

 β-Hydrogenation of porphyrin remarkably enhances (ca. 24 fold) the electrocatalytic HER reactivity of Ni(ii) complexes.
β-Hydrogenation of porphyrin remarkably enhances (ca. 24 fold) the electrocatalytic HER reactivity of Ni(ii) complexes.
Abstract
Fine-tuning of the porphyrin β-periphery is important for naturally occurring metal tetrapyrroles to exert diverse biological roles. Here we describe how this approach is also applied to design molecular catalysts, as exemplified by Ni(ii) porphyrinoids catalyzing the hydrogen evolution reaction (HER). We found that β-hydrogenation of porphyrin remarkably enhances the electrocatalytic HER reactivity (turnover frequencies of 6287 vs. 265 s–1 for Ni(ii) chlorin (Ni-2) and porphyrin (Ni-1), and of 1737 vs. 342 s–1 for Ni(ii) hydroporpholactone (Ni-4) and porpholactone (Ni-3), respectively) using trifluoroacetic acid (TFA) as the proton source. DFT calculations suggested that after two-electron reduction, β-hydrogenation renders more electron density located on the Ni center and thus prefers to generate a highly reactive nickel hydride intermediate. To demonstrate this, decamethylcobaltocene Co(Cp*)2 was used as a chemical reductant. [Ni-2]2– reacts ca. 30 times faster than [Ni-1]2– with TFA, which is in line with the electrocatalysis and computational results. Thus, such subtle structural changes inducing the distinctive reactivity of Ni(ii) not only test the fundamental understanding of natural Ni tetrapyrroles but also provide a valuable clue to design metal porphyrinoid catalysts.
Introduction
Fine-tuning of the porphyrin β-periphery is important for naturally occurring metal tetrapyrroles, which display broad structural similarity, to exert diverse biological functions. 1a–e As a consequence of theoretical and modeling studies on the structural–electronic–functional relationship, β-hydrogenation of porphyrins attracts much attention for lowering the molecular symmetry and altering the energy level of frontier molecular orbitals by subtle structural changes, especially in mimicking light harvesting antennae, 2a,b photosensitizers 3a,b and related optoelectronic materials. 4a–c In contrast to the tremendous progress in the physicochemical properties, applying the structural insights gained from natural tetrapyrroles to catalysis had been long ignored. In fact, β-modification is of equal importance for metalloenzymes, as exemplified by Ni(ii) tetrapyrrole cofactors found in reductases. 5a,b These Ni cofactors such as F430 coenzymes 6a,b and tunichlorin 7a,b possess either corphin or chlorin, and feature at least one reduced pyrrole, other than protoporphyrin (Fig. 1). The interrupted conjugation arising from the reduced pyrrole(s) renders distortions of the porphyrin ring, a larger binding cavity and a more electron rich Ni center after one or two electron reduction, as revealed by electrochemical studies on β-hydroporphyrin models. 8a–d Encouraged by these pioneering studies, we herein report the first example showing that β-hydrogenation of porphyrinoids significantly enhances the reactivity of nickel(ii) in the hydrogen evolution reaction (HER), which demonstrates the importance of bioinspired β-modification in designing molecular catalysts.
Fig. 1. Selected examples of natural tetrapyrroles and synthetic models.

The hydrogen evolution reaction (HER) is a fundamental process in water splitting to produce hydrogen energy as a potential alternative to traditional fossil fuels. 9a–d On the route to seek earth-abundant metals to replace noble metals, 10a–f nickel(ii) complexes of porphyrins have attracted increasing attention due to the combination of the pivotal role of Ni(ii) in [NiFe] hydrogenases 11a,b and the sophisticated porphyrin chemistry. 12a–e Recently Cao and co-workers proposed a one-electron reduction and bimetallic homolysis mechanism based on a Ni(ii) porphyrin with meso-pentafluorophenyl substituents.13 Nocera and co-workers constructed a “2nd coordination sphere” around the Ni(ii) center using a hangman type porphyrin, which demonstrated a hydrogen bond facilitating proton coupled electron transfer process.14 More importantly, the same group found a Ni(ii) phlorin intermediate during HER, in which a proton was added to the meso-carbon appended to the hangman moiety.15 The appearance of the phlorin intermediate indicated that it was necessary to investigate the hydroporphyrin with interrupted conjugation.
As part of our continued effort to unravel the effect of β-modification on the functions of porphyrins, 16a–d in the present study we report the synthesis, electrochemical characterization and HER reactivity of nickel complexes of porphyrin (Ni-1), chlorin (Ni-2), porpholactone (Ni-3) and β-hydroporpholactone (Ni-4). Importantly, as a result of β-hydrogenation, Ni-2 and Ni-4 exhibited significantly enhanced electrocatalytic HER reactivity (determined by turnover frequencies, TOFs) which was 24 and 5 times that of the corresponding porphyrin (Ni-1) and porpholactone (Ni-3), respectively. More importantly, DFT calculations suggest that β-hydrogenation renders more electron rich Ni centers in the two-electron reduced [Ni-2]2– and [Ni-4]2– than the analogues [Ni-1]2– and [Ni-3]2–, and thus tends to form active nickel hydride intermediates. This is supported by the titration of trifluoroacetic acid (TFA) to two-electron reduced nickel complexes, in which [Ni-2]2– reacts ca. 30 times faster than [Ni-1]2–. Therefore, this work demonstrates that such a subtle structural modification inspired by nature is instrumental to the observation and investigation of catalytic properties of metal porphyrinoid complexes.
Results and discussion
Synthesis and characterization
In this work, free-base porphyrinoids 1–4 were synthesized in good yields according to the literature. 16c,17 Nickel complexes (Scheme 1) were prepared by refluxing excess nickel(ii) acetate (30 equiv.) with the corresponding porphyrinoid ligands in dimethylformamide (DMF) under a N2 atmosphere. Four nickel complexes were fully characterized by UV-vis absorption, and 1H, 19F, 13C-NMR and HR ESI-MS spectroscopes and spectrometers (ESI†). Ni-2 and Ni-4 were diamagnetic for the low spin d8 electronic structure and showed proton signals at 3.93 and 3.77–3.65 ppm, respectively, which can be assigned to the hydropyrrole moiety. Compared to Ni-1 and Ni-3, the β-hydro-analogues Ni-2 and Ni-4 exhibited two bathochromic Q-bands centered at 576 or 565 and 617 or 613 nm in acetonitrile, which is also the characteristic absorption pattern of metal chlorins arising from lower molecular symmetry.
