

Abstract
Major depressive disorder (MDD) patients display a common, but often variable set of symptoms making successful, sustained treatment difficult to achieve. Separate depressive symptoms may be encoded by differential changes in distinct circuits in the brain, yet how discrete circuits underlie behavioral subsets of depression and how they adapt in response to stress has not been addressed. We identify two discrete circuits of parvalbumin-positive (PV) neurons in the ventral pallidum (VP) projecting to either the lateral habenula or ventral tegmental area contributing to depression. We find that these populations undergo different electrophysiological adaptations in response to social defeat stress, which are normalized by antidepressant treatment. Furthermore, manipulation of each population mediates either social withdrawal or behavioral despair, but not both. We propose that distinct components of the VP PV circuit can subserve related, yet separate depressive-like phenotypes in mice which could ultimately provide a platform for symptom-specific treatments of depression.
In brief
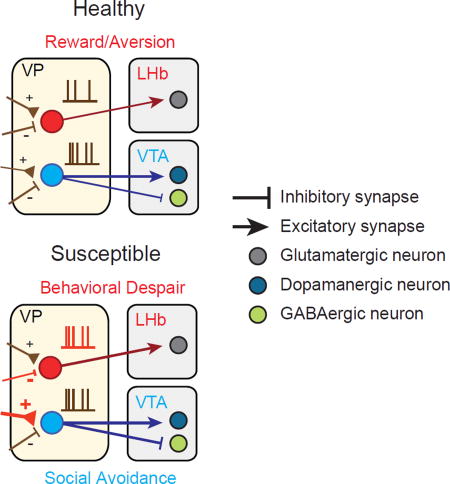
Distinct symptoms of depression are encoded in discrete circuits of the ventral pallidum, that project to separate brain regions contributing to depression.
Introduction
Multiple lines of evidence show that depression results from long term adaptations to stress in the brain and elicits a broad range of behavioral symptoms from anhedonia and feelings of helplessness to social withdrawal (Russo and Nestler, 2013). This diversity of behavioral symptoms suggests that discrete neural circuits may mediate separate depressive-like phenotypes. Studies have reported maladaptive plasticity of various circuit elements in animal models of depression; however, it remains unknown how these areas are interrelated or how different circuits contribute to discrete behavioral domains of depression (Lim et al., 2012).
The ventral pallidum (VP) is an important convergent point at the interface of the motivational and reward circuitry implicated in depression (Haber et al., 1985; Heimer et al., 1982; Smith et al., 2009). The VP receives dense inputs from the nucleus accumbens (NAc), a major component of the mesolimbic reward pathway, and transmits this information to downstream targets such as the ventral tegmental area (VTA) and the lateral habenula (LHb) (Root et al., 2015). While cellular and molecular changes in the NAc, LHb, and VTA have been identified as hallmarks of depression, identifying a link between these areas has remained elusive (Bagot et al., 2015; Friedman et al., 2014; Proulx et al., 2014). Given its connections to these regions, the VP is a likely target capable of integrating and transmitting depression-relevant signals across the brain. Recent studies have shown selective decreases in VP serotonin receptor binding in patients with MDD, and that the VP is critical for the antidepressant effects of ketamine (Murrough et al., 2011; Yamanaka et al., 2014). However, it is still unknown how the VP circuitry contributes to depression, and whether specific cell types within the VP are responsible for these effects.
The VP exhibits considerable cellular heterogeneity (Root et al., 2015), yet few studies have parsed the functional and anatomical organization of VP neuronal cell-types. A significant population of parvalbumin-positive (PV) neurons projecting to the NAc and VTA has been previously described in the VP (Gaykema and Zaborszky, 1997; Kuo and Chang, 1992). PV neurons have also been reported to be plastic in response to the valence of the environment in other brain areas (Donato et al., 2013), yet their function in the VP is, to date, unknown.
Here, we investigate the role of VP PV neurons underlying social defeat stress induced depressive behaviors. Specifically, we find that increased activity of VP PV neurons is a hallmark of depressed animals and can be reversed by chronic antidepressant treatment. Furthermore, we report that distinct VP PV neuronal projections to the LHb and VTA subserve different depressive behaviors: behavioral despair and social withdrawal, respectively. Both chronic and acute silencing of LHb-projecting VP PV neurons attenuates behavioral despair depressive-like phenotypes, while the same manipulation of the VTA-projecting population only rescues social withdrawal. Taken together, we suggest that increased VP PV neuronal activity is a salient feature of depression and that discrete components of the VP PV neuronal circuit contribute to specific behavioral subdomains of depression.
