

Abstract
C–H functionalization is a very active research field that has attracted the interest of scientists from many disciplines. This Outlook describes the collaborative efforts within the NSF CCI Center for Selective C–H Functionalization (CCHF) to develop catalyst-controlled selective methods to enhance the synthetic potential of C–H functionalization.
Short abstract
This manuscript describes the collaborative efforts within the NSF CCI Center for Selective C−H Functionalization (CCHF) to bring C−H functionalization into the mainstream of organic chemistry.
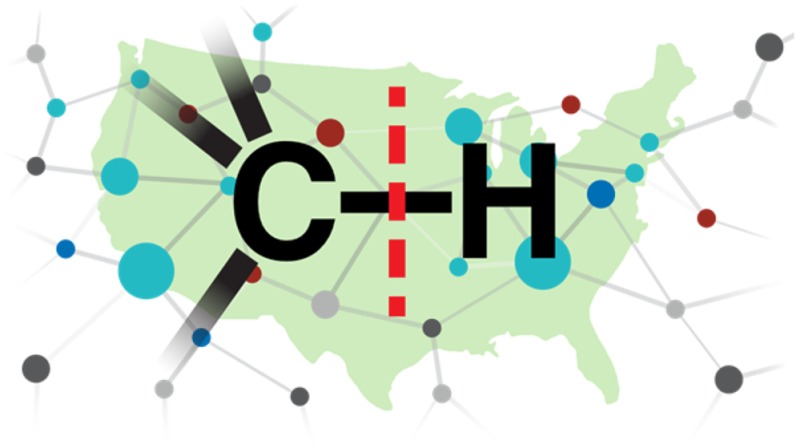
C–H functionalization has become an exciting field of research with the potential of imparting a paradigm shift in how chemicals are made.1 The ability to selectively functionalize C–H bonds has been recognized as an attractive challenge, with much of the early progress relying on free radical reactions. In the 1980s organometallic chemists became interested in designing complexes capable of activating C–H bonds,1 and soon thereafter, the development of new strategies for synthesis using C–H functionalization became an ambitious research field for the organic synthesis community.1−11 As the aspirations for the field heightened, it was quickly recognized that scientists with a diverse range of skill sets were required to bring C–H functionalization to its full synthetic potential. In 2009, we initiated the NSF Center for Selective C–H Functionalization (CCHF) to address this challenge, and it now encompasses 25 professors and their research groups from 16 different universities (Figure 1). This Outlook describes the development of this collaborative community and highlights some of its recent accomplishments.
Figure 1.

Geographic and expertise distribution of the CCHF.
An introductory challenge for the Center was to develop effective methods for communication and the promotion of a culture of collaborative research. The typical approach to collaboration in organic synthesis is in an interdisciplinary fashion, such that individual organic chemistry groups connect with scientists from other fields but rarely with each other. The Center, comprising of synthetic methodologists, synthetic strategists, physical organic chemists, catalyst developers, enzymologists, computational chemists, chemical engineers, material scientists, and several partners in the pharmaceutical industry needed to develop a culture in which not just results but nascent ideas could be shared. Early on the Center organized a number of face-to-face meetings to establish the guidelines for broad collaborative engagement and to brainstorm the most promising research areas. A program of weekly videoconference meetings was established at which 20–25 groups and subgroups are regularly in attendance. These virtual meetings have generated a highly synergistic and collaborative community (Figure 2). The Center encourages wide-ranging collaborations within the Center and with the broader scientific community, as illustrated by the numerous joint publications stemming from within the Center and with other academic, industrial, and international partners. Of the Center’s 207 publications to date, 121 are collaborative, 98 have two senior investigators as coauthors, 13 have three, 8 have four, 1 has five, and 1 has eight (see Supporting Information for details).
Figure 2.

CCHF collaborative research interactions based upon published articles. This visualizes work from 2009 to the present. Each interaction is represented by a connective ribbon, and the size of each ribbon is directly proportional to the number of interactions.
The Center research efforts were initially organized around six thematic areas related to C–H functionalization: mechanistic understanding, catalyst and methodology development, strategic disconnections, late stage C–H functionalization, pharmaceutical applications, and materials science applications. The thematic leaders organize the discussions in the videoconference meeting and focus their teams on the most pressing challenges in the specific area. When we initiated the CCHF, we had some experts in C–H functionalization methodology development, but the rest of the group, although they had very valuable skill sets, had limited prior engagement in C–H functionalization. Early on, developing strong mechanistic understanding was crucial. The combined computational and experimental mechanistic studies have been extremely informative in guiding us to move away from empirical to design-led approaches for new catalyst and methodology development. We also have the expertise to demonstrate the synthetic applications of C–H functionalization to complex target synthesis, including pharmaceutical and materials science applications. As the Center matured, it was recognized that in order to make a lasting impact on the field more broadly the Center should shift the focus of its investigations from the general application of the technology to answering the big challenge in this field: selectivity. To this end three strategies were outlined that seek to move the controlling factors away from the inherent reactivity of a given substrate and instead employ catalysts to define the product distribution (Figure 3). The first uses the catalyst/reagent control, the second uses prior coordination to the substrate but still catalyst control, and the third uses bioinspired approaches for site selection. The next section of this article will describe an example from each of these catalyst control approaches and illustrate how they have evolved within the Center, and how the development of these fields informed the reorganization of the Center’s research to establish a program that propels the field forward.
Figure 3.