Scheme 1. The synthetic procedures for Ni-1, Ni-2, Ni-3 and Ni-4.

Crystals of Ni-2, Ni-3 and Ni-4 were obtained by slow evaporation of a dichloroethane/n-hexane solution. As shown in Fig. 2, the crystal structure of Ni-2 (CCDC: ; 1528290†) displays the typical ruffling distortion with the Ni atom centered at the N4 planar squares. Although it is difficult to distinguish the hydropyrrole ring from the other ones due to the symmetry condition of the I42d space group, the average pyrrole C–C bond length (C2–C3, 1.383 Å) of Ni-2 is 0.037 Å longer than that of Ni-1 (C11–C13, 1.346 Å).13 Similarly, the hydropyrrole ring on Ni-3 (CCDC: ; 1554494†) can be distinguished through comparison of the C–C bond length with Ni-4 (CCDC: ; 1554445†) (C1–C(O)2, 1.338 Å and C3–C4, 1.341 Å for Ni-3 and C1–C(O)2, 1.404 Å for Ni-4). In addition, Ni-4 displays the typical ruffling distortion with the Ni–N bond measuring 1.921 Å, however Ni-3 displays a planar construction with a Ni–N bond of 1.950 Å. The shorter Ni–N bond induces steric strain on Ni-4, the release of which is the driving force for easier reduction of the nickel complexes.1b
Fig. 2. ORTEP drawings of Ni-2, Ni-3 and Ni-4. The hydrogen atoms and solvent molecules are omitted for clarity. Thermal ellipsoids are drawn with 50% probability. The lactone ring in Ni-3 shows disorder at the oxazolone and the opposite position while the Ni-4 and Ni-2 show disorder at all four β-peripheries.

Electrochemistry
Cyclic voltammograms of Ni complexes in acetonitrile using 0.10 M n Bu4NPF6 as the supporting electrolyte are shown in Fig. S18.† Both Ni-1 and Ni-2 display two distinct reversible one-electron diffusion controlled reductions (–1.27 and –1.83 V for Ni-1, and –1.28 and –1.77 V for Ni-2), referenced to the standard reduction potential of Fc+/Fc. According to previous reports, 18a,18b the insertion of an electron deficient oxazolone moiety lowers the energy levels of the ligand LUMOs and thus we could observe the positive shift of the reduction potentials of Ni-3 to Ni-1. However, Ni-4 displayed more negative reduction potentials than Ni-3 (–0.99 and –1.59 V for Ni-3, and –1.14 and –1.74 V for Ni-4), indicating that β-hydrogenation increases the energy levels of LUMOs in porpholactones.
Spectroelectrochemistry of the Ni(ii) complexes was performed to monitor the spectral changes during the reduction process. As shown in Fig. 3, one-electron reduction (ca. –1.3 V) of Ni-1 and Ni-2 caused a slight decrease and hypsochromic shift of the Soret and Q bands, suggesting the formation of a formal NiII/I couple according to previous studies by Savéant19, Brückner20 and others. 21a,b When ca. –1.85 V was applied, the Soret and Q bands of [Ni-1]2– decreased in intensity, accompanied by the appearance of two broad bands at 621 and 850 nm, thus indicating the formation of a typical porphyrinic dianion. 8a,22 For [Ni-2]2–, the broad band centered at 752 nm is assigned to the porphyrin π-anion radical. 8a,23a,b This assignment is also supported in the context of DFT calculation results. For the porpholactone analogues Ni-3 and Ni-4, it is difficult to distinguish the electronic structures from the UV-vis spectra. When they were reduced at ca. –1.2 or –1.85 V, the intensity of the Soret and Q bands decreased with red-shift and an absorption peak centered at 750 nm appeared, indicating that the electrons were delocalized on the macrocycle. Definitive assignment of the electronic structures of Ni complexes will be further discussed in the context of DFT calculations.
Fig. 3. UV-vis absorption spectra of (a): Ni-1 (black), [Ni-1]– (blue), and [Ni-1]2– (red), (b): Ni-2 (black), [Ni-2]– (blue), and [Ni-2]2– (red) (c): Ni-3 (black), [Ni-3]– (blue), and [Ni-3]2– (red) and (d): Ni-4 (black), [Ni-4]– (blue), and [Ni-4]2– (red) in acetonitrile using thin-layer spectroelectrochemistry. Insets: enlarged views of the absorbance at 750–850 nm for Ni-1 and Ni-2, and 700–840 nm for Ni-3 and Ni-4.

Electrocatalysis hydrogen production
The HER activities of the four nickel(ii) complexes were evaluated by successive cyclic voltammograms in acetonitrile using TFA (pK a = 12.7 in CH3CN) as a proton source. Electrocatalytic production of H2 was confirmed by gas chromatographic analysis, and the faradaic yields of the four complexes were almost above 90% based on the amount of H2 produced (Fig. S21†). There was no observable decomposition of the Ni complexes during HER, according to LDS (Fig. S22†), TEM (Fig. S23†) and UV-vis absorption spectra (Fig. S26†) of the reaction solution before and after electrolysis. In addition, SEM and EDX (Fig. S25†) of the glassy carbon working electrode before and after the HER reaction showed no nickel film deposition on the electrode and thus exclude the possibility of free Ni(ii) ions from the Ni complexes being active species. The HER experiments using the dissolved Ni(ii) salts of Ni(OTf)2 (Fig. S24†) showed a relatively high overpotential and low catalytic activity, thus we excluded the possibility of the dissolved Ni(ii) salts being active intermediates.