RESULTS
VP PV neurons projecting to the LHb or VTA represent independent subpopulations
Consistent with previous reports, we find a substantial population of PV neurons in the VP (Brauer et al., 1993; Zahm et al., 1996) using a Cre-dependent tdTomato (tdT) reporter line (Ai14-LSL-tdT) crossed with a PV-Cre mouse (Figure 1A; Figures S1A and S1B). To identify projection targets of VP PV neurons, we labeled VP PV axons and presynaptic terminals by injecting AAV-DIO-mRuby-T2A-Synaptophysin-eGFP into the VP of PV-Cre mice (Figure 1B). Unlike their cortical and striatal local-projecting counterparts, VP PV neurons are long-range projection neurons demonstrated by dense innervation in several brain areas including the LHb, VTA (lateral part), and others (Figures 1C–1E and Figure S1D).
Figure 1. Distinct subpopulations of VP PV neurons project to the LHb and VTA.

(A) PV neuronal localization in VP. Substance P (SP)-immunostaining denotes boundaries of VP. Scale bars: 100 µm; inset, 50 µm.
(B) Schematic of tracing strategy. Cell bodies and efferent fibers in red and presynaptic sites in green using AAV-DIO-mRuby2-T2A-Synaptophysin-eGFP.
(C) Representative image of injection site in VP. Scale bar: 80 µm.
(D and E) Fibers and synaptic puncta in LHb and VTA from VP PV neurons. Scale: 200 µm; inset, 20 µm.
(F) Two possible projection patterns: VP PV neurons send collateralized axons to both the LHb and VTA (top), or independent subpopulations send axons exclusively to either LHb or VTA (bottom).
(G and H) Schematic of viral strategy. EIAV is pseudotyped with a fusion protein containing rabies virus glycoprotein and VSVG transmembrane (RG-EIAV; G). Injection of RG-EIAV-DIO-Flp into target area and AAV-fDIO-eGFP into VP of PV-Cre mice.
(I) Images of injection site and target areas showing cell-type and projection-specific labeling. VP PV neurons selectively project to either the LHb or VTA, respectively, but not to both. Scale: 200 µm.
(J) Light-evoked synaptic responses in LHb or VTA. EPSCs measured at −70 mV, IPSCs at 0 mV. Pie charts indicate whether absolute amplitude was greater for evoked EPSC, IPSC, or no response (individual responses in Figure S4). Top row, LHb neurons; bottom row, VTA neurons distinguished as either GABAergic or dopaminergic. Scales: 50pA, 100ms.
(K) Strategy using PV-Flp×Ai14 mouse for labeling VP PV neurons in projection specific manner. RG-EIAV-fDIO-Cre is injected into either LHb (top) or VTA (bottom).
(L) mRNA labeling of PVVP→LHb neurons (top panel) and PVVP→VTA neurons (bottom panel). Green arrowheads show VGAT+ cells, white arrowheads VGluT2+ cells. Scale: 10 µm. Pie charts indicate percentage of tdTomato-mRNA+ neurons projecting to LHb or VTA colocalizing with probes to VGAT or VGluT2 (n = 154 tdT+ cells for VTA-projecting, n = 24 cells for LHb-projecting).
EIAV, equine infectious anemia virus; RV G, rabies virus glycoprotein; ac, anterior commissure; SNr, substantia nigra reticulata; MHb, medial habenula; CPu caudate putamen.
The VTA is primarily thought to mediate reward while the LHb encodes aversive signals (Pascoli et al., 2015; Shabel et al., 2012). These opposing functions led us to distinguish between two possible circuit organizations: 1) Do individual VP PV neurons send collateralized axons to target structures and coordinately regulate activity, or 2) Do LHb- and VTA-projecting VP PV neurons represent distinct subpopulations capable of independently regulating target activity (Figure 1F)? To define neurons by both their cell type and projection target, we developed and adapted a novel viral vector capable of neuron-specific retrograde infection by pseudotyping equine infectious anemia lentivirus (EIAV) with a fusion protein (FuG-B2) carrying extracellular and transmembrane domains of the rabies glycoprotein connected to the cytoplasmic domain of vesicular stomatitis virus glycoprotein (RG-EIAV, Figure 1G) (Cetin and Callaway, 2014; Kato et al., 2011). RG-EIAV confers stable, retrograde expression of transgene suitable for long-term studies without cytotoxicity.
In PV-Cre mice, we injected RG-EIAV inducing Flp recombinase in a Cre-recombinase-dependent manner (RG-EIAV-DIO-Flp) into either the LHb or VTA concurrently with an adeno-associated virus expressing eGFP in a Flp-dependent manner (AAV-fDIO-eGFP) into the VP (Figure 1H). In this manner, only PV neurons that project to the injection site of RG-EIAV-DIO-Flp (VTA or LHb) will be labeled. Examination of axonal fibers originating from LHb- or VTA-projecting VP PV neurons (PVVP→LHb, PVVP→VTA, respectively) revealed that cells projected primarily to the LHb or VTA, but not both (Figure 1I, and Figures S1E–S1G).
