

Abstract
The lung interfaces with atmospheric oxygen via a large surface area and is perfused by the entire venous return bearing waste products collected from the whole body. It is logical that the lung is endowed with generous anti-oxidative capacity derived both locally and from the circulation. The single-pass pleiotropic alpha-Klotho (αKlotho) protein was discovered when its genetic disruption led to premature multi-organ degeneration and early death. The extracellular domain of αKlotho is cleaved by secretases and released into circulation as endocrine soluble αKlotho protein, exerting wide-ranging cytoprotective effects including anti-oxidation on distant organs including the lung, which exhibits high sensitivity to circulating αKlotho insufficiency. Because circulating αKlotho is derived mainly from the kidney, acute kidney injury (AKI) leads to systemic αKlotho deficiency that in turn increases the risks of pulmonary complications, i.e., edema and inflammation, culminating in the acute respiratory distress syndrome. Exogenous αKlotho increases endogenous anti-oxidative capacity partly via activation of the Nrf2 pathway to protect lungs against injury caused by direct hyperoxia exposure or AKI. This article reviews the current knowledge of αKlotho antioxidation in the lung in the setting of AKI as a model of circulating αKlotho deficiency, an under-recognized condition that weakens innate cytoprotective defenses and contributes to the dysfunction in distant organs.
Keywords: Cytoprotection, Antioxidation, Oxidative stress, Acute respiratory distress syndrome, Hyperoxia, Nrf2 antioxidants
1. Introduction
The Klotho gene was discovered serendipitously when its promoter was disrupted by the insertion of a transgene, resulting in a hypomorphic state of endogenous Klotho deficiency characterized by premature multi-organ degeneration resembling aging [1]. Although initially characterized as an anti-aging protein, Klotho is now considered a pleiotropic cytoprotective and tissue maintenance factor [2,3]. αKlotho is the first member of the three member gene family (α, β, γ) of single-pass transmembrane proteins [4]. Of these, αKlotho has limited organ expression and is highly expressed in the kidney [1,5] The majority of circulating αKlotho is derived from the kidney based on data from renal-specific genetic deletion, renal venous sampling, and organ ablation experiments [6,7]. The extracellular domain of αKlotho is cleaved by proteases and released from the cell (cleaved αKlotho) [6,8–10]. The αKlotho transcript could also be alternatively spliced to generate a shorter form of αKlotho without the transmembrane domain (secreted αKlotho) [11–13]. Collectively, these extracellular polypeptides constitute “soluble αKlotho” which circulates to all organs [6,13–15]. In contrast, βKlotho and γKlotho are not found in the circulation [4].
Transmembrane αKlotho is a co-receptor in conjunction with fibroblast growth factor (FGF) receptor for (FGF)-23 to regulate mineral metabolism [4]. Distinct from its co-receptor function, the extracellular domain of αKlotho is released by secretases into blood, urine, and cerebrospinal fluid as an endocrine soluble αKlotho protein [11,14,16], exerting widespread pleiotropic effects on distant organs [4] (Fig. 1). These actions include anti-oxidation and anti-apoptosis [2,3,17], regulation of growth factors (e.g., insulin-like growth factor-1, fibroblast growth factors) [18,19], signaling molecules (Wnt) [19,20] and ion channels (potassium, transient receptor potential cation channel sub-family V member 5 [TRPV5]) [21,22], mineral and hormone metabolism (calcium, inorganic phosphate, vitamin D) [7,23], stem cell function [24, 25], and tumor suppression [26,27].
Fig. 1.
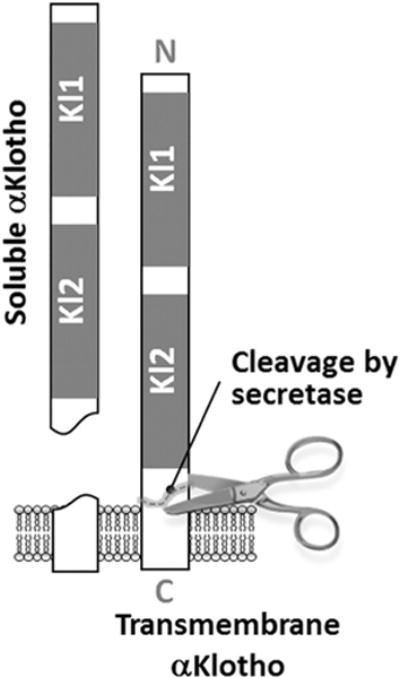
Trans-membrane and soluble αKlotho released from cells. Kl1 and Kl2 are functional domains.
This article focuses on the lung as an example of a systemic organ that is continuously exposed to a high level of oxidative stress, and that depends on endocrine soluble αKlotho for its proper function. The actions of circulating αKlotho on the lung provides a model for probing the systemic actions, and the consequences of genetic and induced perturbations, of kidney-derived circulating αKlotho on an extra-renal organ, and discusses the potential use of targeted αKlotho replacement for the prevention and/or treatment of secondary pulmonary complications in αKlotho deficient states.
2. The lung: an organ experiencing high oxidative stress
The lung interfaces with the atmosphere via a vast surface area and is constantly exposed to fluctuating temperatures, gas pressures, humidity, airborne pollution, toxins, allergens, pathogens, and one of the highest oxygen tensions of any internal organ. The pulmonary alveolar capillary bed is the largest microvascular organ in the body, and the only organ to receive the entire venous return bearing waste products collected from the whole body. Furthermore, the lung parenchyma experiences gross mechanical stress and deformation (strain and shear) with each respiratory cycle, and microvascular distention and shear with each cardiac cycle. All of these factors cause cumulative oxidative stress and tissue damage, contributing to the inexorable degeneration of lung function with age and the heightened susceptibility of lung cells to malignant transformation.
Neonatal lungs are highly sensitive to oxidative stress. Premature-born babies exposed to a high oxygen tension in the intensive care unit develop bronchopulmonary dysplasia, a syndrome of acute lung injury and inflammation that compromised short-term respiratory function and impairs lung growth and maturation leading to long-term respiratory sequelae [28]. In adults, lung function measured from the forced expiratory volume in 1 s (FEV1) and forced expiratory vital capacity (FVC) decline with age with an inverse correlation to the levels of oxygen free radicals and exhaled nitric oxide [29]. The capacity for oxygen transport, commonly measured from the lung diffusing capacity for carbon monoxide (DLCO), increases with maturation in childhood, peaks in young adulthood, then declines steadily thereafter [30] regardless of the level of physical fitness. Age-related changes in the distal lung include enlargement of alveolar air spaces without destruction of alveolar walls, reduced alveolar surface tension, loss of lung elastic recoil leading to a reduction in maximum expiratory air flow rate [31]. An increased rate of oxidative stress such as that related to smoking and pollution exposure leads to oxidant-antioxidant imbalance and accelerated aging-related DNA damage and cancer developmentin lung tissue [32–34] while the capacity to repair oxidative damage diminishes or becomes aberrant [35].
It is logical that the lung is endowed with generous anti-oxidative capacity [36–38]; some anti-oxidants are endogenous while others are derived from the circulation [39]. The fact that the entire right ventricular cardiac output flows through the pulmonary microcirculation also increases circulatory anti-oxidant delivery to lung tissue. Acute or chronic lung diseases imposes large increases in oxidative stress that often outstrips endogenous anti-oxidant and/or repair capacity. For example, patients with chronic obstructive lung disease [40] or cystic fibrosis [41] are susceptible to oxidative DNA damage, which contributes to more rapid aging-related degeneration and a higher incidence of malignancy in these patients compared to normal age-matched control subjects.
3. αKlotho action in the lung
While endogenous αKlotho expression has been reported in the brain, heart, parathyroid gland [42], breast [18], gonads [43], and bronchial epithelium [44], we are unable to detect native αKlotho protein expression within the alveolar septa by immunohistochemistry or transcript expression by RT-PCR [1,3] (Fig. 2). One report of αKlotho mRNA expression by highly sensitive RT-PCR in alveolar macrophages [45] could not be confirmed from αKlotho protein expression measured by immunoblot [46] or immunohistochemistry. Claims for αKlotho expression in the lung based solely on using reverse transcription-polymerase chain reaction is unreliable as the method is too sensitive and can prime and amplify partially processed transcripts that are never translated [47]. The existence of the short αKlotho protein translated from alternatively spliced transcript is also questionable as this may represent illegitimate slicing as has been found in many other genes [48]. There is little doubt that the αKlotho in the lung is primarily derived from the circulation.
Fig. 2.