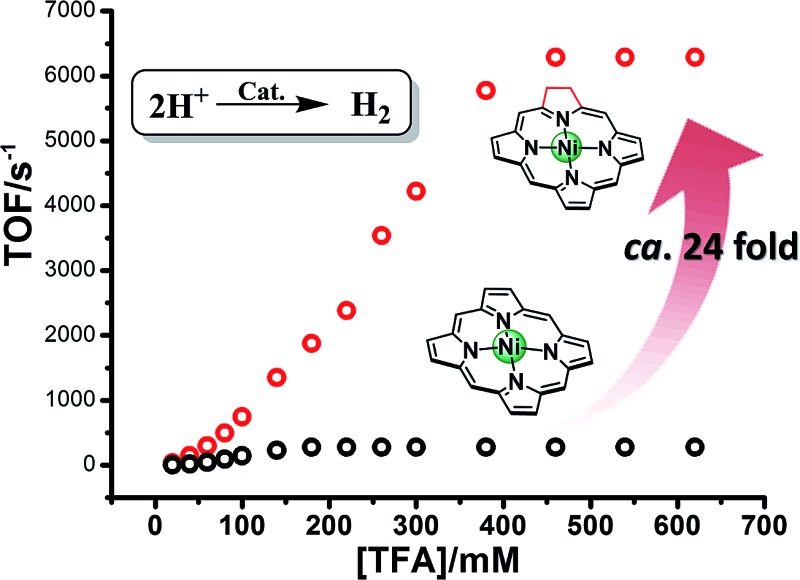
In the presence of TFA, an S-shaped irreversible catalytic response for proton reduction triggered at ca. –1.80 V was observed for the four nickel complexes, as shown in Fig. 4. As shown in Fig. 4c and S19,† Ni-1 reached its maximum turnover frequency when it was applied with 180 mM TFA (Fig. S19†). Here, we roughly estimated the maximum TOF of 265 s–1 using eqn (1) in the ESI† for Ni-1 from the ratio of the maximum catalytic current i cat to the peak current i p(i cat/i p) with TFA (180 mM). Surprisingly, Ni-2 displayed significantly enhanced catalytic reactivity up to a maximum TOF of 6287 s–1 (ca. 24 times greater than that of Ni-1) when the concentration of TFA reached 460 mM. Similar enhancement of the HER reactivity (Fig. S19†) but to a lesser extent was observed by comparison of Ni-3 and Ni-4, in which the latter exhibited a TOF (1737 s–1) almost 5 times greater than that of Ni-3 (342 s–1). The values of the TOF have also been confirmed by the FOWA method (Fig. S27†) 24a,b . The results from the two methods show the same trend that the β-hydrogenation of the pyrrole ring enhances the reactivities of the nickel porphyrinoids, with the TOF of Ni-2 > Ni-1, and Ni-4 > Ni-3. It is worth nothing that replacement of the β-lactone moiety results in comparable reactivity to the porphyrin analogue (Ni-1 vs. Ni-3) but much lower reactivity than the chlorin complex (Ni-2 vs. Ni-4). Thus, these results clearly demonstrate that β-hydrogenation of porphyrin or porpholactone can remarkably enhance catalytic HER reactivity.
Fig. 4. Successive cyclic voltammograms of 1.0 mM Ni-1 (a), Ni-2 (b), Ni-3 (d) and Ni-4 (e) in CH3CN (0.10 M n Bu4NPF6) at increasing concentrations of TFA from 20 mM to 140 mM. Conditions: 3 mm glassy-carbon working electrode; 25 °C; scan rate: 100 mV s–1. Graphs (c) and (f) show the TOFs vs. concentrations of TFA.

With the plateau current of the catalytic S-shaped voltammograms appearing at the second reduction peak of the nickel complexes, we considered that the electrochemical–electrochemical–chemical–chemical (EECC) mechanism (pathway A) might be the most plausible mechanism (Scheme 2).25 However other reaction pathways cannot be excluded. The catalytic onset can appear at the first reduction wave with a stronger acid such as methylsulfonic acid (MsOH) (Fig. S28†). In this situation the acid might be strong enough to hydrogenate [Ni–P]– to [H–Ni–P]0, which can then be reduced to [H–Ni–P]– and react with anther proton to yield hydrogen (pathway B). This phenomenon has been reported by Nocera15 and Cao13. In addition, at a low acid concentration, we can observe another catalytic plateau after the catalytic current increased at the second reduction peak in Ni-3. In this situation, the [H–Ni–P]– might be further reduced to [H–Ni–P]2– and then react with a proton to release H2 and revert back to [Ni–P]– (pathway C), as Savéant and co-workers previously reported.26
Scheme 2. Possible heterolytic reaction pathways in the nickel porphyrin catalyzed HER process.

Electronic structures of Ni-1 to Ni-4
To understand the effect of β-hydrogenation of porphyrins on the reactivity of Ni complexes, DFT calculations were carried out to optimize the electronic structures (see more computational details in the ESI†). The most stable forms of [Ni-1]0, [Ni-2]0 and [Ni-4]0 are saddle-shaped closed-shell singlets, and [Ni-3]0 is most stable in a flat closed-shell singlet, in accordance with the 1H-NMR spectra and crystal structures. The one-electron reduced nickel complexes have the most stable doublet flat geometries with a Mulliken spin density (ρ Ni) of approximately 1.0 on the Ni center, indicating a formal Ni(i) species. Interestingly, the two-electron reduced nickel complexes have different electronic structures as shown in Fig. 5. For [Ni-1]2– and [Ni-3]2–, the flat triplet is lower in energy than the closed-shell bent singlet and two unpaired electron are localized on the porphyrin macrocycle. Small Mulliken spin densities (ρ Nis) of 0.0026 and 0.36 were obtained for [Ni-1]2– and [Ni-3]2–, respectively (Tables S9 and S11†), suggesting a Ni(ii) porphyrin diradical consistent with the above spectroelectrochemistry results. Additionally, the singly occupied molecular orbitals (SOMO) of [Ni-1]2– and [Ni-3]2– also showed that the electron density was almost located on the porphyrin β-periphery (Fig. 5a and c). However, Nocera and his coworker’s reported an analogue of [Ni-1]2– (meso-chlorine atoms were used to replace the meso-pentafluorophenyl substituents of nickel porphyrin in his work) which was a flat triplet species with a Mulliken spin density of almost 1 on the Ni center and almost 1 on the ligands.15 As shown in Fig. S34,† we find that a meso-substitutional group affects the calculation results. In addition, five different DFT functionals have been used (B3LYP, B3P86, BP86, BLYP and M06L) to confirm our results. The above indicated that using 6-31+G(d) as the basis set, LAN2DZ as the pseudopotential and B3LYP as functional is reasonable.