We also injected Cholera toxin subunit B conjugated to Alexa Fluor 488 or 647 into the LHb and VTA of transgenic PV-Cre × Ai14-tdT mice (Figures S1H and S1I) and quantitated double-labeled PV-tdT+ and CTb-positive neurons. We again found largely non-overlapping populations of neurons projecting to the LHb and VTA (Figure S1J), however they did not localize to specific anatomical regions within the VP (Figure S2).
VP PV neurons transmit both excitatory and inhibitory signals
To investigate the transmitter identity of PVVP→LHb and PVVP→VTA neurons, we injected AAV-DIO-ChR2 into the VP of PV-Cre animals and obtained whole-cell patch clamp recordings from neurons in the LHb or VTA. Surprisingly, blue light stimulation of VP PV terminals in the LHb evoked excitatory postsynaptic currents (eEPSCs) in nearly all LHb neurons, with smaller to no inhibitory postsynaptic currents (eIPSCs, Figure 1J top; Figures S3A–S3D).
Since the lateral VTA is known to contain GABAergic and dopaminergic (DAergic) neurons (Tan et al., 2012; Van Zessen et al., 2012), we sought to determine whether VP PV neurons specifically innervated one population or the other. To label GABAergic and DAergic neurons in slice, we crossed GAD67-GFP or TH-GFP mice with PV-Flp mice, a mouse line that expresses Flp-recombinase under control of the endogenous PV promoter (GAD67-GFP×PV-Flp, TH-GFP×PV-Flp, respectively), and injected AAV-fDIO-ChR2-tdT into the VP.
Interestingly, unlike PVVP→LHb neurons, we found that VP PV neurons send exclusively inhibitory projections to GABAergic VTA neurons. Despite being less synaptically connected than GABAergic neurons, DAergic neurons received mixed, but more excitatory inputs (Figure 1J, bottom; Figures S3E–S3J). In total, we see that VP PV neurons transmit exclusively inhibitory signals to GABAergic neurons and predominantly excitatory input to DAergic neurons.
Because PV neurons have long been considered to be GABAergic, we looked to further confirm our electrophysiological results. To precisely label PVVP→LHb or PVVP→VTA neurons, we used a new intersectional approach by crossing PV-Flp mice with Ai14-LSL-tdT reporter animals (PV-Flp×Ai14 hereafter). We then injected RG-EIAV expressing Cre-recombinase in a Flp-dependent manner (RG-EIAV-fDIO-Cre) into either the LHb or VTA such that only PVVP→LHb or PVVP→VTA neurons, respectively, would be selectively labeled with tdT (Figure 1K).
To validate whether RG-EIAV virus expression using this strategy was specific, we employed a multiplexing fluorescent in situ hybridization assay (FISH) to visualize mRNA expression of endogenous PV with tdT from RG-EIAV virus. Quantitation revealed the majority of virally labeled tdT+ neurons co-localized with PV mRNA (Figures S4A and S4B). Furthermore, employing the same labeling strategy we collected mRNA from individual tdT+ neurons from acute brain slices and performed reverse transcription polymerase chain reaction (RT-PCR; Figure S4C). RT-PCR results revealed that all tdT+ neurons collected expressed PV mRNA (Figures S4D–S4F).
Again injecting RG-EIAV-fDIO-Cre into the LHb or VTA of PV-Flp×Ai14 mice, we used FISH to probe mRNA expression of VGAT and VGluT2, markers for GABAergic and glutamatergic neurons, respectively. In line with our physiological recordings, we found extensive colocalization of tdT and VGluT2 mRNA in PVVP→LHb neurons, and mixed VGluT2 and VGAT colocalization for PVVP→VTA neurons (Figure 1L). Cells that co-expressed VGAT and VGluT2 were not found. In contrast, quantitation of all VP PV neurons (non-projection specific) with probes to VGAT and VGluT2 revealed that majority of VP PV neurons are GABAergic (Figures S4G and S4H) (Gaykema and Zaborszky, 1997).
Finally, we crossed PV-Flp animals with mice expressing Cre-recombinase in glutamatergic neurons (PV-Flp×VGluT2-Cre). We then injected a mixture of AAV-fDIO-tdT and AAV-DIO-eGFP into the VP. Colocalization of viral labeling in fixed tissue again revealed a population of VP PV neurons that were glutamatergic (Figure S4I). Immunohistochemistry with synaptic markers in the VTA and LHb also revealed similar trends (Figure S4J).
Upstream and downstream connectivity of PVVP→LHb and PVVP→VTA neurons
Since PVVP→LHb and PVVP→VTA neurons can be distinguished based on their efferent target, we hypothesized that they may receive distinct inputs as well. To test this, we probed the input-output organization of VP PV neurons by cell-type and projection-specific trans-synaptic tracing. First, we injected RG-EIAV-DIO-Flp into either the LHb or VTA, then Flp-dependent AAVs expressing TVA receptor and an optimized rabies glycoprotein into the VP (AAV-fDIO-mRuby-TVA, AAV-fDIO-oPBG, respectively) (Kim et al., 2016). We then delivered EnvA-pseudotyped, glycoprotein-deleted rabies virus (EnvA-RVΔG-eGFP) into the VP to map monosynaptic inputs to these two populations (Figure 2A) (Wickersham et al., 2007).
