

Knocking down clathrin in PC12 cells not only affects the maturation of dense core vesicles, it also renders them essentially incapable of secretagogue-induced exocytosis.
Abstract
The dense core vesicles (DCVs) of neuroendocrine cells are a rich source of bioactive molecules such as peptides, hormones, and neurotransmitters, but relatively little is known about how they are formed. Using fractionation profiling, a method that combines subcellular fractionation with mass spectrometry, we identified ∼1200 proteins in PC12 cell vesicle-enriched fractions, with DCV-associated proteins showing distinct profiles from proteins associated with other types of vesicles. To investigate the role of clathrin in DCV biogenesis, we stably transduced PC12 cells with an inducible short hairpin RNA targeting clathrin heavy chain, resulting in ∼85% protein loss. DCVs could still be observed in the cells by electron microscopy, but mature profiles were approximately fourfold less abundant than in mock-treated cells. By quantitative mass spectrometry, DCV-associated proteins were found to be reduced approximately twofold in clathrin-depleted cells as a whole and approximately fivefold in vesicle-enriched fractions. Our combined data sets enabled us to identify new candidate DCV components. Secretion assays revealed that clathrin depletion causes a near-complete block in secretagogue-induced exocytosis. Taken together, our data indicate that clathrin has a function in DCV biogenesis beyond its established role in removing unwanted proteins from the immature vesicle.
INTRODUCTION
Dense core vesicles (DCVs) are a type of regulated secretory granule packed with bioactive molecules such as hormones, neuropeptides, and catecholamines, which are found mainly in neurons and neuroendocrine cells. Although there has been interest in how molecules are trafficked to DCVs for more than 30 yr (Gumbiner and Kelly, 1982; Moore et al., 1983), our knowledge of sorting signals and machinery for DCV trafficking is still relatively limited (Dikeakos and Reudelhuber, 2007). A role for clathrin was first proposed by Tooze and Tooze in 1986, on the basis of their observations that clathrin coats are frequently associated with immature DCVs in AtT-20 cells, an anterior pituitary-derived cell line (Tooze and Tooze, 1986). Subsequent work by Klumperman and colleagues, on both DCVs and other types of secretory granules, showed that these clathrin-coated profiles were also positive for AP-1, the major adaptor for intracellular clathrin-mediated trafficking, as well as for AP-1–dependent cargo proteins such as mannose 6-phosphate receptors, hydrolases, furin, and syntaxin 6, all of which are absent from the mature granule (Klumperman et al., 1998; Dittié et al., 1999). These findings led to the proposal that the role of the clathrin coat is to remove unwanted proteins from the nascent secretory granule, so that they can then be redirected to other parts of the cell.
Although there is no doubt that this is part of the story, several observations suggest that the situation may be somewhat more complicated. For instance, one of the few membrane proteins known to be associated with DCVs is phogrin, which has a dileucine sorting signal that binds to AP complexes, including AP-1. Mutating this sorting signal causes phogrin to be either retained in the Golgi region or mislocalized to the plasma membrane (Torii et al., 2005), suggesting that APs and clathrin may be involved not only in trafficking away from DCVs, but also in trafficking to DCVs. A further complication is that some of the cells that make DCVs also make synaptic-like microvesicles (SMVs). Like DCVs, SMVs are secreted in a regulated manner, and again clathrin has been invoked, although most likely in an endocytic capacity (Hannah et al., 1999). In addition, depleting clathrin and/or AP-1 in two other types of cells has been shown to completely abolish the formation of regulated secretory organelles (Lui-Roberts et al., 2005; Burgess et al., 2011). Thus there is still a lack of understanding of the precise role of clathrin in DCV biogenesis.
One of the reasons progress has been slow is that it is difficult to carry out knockdowns in neuroendocrine cell lines, which are the best model system for investigating DCV formation and function. For instance, in a recent study by Eipper and colleagues on AtT-20 cells, short hairpin RNA (shRNA) was used to deplete the medium (μ1A) subunit of the AP-1 adaptor complex, resulting in a ∼50% loss (Bonnemaison et al., 2014), as compared with the >90% protein loss routinely obtained in other cell types (Elbashir et al., 2001). However, even with only a 50% loss, there was a clear phenotype, with secretagogue-induced release of DCV proteins also down by ∼50% (Bonnemaison et al., 2014). Other groups have made use of dominant negative constructs to investigate the function of clathrin and associated proteins in DCV biogenesis. Overexpressing the clathrin “hub” in pancreatic beta cells caused an increase in the proteolysis of insulin, consistent with a defect in the removal of unwanted lysosomal hydrolases from immature vesicles (Molinete et al., 2001); and overexpressing a truncated version of GGA1 (another clathrin adaptor) in PC12 (adrenal medulla–derived) cells also strongly affected DCV maturation (Kakhlon et al., 2006). However, one problem with dominant negative experiments is that the phenotype may be indirect. For instance, the truncated forms of clathrin and GGA1 that were used in the two studies bind to Hip1R and to Arf1, respectively (Bennett et al., 2001; Puertollano et al., 2001), so it is possible that the block in DCV maturation might have been due to a lack of Hip1R or Arf1, rather than because clathrin or GGA1 are directly involved.
We decided to revisit the role of clathrin in DCV biogenesis using two new approaches: fractionation profiling and induced shRNA knockdown. Fractionation profiling was developed by our laboratory as a means of identifying proteins associated with different types of subcellular structures (Borner et al., 2014). Our aim was first to apply this method to PC12 cells to find out whether we could distinguish between clathrin-coated vesicles (CCVs), DCVs, and SMVs, and then to look for changes in the behavior of the different types of proteins when clathrin was depleted.
RESULTS
Fractionation profiling of PC12 cells
We began by developing a protocol for fractionation profiling in PC12 cells, shown schematically in Figure 1A. Briefly, cells were grown in stable isotope labeling with amino acids in cell culture (SILAC) “light” or SILAC “heavy” medium for 3 wk, by which time incorporation of heavy amino acids into proteins was found to be ∼94%. Both sets of cells were homogenized and a vesicle-enriched fraction was obtained by differential centrifugation (see Materials and Methods). Then the “light” vesicle-enriched fraction was subjected to three successive centrifugation steps, collecting the pellets at the end of each spin and then recentrifuging the supernatants at a higher g-force. The “heavy” vesicle-enriched fraction was only centrifuged once, at a high g-force, to provide a reference pellet to compare with the three “light” pellets.