Fig. 5. The calculated (B3LYP) SOMO+1 (top) and SOMO (bottom) of [Ni-1]2– (a), [Ni-2]2– (b), [Ni-3]2– (c) and [Ni-4]2– (d) showing the different electronic structures.

In sharp contrast, the two-electron reduced [Ni-2]2– and [Ni-4]2– possess the most stable flat triplet with Mulliken spin densities on the nickel center of ρ Ni = 1.22 and 0.95 (Tables S10 and S12†), indicating that one unpaired electron is localized on the nickel center and another on the macrocycle. The calculated SOMOs of [Ni-2]2– and [Ni-4]2– also showed that one unpaired electron is localized on the σ*(d x 2–y 2 ) orbital of the Ni center, and another is delocalized on the porphyrin, indicating a Ni(i) porphyrin radical (Fig. 5b and d).
DFT calculated formation of [Ni-1–H]– and [H–Ni-2]–
Since HER occurred at the second reduction peak experimentally, we further used the relative free energies to compare the formation and reactivity of the hydride intermediate from [Ni-1]2– and [Ni-2]2–, as shown in Fig. 6. Protonation of [Ni-1]2– by TFA was found to be thermodynamically downhill by 7.7 kcal mol–1 (Fig. 6a) but protonation of [Ni-1]– was thermodynamically uphill by 32.2 kcal mol–1 (Fig. S36†). Thus, starting from [Ni-1]2– is thermodynamically preferred, which is consistent with the experimental CVs in that the HER occurred at ca. –1.80 V. The resulting exothermal product, [Ni-1–H]–, was formed through a stretching vibration between the H and N transition state TS1 with an energy barrier of 18.5 kcal mol–1. To find the protonation intermediate, four protonation sites were tested: the meso-carbon (metallophlorin intermediate), the β-carbon, the pyrrolic nitrogen and the nickel center (metal hydride), and we chose the most thermodynamically favored pyrrolic nitrogen protonated one as the intermediate. In addition, for the pyrrolic nitrogen we could find a transition state (Fig. S35c†). The IRC path showed that the H-atom went through a stretching vibration and the porphyrin ring changed from being flat to ruffled. This phenomenon indicates that the protonation of [Ni-1]2– to [Ni-1–H]– occurred, since [Ni-1]2– is a flat triplet and [Ni-1–H]– is a bent singlet. Then [Ni-1–H]– could react with another TFA to release H2 exothermically by 22.3 kcal mol–1, regenerating the neutral [Ni-1]0. For Ni-2, the protonations of [Ni-2]– and [Ni-2]2– were both thermodynamically uphill by 29.5 and 4.8 kcal mol–1, respectively, and [Ni-2]2– was thermodynamically favourable (Fig. S37†). To find the protonation intermediate for Ni-2, the four protonation sites were also tested and the most thermodynamically favored metal hydride was chosen. We could also find a transition state (Fig. S35d†) and the IRC path showed that the generation of [H–Ni-2]– is through a transition state (TS2) with an energy barrier of 15.2 kcal mol–1, which is 3.3 kcal mol–1 lower than TS1. The reaction of [H–Ni-2]– with another proton was also exothermic by 31.9 kcal mol–1.
Fig. 6. (a) Free energy diagram for the formation of [Ni-1–H]– (black line) and [H–Ni-2]– (red line) intermediates during the HER process. Free-energy values are given in kcal mol–1. The geometric conformation of [Ni-1–H]– (b) and [H–Ni-2]– (c) active intermediates during the HER process.

Interestingly, for [Ni-2]2–, the proton of TFA approaches the electron rich nickel center at a distance of 1.49 Å, positioned obliquely to the pyrrolic nitrogen in the porphyrin ring, indicating a metal hydride-like intermediate [H–Ni-2]– (Fig. 6c).
However, in [Ni-1]2–, the proton bonds to the pyrrolic nitrogen and forms an N–H bond with a length of 1.02 Å. Further insight into the bonding characteristics of the proton adducts was gained from analysis of the Mulliken atomic charges of [Ni-1–H]– (0.397) and [H–Ni-2]– (–0.016) (Fig. S33†). This demonstrated that the hydrogen atom of [Ni-2–H]– is more electronegative than the hydride of [H–Ni-1]–, and might prefer to react with another TFA molecule via a heterolytic pathway.
Spectroelectrochemical TFA titration of [Ni-1]2– and [Ni-2]2–
To verify the reactivity of the two intermediates, [Ni-1]2– and [Ni-2]2–, we carried out redox titrations of the nickel complexes in acetonitrile solution using Co(Cp*)2 (E ox = –1.90 V vs. Fc+/Fc) as a chemical reductant. The one-electron oxidation potential of Co(Cp*)2 is lower than the two-electron reduction potential of Ni-1 and Ni-2. Thus, the addition of Co(Cp*)2 led to the formation of [Ni-1]2– or [Ni-2]2–, which is evidenced by the similarity of the UV-vis absorption spectra to those obtained via spectroelectrochemistry (Fig. S29 and S30†). The addition of excess TFA to the solution of [Ni-1]2– or [Ni-2]2– resulted in fast conversion to Ni-1 or Ni-2 (Fig. 7a), accompanied by the formation of H2 bubbles. The overall two-step kinetics reaction mechanisms of H2 production are shown in Scheme 2 (pathway A). The first proton addition step might be very fast and the second step might be the rate-determining step. 27a–c As shown in Fig. 7a, [Ni-2]2– displayed much larger k obs (0.27 s–1) than [Ni-1]2– (6.2 × 10–3 s–1) (pseudo-first-order kinetic model), as exemplified in the presence of 100 equiv. of TFA at 10 °C. Secondary reaction kinetics constants k 2s for [Ni-1]2– and [Ni-2]2– of 9.17 × 10–2 and 3.03 M–1 s–1, respectively, were obtained by linearly fitting k obss vs. TFA concentration (Fig. 7b). Similar enhancement of the titration reaction rates to electrocatalytic HER clearly demonstrated the importance of disrupted conjugation on the electronic structures and the reactivity of reduced Ni complexes, which is consistent with DFT calculation results.
