

The opening line of Tolstoy’s famous Anna Karenina, “All happy families are alike; each unhappy family is unhappy in its own way”, has many parallels to allosteric drugs.1 Traditionally, drugs targeting enzyme active sites or receptor orthosteric sites in some sense have similar mechanisms of action—that is, they “act alike”. However, allosteric drugs, or drugs that alter target activity by binding at a site that is distinct from the active or orthosteric site, often act in vastly different, and even unexpected, ways (Figure 1). Understanding “the ways” of allosteric drugs continues to be a central challenge in drug design programs, and the work of La Sala et al.2 offers new possibilities for disentangling their mysteries.
Figure 1.
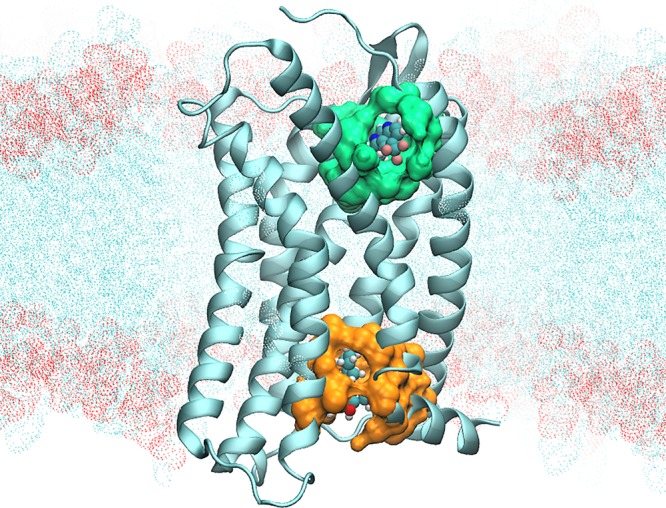
Allosteric drugs work by manipulating binding pockets away from active sites to effect global conformational changes, making them harder to predict.
The uniqueness of allosteric drugs is one of their most promising attributes, as it offers the tantalizing possibility of developing highly selective molecules for challenging targets such as kinases or G-protein coupled receptors. Allosteric drugs may impart “absolute subtype specificity” and thus are more likely to be good clinical candidates due to reduced off-target effects.3 By acting at a site (or receptor state) that is only open to binding (or existing) during particular times in the activation or signaling process (e.g., in the presence of an endogenous ligand), allosteric drugs can also bring spatiotemporal specificity.4 Such features may enable therapies to target specific tissues at certain times, slowing the process of desensitization.5
At the same time, the uniqueness of allosteric drugs brings a number of intense challenges from the standpoint of standard drug discovery programs, including, in particular, rational design.6 Rational drug design programs have primarily centered around the high throughput determination of static X-ray crystallographic structures of targets in complex with ligands, and, generally speaking, biochemical and biophysical assays that have been developed with orthosteric sites in mind. This enables the highly optimized application of computational modeling and simulation methods to aid in the drug design process, assisting in “hit discovery”, a more complete understanding of mechanism of action, and the ability to prospectively design ligands with improved target binding affinity (a.k.a. hit-to-lead optimization). Such efforts can bring significant efficiencies to drug design programs, but are not yet routinely relied on for the rational design of allosteric drugs.
A key aspect of unlocking allosteric mechanisms is the universal requirement to understand each target’s dynamic conformational landscape, and in a way that is detailed and accurate enough to impact the design of chemical matter. One of the best ways researchers have to explore the detailed, time dependent dynamics of drug targets is with molecular dynamics simulations—which allow an atomic-level view of biomolecular systems, similar to a computational microscope.7 The convergence of consistent gains in computer power with improvements in the algorithms and software used to run the simulations (as well as analyze the vast amount of data that these simulations generate) now allows researchers to routinely build fairly complex, biomedically relevant systems in silico and then sample their time dependent dynamics on biologically relevant time scales. These simulations can be used to predict the atomic-level motion of drug targets and more accurately quantify their interactions with small molecules, including the effects of allosteric modulators. Molecular dynamics based methods, for example, have been used to discover previously unknown druggable binding sites,8 quantify kinetics of ligand binding,9 and even describe the kinetic and thermodynamic landscape of protein–protein interactions at time scales approaching minutes.10
The work by La Sala et al.2 offers a broadly applicable automated technology for detecting potential allosteric mechanisms. To do this, they combine elegant and efficient mathematical methods with long time scale simulations to provide a detailed view of the many dynamic pockets found within drug targets. Their tool allows researchers not only to discover pockets possibly previously undetected in X-ray crystallographic structures but also to characterize the opening and closing of various pockets and their time dependent dynamics, and, in some instances, detect pocket crosstalk. The novel method they present provides researchers a tool capable to reveal hidden allosteric networks, or possible communication pathways, between disparately located sites on a drug target, and, in doing so, may aid the prospective design of allosteric drugs.
References
- L. Tolstoy graf, 1828–1910. Anna Karenina [Internet]; Oxford University Press: Oxford; New York, 1980. Available from https://search.library.wisc.edu/catalog/999776789402121. [Google Scholar]
- La Sala G.; Decherchi S.; De Vivo M.; Rocchia W. Allosteric communication networks in proteins revealed through pocket crosstalk analysis. ACS Cent. Sci. 2017, 10.1021/acscentsci.7b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May L. T.; Leach K.; Sexton P. M.; Christopoulos A. Allosteric Modulation of G Protein-Coupled Receptors. Annu. Rev. Pharmacol. Toxicol. 2015, 47 (1), 1–51. 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- Nussinov R.; Tsai C.-J. The Different Ways through Which Specificity Works in Orthosteric and Allosteric Drugs. Curr. Drug Metab. 2012, 18 (9), 1311–1316. 10.2174/138920012799362855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenthur C. J.; Gentry P. R.; Mathews T. P.; Lindsley C. W. Drugs for Allosteric Sites on Receptors. Annu. Rev. Pharmacol. Toxicol. 2014, 54 (1), 165–184. 10.1146/annurev-pharmtox-010611-134525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner J. R.; Lee C. T.; Durrant J. D.; Malmstrom R. D.; Feher V. A.; Amaro R. E. Emerging Computational Methods for the Rational Discovery of Allosteric Drugs. Chem. Rev. 2016, 116 (11), 6370–6390. 10.1021/acs.chemrev.5b00631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. H.; Hsin J.; Sotomayor M.; Comellas G.; Schulten K. Structure 2009, 17 (10), 1295–1306. 10.1016/j.str.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassman C. D.; Baronio R.; Demir Ö; Wallentine B. D.; Chen C.-K.; Hall L. V. Computational Identification of a Transiently Open L1/S3 Pocket for Reactivation of Mutant p53. Nat. Commun. 2013, 4, 1407. 10.1038/ncomms2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwary P.; Limongelli V.; Salvalaglio M.; Parrinello M. Kinetics of Protein-Ligand Unbinding: Predicting Pathways, Rates, and Rate-Limiting Steps. Proc. Natl. Acad. Sci. 2015, 112 (5), E386–E391. 10.1073/pnas.1424461112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner N.; Doerr S.; De Fabritiis G.; Noé F. Complete protein–protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling. Nat. Chem. 2017, (June), 1–7. 10.1038/nchem.2785. [DOI] [PubMed] [Google Scholar]