

Summary
Background
Bronchopulmonary dysplasia (BPD) results from alveolar simplification and abnormal development of alveolar and capillary structure. Survivors of BPD display persistent deficits in airflow and membrane and vascular components of alveolar gas diffusion. Despite being the defining feature of BPD, various neonatal hyperoxia models of BPD have not routinely assessed pulmonary gas diffusion.
Methods
To simulate the most commonly-utilized neonatal hyperoxia models, we exposed neonatal mice to room air or ≥90% hyperoxia during key stages of distal lung development: through the first 4 (saccular), 7 (early alveolar), or 14 (bulk alveolar) postnatal days, followed by a period of recovery in room air until 8 weeks of age when alveolar septation is essentially complete. We systematically assessed and correlated the effects of neonatal hyperoxia on the degree of alveolar–capillary structural and functional impairment. We hypothesized that the degree of alveolar–capillary simplification would correlate strongly with worsening diffusion impairment.
Results
Neonatal hyperoxia exposure, of any duration, resulted in alveolar simplification and impaired pulmonary gas diffusion. Mean Linear Intercept increased in proportion to the length of hyperoxia exposure while alveolar and total lung volume increased markedly only with prolonged exposure. Surprisingly, despite having a similar effect on alveolar surface area, only prolonged hyperoxia for 14 days resulted in reduced pulmonary microvascular volume. Estimates of alveolar and capillary structure, in general, correlated poorly with assessment of gas diffusion.
Conclusion
Our results help define the physiological and structural consequences of commonly-employed neonatal hyperoxia models of BPD and informtheir clinical utility.
Keywords: bronchopulmonary dysplasia, pulmonary diffusion capacity, lung function, neonatal, hyperoxia
INTRODUCTION
Bronchopulmonary dysplasia (BPD) is the most prevalent long-term consequence of extremely preterm birth.1 BPD is strongly associated with adverse neurodevelopmental and pulmonary outcomes with respiratory morbidities lasting well beyond childhood.2–4 The mechanical ventilation and supplemental oxygen required to support extremely preterm infants has been shown in human and animals studies to disrupt normal alveolar development leading to enlarged airspaces, reduced pulmonary microvasculature, and a diminished surface area for gas exchange.5–9 Despite improvement in clinical respiratory symptoms with time, BPD survivors have a reduced diffusing capacity of the lung for carbon monoxide (DLCO) with normal alveolar volume (VA) suggesting larger, more simplified alveoli.5,10
While animal models of BPD have assessed the structural consequences of neonatal lung injury, the functional consequences have not been well described. Various durations and degrees of neonatal hyperoxia have been used to effectively inhibit both alveolar and capillary development, resulting in reduced alveolar surface area and capillary density, but they have not assessed systematically the impact on alveolar–capillary function.11–14 Because BPD is defined clinically as dependence on supplemental oxygen,15,16 it would be beneficial to assess not only the structural, but also the functional consequences resulting from commonly-employed neonatal hyperoxia models to inform their physiological relevance.
The pulmonary diffusing capacity, a functional measure of the alveolar capillary unit, is determined primarily by the alveolar surface area and the alveolar capillary volume.17–19 When a gas molecule enters the alveolar space, it first diffuses across the alveolar membrane (membrane component), then enters the capillary space and reacts with red blood cell hemoglobin (vascular component). We and others have utilized a simple method to assess pulmonary gas diffusion per alveolar volume in rodents, termed the diffusion factor for carbon monoxide (DFCO), which is the relation between the uptake of carbon monoxide (CO) and the dilution of neon (Ne) during a single breath hold.20,21 Using a model of neonatal hyperoxia exposure for the first 7 postnatal days, we recently demonstrated that, despite recovery in room air, there are persistent structural changes induced by hyperoxia, consisting of larger, more simplified airspaces with normal alveolar volume, that are accompanied by functional impairment as evidenced by significant reductions in adult DFCO.21
The purpose of this study was to relate the structural changes resulting from differing durations of hyperoxia exposure commonly utilized in the literature, which correspond to lung injury during critical stages of pulmonary development, to functional changes as measured by DFCO. We hypothesized that increasing the duration of initial neonatal hyperoxia exposure would result in more severe alveolar–capillary simplification, as evidenced structurally by reduced alveolar surface area and capillary volume, and physiologically by impaired pulmonary diffusion. Given that pulmonary diffusion is determined predominately by the surface area of the alveolar membrane and the pulmonary capillary volume,19,22 we focused our assessment of lung structure on alveolar surface area and estimates of pulmonary microvascular volume. The mean linear intercept (MLI), in the context of lung volume, estimates alveolar surface area, and assuming that changes in alveolar wall thickness are minimal, the volume fraction of the alveolar wall correlates roughly with alveolar complexity. We previously demonstrated that these estimates of alveolar–capillary structural development and DFco are correlated.21 Therefore, we predicted that the degree of alveolar–capillary simplification, as measured by the aforementioned stereological estimates of alveolar surface area and alveolar microvascular volume, would correlate strongly with the degree of diffusion impairment.