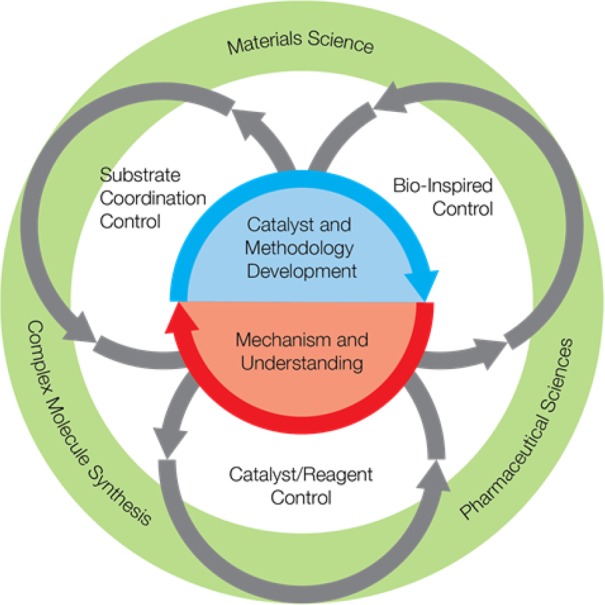
Current research focus of the CCHF.
Catalyst/Reagent Control Development
At the outset of the Center, group transfer reactions were recognized by the C–H functionalization community as a complementary strategy to the use of directing groups for achieving site selective transformations. The Davies and Du Bois groups had conducted extensive studies on the metal-catalyzed intermolecular C–H insertion reactions of carbenes and nitrenes2 and demonstrated their utility in transformations of strategic importance and as streamlining technologies in the total synthesis of complex targets.12,13
However, the chemistry was primarily under substrate control, and if the substrate did not give a clean reaction, enhancing the chemistry through catalyst selection was limited. In many instances optimization was required on a substrate-by-substrate basis. To overcome this limitation a team within the Center came together, including the Davies and Du Bois groups collaborating with physical organic and mechanistic chemists (Blackmond, TSRI; Sigman, Utah; Zare, Stanford), computational chemists (Houk, UCLA; Musaev, Emory), and inorganic chemists (Berry, Wisconsin) to better understand the mechanistic aspects and controlling influences of the catalysts. In parallel to these studies a Center-led team (Sorensen, Princeton; Yu, TSRI; Blakey, Emory; Davies, Emory), with international collaborators (Itami, Nagoya) and industrial collaborators (at Novartis and AbbVie), has contributed to the demonstration of the practical strategic opportunities offered by this chemistry. Highlights of the research accomplishments in group transfer reaction from the past few years as illustrated in Figure 4 include the following:
Demonstration that dirhodium catalysts are capable of over 1,000,000 turnover numbers in carbene chemistry.14
Capture and identification of short-lived intermediates in the dirhodium catalyzed C–H amination reaction.15
Isolation and characterization, for the first time, of the reactive dirhodium carbene intermediate.16
Understanding the role of Rh(II)Rh(III) intermediates in rhodium nitrene chemistry.17
Intermolecular C–H amination of elaborate substrates at tertiary sites.18
Generation of predictive models for group transfer C–H functionalization.19,20
Development of a chiral iridium complex for enantioselective C–H functionalization with acceptor carbenes21
Development of a new class of chiral ligands, the triarylcyclopropane caraboxylates (TPCP), for dirhodium catalysts.22
Application of group transfer reactions to the functionalization of pharmaceutically relevant targets with heterocyclic functionality.23,24
Application of sequential C–H functionalization strategies in complex target synthesis.25−27
Figure 4.
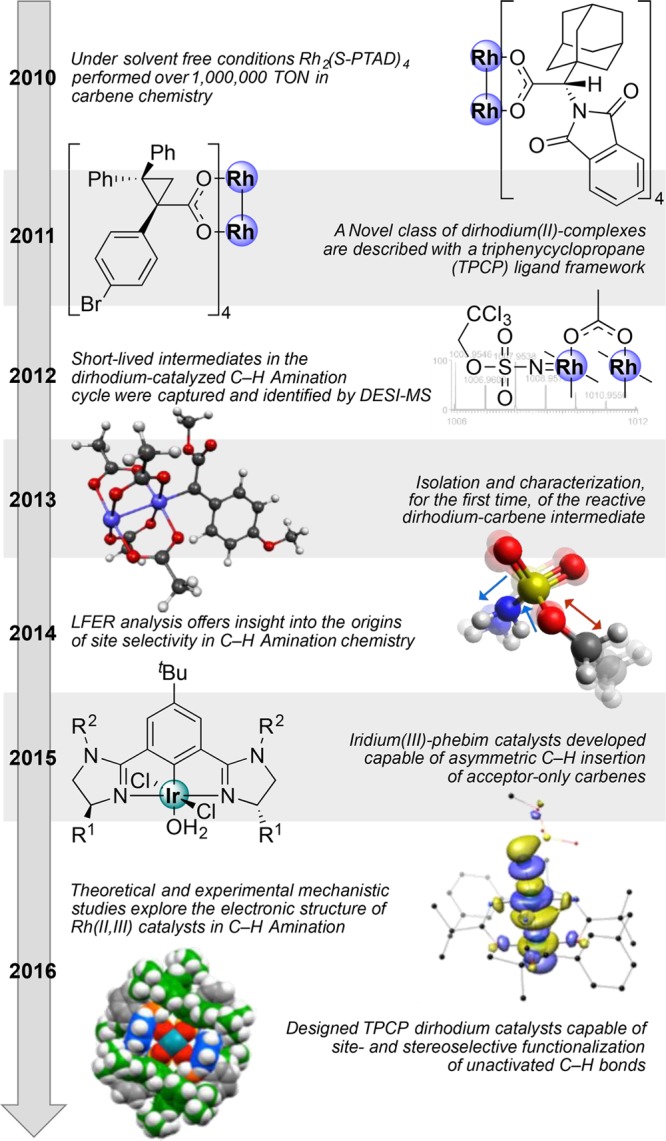
Timeline of CCHF-led group transfer C–H functionalization research.