Figure 2. Input-output mapping reveals anatomically and molecularly distinct inputs of PVVP→LHb and PVVP→VTA neurons.

(A) Injection regiment to map pseudotype rabies-mediated monosynaptic inputs to either LHb-projecting or VTA-projecting VP PV neurons (top). Starter cell localization of AAV-fDIO-mRuby2-TVA, AAV-fDIO-oPBG, and EnvA-RVΔG-eGFP (bottom, yellow arrows). Scale: 200 µm.
(B) Whole-brain quantitation of inputs to PVVP→LHb and PVVP→VTA neurons. Data presented as mean ± SEM, percentage of total cells in a given brain area relative to total number of brain-wide inputs. Unpaired t-tests used for individual brain regions, * P < .05, ** P < .01, n = 4 for each condition.
(C) Representative images of inputs in select brain areas. Scale bars: 200 µm.
(D) Molecular characterization of CeA inputs to PVVP→VTA neurons via FISH assay. Quantitation (top) and representative images of mRNA labelling (bottom). n = 257 cells from 2 animals. Scale: 100 µm.
(E) Images of CeA neurons sending input to PVVP→VTA neurons with cell-type specific markers. Colored arrows represent colocalization between rabies+ input cells and mRNA for specified probe. Scale: 10 µm.
(F) Quantitation of NAc D1R- and D2R-expressing MSN inputs to PVVP→LHb and PVVP→VTA neurons. Scale: 100 µm.
(G) Representative images in NAc showing colocalization of D1R or D2R in neurons projecting to PVVP→LHb and PVVP→VTA neurons. Red arrows denote D1R colocalization, white arrows D2R, yellow arrows co-express D1R and D2R. N = 1505 cells from 2 animals each. Scale: 10 µm.
mPFC, medial prefrontal cortext; BNST, bed nucleus stria terminalis; LH, lateral hypothalamus; LGP, lateral globus pallidus; AHA/VMH, anterior hypothalamic area/ventromedial hypothalamus; PAG, periaqueductal grey; RMTg, rostromedial tegmental nucleus; DRN, dorsal raphe nucleus. SST, somatostatin; PENK, preproenkephalin; CRH, corticotropin releasing hormone; PKCδ, protein kinase C delta.
Whole-brain quantitation of eGFP-labeled neurons revealed significant differences in inputs to PVVP→LHb and PVVP→VTA neuronal populations. In particular, while PVVP→LHb neurons receive proportionally more input from the bed nucleus of the stria terminalis (BNST) and medial shell of NAc, PVVP→VTA cells receive significantly more input from areas such as the NAc lateral shell (NAc lSh), subthalamic nucleus (STN), and central amygdala (CeA) (Figures 2B and 2C).
Since the CeA to VP connection has been poorly defined, we combined input-output viral tracing with FISH to investigate the molecular identity of CeA input neurons. Amongst the major cell types in the CeA (Haubensak et al., 2010), we found the most extensive colocalization of rabies-eGFP+ CeA neurons with preproenkephalin (PENK) (Figures 2D and 2E).
Finally, recent work has shown that contrary to canonical direct-indirect pathway notions of accumbal projections to the VP, both D1- and D2-receptor (D1R and D2R) expressing medium spiny neurons (MSNs) project to the VP (Creed et al., 2016; Kupchik et al., 2015). As such, we investigated whether PVVP→LHb and PVVP→VTA received preferential input from D1R or D2R MSNs. Quantitation revealed that both PVVP→LHb and PVVP→VTA neurons receive proportionally more input from D2R-expressing MSNs than D1R-expressing MSNs, with a smaller fraction of input neurons co-expressing D1R and D2R (Figures 2F and 2G).
We next sought to determine the downstream connectivity of LHb and VTA neurons receiving input from VP PV neurons. We used a multi-virus strategy by injecting AAV-DIO-Synaptophysin-eGFP into the VP of PV-Cre mice and glycoprotein-deleted rabies viruses encoding different fluorescent markers (RVΔG-tdT, RVΔG-mTagBFP2, RVΔG-iRFP670) into common output regions of the VTA and LHb (Figure S5A). This approach revealed that LHb neurons receiving inputs from VP PV neurons project to the RMTg while VTA neurons project predominantly to the NAc lSh (Figures S5B–S5D).
Since we were limited by modest expression of RV-iRFP670 and RV-mTagBFP, we leveraged the development of rabies virus expressing spaghetti monster fluorescent proteins (RV-smFPs) which combines the advantages of epitope tagging and traditional fluorescence to robustly label LHb and VTA outputs (Figure S5E; Viswanathan et al., 2015). Somatic and dendritic quantitation of smFP-synaptophysin+ synaptic contacts again revealed that VTA neurons receiving input from VP PV neurons primarily projected to NAc lSh while LHb neurons preferentially project to the RMTg (Figures S5F–S5I).