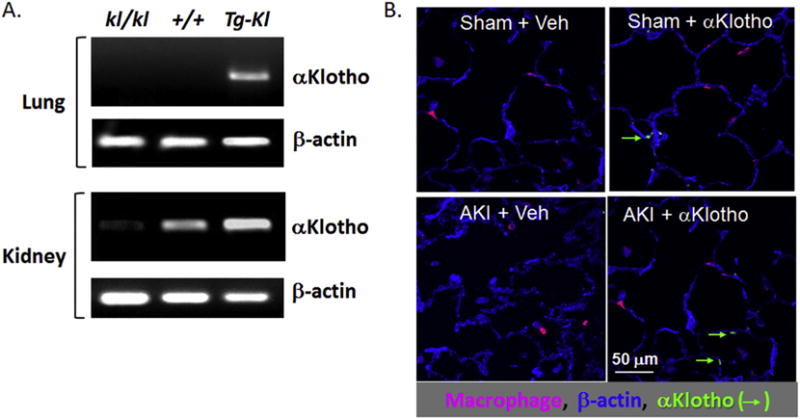
Lack of native αKlotho expression in resident lung cells. A. Non-quantitative αKlotho transcript detection by reverse transcription-polymerase chain reaction (RT-PCR) using primers that cross several exons are shown, detecting only the mature transcript in lung and kidney tissue from three different strain of mice. kl/kl = homozygous αKlotho hypomorphs. +/+: wild type. Tg-Kl: transgenic αKlotho overexpressing mice, driven by the ubiquitous elongation factor promoter. There is no native αKlotho expression in the lung in kl/kl or WT mice. The Tg-Kl mice with ectopic lung αKlotho expression serve as a positive control. The kidney expression of αKlotho is as expected and compatible with the literature. Data reproduced from reference [3]. B. Staining of lung parenchyma for β-actin, macrophages, and αKlotho under four conditions. AKI: Acute kidney injury induced by renal artery ischemia-reperfusion. Sham: Control with same length of time of operation and handling of the kidneys but no renal artery cross-clamp. αKlotho: recombinant αKlotho injected after AKI induction. Veh: Vehicle injection. There is no detectable αKlotho in lung epithelial cells or macrophages without exogenous administration. Exogenously injected αKlotho is detected in alveolar epithelial cells but not macrophages.
3.1. αKlotho deficiency alters lung structure and function
Disruption of the αKlotho gene (kl/kl) encoding the single-pass large protein (~1000 amino acids) leads to premature multi-organ degeneration and early death [1]. We examined the lungs of wild type (WT), haplo-insufficient (kl/+), and homozygous Klotho hypomorphic (kl/kl) mice. αKlotho expression is not found in resident lung cells in any of the mice [3]. The kl/kl mice have no circulating αKlotho; these mice are small but otherwise phenotypically normal until ~4–6 weeks of age; followed by the development of rapid multi-organ degeneration and premature death by 9–12 weeks of age. At necropsy their lungs are abnormally friable with grossly enlarged air spaces. The hemizygous αKlotho haplo-insufficient (kl/+) mice with 50% reduced circulating αKlotho level are grossly normal at baseline except the lung exhibits subclinical though definite age-exacerbated degeneration with air space enlargement, elevated compliance, and increased apoptosis [49, 50]. These abnormalities were initially described as “emphysemalike”, with one major distinction that inflammation is absent. In these genetically altered mice there is a strong inverse correlation between circulating αKlotho level (+/+ > kl/+ > kl/kl) and oxidative DNA damage measured by 8-hydroxy-guanosine (8-OHdG) in lung tissue at baseline [3], suggesting high sensitivity of the lung to circulating αKlotho (Fig. 3).
Fig. 3.

Murine models of αKlotho deficiency. Distal lung abnormalities in three genotypes of mice at baseline. +/+ Normal. kl/+: Klotho-haploinsufficient. kl/kl: Klotho-deficient. Histology shows air space enlargement (left micrographs, bar = 200 μm), and elevated oxidative DNA damage in kl/kl mice (right panel, 8-hydroxy-deoxyguanosine, 8-OHdG), p < 5 vs. +/+, # vs. kl/+. Data are from [3].
3.2. αKlotho protects lung cells in vitro
αKlotho is known to protect against oxidative stress in extrapulmonary tissues [17,19,51–54] but little data existed on the lung. We imposed ambient hyperoxia (95% O2) on human adenocarcinoma-derived alveolar epithelial (A549) cells (Fig. 4) and human primary alveolar type I epithelial (AT1) cells. In both cell types, exogenous αKlotho administration by cDNA transfection or addition of protein alleviates oxygen toxicity measured from cell injury (lactate dehydrogenase, LDH), apoptosis (caspase-8), and oxidative DNA damage (8-hydroxy-deoxyguanosine, 8-OHdG); these changes are associated with enhanced endogenous total anti-oxidative capacity measured by copper (Cu)-reducing equivalents, and activation of the antioxidant response element (ARE) of target genes in the nuclear factor erythroid derived 2 transcriptional factor (Nrf2) pathway measured by the firefly luciferase assay (Fig. 4) [3]. In addition, exogenous αKlotho protein protects lung epithelial cells against hydrogen peroxide-induced oxidative damage to DNA, protein and lipid, also via activation of the ARE of target antioxidant proteins in the Nrf2 pathway, in a dose-dependent manner [55]. Thus, the cytoprotective effect of αKlotho on the alveolar epithelium is at least partly mediated via activation of genes that increase endogenous antioxidant capacity [3].
Fig. 4.
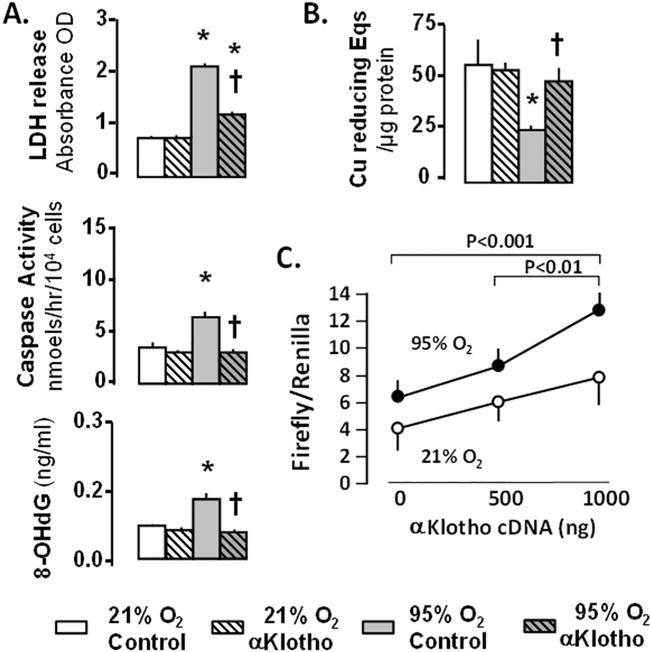
A549 lung epithelial cells exposed to 21% or 95% O2 with/without αKlotho. A. Cell death measured by lactate dehydrogenase (LDH); apoptosis measured by caspase-8; oxidative damage measured by 8-hydroxy-deoxyguanosine (8-OHdG). B. Antioxidant capacity measured by copper (Cu) reducing equivalents (Eqs). * 95% vs. 21% O2 at the same αKlotho state; † vs. Control treatment at the same O2. C. A549 cells transfected with αKlotho cDNA: Luciferase antioxidant response element (ARE) reporter assay. Data are from [3].
3.3. αKlotho protects against direct lung injury in vivo
To study the cytoprotective effects of aklotho in vivo, we used a model of hyperoxia-induced pulmonary injury. Intraperitoneal injection of αKlotho-containing media reduces hyperoxia-induced histologic changes in the distal lung, and attenuates lung apoptosis, oxidative DNA damage, and edema (Fig. 5) [3]. In normal rats, baseline serum αKlotho level did not change during hyperoxia exposure for 3 days. Thus, increasing circulating αKlotho level protects against acute lung injury caused by oxidant exposure.
Fig. 5.
In vivo lung protection by exogenous circulating αKlotho. Rats were given control or αKlotho-containing media by intra-peritoneal injection and exposed to acute hyperoxia (90% O2) or normoxia (21% O2) for 3 days. *p < 0.5 vs. normoxia, #p < 0.5 vs. control (saline injection). Lungs were assayed for: Apoptosis (caspase-8), oxidative DNA damage (8-OHdG), and edema (sodium-to-dry weight ratio). Data from [3].
3.4. αKlotho protects against lung injury complicating acute kidney injury
In contrast to hyperoxia that causes direct redox imbalance leading to lung injury in the presence of a normal serum αKlotho level, renal disease leads to systemic circulating αKlotho deficiency [56,57]. Acute kidney injury (AKI) [58–62], a common clinical disease, represents a state of heightened oxidative stress [63] and a model of titratable endogenous “pan-αKlotho deficiency” where renal, serum, and urinary αKlotho levels all decrease by up to >90% [15,55,64,65]. Thus, AKI is a useful model for studying the pulmonary complications that develop secondary to systemic organ failure. Pulmonary dysfunction develops frequently and to different degrees in clinical and experimental AKI; this under-appreciated complication of AKI could potentially culminate in the acute respiratory distress syndrome (ARDS) [58–62]. Typical clinical manifestations of incipient ARDS include decreased lung diffusing capacity, forced vital capacity and maximal ventilation [66], overt inflammation [67,68], increased alveolar-capillary permeability [69,70], leading to low-pressure alveolar edema arterial hypoxemia and impaired carbon dioxide excretion [71]. Severe pulmonary dysfunction requiring ventilator support in the setting of AKI increases mortality from 29% to 81% even after multivariate adjustment [72]. Conversely, the presence of AKI gravely impacts outcome in ventilated critically ill patients [73].