MATERIALS AND METHODS
Murine Neonatal Hyperoxia Exposure Model of BPD
All procedures were approved by the Institutional Animal Care and Use Committee of the Indiana University School of Medicine (protocol 10897) and were previously described.21 For each individual experimental exposure, two or more litters of wild-type C57BL/6J mice <12 hr of age and born <12 hr apart were pooled and separated into two equal groups of up to eight mice. Half of the pups were placed in a 30″ × 20″ × 20″ propylene chamber in which the oxygen concentration was maintained at ≥90% O2 (BioSpherix, Lacona, NY), and the other half were maintained in room air (RA; 21% O2). Nursing dams were rotated between groups every 48 hr to prevent oxygen toxicity to the dams. Humidity and carbon dioxide levels were maintained within the ambient range of the facility. To determine the effect of hyperoxic injury during different stages of lung development, pups were continuously exposed to hyperoxia for the first 4, 7, or 14 postnatal (P) days, which corresponds to the murine saccular phase (P0-P4), early alveolarization (P7), and bulk alveolarization (P14). To simulate the period of clinical recovery typically observed in preterm infants through childhood and adolescence, mice were allowed to recover in RA until 8 weeks of age when alveolar septation is essentially complete,23 at which time all structural and functional analyses were performed.
Assessment of Diffusing Factor for Carbon Monoxide (DFCO)
DFCO was assessed with a 3000 Micro GC bench top gas chromatographer (INFICON, East Syracuse, NY) using 0.8 ml of test gas (0.5% Ne; 0.5% CO; 21% O2; balance nitrogen) to inflate the lungs over a 6-second breath hold as previously described.21,24 The 6-second breath hold was used to minimize the potential for bradycardia which would potentially impact cardiac output. Although rare in our experience, any subject that experienced significant bradycardia (>10% of baseline) was excluded from analysis. Mice were anesthetized with 50 mg/kg pentobarbital i.p., an 18g, 0.05-inch catheter was placed in the mid trachea via tracheostomy and secured with suture, and mice were ventilated with a flexiVent (SCIREQ, Montreal, Canada) using 21% O2, tidal volume of 10 ml/kg at a rate of 150 breaths/min, and positive end expiratory pressure of 3 cm H2O. Alveolar volume was estimated by the degree of neon dilution in the 0.8 ml test gas solution, following the 6-second breath hold, as per the American Thoracic Society/European Respiratory Society Task Force 19 using the equation VA=VI (FI,Tr/FA,Tr) where VI is the volume of the inhaled test gas and FI,Tr/FA,Tr is the ratio of neon concentration before and after dilution of the test gas in the alveolar space. Although the volume of the conducting airways (the anatomical dead space) would certainly contribute to neon dilution, for simplicity we disregarded it in our calculations as we assumed anatomical dead space amongst all treatment groups was similar. Each measurement was separated by a 150 sec period of mechanical ventilation to ensure disappearance of carbon monoxide and neon from the previous measurement. After measuring the DFCO, a whole blood sample was obtained to determine hemoglobin and hematocrit concentration using a Hemavet (Drew Scientific, Waterbury, CT).
Lung Histology and Morphometry
Immediately following functional analyses, the lungs were inflation-fixed, processed, and embedded in paraffin exactly as previously described.6 Morphometric analyses were performed on 7-μm thick transverse sections from the left lung stained with hematoxylin and eosin. Shrinkage effects due to paraffin embedding were assumed to be similar amongst the experimental groups, and the use of a reference volume and point-counting were employed to minimize its confounding effects. From 4–6 randomly-selected sections representative of the entire length of the lung, a minimum of 6–8 random total fields were photographed with a 5× objective and 12 total random fields that contained distal airspaces were photographed with a 20× objective on a Zeiss Axioshop2Plus microscope (Carl Zeiss Inc., Thornwood, NY). Images used for morphometric analysis and microvascular volume determination, taken at 20× objective, consisted of fields containing alveolar structures, avoiding large vessels, or conducting airways. The STEPanizer stereology tool, version 1.0 was used to determine alveolar surface area and calculate MLI as previously described.25,26 Pulmonary microvasculature was identified by immunohistochemical staining for the endothelial marker, von Willebrand factor (vWF; rabbit polyclonal anti-human vWF; dilution 1:2500;[A0082]; Dako, Carpinteria, CA), using the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA) with diaminobenzidine (Vector Laboratories) as previously described,6 and alveolar microvascular volume was estimated via the STEPanizer tool.
Statistical Analysis
Statistical analyses were performed using Prism software version 6.04 (GraphPad Inc., San Diego, CA). Comparisons between experimental groups (RA vs. hyperoxia) were made with one way ANOVA using Tukey’s multiple comparison post-test analysis. All data are presented as means±SEM (with n≥9 mice per group) and are representative of at least 2–3 independent hyperoxia exposures per time-point. A P-value <0.05 was considered statistically significant.
RESULTS
Neonatal Hyperoxia Impairs Alveolar–Capillary Diffusion
Analysis of adult alveolar–capillary function following neonatal hyperoxia demonstrated persistently impaired diffusion. Remarkably, impaired diffusion was evident with even the shortest duration of oxygen exposure (Fig. 1). Compared to RA controls, DFCO was similarly reduced following neonatal hyperoxia exposure from P0-4 to P0-7 (Fig. 1A). Animals exposed to neonatal hyperoxia from P0-14 demonstrated a reduction in DFCO that was significantly more severe compared to shorter durations of oxygen exposure. As calculated by neon dilution, neonatal hyperoxia from P0-4 to P0-7 resulted in similar small (~15%) but statistically significant increases in alveolar volume (VA) (Fig. 1B). However, VA was significantly increased by ~50% in animals exposed to prolonged (P0-14) hyperoxia (Fig. 1B). Hemoglobin values were not significantly different than RA controls following any duration of hyperoxia exposure (14.0 ± 0.3 g/dl (RA) vs. 13.2±0.4 g/dl (O2 P0-4) vs. 14.5±0.4 g/dl (O2 P0-7) vs. 13.6 ± 0.3 g/dl (O2 P0-14); P > 0.05 vs. RA), verifying that differences in DFCO did not result from decreases in hemoglobin concentration.