PVVP→LHb neurons mediate reward and aversion
Since our tracing data suggests that PVVP→LHb and PVVP→VTA neurons represent discrete subpopulations, we hypothesized that they may have different roles in reward-related behaviors. To this end, we used optogenetics to test whether PVVP→LHb and PVVP→VTA neurons mediate reward and aversion by subjecting mice to a three-day context dependent conditioned place preference/aversion (CPP/CPA) protocol (Figures S6A–S6C)(Lammel et al., 2012a).
In line with previous studies indicating that increases in LHb activity are aversive (Shabel et al., 2012), silencing PVVP→LHb glutamatergic projections induced a robust preference on day 3 of testing while stimulation of this projection induced aversion and even blocked cocaine-induced preference (Figures S6D–S6H). Surprisingly, we observed no change in preference or aversion after PVVP→VTA pathway manipulation suggesting that PVVP→LHb and PVVP→VTA neurons have differential effects on reward and can mediate separate behaviors.
Stress-induced hyperactivity of PVVP→LHb neurons
The differential effects of projection-specific manipulation observed in CPP/CPA in conjunction with our tracing data strongly suggest that these populations are separately involved in motivation-related behaviors. Since elevated activity in the LHb and VTA have been previously described in maladapted behavioral states such as depression (Cao et al., 2010; Li et al., 2011), we next reasoned that PVVP→LHb and PVVP→VTA may exhibit aberrant physiological adaptations in depression as well.
To mimic depression in mice, we used the chronic social defeat stress (SDS) model of depression (Figure 3A and 3B) (Golden et al., 2011). SDS reliably induces an array of depressive-like phenotypes that parallel those seen in humans. Additionally, SDS exhibits robust predictive validity; chronic but not acute antidepressant treatment reverses its behavioral effects (Berton et al., 2006, Figure 3C). After ten days of SDS, mice can be separated into those that develop depressive-like behaviors (susceptible) and those that actively respond and cope with stress and do not display depressive-like symptoms (resilient)(Franklin et al., 2012).
Figure 3. PVVP→LHb and PVVP→VTA neurons from susceptible animals exhibit distinct electrophysiological adaptations to SDS and can be reversed by chronic fluoxetine.

(A and B) Behavioral paradigm and experimental timeline of repeated chronic social defeat stress (SDS) experiments.
(C) Chronic fluoxetine (FLX) treatment ameliorates SDS-induced social withdrawal. Red line indicates bound between susceptible (< 1) and resilient animals (> 1).
(D) Schematic for labeling PVVP→LHb neurons in acute slices.
(E and F) Spikes elicited after 100 pA current injection in PVVP→LHb neurons. One-way ANOVA F3,34 = 9.088, P < .001. Tukey post-test; *** P < .001, ** P < .01, * P < .05; n = 9, 11, 10, 8 cells from 4 animals each in control, resilient, susceptible, and FLX groups, respectively. Example traces in (F), blue bars represent duration of current injection (500 ms). For full responses see Fig S7A.
(G–I) Evoked excitatory and inhibitory inputs (E/I) onto PVVP→LHb neurons in healthy and SDS groups. Absolute amplitudes (G), ratio of E/I inputs (E/I ratio; H), and example traces (I) of cells recorded. One-way ANOVA F3,42 = 5.499, P < .01; Tukey post-test: ** P < .01; n = 8, 9, 13, 16 cells from 5 animals each in control, resilient, susceptible, FLX groups, respectively.
(J) Schematic for labeling PVVP→VTA neurons in acute slices.
(K–L) Same as in (E and F), but PVVP→VTA neurons. One-way ANOVA F3,36 = 8.770, P < .001. Tukey post-test; *** P < .001, ** P < .01, * P < .05; n = 12, 10, 10, 8 cells from 7, 4, 4, and 3 animals in control, resilient, susceptible, and FLX groups, respectively. For full responses see Fig S7B.
(M–O) PVVP→VTA neurons have a significant increase in E/I ratio in susceptible animals that is normalized by FLX. Absolute amplitudes (M), E/I ratio quantitation (N), and example traces (N).
One-way ANOVA F3,50 = 9.585, P < .001; n = 11, 15, 13, 15 cells from 5 animals each in control, resilient, susceptible, FLX groups, respectively. Tukey post-test: *** P < .001.
Scale for (F and L): 100 ms, 10 mV; (I and O): 200 pA, 100 ms. Data presented as average mean ± SEM.