FIGURE 1:

Fractionation profiling of PC12 cells. (A) Schematic diagram of the protocol. Cells that had been grown for 3 wk in either SILAC light or SILAC heavy medium were homogenized and a vesicle-enriched fraction was prepared. The vesicle-enriched fraction from cells grown in light medium was then subjected to three successive centrifugation steps at increasing g-force to generate three separate pellets. The vesicle-enriched fraction from cells grown in heavy medium was centrifuged only once, at high g-force, to provide the reference pellet. (B) Electron micrographs of the three SILAC light pellets. Pellet 1 contains the majority of the clathrin-coated vesicles (CCVs; red arrows) and the dense core vesicles (DCVs; blue arrows). Scale bar: 200 nm. (C) Each of the three pellets was mixed with a third of the reference pellet and analyzed by mass spectrometry to quantify the light-to-heavy ratio for every protein; then the data were subjected to principal component analysis (PCA; the axes show principal components 1 and 2). Proteins associated with the three types of vesicles form distinct clusters because of their similar vesicle-specific and consistent fractionation profiles. Individual protein identities are shown in Supplemental Figure S1.
Electron micrographs of pellets 1, 2, and 3 are shown in Figure 1B. Both CCVs (red arrows) and DCVs (blue arrows) can be easily identified in the first pellet, whereas the second and third pellets contain relatively few CCVs or DCVs and instead are enriched in smaller vesicles and other particles such as ribosomes and proteasomes.
To investigate the relative abundance of individual proteins in the three fractions, each of the three pellets was mixed with a third of the reference pellet and subjected to SDS–PAGE, then mass spectrometry was used to quantify the light-to-heavy ratio for every protein. In this way, individual distribution profiles of each protein across the subfractions were obtained. Because a visual representation of high-dimensional data is impractical to illustrate, we used principal component analysis (PCA) to achieve dimensionality reduction. Briefly, PCA derives linear combinations (“principal components”) of the original variables that preserve a maximum of the information contained in the data, while reducing the overall number of variables. When applied to the fractionation profiling data set, principal components 1 and 2 accounted for 95% of the data variability. The structure of the data could hence be visualized in a 2D scatter plot (Figure 1C and Supplemental Figure S1). As we expected, proteins known to be associated with CCVs, DCVs, and SMVs had similar profiles, and formed distinct clusters. Therefore fractionation profiling is a suitable and sensitive method for analyzing several different types of PC12 cell vesicles simultaneously.
Clathrin knockdown in PC12 cells
Having established a fractionation profiling protocol for PC12 cells, we next investigated the effects of clathrin heavy chain depletion. The system that worked best in our hands was SMARTvector lentiviral shRNA, in which the shRNA is inducibly coexpressed with green fluorescent protein (GFP) (Figure 2). Stably transduced cells were initially selected on the basis of puromycin resistance, then coexpression of GFP and shRNAs was induced by an overnight incubation with doxycycline. High GFP expressors were selected by flow cytometry, after which the cells were maintained in the absence of doxycycline because clathrin depletion compromises cell growth (Figure 2A). When doxycycline was added to the cells for 5 d, both immunofluorescence and Western blotting showed a robust loss of clathrin in the doxycycline-treated cells when compared with the mock-treated cells (Figure 2B). There was an even more dramatic loss of clathrin when we compared vesicle-enriched fractions from the two cell populations (Figure 2B). This is a common phenomenon (e.g., see Motley et al., 2003), and it appears to be because clathrin-depleted cells attempt to form as many CCVs as control cells, but without sufficient clathrin they are unable to form a complete cage and thus vesicle budding is curtailed (Moskowitz et al., 2005; Hinrichsen et al., 2006).
FIGURE 2:

Clathrin depletion from PC12 cells. (A) Cells were transduced with SMARTvector lentiviral shRNA targeting clathrin heavy chain (CHC). Infected cells were initially selected with puromycin. Then, because the shRNA is inducibly coexpressed with GFP, treatment with doxycycline overnight was used to induce expression. The doxycycline was then removed and high expressors were isolated by flow cytometry to provide a stable population of cells harboring inducible CHC shRNA. (B) To investigate the effects of clathrin depletion, the cells were either mock-treated or treated for 5 d with doxycycline. Immunofluorescence and Western blotting both show a robust loss of clathrin. EF2 was used as a loading control. Scale bar: 10 μm.
What happens to DCVs in the clathrin-depleted cells? Electron microscopy showed that DCVs are still present (Figure 3A), but mature DCVs are relatively scarce (Figure 3, B and C). For instance, the most mature “type 1″ structures, which have only a dense core and no electron-lucent space, were depleted approximately fourfold in the knockdown cells, while the least mature “type 4″ structures, which consist mainly of an electron-lucent space with a relatively light core, were increased approximately twofold. The clathrin-depleted cells also contained unusual structures that we classified as “type 5.” The type 5 structures were somewhat larger than even immature DCVs, mainly electron-lucent, and contained small intralumenal inclusions that resembled the intralumenal vesicles of multivesicluar bodies (MVBs). However, the type 5 organelles were smaller than MVBs, and unlike MVBs they were absent in the mock-treated cells. We suspect that they may be similar to the vacuoles observed by Eipper and coworkers in μ1A-depleted AtT-20 cells (Bonnemaison et al., 2014).
FIGURE 3:

Electron microscopy of mock-treated and clathrin-depleted cells. (A) Representative images of the mock-treated and clathrin-depleted cells, with CCVs indicated in red and DCVs in blue. Scale bar: 500 nm. (B) Classification of different types of DCVs. Type 1 are the most mature and type 4 are the least mature. The type 5 vesicles were only seen in clathrin-depleted cells. Scale bar: 200 nm. (C) Quantification of the different vesicle types. Type 1 vesicles were approximately fourfold reduced in the clathrin-depleted cells. Total vesicle counts were 126 for control and 133 for clathrin knockdown.
Comparative proteomics
To look for differences in the protein composition of DCVs in clathrin-depleted cells, we first analyzed total homogenates and vesicle-enriched fractions from SILAC-labeled mock-treated and doxycycline-treated cells. Consistent with our Western blotting results (Figure 2B), clathrin heavy chain was more strongly depleted from the vesicle-enriched fractions than from the cells as a whole (∼13-fold vs. ∼7-fold). Other CCV components such as AP-1 were not depleted from the cells as a whole, only from the vesicle-enriched fractions, consistent with our work on HeLa cells (Borner et al., 2006).
Interestingly, however, when we compared DCV proteins, we saw a modest but reproducible depletion from the cells as a whole (∼1.5 to 2-fold), as well as a more pronounced loss from the vesicle-enriched fractions (∼4- to 6-fold). These results indicate that at steady state, in the clathrin-depleted cells DCV proteins are not only less abundant but also differentially localized. SMV proteins were unaffected in the cell as a whole, but ∼1.5- to 2-fold depleted in the vesicle-enriched fractions.