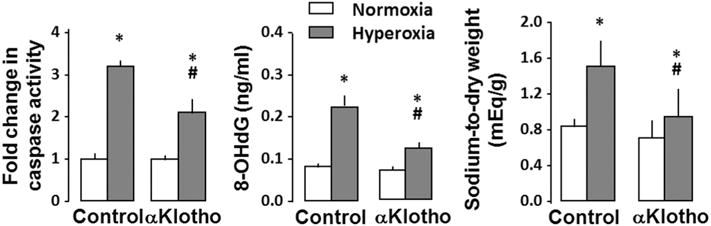
Development of ARDS in clinical and experimental AKI involves complex pathophysiology and a host of “reno-pulmonary” interactive pathways [58–62,74,75]. Clinicians often attribute the pulmonary complications to volume overload, heart failure, metabolic acidosis, and sepsis that are prevalent in AKI. Experimentalists point to chemokine accumulation, macrophage infiltration, and deranged ions channels as causes for an increased permeability contributing to an “inflamed, leaky, and wet” lung [67,76–78]. Independent from the above, derangement in kidney-derived factors per se such as αKlotho can also cause pulmonary dysfunction but are under-recognized. In rats with AKI due to ischemia-reperfusion injury [15,65,79] and severely reduced serum αKlotho level, overt alveolar septal thickening, interstitial edema and exudation develop within 3 days and are associated with elevated tissue oxidative damage and reduced endogenous total antioxidant capacity (Fig. 6) [55]. Systemic injection of αKlotho increases total antioxidant capacity and ameliorates the lung damage and edema without changing peak plasma creatinine, i.e., no change in AKI severity. Thus, αKlotho repletion in AKI alleviates secondary pulmonary complications independent from its renal effect on alleviation of AKI.
Fig. 6.
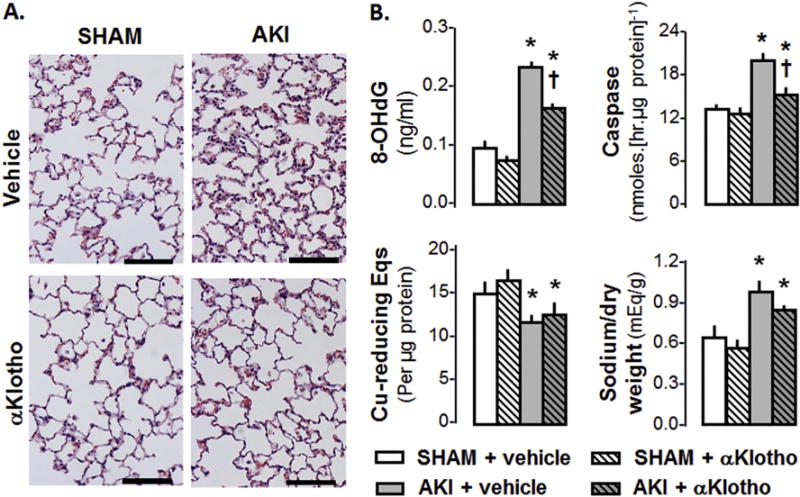
AKI from ischemia reperfusion injury (IRI) in the rat. αKlotho protein given by intra-peritoneal injection 6 h after IRI when renal damage was already established alleviates lung damage measured 3 d late. A: Lung histology. Bar = 100 pm. B: Lung oxidative DNA damage (8-OHdG), apoptosis (caspase-8 activity), endogenous antioxidant capacity (copper [Cu]-reducing equivalents [Eqs]) and edema (sodium/dry weight ratio). P < 0.001: * vs. SHAM at the same αKlotho state; † vs. AKI + vehicle. Based on data from [55].
4. Trafficking of αKlotho in kidney and lung
To act on the lung epithelium, circulating αKlotho must exit the capillaries as a 130 kD glycoprotein. αKlotho exhibits rapid kinetics, leaving the renal peritubular capillary followed by uptake by the proximal tubule and urine excretion (Fig. 7A–C) [6]. Within 30 min of an intravenous injection of fluorescent or FLAG-tagged αKlotho, the protein is clearly visible within and outside the alveolar capillary, in the extracellular space, and in epithelium (Fig. 7D). The ability of αKlotho to freely exit the vascular space is a generalized property of this protein.
Fig. 7.

αKlotho trafficking in kidney and lung. A. Kidney. Red TAMRA dye-labeled FLAG-tagged αKlotho was injected intravenously as a bolus and surface nephrons were monitored by dual laser intravital fluorescent microscopy. Nuclei (green) highlight the tubules. B. At 30 min, kidneys were fixed and stained with anti-FLAG (red, for αKlotho) and phalloidin (green, for actin). Anti-aquaporin-2 (AQP2, blue) identifies collecting ducts. αKlotho was present in proximal tubules (PT) and distal tubules (DT) and not glomeruli (G). C. Exogenous FLAG-αKlotho tracked with time in the plasma and urine after a bolus intravenous injection. D. FLAG-αKlotho was injected into normal mice and the lungs perfusion-fixed and stained for FLAG-αKlotho (red), endothelium (CD31, green) and nuclei (SYTO, blue). Differential interference contrast (Top) and fluorescent microscopy (bottom) of the same image are shown [55]. 1 – intravascular. 2 – extravascular. 3 – non-vascular cells including type 2 alveolar epithelial cells.
Circulating αKlotho is known to interact with the endothelium and the interaction has been postulated to protect the endothelium [80–83]. Transcellular transport across the endothelium can occur via pores across the endothelial cell, e.g. vesiculovacuolar organelles, fenestrae, or transendothelial channels [84,85]. Endothelial vesicular endocytosis can proceed via several mechanisms: clathrin-mediated endocytosis, caveolae-mediated endocytosis, clathrin- and caveolae-independent internalization, or macro-pinocytosis [86–89]. Although not yet definitively proven, fibroblast growth factor receptor 1 (FGFR1) may provide initial docking of soluble αKlotho on endothelial cells, triggering transcytosis pathways that shuttle αKlotho to epithelium. αKlotho binds to FGFR1 [90], which is abundant on the apical surface of endothelium [91]. Most transmembrane proteins that shuttle transport-vesicle cargo are recognized by short, linear amino-acid motifs in their cytoplasmic tails by vesicle adaptor proteins [92]. The FGFR1 C-terminus has 3 motifs [2 YXXØ (Ø = F,I,L,M,V) and 1 acidic dileucine EEXXXLL] that can bind to adaptor protein 2 (AP2), which could provide the linkage to initiate clathrin-mediated endocytosis.
5. Mechanisms of αKlotho cytoprotection in the lung
Nearly 20 years after its discovery [1], the mechanism of αKlotho action remains poorly understood despite a large list of downstream effects in many organs [4]. The amino acid sequence of αKlotho predicts glycosidase activity [1] but biochemically αKlotho functions as a glucuronidase [65,93] or sialidase [94], and it binds TGF-β [95], the transient receptor potential channel 1, and VEGF receptor-2 [80]. If and how these molecular actions translate into cytoprotection remain to be elucidated.
Among the pleiotropic actions of αKlotho, anti-oxidative cytoprotection plays a prominent role [19]. Transgenic αKlotho overexpression confers resistance to paraquat-induced oxidative damage [17]. αKlotho activates the Forkhead box class O (FoxO’s) transcriptional factors and induces superoxide dismutase-2 expression [17,52] in human umbilical vein endothelial cells [96]. αKlotho overexpression drives Nrf2 localization to the nucleus in cells [97]. Pretreatment of neurons with recombinant αKlotho protects the cells from amyloid β-induced cytotoxicity, and anti-oxidative stress array analysis showed that αKlotho increased the thioredoxin and peroxiredoxin expression system that constitutes downstream effectors of the Nrf2 pathway [98,99].