The ultimate goal of this program is to override the influence exerted by the inherent properties of the substrate and develop a suite of catalysts that can control the site selectivity at will. An illustration of what can be achieved was recently reported, which described how a new TPCP dirhodium catalyst influences the insertion of a donor/acceptor carbene to such an extent that it occurs cleanly at the most accessible methylene position in n-alkanes and terminally substituted n-alkanes (Figure 5).28 In other words, the catalyst/reagent system distinguishes between a methylene site with a methyl substituent and any other methylene containing larger alkyl substituents. The future plans of the CCHF will be to build on these advances and generate a tool-box of chiral catalysts that enable practitioners to select which site in a molecule will be functionalized, not based on the properties of the substrate but rather determined by the catalyst choice.
Figure 5.
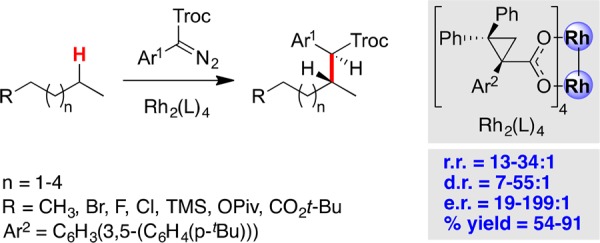
Catalyst-controlled C–H functionalization at the most accessible methylene group.
Coordination-Assisted Control
Arguably the most effective strategy to date for selective product formation in C–H activation is through the use of so-called directing groups: functionality present on the substrate molecule that can chelate to the metal complex catalyst, preorienting the reactive species to a specific site on the molecule, reliably furnishing single product formation. This has proven to be a highly effective class of transformations for C–H activation, capable of introducing a range of different atoms and functional groups, especially in the functionalization of aromatic C–H bonds.3,29 The Yu group are leaders in this field, pioneering the broad use of weakly coordinating directing groups,4 an advance that fundamentally changed the efficiency and scope of this strategy. In order for this transformation to maintain a high level of site selectivity, careful consideration of the ligand environment surrounding the palladium catalyst was required. Early reports from the Yu group detailed the use of monoprotected amino acids (MPAA), ligand frameworks as effective choices for this system, though the origins of how this ligand framework struck this subtle balance were poorly understood.30 It was around this challenge that a Center team united.
Development of this strategy required insight into the mechanistic underpinnings of this chemical assembly; to this end the Yu group collaborated with a team of computational theoreticians (Houk, UCLA; Musaev, Emory), chemical engineers (Blackmond, TSRI), and physical organic chemists (Sigman, Utah), to systematically probe the factors that determine site selection. At the same time collaborations with Center members (Davies, Emory; Movassaghi, MIT; Sorensen, Princeton), international collaborators (Dai, SIOC; Wu, HKUST), and industrial partners (BMS, NIBR, Pfizer, Merck), challenged and drove the application of this strategy to new innovations. Highlights of the CCHF research accomplishments in weakly coordinated directing group chemistry from the past few years, as highlighted in Figure 6, include the following:
Use of RPKA analysis to determine rate dependence on catalyst loading, identification of off-cycle catalyst reservoirs, and insights into the role of the MPAA ligands.31
Calculation of the key intermediates involved in these transformations and a theoretical outline of the elemental steps, offering insight into how the ligands surrounding palladium engage in the reaction.32
DFT calculations that describe the spatial arrangement of the ligand framework around the core, aiding understanding of stereochemical induction.33
The development of this strategy to employ new directing group functionalities, expanding both scope and product versatility.34,35
Substitution of palladium complexes for copper(II) complexes as the reactive center, enabled by the design of an oxazole-based directing group.36−40
Application to pharmaceutically relevant targets.41,42,25,43,27,44
Development of templated tether-like directing groups that enabled site selection at more remote C–H bonds.45,46
Figure 6.
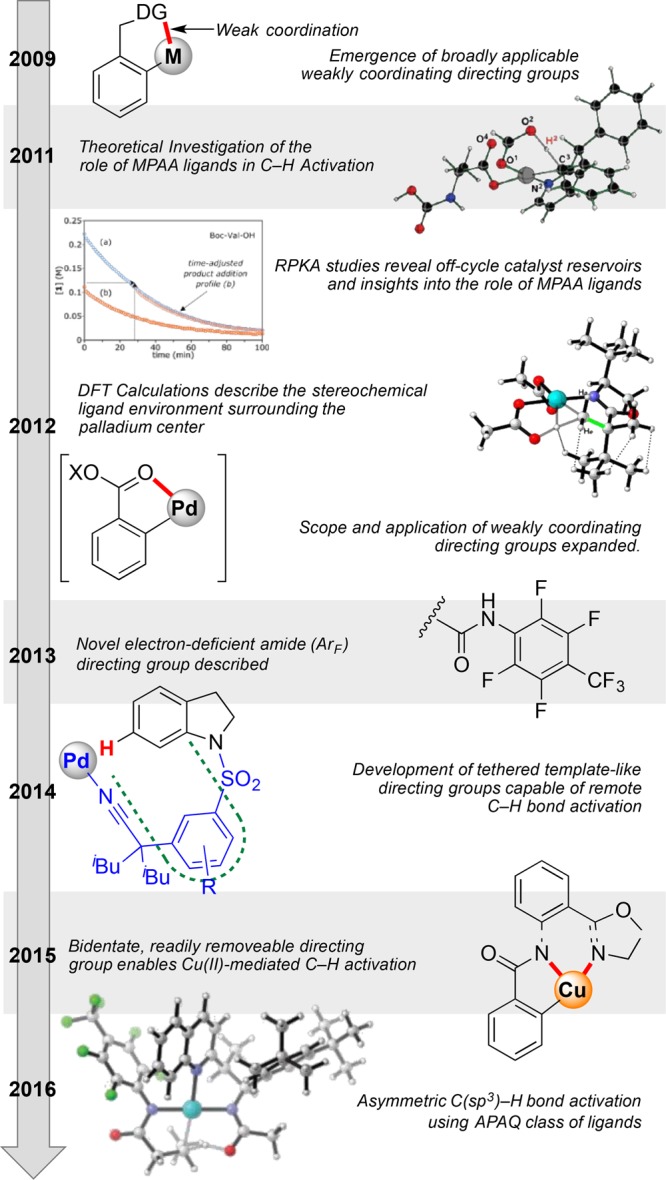
Timeline of the key CCHF-led investigations into the use of weakly coordinating directing groups.