To label only PVVP→LHb or PVVP→VTA neurons for electrophysiological recordings, we again employed the intersectional strategy using PV-Flp×Ai14 mice injected with RG-EIAV-fDIO-Cre into the LHb or VTA (Figure 3D and 3J). Ex vivo acute brain slices were prepared and whole-cell patch clamp recordings obtained from healthy animals and a separate cohort subjected to SDS and separated into resilient and susceptible animals. We also treated a cohort of susceptible animals to two weeks of daily antidepressant treatment (fluoxetine (FLX); Figure 3B).
Interestingly, we observed an increase in intrinsic excitability of PVVP→LHb neurons in both susceptible and resilient animals following SDS, yet animals that were previously susceptible but received chronic treatment of FLX exhibited excitability levels similar to controls (Figure 3E and 3F; Figure S7A). We also examined changes in synaptic inputs by measuring the ratio of evoked EPSCs and IPSCs (E/I ratio). We observed that susceptible animals had significantly greater E/I ratios compared to resilient and FLX-treated animals (Figures 3G–3I), suggesting that FLX-treated animals have an afferent profile that closely resembles resilient and healthy animals. This maintained, reduced E/I ratio observed in resilient animals may serve to counteract the increase in SDS-induced excitability.
To understand whether the increase in E/I ratio observed in susceptible animals was due to an increase in excitatory input, a reduction in inhibitory input, or both; we measured the frequency and amplitude of spontaneous IPSCs and EPSCs (sIPSCs and sEPSCs, respectively). We found a decrease in the frequency and amplitude of sIPSCs in PVVP→LHb neurons in susceptible animals, yet no changes in sEPSCs. The reduction in sIPSC frequency persisted in animals treated with FLX, but the amplitude of sIPSCs returned to the level of control group. (Figures S7F–S7I). Taken together, the increase in intrinsic excitability coupled with the decrease in inhibitory drive onto susceptible PVVP→LHb neurons has a net increase in glutamatergic PVVP→LHb activity.
It is possible that stress-induced alterations in synaptic input may cause the observed excitability changes. To isolate excitability measures from synaptic input, we measured intrinsic excitability in the presence of blockers for excitatory and inhibitory synaptic currents, NBQX and PTX, respectively. We also quantified membrane and action potential (a.p.) properties to gain an in-depth understanding of how excitability changes may arise (Figure S8A). We again find increased excitability in both resilient and susceptible animals, suggesting that changes in intrinsic excitability are not a secondary effect of synaptic alterations (Figure S8B). Furthermore, we find that the rheobase, the minimum current required to elicit an a.p., as well as the a.p. threshold and half-width, is significantly reduced in susceptible PVVP→LHb neurons relative to healthy controls (Figures S8C–S8L).
Related, but distinct SDS-driven cellular changes in PVVP→VTA neurons
We next examined whether PVVP→VTA neurons also displayed SDS-driven physiological changes. In contrast to PVVP→LHb neurons, PVVP→VTA neurons in susceptible animals displayed no changes in intrinsic excitability compared to healthy controls. However, a significant decrease in excitability was observed in both resilient and FLX-treated groups (Figures 3K and 3L, Figure S8B) representing possible compensatory changes functioning to maintain low levels of activity in these cells.
Blunted excitability of PVVP→VTA neurons in resilient animals was also accompanied by a significant increase in rheobase and action potential half-width (Figures S8N and S8Q). We also observed significant stress-induced changes in membrane properties. Namely, neurons from resilient animals displayed increases in capacitance and corresponding reductions in membrane resistance (Figures S8U and S8V).
Similar to the PVVP→LHb population, PVVP→VTA neurons in susceptible animals displayed a significant increase in E/I ratio relative to healthy and resilient animals that was reversed by FLX treatment (Figures 3M–3O). This reversal was not due to passive recovery during the two week FLX treatment period post SDS, as the E/I ratio remained elevated after chronic saline injections (Figures S7C–S7E) (Krishnan et al., 2007).
We additionally found an increase in both frequency and amplitude of sEPSCs, but not sIPSCs, in PVVP→VTA neurons in susceptible groups relative to healthy controls which was reversed by FLX (Figures S7J–S7M). The increase in excitatory synaptic input to VP PV neurons via enhanced E/I ratio and sEPSC frequency and amplitude selectively observed in susceptible animals suggests that, similar to PVVP→LHb neurons, increased PVVP→VTA activity is a hallmark of depressed mice. Interestingly, although both populations exhibit enhanced activity, the cellular adaptations by which this altered susceptible state is achieved are different.
Overall, we find that enhanced VP PV neuronal activity is a salient characteristic of susceptible animals which can be normalized by an antidepressant commonly prescribed to human patients with MDD. Furthermore, animals that have undergone SDS but are resilient exhibit neither the social avoidance behavioral phenotypes nor the physiological changes seen in susceptible animals.