By combining the fractionation profiling and clathrin knockdown data and analyzing the entire data set by PCA (Figure 4B), it was possible to identify other proteins that behaved in a similar manner to known DCV proteins, making them good candidates for additional DCV components. For instance, Emilin-1 (elastin microfibril interface–located protein 1) is a secreted protein not found in HeLa cells, which cofractionated with DCVs and was strongly affected by clathrin knockdown. Other novel candidate DCV proteins include Igfbp6 (insulin-like growth factor binding protein 6), Hapln4 (hyaluronan and proteoglycan link protein 4), Tprg1L (tumor protein p63–regulated 1–like), and Pnma2 (paraneoplastic antigen Ma2) (Supplemental Figure S2).
FIGURE 4:
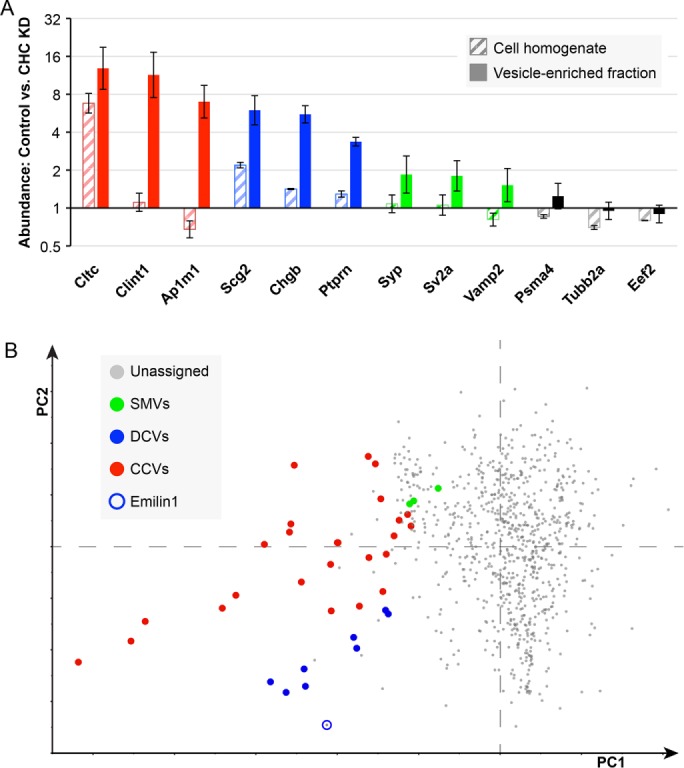
Effect of clathrin depletion on different types of vesicles. (A) Mass spectrometry was carried out on SILAC-labeled mock-treated and clathrin-depleted cell homogenates and vesicle-enriched fractions to identify proteins with altered behavior. More than 2500 proteins were quantified, of which a few examples are shown here. CCV-associated proteins (shown in red) were depleted from the vesicle-enriched fraction, but with the exception of clathrin they were not depleted from the cell homogenate. DCV-enriched proteins (shown in blue) were depleted 1.5- to 2-fold from the cell homogenate and 4- to 6-fold from the vesicle-enriched fraction, while SMV proteins (green) were unaffected in the homogenate but moderately depleted from the vesicle-enriched fraction. Other proteins (shown in black) were generally unaffected. Proteins are indicated by their gene names (Cltc = clathrin heavy chain; Clint1 = epsinR; Ap1m1 = AP-1 μ1; Scg2 = secretogranin II; Chgb = chromogranin B; Ptprn = receptor-type tyrosine-protein phosphatase-like N; Syp = synaptophysin; Sv2a = synaptic vesicle glycoprotein 2A; Vamp2 = VAMP2; Psma4 = proteasome subunit alpha 4; Tubb2a = tubulin beta 2A; Eef2 = eukaryotic translation elongation factor 2). Error bars show SD (n = 3). (B) Principal component analysis combining the fractionation profiling data shown in Figure 1C and the data shown above on fold-depletion in the vesicle-enriched fraction after clathrin knockdown (three replicates). CCVs and DCVs are well separated, and new DCV components can be predicted, such as Emilin (circled). The identities of the protein are shown in Supplemental Figure S2.
Fate of DCV components in clathrin-depleted cells
What happens to DCV proteins when clathrin is depleted? To investigate whether they are still secreted in a regulated manner, we collected culture medium from cells that had been washed and then treated for 15 min either with or without BaCl2, a standard secretagogue. Both the collected medium and the cell homogenates were then subjected to Western blotting and probed with an antibody against chromogranin B (CgB) (Figure 5A). These experiments showed a dramatic decrease in BaCl2-induced release of CgB in the clathrin-depleted cells, while the basal or constitutive release (i.e., in the absence of BaCl2) looked roughly similar, indicating that regulated secretion is severely impaired in the clathrin-depleted cells.
FIGURE 5:

Secretion of chromogranin B (CgB) in mock-treated and clathrin-depleted cells. A. Western blot of mock-treated and clathrin-depleted cells incubated for 15 min in the presence or absence of the secretagogue BaCl2, after which both the cells and the culture supernatants were probed with anti-CgB. Clathrin depletion strongly reduces induced secretion of CgB. (B) Both mock-treated and clathrin-depleted cells were grown in both SILAC heavy and SILAC light medium; then the heavy and light cells were incubated for 15 min with or without BaCl2, respectively. Heavy and light media were pooled, immunoprecipitated with anti-CgB, and analyzed by mass spectrometry. In the mock-treated cells, BaCl2 caused a (9.59 ± 1.47)-fold increase in CgB secretion. However, in the clathrin-depleted cells, the increase was negligible (1.16 ± 0.11). The indicated p value is derived from a two-tailed Student’s test (four biological replicates).
To obtain more quantitative data, we turned to comparative proteomics. Cells were grown in either heavy or light medium for 3 wk, and for the last 5 d doxycycline was added to half of the “heavy” cells and half of the “light” cells in order to deplete clathrin (Figure 5B). All of the cells were washed, and then the heavy cells were incubated for 15 min with BaCl2 and the light cells without BaCl2. The four sets of culture supernatants were collected, and the two supernatants from the mock-treated cells were pooled, as were the two supernatants from the clathrin-depleted cells. CgB was then immunoprecipitated from the mixed supernatants. Using mass spectrometry to calculate the heavy-to-light ratios, we were able to determine the relative amounts of constitutive versus regulated secretion in mock-treated and clathrin-depleted cells. In the mock-treated cells, there was ∼10 times as much CgB secreted into the culture supernatant in the presence of BaCl2 as in the absence of BaCl2. However, in the clathrin-depleted cells, there was almost no increase in CgB secretion in the presence of BaCl2 (1.164 ± 0.113). This indicates that the block in DCV maturation into functional organelles is much more severe than that indicated by electron microscopy, where morphologically recognizable mature DCVs were reduced fourfold. The secretion experiments suggest that, in fact, almost none of the DCVs are capable of regulated exocytosis.