This accumulation of knowledge around the elemental steps of the palladium mechanism and the conformational preference of the ligand and directing group structures has enabled the most recent advance in this field, the development of a new class of bidentate ligands that outcompete the background reaction driven by substrate-directed cyclopalladation, enabling the asymmetric palladium insertion into prochiral C–H bonds on a single methylene sp3 center (Figure 7).47 The development of reactions that employ the acetyl-protected aminomethyl quinoline (APAQ) class of ligands marks the path forward for Center research in this area, one in which this strategy moves to regio- and stereoselectively functionalize sp3 C–H bonds in a general and efficient manner.
Figure 7.
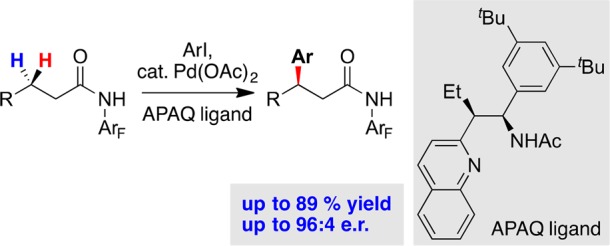
APAQ-ligand accelerated asymmetric C–H activation of C(sp3)–H bonds.
Re-Engineering Nature’s C–H Functionalization Machinery
The defining characteristic of enzymes from secondary metabolic pathways is the pairing of powerful reactivity with exquisite site selectivity. However, the application of these biocatalytic tools in organic synthesis remains underutilized, in large part due to limitations in substrate scope and scalability. Many enzymes employ selective C–H oxidation as a mechanism for streamlined molecule synthesis, inspiring Center researchers to assemble a team around addressing the challenges outlined in bringing this technology into the mainstream. Expertise from across a broad spectrum was connected, including biochemists (Sherman, U-Michigan), synthetic organic chemists (Montgomery, U-Michigan), structural biologists (Podust, UC-SF (external to CCHF)), and theoretical chemists (Houk, UCLA), to shape a multifaceted approach that brought a combination of computationally guided protein and substrate engineering to the application of the oxidation of unactivated methylene C–H bonds.
The system chosen for these studies was the cytochrome P450 monooxygenase PikC, from the pikromycin natural product biosynthetic pathway, and the timeline of the developmental studies within the center is summarized in Figure 8. PikC has an unusually high level of substrate promiscuity, which is rare in secondary metabolic pathways.
Figure 8.
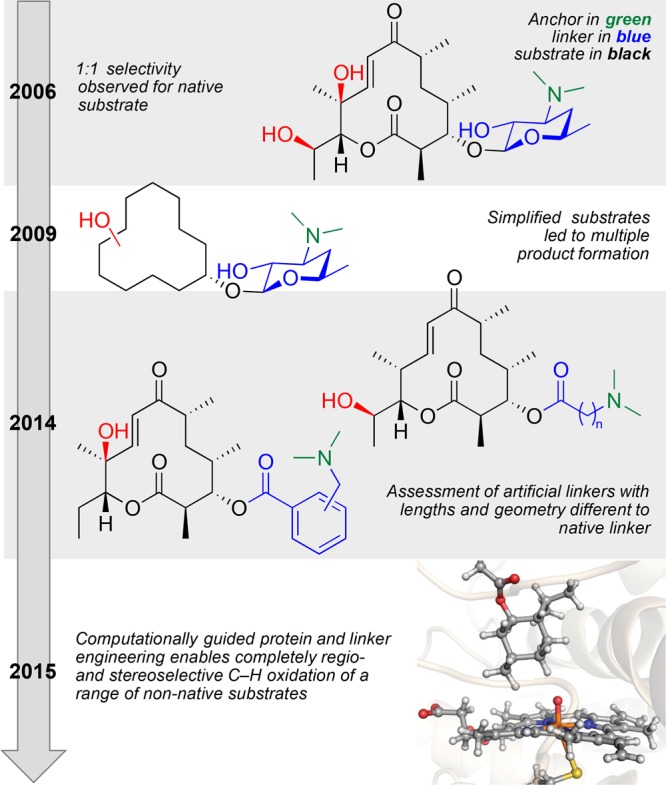
Timeline of the investigations into the development of PikC as a broadly applicable biocatalyst for C–H oxidation.