We know that αKlotho acts directly on epithelial cells [4,51,65,93] including in the lung [3], increases endogenous anti-oxidant capacity in lung cells [3] and directly activates antioxidant responsive elements in Nrf2-related endogenous antioxidants (Fig. 4) [55]. The “Cap and Collar” family of transcription factors consists of four members: Nrf (nuclear factor erythroid 2-related factor)1, Nrf2, Nrf3, and p45 NF-E2 (nuclear factor erythroid-2) [100]. Nrf1 is believed to combat oxidative stress under basal states while Nrf2 mediates response to pathologic challenges such as reactive oxygen species, inflammatory cytokines, and endoplasmic reticulum stress [101–104]. Nrf2 heterodimerize with small Maf (musculoaponeuroticfibrosarcoma) proteins and bind to the ARE’s found in the transcriptional regulatory regions of many antioxidant and xenobiotic-metabolizing enzyme genes [100,105]. Instead of relying on any single antioxidant enzyme, Nrf-2 activation leads to orchestrated up-regulation of many protective proteins to achieve coordinated detoxification. Consistent with this view is the finding of severely impaired activation of many antioxidant and detoxification enzyme genes in Nrf2-null mutant mice [106]. The signaling pathways from aklotho to Nrf2 remain to be established.
In the lung, αKlotho increases endogenous anti-oxidant capacity during oxidative stress challenge, and reduces hyperoxia-induced edema and oxidative damage to DNA, protein, and lipids [3]. Addition of recombinant purified αKlotho to lung cells activates the ARE promoter and increases endogenous total anti-oxidant capacity measured by either iron- or Copper-based assays [3]. αKlotho increases protein levels of several antioxidants in the Nrf2 network (Fig. 8). Thus, Nrf2 is likely to be a major pathway by which αKlotho protects the lung.
Fig. 8.
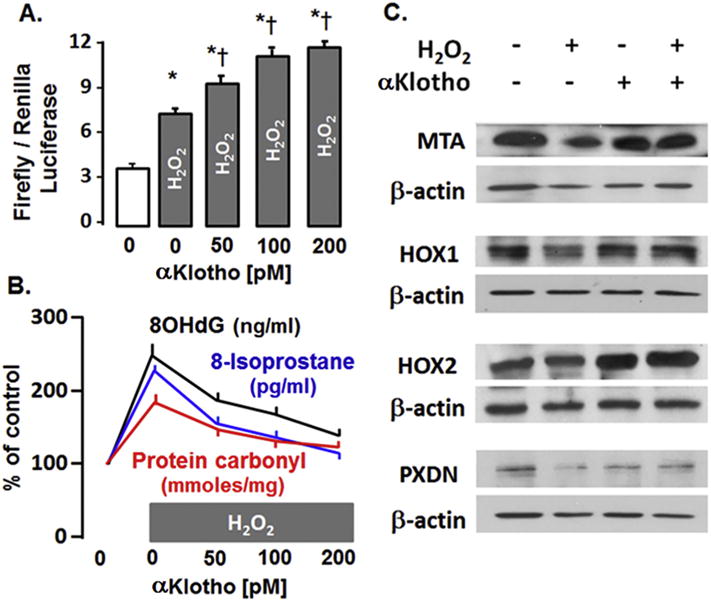
αKlotho increases endogenous antioxidants in A549 lung epithelial cells. A. Hydrogen peroxide (H2O2) activates and αKlotho further activates the ARE reporter in a dose-dependent manner measured by firefly/renilla luciferase assay. P < 0.05: * vs. 0 αKlotho. Ɨ vs. (H2O2 + 0 Klotho). B. Addition of αKlotho ameliorates DNA (8-OHdG), protein (carbonyl) and lipid (8-isoprostane) oxidative damage caused by H2O2. All data points are statistically different from baseline (H2O2 + 0 αKlotho). C. Immunoblots of selected candidates of the endogenous Nrf2 antioxidant network. MTA: methalothionine, HOX1 or 2: Heme oxygenase 1 or 2, PXDN: peroxidasin. Based on data from [55].
Besides augmenting endogenous anti-oxidation, αKlotho upregulates signaling via the paracrine erythropoietin receptor (EpoR) in the kidney [2]. EpoR is a cytoprotective, anti-apoptotic, and pro-angiogenic pathway that protects many organs including the lung against injury [107]. αKlotho has also been reported to activate autophagy in the kidney to mitigate acute ischemic damage, accelerate recovery, reduce fibrosis, and retard the progression from acute injury to chronic kidney disease [64]. Autophagy is a fundamental defense mechanism of lysosomal degradation or recycling of cellular components to maintain homeostasis. It remains to be determined whether Klotho activates EpoR or autophagy flux in the lung, and how these different pathways intersect.
6. Potential for targeted pulmonary αKlotho replacement in AKI
Oxidative stress occurs when the production of oxidants or reactive oxygen species exceeds local antioxidant capacity. With this imbalance, increased oxidation of biologic macromolecules including proteins, lipids, carbohydrates, and DNA lead to tissue damage. AKI represents a state of generalized heightened oxidative stress [108]. In the PICARD (Program to Improve Care in Acute Renal Disease) Study, plasma protein thiol oxidation and carbonyl content increases dramatically in critically ill patients with AKI [109]. These parameters improve following dialysis with quick interval re-accumulation between dialysis sessions; plasma pro-inflammatory cytokine levels also increase in parallel [109], indicating that the state of heightened systemic oxidative stress in human AKI is amenable to therapy.
There has been significant progress in the management of ARDS; however, except for the use of antibiotics for infections, the mainstay treatment modalities remain supportive with “hopeful waiting” for innate healing and recovery to occur. Other than the supportive fluid and electrolyte replacement and dialysis, no specific therapy for ARDS in AKI has improved clinical outcome. At the same time, positive pressure mechanical ventilation with exposure to a high oxygen concentration can induce barotrauma and oxidant stress that further aggravate existing lung injury and increase morbidity and mortality. Survivors of ARDS often endure long-term disability such as lung fibrosis and respiratory insufficiency. There is a definite need for novel treatment modalities to minimize lung damage and accelerate recovery.
Given the added oxidative stress in the face of circulating αKlotho deficiency in AKI, and that the lung requires circulating αKlotho for antioxidation, targeted pulmonary αKlotho replacement may have therapeutic value in preventing or alleviating ARDS in AKI. There are several reasons for considering such an approach: 1. Upon induction of lung injury, an immediate increase in demand for local cytoprotection is not met by a corresponding increase in circulating αKlotho, creating a state of relative Klotho insufficiency, which could exacerbate redox imbalance and perpetuate tissue injury. Targeted exogenous αKlotho delivery to augment local pulmonary αKlotho level as prophylaxis (preinjury) or treatment (post-injury) could reduce lung damage and improve outcome. 2. In terms of understanding pathophysiologic mechanisms, intravenous αKlotho administration may improve various systemic factors that secondarily improve lung function, thereby confounding the ability to conclude how αKlotho exerts direct in vivo effects on the lung. Localized restoration of αKlotho level will address this issue directly. 3. There may be differential needs for αKlotho replacement in different organs, i.e. the lung may require greater αKlotho delivery as it is exposed to higher basal levels of oxidative stress than other internal organs and therefore more sensitive to αKlotho insufficiency. It may be possible to engineer lung cells to express αKlotho locally as well as simultaneously release a certain amount of soluble αKlotho to the systemic circulation. 4. Inhalational delivery of cDNA is an excellent vehicle to attain sustained protein expression in the lung [107] compared to the delivery of protein. 5. Inhalational therapy can be generalized to deliver additional compounds such as cDNA, silencing RNA, microRNA, mixed biologic extracts, proteins, and other pharmaceutical agents that may have beneficial effects on the lung in AKI.
We recently validated the technique of inhalational delivery of aerosolized nanoparticles containing protein or DNA [107,110]. We screened common natural and synthetic polymeric nanoparticle preparations to determine the most promising formulations for pulmonary delivery and uptake by distal lung cells. Poly(lactic co-glycolic acid) (PLGA) nanoparticles are more effectively retained in the distal lung under physiological conditions and hence judged to be promising carriers for pulmonary applications [110]. Delivery of nebulized PLGA nanoparticles encapsulating EpoR cDNA to the lung results in rapid uptake by alveolar septal cells with sustained upregulation of EpoR signal transduction in lung tissue that peaks by 10 days and persists for 21 days following a single treatment. This treatment effectively enhances cytoprotection against acute hyperoxic lung injury [107]. A similar approach could be developed for αKlotho replacement to potentially prevent pulmonary complications in the setting of AKI.
Acknowledgments
The author thanks Orson Moe for helpful suggestions and reading of the manuscript. This work is supported by the National Heart, Lung and Blood Institute Grants R01-HL40070, R01-HL134373, and U01-HL111146. The contents of this article are solely the responsibility of the author and do not necessarily represent the official views of the National Heart, Lung, and Blood Institute or of the National Institutes of Health.