Norepinephrine uptake and release
A well-established assay for DCV function is to measure the uptake and release of a catecholamine such as norepinephrine (Taupenot, 2007). The DCV membrane contains a monoamine transporter, VMAT1, which efficiently concentrates catecholamines, and the catecholamines are then released into the medium when the cells are treated with a secretagogue such as BaCl2. To investigate whether clathrin depletion alters catecholamine uptake and/or release, we first incubated cells for 3 h in medium containing [3H]norepinephrine (Figure 6A). The cells were then washed and incubated for 15 min either with or without BaCl2, the culture supernatants were collected, the cells were lysed, and the radioactivity present in both supernatants and lysates was quantified by scintillation counting. Adding the values together provided a measure of total [3H]norepinephrine uptake under each condition.
FIGURE 6:

Uptake and secretion of [3H]norepinephrine in mock-treated and clathrin-depleted cells. (A) Schematic diagram of the protocol. Mock-treated and clathrin-depleted cells were incubated for 3 h with [3H]norepinephrine, then washed and incubated for 15 min either with or without BaCl2. Radioactivity in the cells and in the culture supernatant was determined by scintillation counting. (B) Adding up the counts in both cells and medium showed that there was approximately fivefold more uptake in the mock-treated cells. **Indicates statistical significance at the p < 0.005 level assessed by two-tailed Student’s t test (three biological replicates). (C) In the mock-treated cells, release of [3H]norepinephrine was strongly dependent on BaCl2, but in the clathrin-depleted cells, the effect of BaCl2 was insignificant. **Indicates statistical significance at the p < 0.005 level assessed by analysis of variance with Tukey’s post-hoc test; n.s. indicates lack of statistical significance at the p < 0.05 level (three biological replicates).
Uptake of [3H]norepinephrine was found to be approximately fivefold reduced in the clathrin-depleted cells when compared with equal numbers of mock-treated cells (Figure 6B). In the absence of BaCl2, ∼4% of the [3H]norepinephrine was released from mock-treated cells, while BaCl2 treatment caused a ∼12-fold increase in [3H]norepinephrine release (Figure 6C). In contrast, in the clathrin-depleted cells, ∼10% of the [3H]norepinephrine was released into the culture supernatant in the absence of BaCl2, and there was no significant increase when cells were incubated in the presence of BaCl2. Thus clathrin depletion causes PC12 cell DCVs to become essentially incapable of secretagogue-induced catecholamine release.
To ensure that the phenotype we observed was not an off-target effect, we first tried stably transfecting our inducible cell line with a tagged shRNA-resistant clathrin heavy chain construct. Unfortunately, even our most highly expressing cells were not making enough clathrin for a rescue (Supplemental Figure S3, A–D). Therefore we tried an alternative approach: use of a second independent shRNA, shRNA-B. Although the knockdowns were not as effective as with our other shRNA (shRNA-A), we observed qualitatively similar effects on [3H]norephrine uptake and release. We think that the reason we got only a partial block in secretagogue-induced exocytosis, instead of a complete block, can be explained by the cooperative nature of clathrin assembly. Whereas shRNA-A most likely reduced the amount of clathrin to below the critical concentration needed to form a CCV (Moskowitz et al., 2005), shRNA-B reduced it to the extent that CCV formation was impaired but not abolished.
DISCUSSION
Clathrin has long been implicated in DCV biogenesis, but there are still questions about its precise role. In the present study, we have combined fractionation profiling, inducible clathrin knockdown, and morphological and biochemical assays, to try to understand how clathrin contributes to the formation and function of DCVs in PC12 cells.
Fractionation profiling is a method we originally developed in HeLa cells and then applied to Drosophila S2 cells (Borner et al., 2014). It was designed to optimize the segregation of CCVs from other subcellular particles, but the present study shows that it also provides good resolution of DCVs, which like CCVs are fairly uniform in size and density. Even though CCVs and DCVs are both most concentrated in fraction 1 (see Figure 1), their distinct profiles mean that they can be well separated by PCA.
We were able to optimize clathrin knockdown using a lentiviral system that allows inducible coexpression of shRNA and GFP. This system not only works much better on PC12 cells than siRNA transfection, it also has several other advantages. First, because the cells are stably transduced, scaling up for large-scale biochemistry experiments is much cheaper than when oligonucleotides and transfection reagents are used. Second, because expression of the shRNA is inducible, there is less of a problem with cell viability (always a concern when knocking down clathrin). Third, because the starting populations of cells are identical, variability between control and knockdown cells is minimized. Fourth, because the shRNA is coexpressed with GFP, efficiency of induction can be monitored by fluorescence microscopy of living cells. We found that we were routinely getting >95% of the cells expressing GFP.
One outcome of combining fractionation profiling and clathrin depletion was that it allowed us to identify new candidate DCV proteins, because these proteins not only had similar profiles to known DCV proteins, but also were strongly depleted from the vesicle-enriched fraction after clathrin knockdown. One such protein, Emilin-1, has been shown to be an inhibitor of TGF-β signaling, and mice lacking this protein are susceptible to high blood pressure, similar to CgA- and CgB-deficient mice (Litteri et al., 2012). Furthermore, polymorphisms in the human EMILIN1 gene have been found to correlate with hypertension, suggesting that Emilin-1 may be a DCV cargo protein with clinical relevance (Shimodaira et al., 2010).
When we examined our cells by electron microscopy, we observed more immature and fewer mature DCVs in clathrin-depleted PC12 cells, consistent with the well-documented role for clathrin in DCV maturation. However, although typical-looking mature (“type 1″) DCVs could be observed by electron microscopy in clathrin-depleted cells (albeit at a fourfold reduced frequency), our results indicate that these DCVs are highly impaired functionally. Secretagogue-induced release of both CgB and norepinephrine was essentially abolished in the clathrin-depleted cells. Norepinephrine uptake was also reduced, by approximately fivefold, even though there was only approximately a twofold reduction in VMAT (the monoamine transporter) in the vesicle-enriched fraction. This observation suggests that there is likely to be missorting of other proteins involved in the sequestration of norepineprine into DCVs as well. Furthermore, the near-complete insensitivity of the cells to BaCl2 indicates that there is also mislocalization of DCV-associated proteins involved in regulated exocytosis. For instance, there might be insufficient amounts of machinery such as SNAREs and synaptotagmins. One caveat is that regulated exocytosis involves the plasma membrane as well as the DCV, so at present we cannot rule out the possibility that the resistance to secretagogue in the clathrin-depleted cells may have been due at least in part to changes in the composition of the plasma membrane. Similarly, the relatively modest but reproducible loss of SMV proteins from the vesicle-enriched fraction after clathrin knockdown could be due to effects on clathrin-mediated endocytosis rather than intracellular trafficking (Saheki and De Camilli, 2012).