This indifference to specific substrate structure is explained by the mechanism in which natural substrates bind within the active site. Extensive structural studies revealed salt-bridge interactions between the dimethylamino group of the substrate desosamine sugar and an exposed carboxylate moiety within the active site. Essentially the amine acts as an anchor, holding the substrate in a specific location, and the sugar acts as a linker, controlling the specific orientation of the substrate with respect to the active oxidant. Thus, it is a combination of the active site, anchor, and linker controlling factors that dictate the site-selective reaction of the range of natural substrates. Inspired by the “mix-and-match” nature of this system, the collaborative team recognized the unusual and exciting opportunity to bring both protein engineering (modifying the enzyme active site) and synthetic organic chemistry (synthesis of novel anchor and linker combinations) together to expand the system’s scope to un-natural and synthetically significant substrates. However, in order to understand, and predict, the impact of the permutations possible in varying the active site, anchor, and linker, a unifying theory was required, one that was provided through computational modeling and prediction. Highlights of the CCHF research accomplishments developing biocatalysts as tools for C–H functionalization include the following:
Development of an advanced understanding of the PikC active site and the roles of the anchor and linker in site-selective C–H oxidation.48
Using theoretical modeling of the PikC active site to unify protein and substrate engineering to furnish systems capable of complete regio- and stereoselective C–H oxidation on a range of non-native substrates.49
The design and synthesis of novel linker classes, intended to enable high-throughput screening of product distribution in order to keep pace with the theoretical modeling and protein engineering.49
Development of molecular dynamics simulations (MDS) capable of accurately and reliably modeling the PikC active site and surrounding geometry.49
The in silico simulation of these systems is a tool that will be truly enabling in this line of research. A crystal structure, or a docking simulation, offers a snapshot of how an active site in an enzyme might be organized with respect to the substrate. Enabled by advances in computing power, MD simulations can now offer an insight into the orientation and spatial location, as a function of time, of the key functionalities involved in positioning substrates in the active site of this large and multifaceted system. The information gained from these studies has already begun to make a significant impact on this research as illustrated in Figure 9 by the C–H oxidation products that can be selectively formed,49 and as our research moves forward, this tool will prove invaluable to the investigation of new enzymatic systems.
Figure 9.
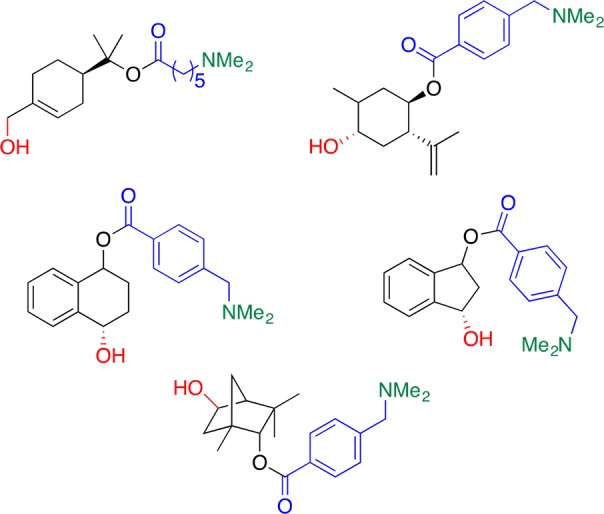
Site-selective C–H oxidation with PikC-RhFRED of various cycloalkanes.
As the Center has matured and its participants became more familiar with the large-scale collaborative engagement, researchers have begun to explore new initiatives in order to enhance our industrial and global engagement and connection with the broader C–H functionalization community. Members of the Center had already been involved in organizing C–H functionalization symposia at ACS and Pacifichem meetings and editing books and thematic issues on the topic, but we wanted to reach a broader audience. In 2015 we started the Virtual Symposium on C–H Functionalization, in which four speakers, two from the Center and two from the broader C–H functionalization field, deliver short, easily digestible talks on recent advances within the C–H functionalization discipline. We have three of these symposia each year, and they draw on average over 1,000 participants, joining from lecture halls across the globe, with audiences coming from up to 45 countries. We have also developed an international exchange and collaborative research network with C–H functionalization focused endeavors in Nagoya, Japan, and KAIST, South Korea, and with Cambridge University, U.K. This relationship has been built through large-scale videoconferences, and over 30 students and postdocs have participated in international research exchanges.
One of the goals of the Center is to facilitate the broad industrial application of C–H functionalization, and the Center has developed numerous collaborative interactions with pharmaceutical companies to achieve this goal. In addition to the face-to-face meetings with our industrial partners, they engage with us in large-scale videoconference meetings providing overviews of recent advances from the CCHF, collaborative research projects, short courses on pharmaceutical research, and career development discussions with our students. The collaborative research programs cover a wide range of projects, including library generation,44 late stage C–H functionalization,24 enhanced heterocycle compatibility studies,23 and bioinspired transformations.50 The cross-communication has been beneficial for both groups, and the logic of C–H functionalization is significantly impacting the synthesis of therapeutic agents.51
In summary, the collaborative and synergistic relationship that has been built in the CCHF has enabled us to have a far greater impact on our science and the chemical community as a whole than would be possible as individual investigators. As a collective, we can demonstrate the value of basic research in modern organic synthesis. We can illustrate the positive impact of collaboration and demonstrate how to develop productive precompetitive research with industry. Effective virtual communication has been critical to this venture, and it has enhanced our domestic and international engagement. We continue to explore new methods of communication and engagement, to ensure that the center remains vibrant and exciting for all our members. Overwhelmingly, it is such an honor as scientists to be able to share ideas with other experts in one’s field, and within a trusting and sharing environment it is possible for everyone to enrich their research experience and program.