Fig. 1.

Impact of neonatal hyperoxia on pulmonary diffusion and alveolar volume. (A) Assessment of the diffusion factor for carbon monoxide (DFCO) at 2 months of age in animals continuously raised in room air (RA), or initially exposed to neonatal hyperoxia (≥90% O2) for the first 4, 7, or 14 days after birth followed by recovery in RA. (B) Assessment of alveolar volume by neon gas dilution. Values are expressed as means±SEM. ***P < 0.001 versus RA, **P < 0.01 versus RA, #P < 0.05 versus O2 P0-4, ###P < 0.001 versus O2 P0-4, $$$P < 0.001 versus O2 P0-7 by one way ANOVA using Tukey’s multiple comparison post-test analysis. Results are representative of three independent experiments. n = 10 (RA), 10 (O2 P0-P4), 18 (O2 P0-P7), 9 (O2 P0-P14).
Neonatal Hyperoxia Durably Reduces Alveolar Surface Area
Given the significant reductions we observed in alveolar–capillary diffusion capacity, we more closely examined alveolar surface area following neonatal hyperoxia. Representative adult lung histology is shown in Figure 2. Compared to mice raised in RA, mice previously exposed to hyperoxia from P0-4, P0-7, to P0-14 demonstrated increasing alveolar simplification (Fig. 2A–D). Compared to RA controls, mean linear intercept (MLI) of mice exposed to hyperoxia showed an almost stepwise increase proportionate to the duration of neonatal hyperoxia (Fig. 2E). Unlike estimates of VA, exposure to hyperoxia from either P0-4 or P0-7 had no significant effect on lung volume as measured by water displacement (Fig. 2F). However, exposure to hyperoxia from P0-14 resulted in a significant 29% increase in lung volume compared to RA control mice (Fig. 2F). Estimates of VA by neon dilution correlated reasonably well with estimates of lung volume by water displacement (r2 = 0.599, P < 0.0001). Consequently, compared to RA controls, the total alveolar surface area was similarly and significantly reduced in all hyperoxia-exposed animals (Fig. 2G). Likewise, following any exposure to hyperoxia, the volume of the alveolar wall (Vaw) was similarly and significantly decreased by at least 20% compared to RA controls (0.062±0.002 ml [RA] vs. 0.046±0.002 ml [O2 P0-4] vs. 0.050±0.003 ml [O2 P0-7] vs. 0.046±0.003 ml [O2 P0-14]; P < 0.0001 vs. RA) (not shown). Therefore, despite having significantly larger distal airspaces compared to animals exposed to shorter durations of hyperoxia, animals exposed to neonatal hyperoxia from P0-14 had similar alveolar wall volumes and a relative preservation of alveolar surface area owing to the significant increase in lung volume.
Fig. 2.

Development of Alveolar surface area following neonatal hyperoxia. (A–D) Representative hematoxylin/eosin stained lung sections at 2 months of age in animals continuously raised in (A) RA, or initially exposed to neonatal hyperoxia (≥90% O2) from (B) P0-4, (C) P0-7, or (D) P0-14, followed by recovery in RA. Assessment of (E) mean linear intercept (MLI), (F) left lung volume by water displacement, and (G) alveolar surface area. Original images obtained with 20× objective. Scale bar = 50μm. Values are expressed as means±SEM. ***P < 0.001 versus RA, ###P < 0.001 versus O2 P0-4, $$$P < 0.001 versus O2 P0-7) by one way ANOVA using Tukey’s multiple comparison post-test analysis. Results are representative of three independent experiments. n = 19 (RA), 10 (O2 P0-P4), 12 (O2 P0-P7), 9 (O2 P0-P14).
Assessment of pulmonary microvascular volume is shown in Figure 3. Although there was a downward trend in pulmonary microvascular volume after neonatal hyperoxia exposure from P0-4 to P0-7, it was not statistically significant compared to RA. Only hyperoxia exposure from P0-14 resulted in a significantly decreased pulmonary microvascular volume when compared to RA controls.
Fig. 3.
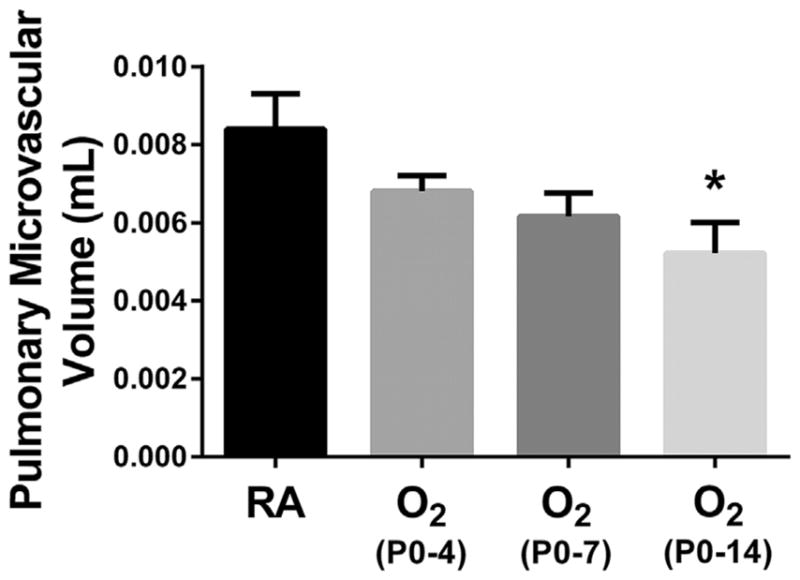
Effect of Neonatal hyperoxia on pulmonary microvascular development. Pulmonary microvascular volume was assessed using stereological analysis on von Willebrand-stained lung sections to identify vessels 20–50μm in diameter. Values are expressed as means±SEM. *P < 0.05 versus RA by one way ANOVA using Tukey’s multiple comparison post-test analysis. Results are representative of three independent experiments. n = 11 (RA), 10 (O2 P0-P4), 12 (O2 P0-P7), 9 (O2 P0-P14).