Abbreviations
- 8-OHdG
8-hydroxy-′-deoxyguanosine
- AKI
Acute kidney injury
- ARDS
Acute respiratory distress syndrome
- ARE
Antioxidant response element
- EpoR
Erythropoietin receptor
- FGF
Fibroblast growth factor
- FGFR1
Fibroblast growth factor receptor-1
- IGF-1
Insulin-like growth factor-1
- PLGA
Poly(lactic co-glycolic acid)
- Nrf1 and 2
Nuclear factor erythroid 2-related factors 1 and 2
References
- 1.Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima Y-i. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. doi: 10.1038/36285. [DOI] [PubMed] [Google Scholar]
- 2.Hu MC, Shi M, Cho HJ, Zhang J, Pavlenco A, Liu S, Sidhu S, Huang LJ, Moe OW. The erythropoietin receptor is a downstream effector of Klotho-induced cytoprotection. Kidney Int. 2013;84(3):468–481. doi: 10.1038/ki.2013.149. http://dx.doi.org/10.1038/ki.2013.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ravikumar P, Ye J, Zhang J, Pinch SN, Hu MC, Kuro-o M, Hsia CC, Moe OW. alpha-Klotho protects against oxidative damage in pulmonary epithelia. Am J Phys Lung Cell Mol Phys. 2014;307(7):L566–L575. doi: 10.1152/ajplung.00306.2013. http://dx.doi.org/10.1152/ajplung.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–533. doi: 10.1146/annurev-physiol-030212-183727. http://dx.doi.org/10.1146/annurev-physiol-030212-183727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kato Y, Arakawa E, Kinoshita S, Shirai A, Furuya A, Yamano K, Nakamura K, Iida A, Anazawa H, Koh N, Iwano A, Imura A, Fujimori T, Kuro-o M, Hanai N, Takeshige K, Nabeshima Y-I. Establishment of the anti-klotho monoclonal antibodies and detection of klotho protein in kidneys. Biochem Biophys Res Commun. 2000;267(2):597–602. doi: 10.1006/bbrc.1999.2009. http://dx.doi.org/10.1006/bbrc.1999.2009. [DOI] [PubMed] [Google Scholar]
- 6.Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, Ravikumar P, Gillings N, Bian A, Sidhu SS, Kuro-o M, Moe OW. Renal production, uptake, and handling of circulating αKlotho. J Am Soc Nephrol. 2016;27(1):79–90. doi: 10.1681/ASN.2014101030. http://dx.doi.org/10.1681/asn.2014101030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lindberg K, Amin R, Moe OW, Hu MC, Erben RG, Ostman Wernerson A, Lanske B, Olauson H, Larsson TE. The kidney is the principal organ mediating Klotho effects. J Am Soc Nephrol. 2014;25(10):2169–2175. doi: 10.1681/ASN.2013111209. http://dx.doi.org/10.1681/ASN.2013111209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C-D, Tung TY, Liang J, Zeldich E, Tucker Zhou TB, Turk BE, Abraham CR. Identification of cleavage sites leading to the shed form of the anti-aging protein Klotho. Biochemistry. 2014;53(34):5579–5587. doi: 10.1021/bi500409n. http://dx.doi.org/10.1021/bi500409n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104(50):19796–19801. doi: 10.1073/pnas.0709805104. http://dx.doi.org/10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bloch L, Sineshchekova O, Reichenbach D, Reiss K, Saftig P, Kuro-o M, Kaether C. Klotho is a substrate for α-, β- and γ-secretase. FEBS Lett. 2009;583(19):3221–3224. doi: 10.1016/j.febslet.2009.09.009. http://dx.doi.org/10.1016/j.febslet2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsumura Y, Aizawa H, Shiraki-Iida T, Nagai R, Kuro-o M, Nabeshima Y-i. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem Biophys Res Commun. 1998;242(3):626–630. doi: 10.1006/bbrc.1997.8019. http://dx.doi.org/10.1006/bbrc.1997.8019. [DOI] [PubMed] [Google Scholar]
- 12.Shiraki-Iida T, Aizawa H, Matsumura Y, Sekine S, Iida A, Anazawa H, Nagai R, Kuro-o M, Nabeshima Y. Structure of the mouse klotho gene and its two transcripts encoding membrane and secreted protein. FEBS Lett. 1998;424(1–2):6–10. doi: 10.1016/s0014-5793(98)00127-6. [DOI] [PubMed] [Google Scholar]
- 13.Tohyama O, Imura A, Iwano A, Freund JN, Henrissat B, Fujimori T, Nabeshima Y. Klotho is a novel beta-glucuronidase capable of hydrolyzing steroid beta-glucuronides. J Biol Chem. 2004;279(11):9777–9784. doi: 10.1074/jbc.M312392200. http://dx.doi.org/10.1074/jbc.M312392200. [DOI] [PubMed] [Google Scholar]
- 14.Imura A, Iwano A, Tohyama O, Tsuji Y, Nozaki K, Hashimoto N, Fujimori T, Nabeshima Y. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565(1–3):143–147. doi: 10.1016/j.febslet.2004.03.090. http://dx.doi.org/10.1016/j.febslet.2004.03.090 (S0014579304003990 [pii]) [DOI] [PubMed] [Google Scholar]
- 15.Hu MC, Shi M, Zhang J, Quinones H, Kuro-o M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia-reperfusion injury and its replacement is protective. Kidney Int. 2010;78(12):1240–1251. doi: 10.1038/ki.2010.328. (ki2010328 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu MC, Kuro-o M, Moe OW. Secreted klotho and chronic kidney disease. Adv Exp Med Biol. 2012;728:126–157. doi: 10.1007/978-1-4614-0887-1_9. http://dx.doi.org/10.1007/978-1-4614-0887-1_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, Kurosu H, Miyoshi M, Ogawa Y, Castrillon DH, Rosenblatt KP, Kuro-o M. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem. 2005;280(45):38029–38034. doi: 10.1074/jbc.M509039200. http://dx.doi.org/10.1074/jbc.M509039200 (M509039200 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolf I, Levanon-Cohen S, Bose S, Ligumsky H, Sredni B, Kanety H, Kuro-o M, Karlan B, Kaufman B, Koeffler HP, Rubinek T. Klotho: a tumor suppressor and a modulator of the IGF-1 and FGF pathways in human breast cancer. Oncogene. 2008;27(56):7094–7105. doi: 10.1038/onc.2008.292. http://dx.doi.org/10.1038/onc.2008.292. [DOI] [PubMed] [Google Scholar]
- 19.Kuro-o M. Klotho as a regulator of oxidative stress and senescence. Biol Chem. 2008;389(3):233–241. doi: 10.1515/BC.2008.028. http://dx.doi.org/10.1515/BC.2008.028. [DOI] [PubMed] [Google Scholar]
- 20.Satoh M, Nagasu H, Morita Y, Yamaguchi TP, Kanwar YS, Kashihara N. Klotho protects against mouse renal fibrosis by inhibiting Wnt signaling. Am J Physiol Ren Physiol. 2012;303(12):F1641–F1651. doi: 10.1152/ajprenal.00460.2012. http://dx.doi.org/10.1152/ajprenal.00460.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cha SK, Hu MC, Kurosu H, Kuro-o M, Moe O, Huang CL. Regulation of renal outer medullary potassium channel and renal K(+) excretion by Klotho. Mol Pharmacol. 2009;76(1):38–46. doi: 10.1124/mol.109.055780. (mol.109.055780 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf MT, An SW, Nie M, Bal MS, Huang CL. Klotho up-regulates renal calcium channel transient receptor potential vanilloid 5 (TRPV5) by intra- and extracellular N-glycosylation-dependent mechanisms. J Biol Chem. 2014;289(52):35849–35857. doi: 10.1074/jbc.M114.616649. http://dx.doi.org/10.1074/jbc.M114.616649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haussler MR, Whitfield GK, Haussler CA, Sabir MS, Khan Z, Sandoval R, Jurutka PW. 1,25-dihydroxyvitamin D and klotho: a tale of two renal hormones coming of age. Vitam Horm. 2016;100:165–230. doi: 10.1016/bs.vh.2015.11.005. http://dx.doi.org/10.1016/bs.vh.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 24.Bian A, Neyra JA, Zhan M, Hu MC. Klotho, stem cells, and aging. Clin Interv Aging. 2015;10:1233–1243. doi: 10.2147/CIA.S84978. http://dx.doi.org/10.2147/CIA.S84978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vadakke Madathil S, Coe LM, Casu C, Sitara D. Klotho deficiency disrupts hematopoietic stem cell development and erythropoiesis. Am J Pathol. 2014;184(3):827–841. doi: 10.1016/j.ajpath.2013.11.016. http://dx.doi.org/10.1016/j.ajpath.2013.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubinek T, Wolf I. The role of alpha-Klotho as a universal tumor suppressor. Vitam Horm. 2016;101:197–214. doi: 10.1016/bs.vh.2016.03.001. http://dx.doi.org/10.1016/bs.vh.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 27.Tang X, Wang Y, Fan Z, Ji G, Wang M, Lin J, Huang S, Meltzer SJ. Klotho: a tumor suppressor and modulator of the Wnt/beta-catenin pathway in human hepatocellular carcinoma. Lab Investig. 2016;96(2):197–205. doi: 10.1038/labinvest.2015.86. http://dx.doi.org/10.1038/labinvest.2015.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baker CD, Alvira CM. Disrupted lung development and bronchopulmonary dysplasia: opportunities for lung repair and regeneration. Curr Opin Pediatr. 2014;26(3):306–314. doi: 10.1097/MOP.0000000000000095. http://dx.doi.org/10.1097/MOP.0000000000000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gramiccioni C, Carpagnano GE, Spanevello A, Turchiarelli V, Cagnazzo MG, Foschino Barbaro MP. Airways oxidative stress, lung function and cognitive impairment in aging. Monaldi Arch Chest Dis. 2010;73(1):5–11. doi: 10.4081/monaldi.2010.307. http://dx.doi.org/10.4081/monaldi.2010.307. [DOI] [PubMed] [Google Scholar]