Acknowledgments
Financial support was provided by NSF under the CCI Center for Selective C–H Functionalization (CHE-1700982). We also wish to thank The Novartis Institutes for Biomedical Research and AbbVie for support of our work on C–H functionalization. We thank the thematic leaders, Simon B. Blakey, Justin Du Bois, Mo Movassaghi, Djamaladdin G. Musaev, David H. Sherman, and Jin-Quan Yu, for their vision and leadership and all of the members of the CCHF community for their tremendous level of engagement and enthusiasm.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.7b00329.
List of publications generated by the CCHF (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Arndtsen B. A.; Bergman R. G.; Mobley T. A.; Peterson T. H. Selective intermolecular carbon-hydrogen bond activation by synthetic metal complexes in homogenous solution. Acc. Chem. Res. 1995, 28, 154–162. 10.1021/ar00051a009. [DOI] [Google Scholar]
- Davies H. M. L.; Manning J. R. Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle K. M.; Mei T. S.; Wasa M.; Yu J. Q. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc. Chem. Res. 2012, 45, 788–802. 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi J.; Yamaguchi A. D.; Itami K. C–H Bond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem., Int. Ed. 2012, 51, 8960–9009. 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]
- Gutekunst W. R.; Baran P. S. C–H Functionalization logic in total synthesis. Chem. Soc. Rev. 2011, 40, 1976–1991. 10.1039/c0cs00182a. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Morton D. Recent advances in C–H functionalization. J. Org. Chem. 2016, 81, 343–350. 10.1021/acs.joc.5b02818. [DOI] [PubMed] [Google Scholar]
- Skubi K. L.; Blum T. R.; Yoon T. P. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 2016, 116, 10035–10074. 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Spring D. R. Arene C-H functionalisation using a removable/modifiable or a traceless directing group strategy. Chem. Soc. Rev. 2014, 43, 6906–6919. 10.1039/C4CS00137K. [DOI] [PubMed] [Google Scholar]
- Doyle M. P.; Duffy R.; Ratnikov M.; Zhou L. Catalytic carbene insertion into C–H bonds. Chem. Rev. 2010, 110, 704–724. 10.1021/cr900239n. [DOI] [PubMed] [Google Scholar]
- Mkhalid I. A. I.; Barnard J. H.; Marder T. B.; Murphy J. M.; Hartwig J. F. C-H Activation for the construction of C-B bonds. Chem. Rev. 2010, 110, 890–931. 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.; Larsen M. A. Undirected, homogeneous C–H bond functionalization: Challenges and opportunities. ACS Cent. Sci. 2016, 2, 281–292. 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinman A.; Du Bois J. A Stereoselective synthesis of (−)-tetrodotoxin. J. Am. Chem. Soc. 2003, 125, 11510–11511. 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]
- Davies H. M. L.; Dai X.; Long M. S. Combined C–H activation/cope rearrangement as a strategic reaction in organic synthesis: Total synthesis of (−)-colombiasin A and (−)-elisapterosin B. J. Am. Chem. Soc. 2006, 128, 2485–2490. 10.1021/ja056877l. [DOI] [PubMed] [Google Scholar]
- Pelphrey P.; Hansen J. H.; Davies H. M. L. Solvent-free catalytic enantioselective C–C bond forming reactions with very high catalyst turnover numbers. Chem. Sci. 2010, 1, 254–257. 10.1039/c0sc00109k. [DOI] [Google Scholar]
- Perry R. H.; Cahill T. J. III; Roizen J. L.; Du Bois J.; Zare R. N. Capturing fleeting intermediates in a catalytic C–H amination reaction cycle. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 18295–18299. 10.1073/pnas.1207600109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornecki K. P.; Briones J. F.; Boyarskikh V.; Fullilove F.; Autschbach J.; Schrote K. E.; Lancaster K. M.; Davies H. M. L.; Berry J. F. Direct spectroscopic characterization of a transitory dirhodium donor-acceptor carbene complex. Science 2013, 342, 351–354. 10.1126/science.1243200. [DOI] [PubMed] [Google Scholar]
- Varela-Álvarez A.; Yang T.; Jennings H.; Kornecki K. P.; Macmillan S. N.; Lancaster K. M.; Mack J. B. C.; Du Bois J.; Berry J. F.; Musaev D. G. Rh2(II,III) Catalysts with chelating carboxylate and carboxamidate supports: Electronic structure and nitrene transfer reactivity. J. Am. Chem. Soc. 2016, 138 (7), 2327–2341. 10.1021/jacs.5b12790. [DOI] [PubMed] [Google Scholar]