Silencing of VP PV neurons induces resilience to SDS
Our physiological results strongly suggest that cellular hyperactivity of PVVP→VTA and PVVP→LHb neurons is a hallmark of animals susceptible to SDS. If heightened activity of VP PV neurons is characteristic of susceptibility to SDS, we hypothesized that reducing VP PV activity may promote resilience to SDS. To achieve this, we expressed Kir2.1, an inward-rectifying potassium channel that reduces cellular activity by hyperpolarizing cells (Rothwell et al., 2014), in VP PV neurons and subsequently subjected mice to SDS (Figure 4A).
Figure 4. Silencing VP PV activity promotes resilience to a subset of depressive-like phenotypes.

(A) Experimental timeline of Kir2.1-mediated silencing.
(B–D) No changes in pre-stress measures of locomotion in open field test (OFT, B), anxiety on elevated plus maze (EPM, C), or social interaction (SI, D). n = 6, 11 for eYFP, Kir2.1, respectively.
(E) No significant difference in measures of anhedonia on sucrose preference test (SPT) after SDS.
(F) Kir2.1 animals show significantly reduced time spent immobile on measure of behavioral despair, the tail suspension test (TST) after SDS. U = 69, P < .001; n = 8, 9 for eYFP and Kir2.1, respectively.
(G) Kir2.1 animals display increased social interaction ratios relative to controls after SDS. U = 13, P < .05; n = 8, 8 for eYFP and Kir2.1, respectively. All data tested on Mann-Whitney U-test and presented as mean ± SEM. * P < .05, ** P < .01.
Silencing of VP PV neuronal activity did not induce changes in baseline measures of locomotion, anxiety, or sociability (Figure 4B–4D). Interestingly, after 10 days of SDS we observed that reduced VP PV neuronal activity significantly attenuated the acquisition of a subset of depressive-like behaviors; behavioral despair/helplessness as measured by the tail suspension test (TST) and social avoidance in social interaction test (SI) (Steru et al., 1985).
On the other hand, no difference was seen in the sucrose preference test (SPT), a measure of anhedonia (Figure 4E–4G). These differences indicate that reducing VP PV neuronal activity promotes a pro-resilient like phenotype in a subset of depressive-like behaviors; social withdrawal and behavioral despair but not anhedonia.
Distinct VP PV neuronal circuitries are involved in different subsets of depressive behaviors
Given that increased activity of VP PV neurons is characteristic of susceptible animals and that silencing of these neurons attenuates the acquisition of behavioral despair and social withdrawal phenotypes, we hypothesized that silencing of VP PV neurons in susceptible animals would acutely reverse depressive-like behaviors.
To specifically manipulate each population in vivo, PV-Cre animals were injected with AAV-DIO-ChR2-eYFP, AAV-DIO-NpHR3.0-eYFP, or AAV-DIO-eYFP into the VP and optic cannulae implanted over the LHb or VTA (Figures 5A and 5B). In SDS-naïve mice, silencing or activating VP PV neuronal terminals in both LHb and VTA showed no difference in baseline social interaction compared to eYFP controls (Figures S5I and S5J). After SDS, we again observed that PVVP→LHb neuronal terminal manipulation had no effect on social interaction. However, NpHR-mediated inhibition of the PVVP→VTA pathway reversed social withdrawal behavior as indicated by a robust increase in social interaction (Figure 5C–5F).
Figure 5. PVVP→LHb and PVVP→VTA neurons mediate discrete symptoms of depression.

(A and B) Schematic of viral injections and optic fiber implantations for optogenetic manipulation in stressed animals.
(C and D) Representative traces of animals expressing NpHR in VP PV neurons in social interaction (SI) test in susceptible animals during light stimulation: off (left panel) and light on (589 nm; right panel). Red rectangle indicates cage in which aggressive CD1 animal was placed, yellow dashed lines outline ‘interaction zone.’ Warmer colors indicate increased time spent.
(E and F) Silencing terminals of VP PV neurons in VTA, but not in LHb, alleviates social withdrawal symptoms in SI test. SI ratio = time interaction zone light ON / light OFF. Dashed line indicates no change. Each line in (E) represents single animal, bolded line indicates average ± SEM. Mann-Whitney U-test U = 80, P < .05. (F) shows population means of data in (E). VTA SI: one-way ANOVA F2,21 = 6.132, P < .01. Tukey post-test: * P < .05; n = 5, 9, 10 and n = 6, 7, 9 animals for eYFP, ChR2, NpHR groups in VTA and LHb conditions, respectively.
(G and H) Modulation of VP PV neuronal terminals in LHb, but not in VTA induces bidirectional effects on the tail suspension test (TST). For (G), LHb-ChR2: Mann-Whitney U-test U = 56, ** P < .01; LHb-NpHR U = 15, *** P < .001. For (H), LHb TST: one-way ANOVA F2,25 = 24.49 P < .001. Tukey post-test: * P < .05; n = 5, 8, 11 and 6, 8, 14 for eYFP, ChR2, and NpHR in VTA and LHb groups, respectively. All data reported as mean ± SEM.