- 30.Paoletti P, Viegi G, Pistelli G, Di Pede F, Fazzi P, Polato R, Saetta M, Zambon R, Carli G, Giuntini C, et al. Reference equations for the single-breath diffusing capacity. A cross-sectional analysis and effect of body size and age. Am Rev Respir Dis. 1985;132(4):806–813. doi: 10.1164/arrd.1985.132.4.806. http://dx.doi.org/10.1164/arrd.1985.132.4.806. [DOI] [PubMed] [Google Scholar]
- 31.Miller MR. Structural and physiological age-associated changes in aging lungs. Semin Respir Crit Care Med. 2010;31(5):521–527. doi: 10.1055/s-0030-1265893. http://dx.doi.org/10.1055/s-0030-1265893. [DOI] [PubMed] [Google Scholar]
- 32.Lee HC, Lim ML, Lu CY, Liu VW, Fahn HJ, Zhang C, Nagley P, Wei YH. Concurrent increase of oxidative DNA damage and lipid peroxidation together with mitochondrial DNA mutation in human lung tissues during aging—smoking enhances oxidative stress on the aged tissues. Arch Biochem Biophys. 1999;362(2):309–316. doi: 10.1006/abbi.1998.1036. http://dx.doi.org/10.1006/abbi.1998.1036. [DOI] [PubMed] [Google Scholar]
- 33.Mooney LA, Perera FP, Van Bennekum AM, Blaner WS, Karkoszka J, Covey L, Hsu Y, Cooper TB, Frenkel K. Gender differences in autoantibodies to oxidative DNA base damage in cigarette smokers. Cancer Epidemiol Biomark Prev. 2001;10(6):641–648. [PubMed] [Google Scholar]
- 34.Peddireddy V, Siva Prasad B, Gundimeda SD, Penagaluru PR, Mundluru HP. Assessment of 8-oxo-7,8-dihydro-2′-deoxyguanosine and malondialdehyde levels as oxidative stress markers and antioxidant status in non-small cell lung cancer. Biomarkers. 2012;17(3):261–268. doi: 10.3109/1354750X.2012.664169. http://dx.doi.org/10.3109/1354750X.2012.664169. [DOI] [PubMed] [Google Scholar]
- 35.Kurundkar A, Thannickal VJ. Redox mechanisms in age-related lung fibrosis. Redox Biol. 2016;9:67–76. doi: 10.1016/j.redox.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poljšak B, Fink R. The protective role of antioxidants in the defence against ROS/RNS-mediated environmental pollution. Oxidative Med Cell Longev. 2014;2014(671539) doi: 10.1155/2014/671539. http://dx.doi.org/10.1155/2014/671539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, inflammation and cancer: respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. Int J Environ Res Public Health. 2013;10(9):3886–3907. doi: 10.3390/ijerph10093886. http://dx.doi.org/10.3390/ijerph10093886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Araneda OF, Tuesta M. Lung Oxidative Damage by Hypoxia. Oxidative Medicine and Cellular Longevity. 2012;2012:856918. doi: 10.1155/2012/856918. http://dx.doi.org/10.1155/2012/856918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pisoschi AM, Pop A. The role of antioxidants in the chemistry of oxidative stress: a review. Eur J Med Chem. 2015;97:55–74. doi: 10.1016/j.ejmech.2015.04.040. http://dx.doi.org/10.1016/j.ejmech.2015.04.040. [DOI] [PubMed] [Google Scholar]
- 40.Tzortzaki EG, Dimakou K, Neofytou E, Tsikritsaki K, Samara K, Avgousti M, Amargianitakis V, Gousiou A, Menikou S, Siafakas NM. Oxidative DNA damage and somatic mutations: a link to the molecular pathogenesis of chronic inflammatory airway diseases. Chest. 2012;141(5):1243–1250. doi: 10.1378/chest.11-1653. http://dx.doi.org/10.1378/chest.11-1653. [DOI] [PubMed] [Google Scholar]
- 41.Brown RK, McBurney A, Lunec J, Kelly FJ. Oxidative damage to DNA in patients with cystic fibrosis. Free Radic Biol Med. 1995;18(4):801–806. doi: 10.1016/0891-5849(94)00172-g. [DOI] [PubMed] [Google Scholar]
- 42.Hu MC, Kuro-o M, Moe OW. Renal and extrarenal actions of Klotho. Semin Nephrol. 2013;33(2):118–129. doi: 10.1016/j.semnephrol.2012.12.013. http://dx.doi.org/10.1016/j.semnephrol.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li SA, Watanabe M, Yamada H, Nagai A, Kinuta M, Takei K. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct Funct. 2004;29(4):91–99. doi: 10.1247/csf.29.91. [DOI] [PubMed] [Google Scholar]
- 44.Gao W, Yuan C, Zhang J, Li L, Yu L, Wiegman CH, Barnes PJ, Adcock IM, Huang M, Yao X. Klotho expression is reduced in COPD airway epithelial cells: effects on inflammation and oxidant injury. Clin Sci (Lond) 2015;129(12):1011–1023. doi: 10.1042/CS20150273. http://dx.doi.org/10.1042/CS20150273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L, Wang Y, Gao W, Yuan C, Zhang S, Zhou H, Huang M, Yao X. Klotho reduction in alveolar macrophages contributes to cigarette smoke extract-induced inflammation in chronic obstructive pulmonary disease. J Biol Chem. 2015;290(46):27890–27900. doi: 10.1074/jbc.M115.655431. http://dx.doi.org/10.1074/jbc.M115.655431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Han X, Li L, Yang J, King G, Xiao Z, Quarles LD. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2 D in macrophages. FEBS Lett. 2016;590(1):53–67. doi: 10.1002/1873-3468.12040. http://dx.doi.org/10.1002/1873-3468.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cooper DN, Berg LP, Kakkar VV, Reiss J. Ectopic (illegitimate) transcription: new possibilities for the analysis and diagnosis of human genetic disease. Ann Med. 1994;26(1):9–14. doi: 10.3109/07853899409147321. [DOI] [PubMed] [Google Scholar]
- 48.Wimmer K, Eckart M, Rehder H, Fonatsch C. Illegitimate splicing of the NF1 gene in healthy individuals mimics mutation-induced splicing alterations in NF1 patients. Hum Genet. 2000;106(3):311–313. doi: 10.1007/s004390051043. [DOI] [PubMed] [Google Scholar]
- 49.Suga T, Kurabayashi M, Sando Y, Ohyama Y, Maeno T, Maeno Y, Aizawa H, Matsumura Y, Kuwaki T, Kuro OM, Nabeshima Y, Nagai R. Disruption of the klotho gene causes pulmonary emphysema in mice. Defect in maintenance of pulmonary integrity during postnatal life. Am J Respir Cell Mol Biol. 2000;22(1):26–33. doi: 10.1165/ajrcmb.22.1.3554. http://dx.doi.org/10.1165/ajrcmb.22.1.3554. [DOI] [PubMed] [Google Scholar]
- 50.Ishii M, Yamaguchi Y, Yamamoto H, Hanaoka Y, Ouchi Y. Airspace enlargement with airway cell apoptosis in klotho mice: a model of aging lung. J Gerontol A Biol Sci Med Sci. 2008;63(12):1289–1298. doi: 10.1093/gerona/63.12.1289. (63/12/1289 [pii]) [DOI] [PubMed] [Google Scholar]
- 51.Hu MC, Zhang MShi,J, Quinones H, Griffith C, Kuro-o M, Moe OW. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22(1):124–136. doi: 10.1681/ASN.2009121311. http://dx.doi.org/10.1681/ASN.2009121311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikushima M, Rakugi H, Ishikawa K, Maekawa Y, Yamamoto K, Ohta J, Chihara Y, Kida I, Ogihara T. Anti-apoptotic and anti-senescence effects of Klotho on vascular endothelial cells. Biochem Biophys Res Commun. 2006;339(3):827–832. doi: 10.1016/j.bbrc.2005.11.094. http://dx.doi.org/10.1016/j.bbrc.2005.11.094. [DOI] [PubMed] [Google Scholar]