Previous studies making use of knockdowns and dominant negative constructs have demonstrated that clathrin and associated proteins play a role in DCV biogenesis (Molinete et al., 2001; Kakhlon et al., 2006; Bonnemaison et al., 2014), but the phenotypes that were reported were less severe than those we have found in the present study, possibly because protein loss was less complete. However, knockdown and dominant negative studies on two other types of regulated secretory organelles, the Weibel-Palade bodies (WPBs) of endothelial cells (Lui-Roberts et al., 2005) and the glue granules of Drosophila salivary gland epithelial cells (Burgess et al., 2011), showed that interfering with clathrin and/or AP-1 completely abolished WPB and glue granule formation. Although these other organelles differ in some respects from DCVs (e.g., the immature WPB is partially or completely wrapped in an AP-1- and clathrin-positive coat [ Lui-Roberts et al., 2005]), nevertheless there are precedents for clathrin having a function in the regulated secretory pathway above and beyond its established role in facilitating organelle maturation by removing unwanted proteins. One drawback of RNAi knockdowns, as well as gene knockouts, is that several days or even weeks elapse between the initiation of depletion or deletion and the analysis of the phenotype, so some of the observed effects may be indirect. Thus more rapid methods for protein removal, such as knocksideways (10 min [ Robinson et al., 2010; Willox and Royle, 2012]) or induced oligomerization (2 h [ Moskowitz et al., 2003; Zlatic et al., 2013]), may provide further insights into the precise role of clathrin in DCV biogenesis.
MATERIALS AND METHODS
PC12 cell culture
Rat pheochromocytoma PC12 cells were a gift from Nitish Mahapatra, IIT Madras. Early passage cells (i.e., no greater than passage number 10) were used for all experiments. For routine maintenance, the cells were grown in T25 flasks (Nunc, USA) in DMEM GlutaMAX (Invitrogen), supplemented with 10% heat-inactivated horse serum, 5% heat-inactivated fetal bovine serum, 100 U/ml penicillin G, and 100 µg/ml streptomycin (Life Technologies, USA). The cells were grown in 5% CO2 at 37°C in a humidified incubator, and were passaged every 4–5 d or when confluent.
SILAC labeling
For SILAC experiments, PC12 cells were cultured in SILAC medium for 3 wk to achieve metabolic labeling. The medium consisted of DMEM (Thermo Fisher) supplemented with 5% (vol/vol) fetal bovine serum (10,000 MW cutoff; Invitrogen), plus 10% (vol/vol) dialyzed horse serum (10,000 MW cutoff; Dundee Cell), and either “heavy” amino acids (l-arginine-13C615N4:HCl (50 mg/l) and l-lysine-13C615N2:2HCl (100 mg/l) or “light” amino acids (Cambridge Isotope Laboratories). The average incorporation efficiency was ∼94%, as determined by mass spectrometry.
Vesicle-enriched fraction
Two confluent 500-cm2 dishes of PC12 cells were scraped into ∼10 ml buffer A (0.1 M MES, pH 6.5 [adjusted with NaOH], 0.2 mM EGTA, 0.5 mM MgCl2). The cells were homogenized with a motorized Potter-Elvehjem homogenizer (25 strokes), syringe-disrupted using a 21G × 2″ (BD Microlance) needle, and centrifuged at ∼4100 × g for 32 min. Supernatants were treated with 50 μg/ml ribonuclease A for 60 min at 4°. Partially digested ribosomes were pelleted by centrifugation (∼4100 × g for 3 min), and discarded. The resultant supernatants were pelleted by centrifugation at 55,000 rpm (209,717 × g RCFmax) for 40 min in an MLA-80 rotor (Beckman Coulter). Pellets were resuspended in ∼800 μl buffer A using a 1 ml Dounce homogenizer, and mixed with an equal volume of FS buffer (12.5% [wt/vol] Ficoll, 12.5% [wt/vol] sucrose, in buffer A). All subsequent centrifugation steps were performed using a TLA-110 rotor (Beckman Coulter). Samples were spun at 20,000 rpm (21,720 × g RCFmax) for 34 min to pellet the bulk of the membranes (pellet discarded). Supernatants were diluted with four volumes of buffer A, and were centrifuged at 35,000 rpm (66,542 × g RCFmax) for 30 min to obtain the vesicle-enriched fraction (pellet). All preparations were performed at 4°C.
Fractionation profiling
Fractionation profiling, a method for combining differential centrifugation with quantitative mass spectrometry to resolve different types of subcellular particles, was carried out essentially as previously described (Borner et al., 2014). A vesicle-enriched fraction was prepared as above, but after diluting the supernatant from the 20,000 rpm spin, samples were first centrifuged at 66,500 × g (35,000 rpm, TLA-110) for 20 min, to obtain the 35K fraction pellet. The supernatant was transferred to a fresh centrifuge tube, mixed by pipetting, and centrifuged at 89,000 × g (45,000 rpm, TLA-110) for 20 min, to obtain the 45K fraction pellet. The supernatant was again transferred to a fresh tube, and centrifuged at 190,000 × g (60,000 rpm, TLA-110) for 40 min to obtain the final 60K fraction pellet. To prepare the reference fraction, the diluted supernatant after the Ficoll/sucrose step was spun at 190,000 × g (60,000 rpm, TLA-110) for 40 min. Pellets were resuspended in 1× NUPAGE LDS buffer, using 20 μl for the pellets from the three successive spins and 60 μl for the reference pellet.
Sample preparation for mass spectrometry
Each of the three subfractions (35K, 45K, and 60K) was combined with one-third of the reference sample, dithiothreitol (DTT) was added to a final concentration of 50 mM, and samples were incubated at 90°C for 3 min. The samples were then subjected to gel electrophoresis, using NUPGAE precast gradient gels (Invitrogen). The gels were stained with SimplyBlue SafeStain (Thermo Fisher), and each lane was cut into eight slices. Thus a routine fractionation profiling experiment yielded 24 samples, which were then analyzed by mass spectrometry.
Mass spectrometry LC-MS/MS and data processing
SDS–PAGE resolved proteins were reduced, alkylated, and digested in-gel with trypsin, and then the resulting peptides were concentrated and desalted using Stage tips (Antrobus and Borner, 2011). Tryptic peptides were analyzed by LC-MS/MS using a Q Exactive coupled to an RSLCnano3000 UHPLC (Thermo Fisher). Peptides were resolved using a 50-cm EASY-Spray PepMap C18 column (Thermo Fisher) with data acquired in a data-dependent acquisition manner. Data were processed using MaxQuant 1.3.0.5 using Andromeda to search a Uniprot Rattus norvegicus database (downloaded 7 April 2014, 27,344 entries) with Homo sapiens AP1G1 (O43747) added. N-terminal protein acetylation and methionine oxidation were set as variable modifications, carbamidomethyl cysteine set as a fixed modification, and both LFQ and iBAQ were enabled. Alternatively, data were processed in Proteome Discoverer 1.4 using Sequest to search a Uniprot R. norvegicus database (downloaded 7 April 2014; 27,344 entries). Oxidized methionine, 13C615N2 lysine, and 13C615N4 arginine were set as variable modifications and carbamidomethyl cysteine as a fixed modification. False discovery rate calculations were performed by Percolator and quantification by the Precursor ion quantifier node. Where indicated, ratios were normalized on protein median. The raw data files were processed using MaxQuant (with requantify and match between runs features enabled). The primary output for each SILAC comparison was a list of identified proteins, a ratio of relative abundance (heavy/light ratio), and the number of quantification events (count). Each MaxQuant output file was formatted in an identical manner: reverse hits, proteins identified only by site, common contaminants, and proteins with no gene name were removed. The SILAC ratios are shown in Supplemental Table S1.
