 10.4049/jimmunol.171.2.1023. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Wilde C, Vogelsgesang M, Aktories K. Rho-specific Bacillus cereus ADP-ribosyltransferase C3cer cloning and characterization. Biochemistry. 2003;42:9694–9702. doi: 10.1021/bi034583b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.