Fig. 7. (a) UV-vis absorption spectra of the process of [Ni-2]2– reacting with TFA. Inset: plot of the absorption of the Soret band vs. time for the reaction of [Ni-1]2– (blue line) and [Ni-2]2– (red line) with 100 equiv. TFA at 10 °C. (b) Plot of k obs vs. concentration of TFA for the reaction of [Ni-1]2– (blue line) and [Ni-2]2– (red line) with TFA.

Conclusion
Taken together, we demonstrated for the first time that β-hydrogenation of porphyrin is important for enhancing the HER reactivity using nickel porphyrinoids. Experimental and theoretical studies showed that β-hydrogenation significantly affects the electronic structures of the two-electron reduced Ni complexes and renders more electron density localized on the Ni center in [Ni-2]2– and [Ni-4]2– than [Ni-1]2– and [Ni-3]2–. Thus, as shown in Scheme 3, protonation of [Ni-2]2– leads to a typical metal hydride intermediate [H–Ni-2]2–, while [Ni-1]2– only affords a ligand centred hydride [Ni–1-H]2–. [H–Ni-2]– prefers to react with another proton via a quick heterolytic pathway as demonstrated by the time course results of reacting with TFA. This work also provides an opportunity to further design metal molecular catalysts with subtle structural ligand changes inspired by natural metal cofactors.
Scheme 3. The proposed mechanism for nickel compound-catalyzed HER.

Experimental section
General procedures
Commercially available solvents were used after being purified by the MBraun SPS-800 Solvent Purification System. Deuterium solvents were stored with 4 Å molecular sieves. Manipulations of air and moisture sensitive materials were performed in a glove box in an atmosphere of argon. CH3CN and dimethylformamide were distilled and degassed before use. Tetrabutylammonium hexafluorophosphate ( n Bu4NPF6) was recrystallized from absolute ethanol. Other reagents were purchased from commercial suppliers and used without purification. UV-vis spectra were recorded on an Agilent 8453 UV-vis spectrometer equipped with an Agilent 89090A thermostat (±0.1 °C). ESI-MS were recorded on a Bruker APEX IV Fourier Transform Ion Cyclotron Resonance Mass Spectrometer using electrospray ionization. 1H, 19F and 13C NMR spectra were recorded on a Bruker-400 MHz NMR. All 1H NMR experiments were reported in δ units, parts per million (ppm), and all coupling constants were in Hz and measured relative to the signal for residual chloroform (7.26 ppm) in the deuterated solvent CDCl3. For the 19F NMR spectra, CF3COOH was used as an external reference at 0 ppm. Transmission electron microscopy was carried out using a JEM-2100F(JEOL). DLS measurements were made using a Laser Light Scattering Spectrometer (ALV/Laser Vertriebsgesellschaft m.b.H), which can detect particle sizes ranging from 1.0 nm to 1.0 μm. SEM and EDX were performed using a Hitachi S-4800 field emission scanning electron microscope at accelerating voltages of 5 and 15 kV, respectively. Elemental analyses were carried out on an Elementar Vario EL CUBE (Germany). Cyclic voltammetry experiments were recorded on Shanghai Chenhua CHI660C electrochemical workstation. A glassy carbon electrode was selected as the working electrode, the auxiliary electrode was a platinum wire electrode and Ag/AgCl was the reference electrode. All samples were recorded in acetonitrile with 0.1 M n Bu4NPF6 as the electrolyte, the scan rate was 0.1 V s–1 and the E 1/2 were calculated based on peak position for the internal standard FeCp2 (E 1/2 = 0.45 V vs. SCE).
Synthesis of free base porphyrinoids 1–4
meso-Tetrakispentafluorophenylporphyrin (1), meso-tetrakispentafluorophenylchlorin (2), meso-tetrakispentafluorophenylporpholactone (3) and adjacent-hydroporpholactone (4) were synthesized according to the literature. 16c,17
Synthesis of Ni-1 to Ni-4
Typical procedure
Porphyrin free base ligand (0.1 mmol) was refluxed with 30 equiv. of Ni(OAc)2 (1 mmol, 180 mg) in acetonitrile under nitrogen for 6 hours. The solvent was removed by adding 100 equivalents of water and the residue was purified through a silica column.
Ni-1
Isolated yields were over 90%. Eluent: ethyl acetate : petroleum ether, 1 : 5. 1H NMR (400 MHz, CDCl3) δ 8.81 (s, 8H). 19F NMR (377 MHz, CDCl3) δ –136.47 (dd, J = 23.2, 7.7 Hz, 8F), –150.17 (m, 4H), –161.32 (dt, J = 22.5, 7.4 Hz, 8F). 13C NMR (101 MHz, CDCl3) δ 142.98 (s), 132.52 (s), 103.09 (s). ESI-MS m/z: calcd for C44H8F20N4Ni 1029.97831, found 1029.97854. Anal. calcd for C44H8F20N4Ni: C, 51.25; H, 0.78; N, 5.43, found C, 51.29; H, 0.92; N, 5.38.
Ni-2
Isolated yields were 75%. Eluent: ethyl acetate : petroleum ether, 1 : 10. 1H NMR (400 MHz, CDCl3) δ = 8.29 (d, J = 4.9 Hz, 2H), 8.14 (s, 2H), 7.95 (d, J = 5.0 Hz, 2H), 3.93 (s, 4H). 19F NMR (377 MHz, CDCl3) δ = –136.94 (dd, J = 23.2, 7.7 Hz, 4F), –137.89 (dd, J = 23.8, 8.3 Hz, 4F), –151.88 (t, J = 20.9 Hz, 2F), –152.10 (t, J = 20.9 Hz, 2F), –160.66 (dt, J = 23.3, 8.0 Hz, 4F), –161.57 (dt, J = 22.6, 7.4 Hz, 4F). 13C NMR (101 MHz, CDCl3) δ 169.03 (s), 152.66 (s), 140.39 (s), 135.31 (s), 132.27 (s), 128.22 (s), 123.62 (s), 106.31 (s), 96.83 (s), 35.31 (s, –CH2–). ESI-MS m/z: calcd for C44H10F20N4Ni 1031.99396, found 1031.98981. Anal. calcd for C44H10F20N4Ni: C, 51.15; H, 0.98; N, 5.42, found C, 51.16; H, 1.14; N, 5.31.