To identify proteins with similar fractionation profiles, PCA was performed as previously described (Borner et al., 2012). Briefly, the ratio data were logarithmically transformed, precentered, and correlated using SIMCAS 14.0 (Umetrics), which was also used to generate the scatter plots.
Proteins were assigned to DCVs, CCVs, or SMVs according to the following literature: Winkler, 1993; Parmer et al., 1997; Wegrzyn et al., 2007 , 2010; Wimalasena, 2011; and Borges et al., 2013, for DCVs; Borner et al., 2012, 2014, for CCVs; and Takamori et al., 2006 for SMVs.
Generation of inducible shRNA-expressing stable cell lines
Clathrin knockdowns were carried out using the SMARTvector rat lentiviral shRNA system (Dharmacon), in which shRNAs targeting a gene of interest are inducibly coexpressed with GFP. PC12 cells were plated in polylysine-coated six-well plates and transduced with virions encoding three different targeting constructs (VSR6432-223531394), according to the manufacturer’s protocol. Cells containing integrated lentiviral sequences were selected using puromycin (5 μg/ml), replacing the medium every 3 d until resistant colonies were observed. Cells were then induced with doxycycline at 1 μg/ml overnight, and highly GFP-positive cells were obtained by flow cytometry. The cells were then maintained in medium containing puromycin but without doxycycline. Out of the three constructs, V2IRNMCG_2797855 (“shRNA-A”) was most effective at depleting clathrin and was used for all subsequent experiments. For the experiments shown in Supplemental Figure S3, we used one of the other shRNAs, V2IRNMCG_2797847 (“shRNA-B”). We also attempted to rescue the cells with an mCherry-tagged version of clathrin heavy chain that contained silent mutations to make it resistant to shRNA-A. Stably transfected cells were selected using 1 mg/ml G418, and high expressors were identified by fluorescence microscopy and flow cytometry. Unfortunately, however, we were unable to get sufficiently high expression for a rescue.
Antibodies, Western blotting, and immunofluorescence
Antibodies used in this study include an in-house rabbit polyclonal antibody against clathrin heavy chain (Simpson et al., 1996), goat antibodies against EF2 and CgB (Santa Cruz Biotechnology), and rat monoclonal anti-RFP (5F8, ChromoTek). For Western blotting, horseradish peroxidase–conjugated secondary antibodies were obtained from Sigma Aldrich, ECL Plus Western Blotting Detection System (GE Healthcare Amersham) was used as the developing agent, the blots were exposed to Super RX film (Fuji, UK), and images were acquired using an Epson Perfection 1650 scanner. For immunofluorescence, Alexa Fluor-conjugated secondary antibodies were obtained from Invitrogen, and images were captured using a LSM880 confocal microscope at 63× magnification.
Electron microscopy
Electron microscopy was carried out on both subcellular fractions and cultured cells. Pelleted fractions were fixed with 2% paraformaldehyde/2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.2, at room temperature for 30 min, washed with 0.1 M cacodylate buffer, and postfixed using 1% osmium tetroxide for 1 h. Pellets were then washed before being incubated with 1% tannic acid in 0.05 M cacodylate buffer, pH 7.2, for 40 min to enhance contrast, and then dehydrated in ethanol and embedded in Araldyte CY212 epoxy resin (Agar Scientific, Stansted, UK). Cultured cells were grown on plastic dishes and fixed using 2% paraformaldehyde, 2.5% glutaraldehyde, and 0.1 M cacodylate buffer, pH 7.2. The cells were then scraped and pelleted, postfixed with 1% osmium tetroxide, incubated with 1% tannic acid to enhance contrast, and dehydrated using increasing concentrations of ethanol before being embedded in epoxy resin in beam capsules and cured overnight at 65°C. For both types of samples, ultrathin (50–70 nm) conventional sections were cut using a diamond knife mounted onto a Reichart ultracut S ultramicrotome and collected onto copper grids. The grids were stained using lead citrate, and sections were viewed on a FEI Tecnai transmission electron microscope at a working voltage of 80 kV.
Quantification of electron microscopy
Pellets of cells ensured random distribution of organelles between cells on each section. High-magnification images were taken of every part of six cells per condition, and the cytosolic area was calculated by removing nuclear area from the total cellular area. Vesicles were categorized by phenotype, where type 1 are most mature and type 4 least mature. The type 5 vesicles were only seen in clathrin-depleted cells. Type 1 vesicles contained a dense and condensed core, with no electron-lucent lumen. Type 2 vesicles contained a dense core but were surrounded by a thin electron-lucent lumen. Type 3 vesicles contained mostly electron-lucent lumens but also contained a small dense patch. Type 4 vesicles were lumenal vesicles with little or no density. Type 5 vesicles were only seen in clathrin-depleted cells and displayed as vesicles with large electron-lucent lumens that occasionally contained small cores. These vesicles were the largest and often appeared in clusters. Clathrin depletion reduced the number of vesicles per area of cytosol (mock cells: 0.587 vesicles/µm2; clathrin-depleted cells: 0.357 vesicles/µm2).
Vesicle-enriched fractions from mock-treated and clathrin-depleted cells
Cells grown in either SILAC heavy or SILAC light medium were plated in two 500 cm2 dishes. One pair of dishes was treated with 1 µg/ml doxycycline for 5 d to induce knockdown, while the other pair of dishes served as the mock-treated control. Label swapping was carried out to ensure that any differences observed were due to the presence or absence of clathrin. Thus, in one set of experiments, the clathrin-depleted cells were incubated in light medium and the mock-treated cells in heavy medium, while in a second set of experiments, the clathrin-depleted cells were incubated in heavy medium and the mock-treated cells in light medium. An additional control experiment was carried out in which clathrin-depleted cells were incubated in heavy medium, and cells transduced with a nontargeting shRNA were incubated in light medium and induced with doxycycline. This was to ensure that effects of doxycycline were due to clathrin depletion rather than to other targets of the drug.