- 53.Nagai T, Yamada K, Kim HC, Kim YS, Noda Y, Imura A, Nabeshima Y, Nabeshima T. Cognition impairment in the genetic model of aging klotho gene mutant mice: a role of oxidative stress. FASEB J. 2003;17(1):50–52. doi: 10.1096/fj.02-0448fje. [DOI] [PubMed] [Google Scholar]
- 54.Papa S, Skulachev VP. Reactive oxygen species, mitochondria, apoptosis and aging. Mol Cell Biochem. 1997;174(1–2):305–319. [PubMed] [Google Scholar]
- 55.Ravikumar P, Li L, Ye J, Shi M, Taniguchi M, Zhang J, Kuro OM, Hu MC, Moe OW, Hsia CC. alphαKlotho deficiency in acute kidney injury contributes to lung damage. J Appl Physiol (1985) 2016;120(7):723–732. doi: 10.1152/japplphysiol.00792.2015. http://dx.doi.org/10.1152/japplphysiol.00792.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu MC, Moe OW. Klotho as a potential biomarker and therapy for acute kidney injury. Nat Rev Nephrol. 2012;8(7):423–429. doi: 10.1038/nrneph.2012.92. http://dx.doi.org/10.1038/nrneph.2012.92 (nrneph.2012.92 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hu MC, Kuro-o M, Moe OW. Klotho and chronic kidney disease. Contrib Nephrol. 2013;180:47–63. doi: 10.1159/000346778. http://dx.doi.org/10.1159/000346778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Doi K, Ishizu T, Fujita T, Noiri E. Lung injury following acute kidney injury: kidney-lung crosstalk. Clin Exp Nephrol. 2011;15(4):464–470. doi: 10.1007/s10157-011-0459-4. http://dx.doi.org/10.1007/s10157-011-0459-4. [DOI] [PubMed] [Google Scholar]
- 59.Faubel S. Pulmonary complications after acute kidney injury. Adv Chronic Kidney Dis. 2008;15(3):284–296. doi: 10.1053/j.ackd.2008.04.008. http://dx.doi.org/10.1053/j.ackd.2008.04.008. [DOI] [PubMed] [Google Scholar]
- 60.Paladino JD, Hotchkiss JR, Rabb H. Acute kidney injury and lung dysfunction: a paradigm for remote organ effects of kidney disease? Microvasc Res. 2009;77(1):8–12. doi: 10.1016/j.mvr.2008.09.001. http://dx.doi.org/10.1016/j.mvr.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seeley EJ. Updates in the management of acute lung injury: a focus on the overlap between AKI and ARDS. Adv Chronic Kidney Dis. 2013;20(1):14–20. doi: 10.1053/j.ackd.2012.10.001. http://dx.doi.org/10.1053/j.ackd.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 62.Yap SC, Lee HT. Acute kidney injury and extrarenal organ dysfunction: new concepts and experimental evidence. Anesthesiology. 2012;116(5):1139–1148. doi: 10.1097/ALN.0b013e31824f951b. http://dx.doi.org/10.1097/ALN.0b013e31824f951b. [DOI] [PubMed] [Google Scholar]
- 63.Sureshbabu A, Ryter SW, Choi ME. Oxidative stress and autophagy: crucial modulators of kidney injury. Redox Biol. 2015;4:208–214. doi: 10.1016/j.redox.2015.01.001. http://dx.doi.org/10.1016/j.redox.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shi M, Flores B, Gillings N, Bian A, Cho HJ, Yan S, Liu Y, Levine B, Moe OW, Hu MC. alphαKlotho Mitigates Progression of AKI to CKD through activation of autophagy. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2015060613. http://dx.doi.org/10.1681/ASN.2015060613. [DOI] [PMC free article] [PubMed]
- 65.Panesso MC, Shi M, Cho HJ, Paek J, Ye J, Moe OW, Hu MC. Klotho has dual protective effects on cisplatin-induced acute kidney injury. Kidney Int. 2014;85(4):855–870. doi: 10.1038/ki.2013.489. http://dx.doi.org/10.1038/ki.2013.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsu HH, Tzao C, Chang WC, Wu CP, Tung HJ, Chen CY, Perng WC. Zinc chloride (smoke bomb) inhalation lung injury: clinical presentations, high-resolution CT findings, and pulmonary function test results. Chest. 2005;127(6):2064–2071. doi: 10.1378/chest.127.6.2064. http://dx.doi.org/10.1378/chest.127.6.2064. [DOI] [PubMed] [Google Scholar]
- 67.Klein CL, Hoke TS, Fang WF, Altmann CJ, Douglas IS, Faubel S. Interleukin-6 mediates lung injury following ischemic acute kidney injury or bilateral nephrectomy. Kidney Int. 2008;74(7):901–909. doi: 10.1038/ki.2008.314. http://dx.doi.org/10.1038/ki.2008.314. [DOI] [PubMed] [Google Scholar]
- 68.Seam N, Meduri GU, Wang H, Nylen ES, Sun J, Schultz MJ, Tropea M, Suffredini AF. Effects of methylprednisolone infusion on markers of inflammation, coagulation, and angiogenesis in early acute respiratory distress syndrome. Crit Care Med. 2012;40(2):495–501. doi: 10.1097/CCM.0b013e318232da5e. http://dx.doi.org/10.1097/CCM.0b013e318232da5e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sibbald WJ, Anderson RR, Reid B, Holliday RL, Driedger AA. Alveolo-capillary permeability in human septic ARDS. Effect of high-dose corticosteroid therapy. Chest. 1981;79(2):133–142. doi: 10.1378/chest.79.2.133. [DOI] [PubMed] [Google Scholar]
- 70.Todisco T, Dottorini M, Rossi F, Baldoncini A, Palumbo R. Pulmonary epithelial permeability in ARDS and cardiogenic pulmonary oedema. Eur Respir J. 1988;1(10):918–922. [PubMed] [Google Scholar]
- 71.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–2740. doi: 10.1172/JCI60331. http://dx.doi.org/10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chertow GM, Christiansen CL, Cleary PD, Munro C, Lazarus JM. Prognostic stratification in critically ill patients with acute renal failure requiring dialysis. Arch Intern Med. 1995;155(14):1505–1511. [PubMed] [Google Scholar]
- 73.Vieira JM, Jr, Castro I, Curvello-Neto A, Demarzo S, Caruso P, Pastore L, Jr, Imanishe MH, Abdulkader RC, Deheinzelin D. Effect of acute kidney injury on weaning from mechanical ventilation in critically ill patients. Crit Care Med. 2007;35(1):184–191. doi: 10.1097/01.CCM.0000249828.81705.65. http://dx.doi.org/10.1097/01.CCM.0000249828.81705.65. [DOI] [PubMed] [Google Scholar]
- 74.Kelly KJ. Distant effects of experimental renal ischemia/reperfusion injury. J Am Soc Nephrol. 2003;14(6):1549–1558. doi: 10.1097/01.asn.0000064946.94590.46. [DOI] [PubMed] [Google Scholar]
- 75.Levy EM, Viscoli CM, Horwitz RI. The effect of acute renal failure on mortality. A cohort analysis. JAMA. 1996;275(19):1489–1494. [PubMed] [Google Scholar]
- 76.Rabb H, Wang Z, Nemoto T, Hotchkiss J, Yokota N, Soleimani M. Acute renal failure leads to dysregulation of lung salt and water channels. Kidney Int. 2003;63(2):600–606. doi: 10.1046/j.1523-1755.2003.00753.x. http://dx.doi.org/10.1046/j.1523-1755.2003.00753.x. [DOI] [PubMed] [Google Scholar]
- 77.Kramer AA, Postler G, Salhab KF, Mendez C, Carey LC, Rabb H. Renal ischemia/reperfusion leads to macrophage-mediated increase in pulmonary vascular permeability. Kidney Int. 1999;55(6):2362–2367. doi: 10.1046/j.1523-1755.1999.00460.x. http://dx.doi.org/10.1046/j.1523-1755.1999.00460.x. [DOI] [PubMed] [Google Scholar]
- 78.Hassoun HT, Lie ML, Grigoryev DN, Liu M, Tuder RM, Rabb H. Kidney ischemia-reperfusion injury induces caspase-dependent pulmonary apoptosis. Am J Physiol Ren Physiol. 2009;297(1):F125–F137. doi: 10.1152/ajprenal.90666.2008. http://dx.doi.org/10.1152/ajprenal.90666.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Di Sole F, Hu MC, Zhang J, Babich V, Bobulescu IA, Shi M, McLeroy P, Rogers TE, Moe OW. The reduction of Na/H exchanger-3 protein and transcript expression in acute ischemia-reperfusion injury is mediated by extractable tissue factor(s) Kidney Int. 2011;80(8):822–831. doi: 10.1038/ki.2011.229. http://dx.doi.org/10.1038/ki.2011.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kusaba T, Okigaki M, Matui A, Murakami M, Ishikawa K, Kimura T, Sonomura K, Adachi Y, Shibuya M, Shirayama T, Tanda S, Hatta T, Sasaki S, Mori Y, Matsubara H. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc Natl Acad Sci U S A. 2010;107(45):19308–19313. doi: 10.1073/pnas.1008544107. http://dx.doi.org/10.1073/pnas.1008544107 (1008544107 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu F, Wu S, Ren H, Gu J. Klotho suppresses RIG-I-mediated senescence-associated inflammation. Nat Cell Biol. 2011;13(3):254–262. doi: 10.1038/ncb2167. http://dx.doi.org/10.1038/ncb2167. [DOI] [PubMed] [Google Scholar]