Ni-3
Isolated yields were over 90%. Eluent: ethyl acetate : petroleum ether, 1 : 5. 1H NMR (400 MHz, CDCl3) δ 8.67 (d, J = 5.1 Hz, 1H), 8.65–8.61 (m, 2H), 8.58 (d, J = 5.1 Hz, 1H), 8.55 (d, J = 4.9 Hz, 2H). 19F NMR (377 MHz, CDCl3) δ –136.69 to –136.96 (m), –138.17 to –138.39 (m), –150.28 to –150.62 (m), –151.20 (q, J = 20.9 Hz), –160.27 to –160.87 (m), –161.37 (qd, J = 22.1, 6.8 Hz). 13C NMR (101 MHz, CDCl3) δ 163.03 (s), 149.75 (s), 144.95 (s), 134.45 (s), 132.47 (d), 131.78 (s), 131.17 (s), 130.34 (s), 120.59 (s). ESI-MS m/z: calcd for C43H6F20N4NiO2 1048.95556, found 1048.95959. Anal. calcd for C43H6F20N4NiO2: C, 49.23; H, 0.58; N, 5.34, found C, 48.66; H, 0.67; N, 5.26.
Ni-4
Isolated yields were over 90%. Eluent: ethyl acetate : petroleum ether, 1 : 5. 1H NMR (400 MHz, CDCl3) δ 7.97 (d, J = 4.8 Hz, 1H), 7.77 (dd, J = 20.5, 4.5 Hz, 2H), 7.54 (d, J = 4.7 Hz, 1H), 3.76 (d, J = 8.4 Hz, 2H), 3.66 (d, J = 8.4 Hz, 2H). 19F NMR (377 MHz, CDCl3) δ –137.63 (d, J = 23.9 Hz), –137.89 (d, J = 15.5 Hz), –138.76 (d, J = 15.8 Hz), –151.16 (d, J = 34.0 Hz), –151.50 (s), –151.79 (s), –160.07 (d, J = 6.2 Hz), –160.31 (d, J = 5.9 Hz), –160.68 (d, J = 6.1 Hz), –161.42 (d, J = 5.9 Hz). 13C NMR (101 MHz, CDCl3) δ 165.72 (s, –C O), 162.06 (s), 156.20 (s), 151.18 (s), 149.93 (s), 141.77 (s), 139.34 (s), 138.75 (s), 133.28 (s), 127.83 (s), 126.19 (s), 115.90 (s), 96.88 (s), 82.19 (s), 34.40 (s, –CH2–), 32.29 (s, –CH2–). Calcd for C43H8F20N4NiO2 [M + H]+ 1050.97586, found 1050.97686. Anal. calcd for C43H8F20N4NiO2: C, 49.13; H, 0.77; N, 5.33, found C, 48.96; H, 0.93; N, 5.20.
Cyclic voltammetry
Cyclic voltammetry was performed with a CHI 660C instrument. A glassy carbon (3 mm diameter) was used as the working electrode, a platinum filament as the counter electrode, and Ag/AgCl as the reference electrode. Solvents were purified by an MBraun SPS-800 Solvent Purification System. All samples were recorded in dichloromethane with 0.1 M n Bu4NPF6 as the electrolyte. Unless otherwise noted, all potentials in this study were adjusted to the ferrocenium/ferrocene (Fc+/0) couple as an internal standard.
Spectroelectrochemistry studies
Spectroelectrochemical measurements were carried out in a quartz cell with an optical path length of 1 cm. A platinum net, platinum filament and Ag/AgCl were used as the working electrode, counter electrode, and reference electrode, respectively. The sample solutions were deaerated with acetonitrile-saturated argon. The spectra were recorded on an Agilent 8453 UV-vis spectrophotometer during electrolysis on a CHI 660C instrument.
Controlled-voltage coulometry experiment
Bulk electrolysis was performed with a CHI 660C instrument with a glassy carbon (1 cm × 1 cm) as the working electrode. The reference electrode was an aqueous Ag/AgCl electrode, and the counter electrode was a platinum wire and 0.1 M n Bu4NPF6 in CH3CN solution. The volume of the electrolysis solution was 20 mL. The reference electrode was directly immersed in the solution to minimize ohmic drop. The gas phase in the head space was analyzed by GC with Ar as the carrier gas. A hydrogen calibration curve was obtained by filling a tube with pure hydrogen gas with a graduated gastight syringe.
Chemistry titration
Acetonitrile solutions of Ni-1 or Ni-2 (1.6 × 10–5 M) and Co(Cp*)2 (9.3 × 10–4 M) were prepared in the glove box and stored in airtight cuvettes and vials respectively. The solutions of Ni-1 or Ni-2 and 25 μL Co(Cp*)2 (1 equivalent) were mixed and the UV-Vis spectra were recorded immediately after the mixing.
Computational details
All geometry optimization calculations were performed using the density functional theory (DFT) functional B3LYP/6-31G+(d) as the basis set and LAN2DZ as the pseudopotential as implemented in the Gaussian 09 software. The solvent effect was considered in all geometry optimizations and property calculations using the conductor-like polarizable continuum model (CPCM). The energies were corrected by single point calculations using B3LYP/6-31+G(d,p) as the basis set. For the absolute solvation free energy of the proton in acetonitrile, a value of –260.2 kcal mol–1 was used.28 The frequency calculations at the same level were carried out to confirm each stationary point to be either a minimum or transition state. Intrinsic reaction coordinate (IRC) paths were calculated to connect each TS to the corresponding reactant and product.
Acknowledgments
We acknowledge financial support from the National Key Basic Research Support Foundation of China (Grant 2015CB856301) and the National Scientific Foundation of China (Grants 21571007, 21271013, 21321001). X. R. He is gratefully thanked for help with the ESI-MS, Z.-C. Huang and H. Zhu are gratefully thanked for help with the DFT calculation parts, T. Huang is gratefully thanked for help with DLS, T.-T. Zuo is gratefully thanked for help with TEM, and Y.-Y. Yuan is gratefully thanked for help with SEM and EDX.