Assessment of alveolar development has largely relied on morphometric and not physiological analysis. We therefore, correlated our structural and functional endpoints to determine if structure alone can estimate functional diffusion (Fig. 4). None of our stereological assessments of alveolar structure were strongly correlated with alveolar function. There was a weak but statistically significant correlation between MLI and DFCO (r2 = 0.282, P < 0.01) (Fig. 4A), as well as between DFCO and the volume fraction of the alveolar septal wall (r2 = 0.224, P < 0.01) (Fig. 4B). Total alveolar surface area correlated poorly with DFCO (r2 = 0.084, P > 0.05) (Fig. 4C), and, likewise, there was no significant correlation between DFCO and pulmonary microvascular volume (r2 = 0.001, P > 0.05) (Fig. 4D). Given that DFCO estimates gas diffusion per lung volume, we also assessed the correlation between DFCO and stereological estimates of lung structure corrected for lung volume. There was a weak but statistically significant correlation between DFCO and alveolar surface area per lung volume (r2 = 0.358, P < 0.001) (Fig. 4E). However, there was no significant correlation between DFCO and pulmonary microvascular volume per lung volume (r2 = 0.036, P > 0.05) (Fig. 4F).
Fig. 4.

Correlation between estimates of distal lung alveolar–capillary simplification and functional impairment. Correlation of mean linear intercept (MLI) (A), volume fraction of the alveolar septal wall (B), alveolar surface area (C), pulmonary microvascular volume (D), alveolar surface area per lung volume (E), and pulmonary microvascular volume per lung volume (F) with DFCO. Analysis performed using simple linear regression.
DISCUSSION
The objective of this study was to correlate commonly-employed measures of distal lung structural development with alveolar–capillary function following increasing durations of neonatal hyperoxia during critical windows of alveolar development. Importantly, we have demonstrated that even brief hyperoxic lung injury, limited to postnatal saccular lung development, is sufficient to significantly impair development of alveolar surface area and reduce functional gas diffusion. Surprisingly, structural estimates of alveolar development and assessments of functional diffusion correlated poorly. Our approach lends clinically significant functional insight into commonly-employed murine neonatal hyperoxia models of BPD, as well as potentially informs pathophysiology of any lung disease that impacts alveolar–capillary function.
The defining feature of BPD following extremely preterm birth is prolonged dependence on supplemental oxygen.15,16 Despite clinical improvement, children, adolescents, and adults recovering from BPD continue to have functional lung impairment with reduced pulmonary diffusing capacity.5,10,27–31 Following neonatal hyperoxia exposure, we demonstrated that adult mice have a reduced DFCO, signifying a similar impairment in pulmonary gas diffusion. Regardless of the duration of hyperoxia, alveolar gas transport was significantly inhibited by neonatal hyperoxia exposure. Although chronic exposure (P0-14) resulted in the largest decline in DFCO, impaired alveolar–capillary diffusion was nearly as severe with brief (P0-4) exposure. The similar reductions in diffusion are likely the result of similar loss of alveolar surface area, as evidenced by similar decreases in alveolar surface area and septal wall volume regardless of the duration of hyperoxia. Mild alveolar fibrosis observed following chronic hyperoxia (not shown) may explain why the efficiency of alveolar–capillary diffusion in these animals, noted especially after 14 days of hyperoxia, was further reduced. Furthermore, we made an assumption that anatomical dead space amongst all exposure groups was similar. However, although anatomical volume was not measured, reports have noted abnormal development and function of the conducting airways following neonatal hyperoxia.32,33 Therefore, it is possible that increased volume of the anatomical dead space could impact our estimates of DFco and alveolar volume and account for some of the differences we observed. However, given the relatively large changes in alveolar volume (which directly correlated with changes in total lung volume), a significant effect due to increased anatomical dead space volume is unlikely. Finally, although we were not able to assess for such here, recent evidence suggests that infants with the most severe forms of BPD can develop pre-capillary arteriovenous anastomoses, which would further impede pulmonary diffusion efficiency.34
Although DFCO was reduced with all durations of neonatal hyperoxia, we were surprised that only prolonged hyperoxia (which may not be developmentally relevant to the clinical scenario) resulted in reduced pulmonary microvascular volume. Likewise, despite capillary volume, by definition, being a major component of total pulmonary diffusion,17 pulmonary microvascular volume and DFCO correlated poorly. It has been recently reported that extremely preterm infants formerly diagnosed with BPD demonstrated proportional reductions in both membrane and vascular components of pulmonary diffusion, consistent with autopsy studies demonstrating larger, fewer alveoli with reduced pulmonary capillary volume.8,9,28 Likewise, in an animal model of BPD, we formerly demonstrated that pulmonary micro-vessels per high-powered field were reduced, which would correlate with a reduction in microvascular density per alveolar volume.21 Our current analysis goes one step forward and provides a stereological approach to quantitate microvascular volume more carefully, which accounts for differences in lung volume that cannot be ascertained by two-dimensional analysis as is commonly employed. One limitation of our assessment of pulmonary capillary volume is the use of von Willebrand staining, which identifies both small venuoles and arterioles, and may not be an appropriate surrogate of the pulmonary capillary bed.35 Although assessment of capillary structure, via direct visualization of capillary structure and/or micro-CT, may