- Roizen J. L.; Zalatan D. N.; Du Bois J. Selective intermolecular amination of C-H bonds at tertiary carbon centers. Angew. Chem., Int. Ed. 2013, 52, 11343–11346. 10.1002/anie.201304238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess E. N.; DeLuca R. J.; Tindall D. J.; Oderinde M. S.; Roizen J. L.; Du Bois J.; Sigman M. S. Analyzing site selectivity in Rh2(esp)2-catalyzed intermolecular C–H amination reactions. J. Am. Chem. Soc. 2014, 136, 5783–5789. 10.1021/ja5015508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess E. N.; Guptill D. M.; Davies H. M. L.; Sigman M. S. Using IR vibrations to quantitatively describe and predict site-selectivity in multivariate Rh-catalyzed C–H functionalization. Chem. Sci. 2015, 6, 3057–3062. 10.1039/C5SC00357A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weldy N. M.; Schafer A. G.; Owens C. P.; Herting C. J.; Varela-Alvarez A.; Chen S.; Niemeyer Z.; Musaev D. G.; Sigman M. S.; Davies H. M. L.; Blakey S. B. Iridium(III)-bis(imidazolinyl)phenyl catalysts for enantioselective C–H functionalization with ethyl diazoacetate. Chem. Sci. 2016, 7, 3142–3146. 10.1039/C6SC00190D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin C.; Boyarskikh V.; Hansen J. H.; Hardcastle K. I.; Musaev D. G.; Davies H. M. L. D2-Symmetric dirhodium catalyst derived from a 1,2,2-triarylcyclopropanecarboxylate ligand: Design, synthesis and application. J. Am. Chem. Soc. 2011, 133, 19198–19204. 10.1021/ja2074104. [DOI] [PubMed] [Google Scholar]
- Malik H. A.; Taylor B. L. H.; Kerrigan J. R.; Grob J. E.; Houk K. N.; Du Bois J.; Hamann L. G.; Patterson A. W. Non-directed allylic C–H acetoxylation in the presence of lewis Basic heterocycles. Chem. Sci. 2014, 5, 2352. 10.1039/c3sc53414f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J.; Hamann L. G.; Davies H. M. L.; Beckwith R. E. J. Late-stage C–H functionalization of complex alkaloids and drug molecules via intermolecular rhodium-carbenoid insertion. Nat. Commun. 2015, 6, 5943. 10.1038/ncomms6943. [DOI] [PubMed] [Google Scholar]
- Wang H.; Li G.; Engle K. M.; Yu J.-Q.; Davies H. M. L. Sequential C–H functionalization reactions for the enantioselective synthesis of highly functionalized 2,3-dihydrobenzofurans. J. Am. Chem. Soc. 2013, 135, 6774–6777. 10.1021/ja401731d. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A. D.; Chepiga K. M.; Yamaguchi J.; Itami K.; Davies H. M. L. Concise syntheses of dictyodendrins A and F by a sequential C–H functionalization strategy. J. Am. Chem. Soc. 2015, 137, 644–647. 10.1021/ja512059d. [DOI] [PubMed] [Google Scholar]
- Bedell T. A.; Hone G. A. B.; Valette D.; Yu J.-Q.; Davies H. M. L.; Sorensen E. J. Rapid construction of a benzo-fused indoxamycin core enabled by site-selective C–H functionalizations. Angew. Chem., Int. Ed. 2016, 55, 8270–8274. 10.1002/anie.201602024. [DOI] [PubMed] [Google Scholar]
- Liao K.; Negretti S.; Musaev D. G.; Bacsa J.; Davies H. M. L. Site selective and stereoselective functionalization of unactivated C–H bonds. Nature 2016, 533 (7602), 230–234. 10.1038/nature17651. [DOI] [PubMed] [Google Scholar]
- Murai S.; Kakiuchi F.; Sekine S.; Tanaka Y.; Kamatani A.; Sonoda M.; Chatani N. Efficient catalytic addition of aromatic carbon-hydrogen bonds to olefins. Nature 1993, 366, 529–531. 10.1038/366529a0. [DOI] [Google Scholar]
- Musaev D. G.; Kaledin A.; Shi B.-F.; Yu J.-Q. Key Mechanistic features of enantioselective C-H bond activation reactions catalyzed by [N-protected amino acid-Pd(II)] complexes. J. Am. Chem. Soc. 2012, 134 (3), 1690–1698. 10.1021/ja208661v. [DOI] [PubMed] [Google Scholar]
- Baxter R. D.; Sale D.; Engle K. M.; Yu J.-Q.; Blackmond G. G. Mechanistic rationalization of unusual kinetics in Pd-catalyzed C–H olefination. J. Am. Chem. Soc. 2012, 134 (10), 4600–4606. 10.1021/ja207634t. [DOI] [PubMed] [Google Scholar]
- Figg T. M.; Wasa M.; Yu J.-Q.; Musaev D. G. Understanding the reactivity of Pd(0)/PR3-catalyzed intermolecular C(sp3)-H bond arylation. J. Am. Chem. Soc. 2013, 135 (38), 14206–14214. 10.1021/ja4053416. [DOI] [PubMed] [Google Scholar]
- Giri R.; Lan Y.; Liu P.; Houk K. N.; Yu J.-Q. Understanding reactivity and stereoselectivity in palladium-catalyzed diastereoselective sp3 C–H bond activation. J. Am. Chem. Soc. 2012, 134 (34), 14118–14126. 10.1021/ja304643e. [DOI] [PubMed] [Google Scholar]
- He J.; Wasa M.; Chan K. S. L.; Yu J.-Q. Palladium(0)-catalyzed alkynylation of C(sp3)–H bonds. J. Am. Chem. Soc. 2013, 135 (9), 3387–3390. 10.1021/ja400648w. [DOI] [PubMed] [Google Scholar]