Footnotes
References
- (a) Kadish K. M., Smith K. M. and Guilard R., Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, World Scientific, 2014. [Google Scholar]; (b) Kraatz H.-B. and Metzler-Nolte N., Concepts and Models in Bioinorganic Chemistry, Wiley-VCh, New York, 2006. [Google Scholar]; (c) Sung Y. M., Oh J., Cha W. Y., Kim W., Lim J. M., Yoon M. C., Kim D. Chem. Rev. 2016;117:2257–2312. doi: 10.1021/acs.chemrev.6b00313. [DOI] [PubMed] [Google Scholar]; (d) Teo R. D., Hwang J. Y., Termini J., Gross Z., Gray H. B. Chem. Rev. 2016;114:2177–2729. [Google Scholar]; (e) Zhang W., Lai W., Cao R. Chem. Rev. 2016;117:3717–3797. doi: 10.1021/acs.chemrev.6b00299. [DOI] [PubMed] [Google Scholar]
- (a) Li L.-L., Diau E. W.-G. Chem. Soc. Rev. 2013;42:291–304. doi: 10.1039/c2cs35257e. [DOI] [PubMed] [Google Scholar]; (b) Wasielewski M. R. Acc. Chem. Res. 2009;42:1910–1921. doi: 10.1021/ar9001735. [DOI] [PubMed] [Google Scholar]
- (a) Ethirajan M., Chen Y., Joshi P., Pandey R. K. Chem. Soc. Rev. 2011;40:340–362. doi: 10.1039/b915149b. [DOI] [PubMed] [Google Scholar]; (b) Castano A. P., Mroz P., Hamblin M. R. Nat. Rev. Cancer. 2006;6:535–545. doi: 10.1038/nrc1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Drain C. M., Varotto A., Radivojevic I. Chem. Rev. 2009;109:1630–1658. doi: 10.1021/cr8002483. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Paolesse R., Nardis S., Monti D., Stefanelli M., Di Natale C. Chem. Rev. 2017;117:2517–2583. doi: 10.1021/acs.chemrev.6b00361. [DOI] [PubMed] [Google Scholar]; (c) Ding Y., Zhu W. H., Xie Y. Chem. Rev. 2016;117:2203–2256. doi: 10.1021/acs.chemrev.6b00021. [DOI] [PubMed] [Google Scholar]
- (a) Kruger M., Meyerdierks A., Glockner F. O., Amann R., Widdel F., Kube M., Reinhardt R., Kahnt R., Bocher R., Thauer R. K., Shima S. Nature. 2003;426:878–881. doi: 10.1038/nature02207. [DOI] [PubMed] [Google Scholar]; (b) Lawton T. J., Rosenzweig A. C. J. Am. Chem. Soc. 2016;138:9327–9340. doi: 10.1021/jacs.6b04568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ellefson W. L., Whitman W. B., Wolfe R. S. Proc. Natl. Acad. Sci. U. S. A. 1982;79:3707–3710. doi: 10.1073/pnas.79.12.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Moore S. J., Sowa S. T., Schuchardt C., Deery E., Lawrence A. D., Ramos J. V., Billig S., Birkemeyer C., Chivers P. T., Howard M. J., Rigby S. E., Layer G., Warren M. J. Nature. 2017;543:78–82. doi: 10.1038/nature21427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Bible K. C., Buytendorp M., Zierath P. D., Rinehart K. L. Proc. Natl. Acad. Sci. U. S. A. 1988;85:4582–4586. doi: 10.1073/pnas.85.13.4582. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Andersson L. A., Huff A., Smith K. M. Biophys. J. 1990;57:A232. [Google Scholar]
- (a) Kadish K., Franzen M., Han B., Araullo-McAdams C., Sazou D. J. Am. Chem. Soc. 1991;113:512–517. [Google Scholar]; (b) Telser J., Fann Y.-C., Renner M. W., Fajer J., Wang S., Zhang H., Scott R. A., Hoffman B. M. J. Am. Chem. Soc. 1997;119:733–743. [Google Scholar]; (c) Atifi A., Ryan M. D. Anal. Chem. 2015;87:12245–12253. doi: 10.1021/acs.analchem.5b03411. [DOI] [PubMed] [Google Scholar]; (d) Lovecchio F. V., Gore E. S., Busch D. H. J. Am. Chem. Soc. 1974;96:3109–3118. [Google Scholar]
- (a) Gray H. B. Nat. Chem. 2009;1:7. doi: 10.1038/nchem.141. [DOI] [PubMed] [Google Scholar]; (b) Gust D., Moore T. A., Moore A. L. Acc. Chem. Res. 2009;42:1890–1898. doi: 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]; (c) Moralesguio C. G., Stern L. A., Hu X. Chem. Soc. Rev. 2014;43:6555–6569. doi: 10.1039/c3cs60468c. [DOI] [PubMed] [Google Scholar]; (d) Jian J. X., Ye C., Wang X. Z., Wen M., Li Z. J., Li X. B., Chen B., Tung C. H., Wu L. Z. Energy Environ. Sci. 2016;9:2083–2089. [Google Scholar]
- (a) Wang M., Chen L., Sun L. Energy Environ. Sci. 2012;5:6763–6778. [Google Scholar]; (b) Du P., Eisenberg R. Energy Environ. Sci. 2012;5:6012–6021. [Google Scholar]; (c) Thoi V. S., Sun Y., Long J. R., Chang C. J. Chem. Soc. Rev. 2013;42:2388–2400. doi: 10.1039/c2cs35272a. [DOI] [PubMed] [Google Scholar]; (d) Mckone J. R., Marinescu S. C., Brunschwig B. S., Winkler J. R., Gray H. B. Chem. Sci. 2014;5:865–878. [Google Scholar]; (e) Ran J., Zhang J., Yu J., Jaroniec M., Qiao S. Z. Chem. Soc. Rev. 2014;43:7787–7812. doi: 10.1039/c3cs60425j. [DOI] [PubMed] [Google Scholar]; (f) Mondal B., Sengupta K., Rana A., Mahammed A., Botoshansky M., Dey S. G., Gross Z., Dey A. Inorg. Chem. 2013;52:3381–3387. doi: 10.1021/ic4000473. [DOI] [PubMed] [Google Scholar]