have provided a more accurate estimate of alveolar–capillary membrane surface area, our objective was to assess commonly-employed methods of assessing pulmonary microvasculature, of which von Willebrand staining has been reported frequently. Moreover, it is the capillary volume, in combination with the membrane surface area, which determines total pulmonary diffusion. Ideally, physiological resolution of both components, as has been shown in infants recovering from BPD,28 would provide the most accurate estimates of their individual contributions. Additionally, given that we previously showed that exposure to neonatal hyperoxia leads to elevations in adult cardiac output as a potential compensatory response to reduced alveolar surface area,36,37 it is possible that smaller reductions in alveolar microvascular volume with shorter durations of neonatal hyperoxia were partially compensated and therefore, were not detected by DFCO. Finally, the loss of microvascular volume may help explain why animals exposed to prolonged neonatal hyperoxia, despite having similar reductions in alveolar surface area compared to shorter durations of neonatal hyperoxia, demonstrated significantly more impairment in gas diffusion.
Many prior studies have examined the effect of various durations of neonatal hyperoxia (most commonly continuing through the first 4–14 postnatal days) on alveolar lung development.11,12,14,33,38–40 However, despite the diagnosis of BPD being based on the physiological need for oxygen, previous models have not adequately assessed the relationship between alveolar–capillary structure and diffusion function. Moreover, it has been common practice to infer loss of alveolar surface area based on an increase in mean linear intercept (MLI), a two-dimensional assessment of lung structure, despite calls to carefully quantitate distal lung structure by stereology.25 Similar to previous reports,14,41 our results demonstrate a stepwise increase in MLI proportionate to cumulative neonatal hyperoxia exposure. However, our stereological estimates of alveolar surface area, which unlike MLI account for differences in lung volume,25,26 clearly reveal a similar decrease in alveolar surface area whether the insult is confined to saccular (P0-4), saccular/early alveolar (P0-7), or the entirety of postnatal saccular and bulk alveolar (P0-14) lung development. Likewise, estimation of the volume of the alveolar wall (V(aw)) following all three hyperoxia exposures was similarly reduced. Despite the significant increase in MLI with chronic neonatal hyperoxia exposure through day 14, an increase in lung volume resulted in the relative preservation of alveolar surface area and septal wall volume compared to those animals exposed to shorter durations of neonatal hyperoxia. Our findings suggest that the effects of neonatal hyperoxia on alveolar septal development are significant very early on, where even relatively brief exposures (P0-4) are sufficient to significantly reduce the surface area for gas exchange. Although our results reinforce the notion that MLI alone may not be adequate to describe the structural changes following hyperoxia exposure, we demonstrate that MLI did correlate significantly with DFCO. Thus, our data would support the notion that MLI may be a reasonable surrogate for functional diffusion. However, it is important to note that none of the measures of distal lung development, including MLI, correlated tightly with DFCO, suggesting that it is imperative to compliment structural assessment with functional measures. Furthermore, given that we only observed loss of pulmonary microvascular volume with prolonged hyperoxia (14 days) and that our assessments of pulmonary microvascular development were not significantly correlated with DFCO, our data highlights the need to more carefully evaluate current methods which assess microvascular development in models of BPD, and develop functional methods to more clearly delineate the contribution of the microvasculature to functional gas exchange.
The strength of the neonatal hyperoxia model is its consistent ability to inhibit alveolar development, and alveolar simplification is a major and consistent finding in human BPD.8,42 However, the murine neonatal hyperoxia model does not recapitulate every aspect of human BPD. Infants dying of BPD demonstrate significant interstitial thickening and inflammation, features that are not consistently present and of varying magnitude in rodent hyperoxia models.8,9,43 Thus, the current model, may not account for ongoing inflammation and alveolar wall thickening, both of which could impact pulmonary diffusion. While human BPD is associated with mechanical ventilation and exposure to supplemental oxygen, the liberal use of oxygen and, therefore, the degree of oxidative stress, has been reduced. Thus, the degree of hyperoxia used in murine models, as compared to current-era practices, may be less clinically relevant. It is important to note that the murine lung, at birth, is designed to develop normally in an environment of 21% oxygen, which is hyperoxic relative to the hypoxic intrauterine environment in which a human lung at the same stage typically develops. Therefore, it is plausible that the neonatal mouse may require much higher degrees of hyperoxia to recapitulate the same degree of oxidative stress. We utilized >90% oxygen as this is the level at which we have documented that despite recovery, alveolar structural and functional changes persist.6,21 Furthermore, the resulting alveolar simplification with minimal-to-mild fibrosis is similar to the histopathological changes in the limited autopsy samples from children with post-surfactant-era, “new BPD.8” Likewise, the impairments in alveolar–capillary function we demonstrate with the current model are similar to those reported in longitudinal studies of children and adolescents recovering from modern-era BPD.10,28,30 Although not without important caveats, the murine neonatal hyperoxia, therefore, continues to be a useful pre-clinical model of the structural and functional consequences of clinical BPD today.