Vesicle-enriched fractions from heavy- and light-labeled cells were prepared as described above, and pellets were resuspended in 50 µl of nonreducing SDS–PAGE buffer (4% [wt/vol] SDS, 10 mM Tris-HCl, pH = 8.0), heated to 65°C for 3 min, and centrifuged at 16,000 × g for 1 min to remove insoluble material. Protein concentrations were estimated with a BCA assay (Pierce). Equal amounts of protein from two complementary samples were then pooled, adjusted with 1:3 volumes of 4× sample buffer (10% [wt/vol] SDS, 40% [vol/vol] glycerol, 8% [vol/vol] 2-mercaptoethanol, 200 mM Tris-HCl, pH = 6.8), heated to 90°C for 3 min, and subjected to SDS–PAGE. In-gel digestion and mass spectrometry were carried out as described above.
Secretion of CgB
Time-dependent secretion experiments were carried out to follow the fate of CgB, both by Western blotting (see above) and by SILAC-based mass spectrometry. For the mass spectrometry experiments, 10-cm dishes of cells were grown in either heavy or light medium, then clathrin knockdown was induced in half of the dishes by treating with doxycycline for 5 d, as described above. The medium was removed and the cells were washed twice with release buffer (150 mM NaCl, 5 mM KCl, 2 mM CaCl2,10 mM HEPES, pH = 7). Then the cells grown in light medium were incubated for 15 min at 37°C in release buffer, while the cells grown in heavy medium were incubated for 15 min at 37°C in stimulation buffer, which has the same composition as release buffer but contains 5 mM BaCl2 and lacks CaCl2. The two supernatants from the mock-depleted cells were then pooled together, and the two supernatants from the clathrin-depleted cells were also pooled together, then each pooled sample was concentrated down to 1 ml using an Amicon filter with a 3 kDa cutoff. Both sets of samples were immunoprecipitated with goat anti-CgB antibody using a standard protocol (Seaman, 2004). Briefly, the samples were precleared and incubated with the antibody overnight (1.6 µg in 24 ml), then the antibody and its bound antigen were captured onto protein G Sepharose beads. The beads were vacuum dried, suspended in 40 µl 2× sample buffer containing 50 mM DTT boiled, and subjected to SDS–PAGE using a MOPS gradient gel. Mass spectrometry was carried out on trypsin-digested gel bands as described above.
Norepinephrine uptake and secretion
PC12 cells grown to 80% confluence in poly-l-lysine–coated six-well plates were incubated with 1 µCi [3H] l-norepinephrine for 3 h, washed twice with release buffer, and then incubated for 15 min at 37°C either in release buffer or in stimulation buffer. The supernatants were then collected, and the cells were lysed in lysis buffer. Both the supernatants and the lysates were mixed with liquid scintillation cocktail, and radioactivity was quantified using a Beckman liquid scintillation counter. The experiments shown in Figure 6 are biological replicates, carried out three times independently, while those in Supplemental Figure S3 are technical replicates, carried out in triplicate on the same day.
Supplementary Material
Acknowledgments
We thank Kamburpola Jayawardena and Yagnesh Umrania for help with the proteomics. We also thank the UMN Imaging Center at the University of Minnesota. B.S.S. acknowledges the members of the Robinson, Bartolomucci, and Mahata laboratories. This work was funded by grants from the Wellcome Trust: 086598 (to M.S.R.), 100140 (Wellcome Trust Strategic Award), and 093026 (for the FEI Tecnai G2 Spirit BioTWIN transmission EM); and by a National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grant (R01DK102496) to A.B.
Abbreviations used:
- AP
adaptor protein
- CCV
clathrin-coated vesicle
- DCV
dense core vesicle
- GFP
green fluorescent protein
- SMV
synaptic-like microvesicle.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-10-0742) on August 16, 2017.
REFERENCES
- Antrobus R, Borner GHH. Improved elution conditions for native co-immunoprecipitation. PLoS One. 2011;23:e18218. doi: 10.1371/journal.pone.0018218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett EM, Chen CY, Engqvist-Goldstein AE, Drubin DG, Brodsky FM. Clathrin hub expression dissociates the actin-binding protein Hip1R from coated pits and disrupts their alignment with the actin cytoskeleton. Traffic. 2001;2:851–858. doi: 10.1034/j.1600-0854.2001.21114.x. [DOI] [PubMed] [Google Scholar]
- Bonnemaison M, Bäck N, Lin YM, Bonifacino JS, Mains R, Eipper B. AP-1A controls secretory granule biogenesis and trafficking of membrane secretory granule proteins. Traffic. 2014;15:1099–1121. doi: 10.1111/tra.12194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges R, Dominguez N, Smith CB, Bandyopadhyay GK, O’Connor DT, Mahata SK, Bartolomucci A. Granins and catecholamines: functional interaction in chromaffin cells and adipose tissue. Adv Pharmacol. 2013;68:93–113. doi: 10.1016/B978-0-12-411512-5.00005-1. [DOI] [PubMed] [Google Scholar]
- Borner GHH, Antrobus R, Hirst J, Bhumbra GS, Kozik P, Jackson LP, Sahlender DA, Robinson MS. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol. 2012;197:141–160. doi: 10.1083/jcb.201111049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner GHH, Harbour M, Hester S, Lilley KS, Robinson MS. Comparative proteomics of clathrin-coated vesicles. J Cell Biol. 2006;175:571–578. doi: 10.1083/jcb.200607164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner GHH, Hirst J, Hein MY, Edgar J, Mann M, Robinson MS. Fractionation profiling: a fast and versatile approach for mapping vesicle proteomes and protein–protein interactions. Mol Biol Cell. 2014;15:3178–3194. doi: 10.1091/mbc.E14-07-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess J, Jauregui M, Tan J, Rollins J, Lallet S, Leventis PA, Boulianne GL, Chang HC, Le Borgne R, Krämer H, Brill JA. AP-1 and clathrin are essential for secretory granule biogenesis in Drosophila. Mol Biol Cell. 2011;22:2094–2105. doi: 10.1091/mbc.E11-01-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikeakos JD, Reudelhuber TL. Sending proteins to dense core secretory granules: still a lot to sort out. J Cell Biol. 2007;177:191–196. doi: 10.1083/jcb.200701024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittié AS, Klumperman J, Tooze SA. Differential distribution of mannose-6-phosphate receptors and furin in immature secretory granules. J Cell Sci. 1999;112:3955–3966. doi: 10.1242/jcs.112.22.3955. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Gumbiner B, Kelly RB. Two distinct intracellular pathways transport secretory and membrane glycoproteins to the surface of pituitary tumor cells. Cell. 1982;28:51–59. doi: 10.1016/0092-8674(82)90374-9. [DOI] [PubMed] [Google Scholar]