- (a) Lubitz W., Ogata H., Ruediger O., Reijerse E. Chem. Rev. 2014;114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]; (b) Layfield J. P., Hammes-Schiffer S. Chem. Rev. 2014;114:3466–3494. doi: 10.1021/cr400400p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Lu H., Zhang X. P. Chem. Soc. Rev. 2011;40:1899–1909. doi: 10.1039/c0cs00070a. [DOI] [PubMed] [Google Scholar]; (b) Che C.-M., Lo V. K.-Y., Zhou C.-Y., Huang J.-S. Chem. Soc. Rev. 2011;40:1950–1975. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]; (c) Kupitz C., Fromme R., Grotjohann I. and Fromme P., in Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, ed. G. C. Ferreira, K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2014, vol. 28, pp. 1–40. [Google Scholar]; (d) Senge M. O., MacGowan S. A., O’Brien J. M. Chem. Commun. 2015;51:17031–17063. doi: 10.1039/c5cc06254c. [DOI] [PubMed] [Google Scholar]; (e) Zhang W., Lai W., Cao R. Chem. Rev. 2017;117:3717–3797. doi: 10.1021/acs.chemrev.6b00299. [DOI] [PubMed] [Google Scholar]
- Han Y., Fang H., Jing H., Sun H., Lei H., Lai W., Cao R. Angew. Chem., Int. Ed. Engl. 2016;55:5457–5462. doi: 10.1002/anie.201510001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bediako D. K., Solis B. H., Dogutan D. K., Roubelakis M. M., Maher A. G., Lee C. H., Chambers M. B., Hammes-Schiffer S., Nocera D. G. Proc. Natl. Acad. Sci. U. S. A. 2014;111:15001–15006. doi: 10.1073/pnas.1414908111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis B. H., Maher A. G., Dogutan D. K., Nocera D. G., Hammes-Schiffer S. Proc. Natl. Acad. Sci. U. S. A. 2016;113:485–492. doi: 10.1073/pnas.1521834112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Ke X.-S., Zhao H., Zou X., Ning Y., Cheng X., Su H., Zhang J.-L. J. Am. Chem. Soc. 2015;137:10745–10752. doi: 10.1021/jacs.5b06332. [DOI] [PubMed] [Google Scholar]; (b) Ke X.-S., Chang Y., Chen J.-Z., Tian J., Mack J., Cheng X., Shen Z., Zhang J.-L. J. Am. Chem. Soc. 2014;136:9598–9607. doi: 10.1021/ja502729x. [DOI] [PubMed] [Google Scholar]; (c) Yu Y., Furuyama T., Tang J., Wu Z.-Y., Chen J.-Z., Kobayashi N., Zhang J.-L. Inorg. Chem. Front. 2015;2:671–677. [Google Scholar]; (d) Ning Y., Ke X. S., Hu J. Y., Liu Y. W., Ma F., Sun H. L., Zhang J. L. Inorg. Chem. 2017;56:1897–1905. doi: 10.1021/acs.inorgchem.6b02481. [DOI] [PubMed] [Google Scholar]
- Yu Y., Lv H., Ke X., Yang B., Zhang J.-L. Adv. Synth. Catal. 2012;354:3509–3516. [Google Scholar]
- (a) Brückner C., McCarthy J. R., Daniell H. W., Pendon Z. D., Ilagan R. P., Francis T. M., Ren L., Birge R. R., Frank H. A. Chem. Phys. 2003;294:285–303. [Google Scholar]; (b) Yu Y., Czepukojc B., Jacob C., Jiang Y., Zeller M., Brückner C., Zhang J.-L. Org. Biomol. Chem. 2013;11:4613–4621. doi: 10.1039/c3ob40758f. [DOI] [PubMed] [Google Scholar]
- Lexa D., Momenteau M., Mispelter J., Savéant J. M. J. Inorg. Biochem. 1989;28:225. [Google Scholar]
- Campbell C. J., Rusling J. F., Brückner C. J. Am. Chem. Soc. 2000;122:6679–6685. [Google Scholar]
- (a) Stolzenberg A. M., Stershic M. T. J. Am. Chem. Soc. 1988;110:6391–6402. [Google Scholar]; (b) Stolzenberg A. M., Stershic M. T. J. Am. Chem. Soc. 1988;110:5397–5403. [Google Scholar]
- Renner M. W., Furenlid L. R., Barkigia K. M., Forman A., Shim H. K., Simpson D. J., Smith K. M., Fajer J. J. Am. Chem. Soc. 1991;113:6891–6898. [Google Scholar]
- (a) Kadish K. M., Franzen M. M., Han B. C., Araullomcadams C., Sazou D. J. Am. Chem. Soc. 1991;113:512–517. [Google Scholar]; (b) Renner M. W., Furenlid L. R., Barkigia K. M., Forman A., Shim H. K., Simpson D. J., Smith K. M., Fajer J. J. Am. Chem. Soc. 1991;113:6891–6898. [Google Scholar]
- (a) Costentin C., Passard G., Savéant J. M. J. Am. Chem. Soc. 2015;137:5461–5467. doi: 10.1021/jacs.5b00914. [DOI] [PubMed] [Google Scholar]; (b) Rountree E. S., McCarthy B. D., Eisenhart T. T., Dempsey J. L. Inorg. Chem. 2014;53:9983–10002. doi: 10.1021/ic500658x. [DOI] [PubMed] [Google Scholar]
- Artero V., Fontecave M. Coord. Chem. Rev. 2005;249:1518–1535. [Google Scholar]
- Bhugun I., Lexa D., Savéant J.-M. J. Am. Chem. Soc. 1996;118:3982–3983. [Google Scholar]
- (a) Kleperis J., Wojcik G., Czerwinski A., Skowronski J., Kopczyk M., Beltowska-Brzezinska M. J. Solid State Electrochem. 2001;5:229–249. [Google Scholar]; (b) Mandal S., Shikano S., Yamada Y., Lee Y. M., Nam W., Llobet A., Fukuzumi S. J. Am. Chem. Soc. 2013;135:15294–15297. doi: 10.1021/ja408080z. [DOI] [PubMed] [Google Scholar]; (c) Eberhardt N. A., Guan H. Chem. Rev. 2016;116:8373–8426. doi: 10.1021/acs.chemrev.6b00259. [DOI] [PubMed] [Google Scholar]
- Chambers C. C., Hawkins G. D., Cramer C. J., Truhlar D. G. J. Phys. Chem. 1996;100:100–16385. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.