The method of using DFco to estimate pulmonary diffusion function may not be as precise as the clinically-determined values of DLco due to the relatively small size of the mouse and the small volume of test gas that can be delivered. The DFco relies on the ratio of carbon monoxide uptake to neon dilution to account for “contamination” with ambient air introduced to the sample during the procedure and via anatomical dead space. Therefore, the DFco more closely approximates pulmonary diffusion per alveolar volume (DLCO/VA), which is conceptually different than DLCO which is an absolute measure of diffusion.19,44 Given the increase in alveolar volume that we observed with increased duration of neonatal hyperoxia, it is important to account for changes in alveolar volumes when comparing CO uptake between groups. Increased alveolar volume results in increased dilution of CO and potentially provides, in a structurally normal lung, increased surface area for diffusion. These important distinctions must be accounted for when attributing differences in DLCO to differences in alveolar structure. Clinically, both DLCO and DLCO/VA are reduced in infants formerly diagnosed with BPD, suggesting reduced efficiency of diffusion per alveolar volume due to decreased surface area and/or reduced capillary volume.5,10,27–30 Despite the limitations of DFCO, it provides a simple, cost-effective, and convenient method to assess pulmonary diffusion in small animals and lends valuable insight into functional impairment.
In summary, we have demonstrated that, whether limited to the saccular or continued throughout saccular and bulk alveolar postnatal lung development, neonatal hyperoxia results in similar reductions of alveolar surface area and alveolar septal volume. Consequently, exposure to neonatal hyperoxia in the saccular period is sufficient to significantly impair functional alveolar–capillary gas diffusion efficiency. However, with prolonged, chronic neonatal hyperoxia (perhaps beyond the clinically-relevant period), there is significantly increased alveolar and total lung volume. Importantly, morphometric estimates of alveolar development and direct assessment of alveolar–capillary function are poorly correlated. Clinically, the majority of infants that develop BPD experience the bulk of lung injury during saccular and early alveolar lung development, suggesting that neonatal hyperoxia models employing exposures beyond the first 4–7 postnatal days may be less clinically relevant.
Acknowledgments
Funding source: James Whitcomb Riley Hospital for Children Foundation.
Footnotes
Conflict of interest: None.
AUTHORS’ CONTRIBUTIONS
A. M. C. contributed to the project implementation, data acquisition and analysis, manuscript preparation. Y. G. contributed to the data acquisition and analysis, manuscript preparation. A. K.T. P. contributed to the data interpretation, manuscript preparation. R. S. T. contributed to the project design, data analysis, manuscript preparation. S. K. A. contributed to the project design and implementation, data analysis, manuscript preparation.
References
- 1.Stoll BJ, Hansen NI, Bell EF, Shankaran S, Laptook AR, Walsh MC, Hale EC, Newman NS, Schibler K, Carlo WA, Kennedy KA, Poindexter BB, Finer NN, Ehrenkranz RA, Duara S, Sanchez PJ, O’Shea TM, Goldberg RN, Van Meurs KP, Faix RG, Phelps DL, Frantz ID, III, Watterberg KL, Saha S, Das A, Higgins RD. For the eunice kennedy shriver national institute of child health and human development neonatal research network. neonatal outcomes of extremely preterm infants from the NICHD neonatal research network. Pediatrics. 2010;126:443–456. doi: 10.1542/peds.2009-2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ehrenkranz RA, Walsh MC, Vohr BR, Jobe AH, Wright LL, Fanaroff AA, Wrage LA, Poole K. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics. 2005;116:1353–1360. doi: 10.1542/peds.2005-0249. [DOI] [PubMed] [Google Scholar]
- 3.Fawke J, Lum S, Kirkby J, Hennessy E, Marlow N, Rowell V, Thomas S, Stocks J. Lung function and respiratory symptoms at 11 years in children born extremely preterm: the EPICure study. Am J Respir Crit Care Med. 2010;182:237–245. doi: 10.1164/rccm.200912-1806OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gough A, Spence D, Linden M, Halliday HL, McGarvey LP. General and respiratory health outcomes in adult survivors of bronchopulmonary dysplasia: a systematic review. Chest. 2012;141:1554–1567. doi: 10.1378/chest.11-1306. [DOI] [PubMed] [Google Scholar]
- 5.Ahlfeld SK, Conway SJ. Assessment of inhibited alveolar-capillary membrane structural development and function in bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol. 2014;100:168–179. doi: 10.1002/bdra.23226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahlfeld SK, Gao Y, Wang J, Horgusluoglu E, Bolanis E, Clapp DW, Conway SJ. Periostin downregulation is an early marker of inhibited neonatal murine lung alveolar septation. Birth Defects Res A Clin Mol Teratol. 2013;97:373–385. doi: 10.1002/bdra.23149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coalson JJ, Winter V, deLemos RA. Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 1995;152:640–646. doi: 10.1164/ajrccm.152.2.7633720. [DOI] [PubMed] [Google Scholar]