- Engle K. M.; Yu J.-Q. Developing ligands for palladium(II)-catalyzed C–H functionalization: Intimate dialogue between ligand and substrate. J. Org. Chem. 2013, 78 (18), 8927–8955. 10.1021/jo400159y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang M.; Wang H.-L.; Sun S.-Z.; Dai H.-X.; Yu J.-Q. Cu(II)-Mediated ortho C–H alkynylation of (hetero)arenes with terminal alkynes. J. Am. Chem. Soc. 2014, 136 (33), 11590–11593. 10.1021/ja507704b. [DOI] [PubMed] [Google Scholar]
- Shang M.; Sun S.-Z.; Wang H.-L.; Laforteza B. N.; Dai H.-X.; Yu J.-Q. Exceedingly fast copper(II)-promoted ortho C–H trifluoromethylation of arenes using TMSCF3. Angew. Chem., Int. Ed. 2014, 53 (39), 10439–10442. 10.1002/anie.201404822. [DOI] [PubMed] [Google Scholar]
- Shang M.; Sun S.-Z.; Dai H.-X.; Yu J.-Q. Cu(OAc)2-catalyzed coupling of aromatic C–H bonds with arylboron reagents. Org. Lett. 2014, 16 (21), 5666–5669. 10.1021/ol5027377. [DOI] [PubMed] [Google Scholar]
- Wang H.-L.; Shang M.; Sun S.-Z.; Zhou Z.-L.; Laforteza B. N.; Dai H.-X.; Yu J.-Q. Cu(II)-catalyzed coupling of Aromatic C–H bonds with malonates. Org. Lett. 2015, 17, 1228. 10.1021/acs.orglett.5b00193. [DOI] [PubMed] [Google Scholar]
- Shang M.; Shao Q.; Sun S.-Z.; Chen Y.; Xu H.; Dai H.-X.; Yu J.-Q. Identification of monodentate oxazoline as a ligand for copper-promoted ortho-C–H hydroxylation and amination. Chem. Sci. 2017, 8, 1469–1473. 10.1039/C6SC03383K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H.-X.; Stepan A. F.; Plummer M. S.; Zhang Y.-H.; Yu J.-Q. Divergent C–H functionalizations directed by sulfonamide pharmacophores: Late-stage diversification as a tool for drug discovery. J. Am. Chem. Soc. 2011, 133 (18), 7222–7228. 10.1021/ja201708f. [DOI] [PubMed] [Google Scholar]
- Rosen B. R.; Simke L. R.; Thuy-Boun P. S.; Dixon D. D.; Yu J.-Q.; Baran P. S. C-H Functionalization logic enables a synthesis of (+)-hongoquercin A and related compounds. Angew. Chem., Int. Ed. 2013, 52 (28), 7317–7320. 10.1002/anie.201303838. [DOI] [PubMed] [Google Scholar]
- Cheng G.; Li T.-J.; Yu J.-Q. Practical Pd(II)-catalyzed C–H alkylation with epoxides: One-step syntheses of 3,4-dihydroisocoumarins. J. Am. Chem. Soc. 2015, 137 (34), 10950–10953. 10.1021/jacs.5b07507. [DOI] [PubMed] [Google Scholar]
- Shang M.; Wang M.-M.; Saint-Denis T. G.; Li M.-H.; Dai H.-X.; Yu J.-Q. Copper-mediated late-stage functionalization of heterocycle-containing molecules. Angew. Chem., Int. Ed. 2017, 56 (19), 5317–5321. 10.1002/anie.201611287. [DOI] [PubMed] [Google Scholar]
- Yang G.; Lindovska P.; Zhu D.; Kim J.; Wang P.; Tang R.-Y.; Movassaghi M.; Yu J.-Q. Pd(II)-Catalyzed meta-C–H olefination, arylation, and acetoxylation of indolines using a U-shaped template. J. Am. Chem. Soc. 2014, 136 (30), 10807–10813. 10.1021/ja505737x. [DOI] [PubMed] [Google Scholar]
- Cheng G.-J.; Yang Y.-F.; Liu P.; Chen P.; Sun T.-Y.; Li G.; Zhang X.; Houk K. N.; Yu J.-Q.; Wu Y.-D. Role of N-acyl amino acid ligands in Pd(II)-catalyzed remote C–H activation of tethered arenes. J. Am. Chem. Soc. 2014, 136, 894–897. 10.1021/ja411683n. [DOI] [PubMed] [Google Scholar]
- Chen G.; Gong W.; Zhuang Z.; Andrä M. S.; Chen Y.-Q.; Hong X.; Yang Y.-F.; Liu T.; Houk K. N.; Yu J.-Q. Ligand-accelerated enantioselective methylene C(sp3)–H bond activation. Science 2016, 353, 1023. 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negretti S.; Narayan A. R. H.; Chiou K. C.; Kells P. M.; Stachowski J. L.; Hansen D. A.; Podust L. M.; Montgomery J.; Sherman D. H. Directing group-controlled regioselectivity in an enzymatic C–H bond oxygenation. J. Am. Chem. Soc. 2014, 136 (13), 4901–4904. 10.1021/ja5016052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan A. R. H.; Jiménez-Osés G.; Liu P.; Negretti S.; Zhao W.; Gilbert M. M.; Ramabhadran R. O.; Yang Y.-F.; Furan L. R.; Li Z.; Podust L. M.; Montgomery J.; Houk K. N.; Sherman D. H. Enzymatic hydroxylation of an unactivated methylene C–H bond guided by molecular dynamics simulations. Nat. Chem. 2015, 7, 653–660. 10.1038/nchem.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andorfer M. C.; Grob J. E.; Hajdin C. E.; Chael J. R.; Siuti P.; Lilly J.; Tan K. L.; Lewis J. C. Understanding flavin-dependant halogenase reactivity via substrate profiling. ACS Catal. 2017, 7 (3), 1897–1904. 10.1021/acscatal.6b02707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernak T.; Dykstra K. D.; Tyagarajan S.; Vachal P.; Krska S. The medicinal chemist’s toolbox for late-stage functionalization of drug-like molecules. Chem. Soc. Rev. 2016, 45, 546–576. 10.1039/C5CS00628G. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.