- Hannah MJ, Schmidt AA, Huttner WB. Synaptic vesicle biogenesis. Annu Rev Cell Dev Biol. 1999;15:733–798. doi: 10.1146/annurev.cellbio.15.1.733. [DOI] [PubMed] [Google Scholar]
- Hinrichsen L, Meyerholz A, Groos S, Ungewickell EJ. Bending a membrane: how clathrin affects budding. Proc Natl Acad Sci USA. 2006;103:8715–8720. doi: 10.1073/pnas.0600312103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakhlon O, Sakya P, Larijani B, Watson R, Tooze SA. GGA function is required for maturation of neuroendocrine secretory granules. EMBO J. 2006;19:1590–1602. doi: 10.1038/sj.emboj.7601067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumperman J, Kuliawat R, Griffith JM, Geuze HJ, Arvan P. Mannose 6-phosphate receptors are sorted from immature secretory granules via adaptor protein AP-1, clathrin, and syntaxin 6-positive vesicles. J Cell Biol. 1998;141:359–371. doi: 10.1083/jcb.141.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litteri G, Carnevale D, D’Urso A, Cifelli G, Braghetta P, Damato A, Bizzotto D, Landolfi A, Ros FD, Sabatelli P, et al. Vascular smooth muscle Emilin-1 is a regulator of arteriolar myogenic response and blood pressure. Arterioscler Thromb Vasc Biol. 2012;32:2178–2184. doi: 10.1161/ATVBAHA.112.254664. [DOI] [PubMed] [Google Scholar]
- Lui-Roberts WW, Collinson LM, Hewlett LJ, Michaux G, Cutler DF. An AP-1/clathrin coat plays a novel and essential role in forming the Weibel-Palade bodies of endothelial cells. J Cell Biol. 2005;170:627–636. doi: 10.1083/jcb.200503054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinete M, Dupuis S, Brodsky FM, Halban PA. Role of clathrin in the regulated secretory pathway of pancreatic β-cells. J Cell Sci. 2001;114:3059–3066. doi: 10.1242/jcs.114.16.3059. [DOI] [PubMed] [Google Scholar]
- Moore HP, Walker MD, Lee F, Kelly RB. Expressing a human proinsulin cDNA in a mouse ACTH-secreting cell. Intracellular storage, proteolytic processing, and secretion on stimulation. Cell. 1983;35:531–538. doi: 10.1016/0092-8674(83)90187-3. [DOI] [PubMed] [Google Scholar]
- Moskowitz HS, Heuser J, McGraw TE, Ryan TA. Targeted chemical disruption of clathrin function in living cells. Mol Biol Cell. 2003;14:4437–4447. doi: 10.1091/mbc.E03-04-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz HS, Yokoyama CT, Ryan TA. Highly cooperative control of endocytosis by clathrin. Mol Biol Cell. 2005;16:1769–1776. doi: 10.1091/mbc.E04-08-0739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MNJ, Robinson MS. Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol. 2003;162:909–918. doi: 10.1083/jcb.200305145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmer RJ, Mahata M, Mahata SK, Sebald MT, O’Connor DT, Miles LA. Tissue plasminogen activator (t-PA) is targeted to the regulated secretory pathway. Catecholamine storage vesicles as a reservoir for the rapid release of t-PA. J Biol Chem. 1997;272:1976–1982. doi: 10.1074/jbc.272.3.1976. [DOI] [PubMed] [Google Scholar]
- Puertollano R, Aguilar RC, Gorshkova I, Crouch RJ, Bonifacino JS. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science. 2001;292:1663–1665. doi: 10.1126/science.1060750. [DOI] [PubMed] [Google Scholar]
- Robinson MS, Sahlender DA, Foster SD. Rapid inactivation of proteins by rapamycin-induced rerouting to mitochondria. Dev Cell. 2010;18:324–331. doi: 10.1016/j.devcel.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheki Y, De Camilli P. Synaptic vesicle endocytosis. Cold Spring Harb Perspect Biol. 2012;4:a005645. doi: 10.1101/cshperspect.a005645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MNJ. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimodaira M, Nakayama T, Sato N, Naganuma T, Yamaguchi M, Aoi N, Sato M, Izumi Y, Soma M, Matsumoto K. Association study of the elastin microfibril interfacer 1 (EMILIN1) gene in essential hypertension. Am J Hypertens. 2010;23:547–555. doi: 10.1038/ajh.2010.16. [DOI] [PubMed] [Google Scholar]
- Simpson F, Bright NA, West MA, Newman LS, Darnell RB, Robinson MS. A novel adaptor-related protein complex. J Cell Biol. 1996;133:749–760. doi: 10.1083/jcb.133.4.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Taupenot L. Analysis of regulated secretion using PC12 cells. Curr Protoc Cell Biol. 2007 doi: 10.1002/0471143030.cb1512s36. doi: 10.1002/0471143030. [DOI] [PubMed] [Google Scholar]
- Tooze J, Tooze SA. Clathrin-coated vesicular transport of secretory proteins during the formation of ACTH-containing secretory granules in AtT20 cells. J Cell Biol. 1986;103:839–850. doi: 10.1083/jcb.103.3.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii S, Saito N, Kawano A, Zhao S, Izumi T, Takeuchi T. Cytoplasmic transport signal is involved in phogrin targeting and localization to secretory granules. Traffic. 2005;6:1213–1224. doi: 10.1111/j.1600-0854.2005.00353.x. [DOI] [PubMed] [Google Scholar]
- Wegrzyn JL, Bark SJ, Funkelstein L, Mosier C, Yap A, Kazemi-Esfarjani P, La Spada AR, Sigurdson C, O’Connor DT, Hook V. Proteomics of dense core secretory vesicles reveal distinct protein categories for secretion of neuroeffectors for cell–cell communication. J Proteome Res. 2010;9:5002–5024. doi: 10.1021/pr1003104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn J, Lee J, Neveu JM, Lane WS, Hook V. Proteomics of neuroendocrine secretory vesicles reveal distinct functional systems for biosynthesis and exocytosis of peptide hormones and neurotransmitters. J Proteome Res. 2007;6:1652–1665. doi: 10.1021/pr060503p. [DOI] [PubMed] [Google Scholar]
- Willox AK, Royle SJ. Stonin 2 is a major adaptor protein for clathrin-mediated synaptic vesicle retrieval. Curr Biol. 2012;22:1435–1439. doi: 10.1016/j.cub.2012.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimalasena K. Vesicular monoamine transporters: structure-function, pharmacology, and medicinal chemistry. Med Res Rev. 2011;31:483–519. doi: 10.1002/med.20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler H. The adrenal chromaffin granule: a model for large dense core vesicles of endocrine and nervous tissue. J Anat. 1993;183:237–252. [PMC free article] [PubMed] [Google Scholar]
- Zlatic SA, Grossniklaus EJ, Ryder PV, Salazar G, Mattheyses AL, Peden AA, Faundez V. Chemical-genetic disruption of clathrin function spares adaptor complex 3–dependent endosome vesicle biogenesis. Mol Biol Cell. 2013;24:2378–2388. doi: 10.1091/mbc.E12-12-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.