- 8.Husain AN, Siddiqui NH, Stocker JT. Pathology of arrested acinar development in postsurfactant bronchopulmonary dysplasia. Hum Pathol. 1998;29:710–717. doi: 10.1016/s0046-8177(98)90280-5. [DOI] [PubMed] [Google Scholar]
- 9.Bhatt AJ, Pryhuber GS, Huyck H, Watkins RH, Metlay LA, Maniscalco WM. Disrupted pulmonary vasculature and decreased vascular endothelial growth factor, flt-1, and tie-2 in human infants dying with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;164:1971–1980. doi: 10.1164/ajrccm.164.10.2101140. [DOI] [PubMed] [Google Scholar]
- 10.Balinotti JE, Chakr VC, Tiller C, Kimmel R, Coates C, Kisling J, Yu Z, Nguyen J, Tepper RS. Growth of lung parenchyma in infants and toddlers with chronic lung disease of infancy. AmJ Respir Crit Care Med. 2010;181:1093–1097. doi: 10.1164/rccm.200908-1190OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balasubramaniam V, Mervis CF, Maxey AM, Markham NE, Abman SH. Hyperoxia reduces bone marrow, circulating, and lung endothelial progenitor cells in the developing lung: implications for the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L1073–L1084. doi: 10.1152/ajplung.00347.2006. [DOI] [PubMed] [Google Scholar]
- 12.Warner BB, Stuart LA, Papes RA, Wispe JR. Functional and pathological effects of prolonged hyperoxia in neonatal mice. Am J Physiol Lung Cell Mol Physiol. 1998;275:L110. doi: 10.1152/ajplung.1998.275.1.L110. [DOI] [PubMed] [Google Scholar]
- 13.Alejandre-Alcazar MA, Kwapiszewska G, Reiss I, Amarie OV, Marsh LM, Sevilla-Perez J, Wygrecka M, Eul B, Kobrich S, Hesse M, Schermuly RT, Seeger W, Eickelberg O, Morty RE. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L537–L549. doi: 10.1152/ajplung.00050.2006. [DOI] [PubMed] [Google Scholar]
- 14.Yee M, Chess PR, McGrath-Morrow SA, Wang Z, Gelein R, Zhou R, Dean DA, Notter RH, O’Reilly MA. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol. 2009;297:L641–L649. doi: 10.1152/ajplung.00023.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163:1723–1729. doi: 10.1164/ajrccm.163.7.2011060. [DOI] [PubMed] [Google Scholar]
- 16.Poindexter BB, Feng R, Schmidt B, Aschner JL, Ballard RA, Hamvas A, Reynolds AM, Shaw PA, Jobe AH. Comparisons and limitations of current definitions of bronchopulmonary dysplasia for the Prematurity and Respiratory Outcomes Program. Ann Am Thorac Soc. 2015;12:1822–1830. doi: 10.1513/AnnalsATS.201504-218OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roughton FJ, Forster RE. Relative importance of diffusion and chemical reaction rates in determining rate of exchange of gases in the human lung, with special reference to true diffusing capacity of pulmonary membrane and volume of blood in the lung capillaries. J Appl Physiol. 1957;11:290–302. doi: 10.1152/jappl.1957.11.2.290. [DOI] [PubMed] [Google Scholar]
- 18.Hughes JM. The single breath transfer factor (Tl, co) and the transfer coefficient (Kco): a window onto the pulmonary microcirculation. Clin Physiol Funct Imaging. 2003;23:63–71. doi: 10.1046/j.1475-097x.2003.00482.x. [DOI] [PubMed] [Google Scholar]
- 19.Macintyre N, Crapo RO, Viegi G, Johnson DC, van der Grinten CP, Brusasco V, Burgos F, Casaburi R, Coates A, Enright P, Gustafsson P, Hankinson J, Jensen R, McKay R, Miller MR, Navajas D, Pedersen OF, Pellegrino R, Wanger J. Standardisation of the single-breath determination of carbon monoxide uptake in the lung. Eur Respir J. 2005;26:720–735. doi: 10.1183/09031936.05.00034905. [DOI] [PubMed] [Google Scholar]
- 20.Fallica J, Das S, Horton M, Mitzner W. Application of carbon monoxide diffusing capacity in the mouse lung. J Appl Physiol. 2011;110:1455–1459. doi: 10.1152/japplphysiol.01347.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahlfeld SK, Gao Y, Conway SJ, Tepper RS. Relationship of structural to functional impairment during alveolar-capillary membrane development. Am J Pathol. 2015;185:913–919. doi: 10.1016/j.ajpath.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forster RE. Exchange of gases between alveolar air and pulmonary capillary blood: pulmonary diffusing capacity. Physiol Rev. 1957;37:391–452. doi: 10.1152/physrev.1957.37.4.391. [DOI] [PubMed] [Google Scholar]
- 23.Mund SI, Stampanoni M, Schittny JC. Developmental alveolarization of the mouse lung. Dev Dyn. 2008;237:2108–2116. doi: 10.1002/dvdy.21633. [DOI] [PubMed] [Google Scholar]
- 24.Limjunyawong N, Fallica J, Ramakrishnan A, Datta K, Gabrielson M, Horton M, Mitzner W. Phenotyping mouse pulmonary function in vivo with the lung diffusing capacity. JoVE. 2015:e52216. doi: 10.3791/52216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tschanz SA, Burri PH, Weibel ER. A simple tool for stereological assessment of digital images: the STEPanizer. J Microsc. 2011;243:47–59. doi: 10.1111/j.1365-2818.2010.03481.x. [DOI] [PubMed] [Google Scholar]