- 82.Nagai R, Saito Y, Ohyama Y, Aizawa H, Suga T, Nakamura T, Kurabayashi M, Kuroo M. Endothelial dysfunction in the klotho mouse and downregulation of klotho gene expression in various animal models of vascular and metabolic diseases. Cell Mol Life Sci. 2000;57(5):738–746. doi: 10.1007/s000180050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saito Y, Nakamura T, Ohyama Y, Suzuki T, Iida A, Shiraki-Iida T, Kuro-o M, Nabeshima Y, Kurabayashi M, Nagai R. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem Biophys Res Commun. 2000;276(2):767–772. doi: 10.1006/bbrc.2000.3470. http://dx.doi.org/10.1006/bbrc.2000.3470 (S0006291X00934703 [pii]) [DOI] [PubMed] [Google Scholar]
- 84.Stan RV. Endocytosis pathways in endothelium: how many? Am J Phys Lung Cell Mol Phys. 2006;290(5):L806–L808. doi: 10.1152/ajplung.00533.2005. http://dx.doi.org/10.1152/ajplung.00533.2005. [DOI] [PubMed] [Google Scholar]
- 85.Stan RV. Structure and function of endothelial caveolae. Microsc Res Tech. 2002;57(5):350–364. doi: 10.1002/jemt.10089. http://dx.doi.org/10.1002/jemt.10089. [DOI] [PubMed] [Google Scholar]
- 86.Kirkham M, Fujita A, Chadda R, Nixon SJ, Kurzchalia TV, Sharma DK, Pagano RE, Hancock JF, Mayor S, Parton RG. Ultrastructural identification of uncoated caveolin-independent early endocytic vehicles. J Cell Biol. 2005;168(3):465–476. doi: 10.1083/jcb.200407078. http://dx.doi.org/10.1083/jcb.200407078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kirkham M, Parton RG. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 2005;1746(3):349–363. doi: 10.1016/j.bbamcr.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 88.McNiven MA. Dynamin in disease. Nat Genet. 2005;37(3):215–216. doi: 10.1038/ng0305-215. http://dx.doi.org/10.1038/ng0305-215. [DOI] [PubMed] [Google Scholar]
- 89.Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell. 2002;2(4):411–423. doi: 10.1016/s1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 90.Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, Kuro-o M. Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem. 2006;281(10):6120–6123. doi: 10.1074/jbc.C500457200. http://dx.doi.org/10.1074/jbc.C500457200 (C500457200 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Presta M, Dell’Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16(2):159–178. doi: 10.1016/j.cytogfr.2005.01.004. http://dx.doi.org/10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 92.Kelly BT, McCoy AJ, Spate K, Miller SE, Evans PR, Honing S, Owen DJ. A structural explanation for the binding of endocytic dileucine motifs by the AP2 complex. Nature. 2008;456(7224):976–979. doi: 10.1038/nature07422. http://dx.doi.org/10.1038/nature07422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW. Klotho: a novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J. 2010;24(9):3438–3450. doi: 10.1096/fj.10-154765. http://dx.doi.org/10.1096/fj.10-154765 (fj.10-154765 [pii]) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cha SK, Ortega B, Kurosu H, Rosenblatt KP, Kuro OM, Huang CL. Removal of sialic acid involving Klotho causes cell-surface retention of TRPV5 channel via binding to galectin-1. Proc Natl Acad Sci U S A. 2008;105(28):9805–9810. doi: 10.1073/pnas.0803223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sugiura H, Yoshida T, Shiohira S, Kohei J, Mitobe M, Kurosu H, Kuro-o M, Nitta K, Tsuchiya K. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am J Physiol Ren Physiol. 2012;302(10):F1252–F1264. doi: 10.1152/ajprenal.00294.2011. http://dx.doi.org/10.1152/ajprenal.00294.2011. [DOI] [PubMed] [Google Scholar]
- 96.Rakugi H, Matsukawa N, Ishikawa K, Yang J, Imai M, Ikushima M, Maekawa Y, Kida I, Miyazaki J, Ogihara T. Anti-oxidative effect of Klotho on endothelial cells through cAMP activation. Endocrine. 2007;31(1):82–87. doi: 10.1007/s12020-007-0016-9. [DOI] [PubMed] [Google Scholar]
- 97.Hsieh CC, Kuro-o M, Rosenblatt KP, Brobey R, Papaconstantinou J. The ASK1-signalosome regulates p38 MAPK activity in response to levels of endogenous oxidative stress in the Klotho mouse models of aging. Aging (Albany NY) 2010;2(9):597–611. doi: 10.18632/aging.100194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zeldich E, Chen CD, Colvin TA, Bove-Fenderson EA, Liang J, Tucker Zhou TB, Harris DA, Abraham CR. The neuroprotective effect of Klotho is mediated via regulation of members of the redox system. J Biol Chem. 2014;289(35):24700–24715. doi: 10.1074/jbc.M114.567321. http://dx.doi.org/10.1074/jbc.M114.567321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tanito M, Agbaga MP, Anderson RE. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic Biol Med. 2007;42(12):1838–1850. doi: 10.1016/j.freeradbiomed.2007.03.018. http://dx.doi.org/10.1016/j.freeradbiomed.2007.03.018. [DOI] [PubMed] [Google Scholar]
- 100.Blank V. Small Maf proteins in mammalian gene control: mere dimerization partners or dynamic transcriptional regulators? J Mol Biol. 2008;376(4):913–925. doi: 10.1016/j.jmb.2007.11.074. http://dx.doi.org/10.1016/j.jmb.2007.11.074. [DOI] [PubMed] [Google Scholar]
- 101.Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283(48):33554–33562. doi: 10.1074/jbc.M804597200. http://dx.doi.org/10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Osburn WO, Karim B, Dolan PM, Liu G, Yamamoto M, Huso DL, Kensler TW. Increased colonic inflammatory injury and formation of aberrant crypt foci in Nrf2-deficient mice upon dextran sulfate treatment. Int J Cancer. 2007;121(9):1883–1891. doi: 10.1002/ijc.22943. http://dx.doi.org/10.1002/ijc.22943. [DOI] [PubMed] [Google Scholar]
- 103.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, Biswal S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun. 2006;351(4):883–889. doi: 10.1016/j.bbrc.2006.10.102. http://dx.doi.org/10.1016/j.bbrc.2006.10.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23(20):7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Motohashi H, O’Connor T, Katsuoka F, Engel JD, Yamamoto M. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 2002;294(1–2):1–12. doi: 10.1016/s0378-1119(02)00788-6. [DOI] [PubMed] [Google Scholar]
- 106.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236(2):313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 107.Ravikumar P, Menon JU, Punnakitikashem P, Gyawali D, Togao O, Takahashi M, Zhang J, Ye J, Moe OW, Nguyen KT, Hsia CC. Nanoparticle facilitated inhalational delivery of erythropoietin receptor cDNA protects against hyperoxic lung injury. Nanomedicine. 2016;12(3):811–821. doi: 10.1016/j.nano.2015.10.004. http://dx.doi.org/10.1016/j.nano.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nath KA, Norby SM. Reactive oxygen species and acute renal failure. Am J Med. 2000;109(8):665–678. doi: 10.1016/s0002-9343(00)00612-4. [DOI] [PubMed] [Google Scholar]
- 109.Himmelfarb J, McMonagle E, Freedman S, Klenzak J, McMenamin E, Le P, Pupim LB, Ikizler TA. The PG, Oxidative stress is increased in critically ill patients with acute renal failure. J Am Soc Nephrol. 2004;15(9):2449–2456. doi: 10.1097/01.ASN.0000138232.68452.3B. http://dx.doi.org/10.1097/01.asn.0000138232.68452.3b. [DOI] [PubMed] [Google Scholar]
- 110.Menon JU, Ravikumar P, Pise A, Gyawali D, Hsia CC, Nguyen KT. Polymeric nanoparticles for pulmonary protein and DNA delivery. Acta Biomater. 2014;10(6):2643–2652. doi: 10.1016/j.actbio.2014.01.033. http://dx.doi.org/10.1016/j.actbio.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]