- 27.Cazzato S, Ridolfi L, Bernardi F, Faldella G, Bertelli L. Lung function outcome at school age in very low birth weight children. Pediatr Pulmonol. 2013;48:830–837. doi: 10.1002/ppul.22676. [DOI] [PubMed] [Google Scholar]
- 28.Chang DV, Assaf SJ, Tiller CJ, Kisling JA, Tepper RS. Membrane and capillary components of lung diffusion in infants with bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2016;193:767–771. doi: 10.1164/rccm.201506-1219OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaplan E, Bar-Yishay E, Prais D, Klinger G, Mei-Zahav M, Mussaffi H, Steuer G, Hananya S, Matyashuk Y, Gabarra N, Sirota L, Blau H. Encouraging pulmonary outcome for surviving, neurologically intact, extremely premature infants in the postsurfactant era. Chest. 2012;142:725–733. doi: 10.1378/chest.11-1562. [DOI] [PubMed] [Google Scholar]
- 30.Welsh L, Kirkby J, Lum S, Odendaal D, Marlow N, Derrick G, Stocks J. The EPICure study: maximal exercise and physical activity in school children born extremely preterm. Thorax. 2010;65:165–172. doi: 10.1136/thx.2008.107474. [DOI] [PubMed] [Google Scholar]
- 31.Caskey S, Gough A, Rowan S, Gillespie S, Clarke J, Riley M, Megarry J, Nicholls P, Patterson C, Halliday HL, Shields MD, McGarvey L. Structural and functional lung impairment in adult survivors of bronchopulmonary dysplasia. Ann Am Thorac Soc. 2016;13:1262–1270. doi: 10.1513/AnnalsATS.201509-578OC. [DOI] [PubMed] [Google Scholar]
- 32.O’Reilly M, Hansbro PM, Horvat JC, Beckett EL, Harding R, Sozo F. Bronchiolar remodeling in adult mice following neonatal exposure to hyperoxia: relation to growth. Anat Rec (Hoboken) 2014;297:758–769. doi: 10.1002/ar.22867. [DOI] [PubMed] [Google Scholar]
- 33.Yee M, White RJ, Awad HA, Bates WA, McGrath-Morrow SA, O’Reilly MA. Neonatal hyperoxia causes pulmonary vascular disease and shortens life span in aging mice. Am J Pathol. 2011;178:2601–2610. doi: 10.1016/j.ajpath.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galambos C, Sims-Lucas S, Abman SH. Histologic evidence of intrapulmonary anastomoses by three-dimensional reconstruction in severe bronchopulmonary dysplasia. Ann Am Thorac Soc. 2013;10:474–481. doi: 10.1513/AnnalsATS.201305-124OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu S, Zhou C, King JA, Stevens T. A unique pulmonary microvascular endothelial cell niche revealed by Weibel-Palade bodies and Griffonia simplicifolia. Pulm Circ. 2014;4:110–115. doi: 10.1086/674879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goss KN, Cucci AR, Fisher AJ, Albrecht M, Frump A, Tursunova R, Gao Y, Brown MB, Petrache I, Tepper RS, Ahlfeld SK, Lahm T. Neonatal hyperoxic lung injury favorably alters adult right ventricular remodeling response to chronic hypoxia exposure. Am J Physiol Lung Cell Mol Physiol. 2015;308:L797–L806. doi: 10.1152/ajplung.00276.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goss KN, Tepper RS, Lahm T, Ahlfeld SK. Increased cardiac output and preserved gas exchange despite decreased alveolar surface area in rats exposed to neonatal hyperoxia and adult hypoxia. Am J Respir Cell Mol Biol. 2015;53:902–906. doi: 10.1165/rcmb.2015-0100LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Randell SH, Mercer RR, Young SL. Neonatal hyperoxia alters the pulmonary alveolar and capillary structure of 40-day-old rats. Am J Pathol. 1990;136:1259–1266. [PMC free article] [PubMed] [Google Scholar]
- 39.Berger J, Bhandari V. Animal models of bronchopulmonary dysplasia. The term mouse models. Am J Physiol Lung Cell Mol Physiol. 2014;307:L936–L947. doi: 10.1152/ajplung.00159.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahlfeld SK, Conway SJ. Aberrant signaling pathways of the lung mesenchyme and their contributions to the pathogenesis of bronchopulmonary dysplasia. Birth Defects Res A Clin Mol Teratol. 2012;94:3–15. doi: 10.1002/bdra.22869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Buczynski BW, Yee M, Paige Lawrence B, O’Reilly MA. Lung development and the host response to influenza Avirus are altered by different doses of neonatal oxygen in mice. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1078–L1087. doi: 10.1152/ajplung.00026.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coalson JJ. Pathology of bronchopulmonary dysplasia. Semin Perinatol. 2006;30:179–184. doi: 10.1053/j.semperi.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Thibeault DW, Truog WE, Ekekezie II. Acinar arterial changes with chronic lung disease of prematurity in the surfactant era. Pediatr Pulmonol. 2003;36:482–489. doi: 10.1002/ppul.10349. [DOI] [PubMed] [Google Scholar]
- 44.Hughes JM, Pride NB. Examination of the carbon monoxide diffusing capacity (DL(CO)) in relation to its KCO and VA components. Am J Respir Crit Care Med. 2012;186:132–139. doi: 10.1164/rccm.201112-2160CI. [DOI] [PubMed] [Google Scholar]