Conversely, on measures of behavioral despair/helplessness in the TST we found that manipulation of PVVP→VTA neuronal terminals had no effect on time spent immobile, yet manipulation of PVVP→LHb neuronal terminals induced a strong bidirectional effect. Inhibition and stimulation caused significant decreases and increases, respectively, in time spent immobile compared to healthy controls (Figure 5G and 5H).
Taken together with the electrophysiological results, it is likely that NpHR-mediated silencing mitigates the elevated activity seen in susceptible PVVP→LHb and PVVP→VTA populations resulting in acute reversal of SDS-induced behavior.
Chemogenetic manipulation recapitulates optogenetic behavior
Recent studies have challenged the efficacy of optogenetic terminal inhibition (Mahn et al., 2016). To circumvent this potential constraint, we utilized chemogenetics by using ‘designer receptors exclusively activated by designer drugs’ (DREADDS) (Armbruster et al., 2007). We expressed the Gi-coupled inhibitory DREADD (hM4Di) in PVVP→LHb or PVVP→VTA neurons by bilaterally injecting RG-EIAV-DIO-Flp into the LHb or VTA in PV-Cre mice, and AAV-fDIO-hM4Di-mCherry or AAV-fDIO-mCherry into the VP (Figure 6A and 6B). Thus, we achieved selective inhibition of PVVP→LHb or PVVP→VTA neurons by injecting clozapine-N-oxide (CNO), an inert ligand specific to hM4Di receptors.
Figure 6. Cell type- and projection-specific inhibitory DREADDs recapitulate optogenetic behavior.

(A) Experimental setup for expression of inhibitory DREADD (hM4Di) or control virus (mCherry) in PVVP→LHb or PVVP→VTA neurons.
(B) Representative images of injection site. hM4Di expression in red, Substance P (SP) in blue to delineate VP.
(C) Inhibition of PVVP→VTA neurons expressing hM4Di by CNO injection promotes increased social interaction in susceptible animals. Mann-Whitney U-test U=6, * P < .05. Individual lines represent individual animals. n = 8, 9 for mCherry, hM4Di, respectively.
(D) Inhibition of PVVP→LHb neurons expressing hM4Di by CNO injection has no effect on social interaction in susceptible animals.
(E) Changes in SI ratio in response to i.p. CNO injection. Mann-Whitney U-test U=5, * P < .05.
(F) No effect of CNO injection in animals expressing hM4Di in PVVP→VTA neurons on behavioral helplessness as measured by change in time immobile with respect to saline injections.
(G) The inhibition of PVVP→LHb neurons expressing hM4Di by CNO injection reduces time spent immobile relative to saline injections in susceptible animals. Mann-Whitney U-test U=40, * P < .05. n = 8, 7 for mCherry, hM4Di, respectively.
(H) hM4Di expression in PVVP→LHb, but not PVVP→VTA neurons, reduces time spent immobile in response to CNO injection. Mann-Whitney U-test U=42, * P < .05. n = 8, 7 for mCherry, hM4Di, respectively.
Consistent with optogenetic data, CNO-mediated inhibition of PVVP→VTA, but not PVVP→LHb neurons increased social interaction in susceptible animals with no effects on locomotion (Figure 6C–6E, data not shown). Additionally, CNO-mediated inhibition of PVVP→LHb, but not PVVP→VTA reduced the amount of time spent immobile on the TST (Figure 6F–6H). Overall, we show that both chemogenetic and optogenetic silencing of PVVP→LHb and PVVP→VTA activity induces symptom-specific anti-depressive effects in susceptible mice.
PVVP→VTA stimulation is sufficient to induce social withdrawal
If a decrease in VP PV activity can promote resilience (Kir2.1 experiments) and both optogenetic and chemogenetic inhibition of PVVP→LHb and PVVP→VTA activity in susceptible mice attenuates depressive-like behaviors, we hypothesized that artificially driving PVVP→LHb or PVVP→VTA activity may be sufficient to recapitulate phenotypes seen in SDS-exposed animals.
To test this, we injected AAV-DIO-ChR2-eYFP into the VP and implanted optic fiber cannula over the LHb and VTA. Twice a day for ten days, animals were subjected to ten minutes light stimulation in their home cage (Figure 7A). Surprisingly, ten days of PVVP→VTA terminal stimulation without any previous exposure to aggressive CD1 mice was sufficient to induce a social avoidance phenotype similar to susceptible animals (Figure 7B). In line with our previous results demonstrating behavioral specificity, PVVP→VTA stimulation did not induce an effect in the TST or SPT (Figure 7C and 7D). On the contrary, PVVP→LHb terminal stimulation had no effect (Figure 7E–7G).
Figure 7. Repeated, chronic stimulation of PVVP→VTA terminals is sufficient to induce social interaction deficits.

(A) Experimental timeline for B–G.
(B–D) Repeated stimulation of VP PV terminals in VTA is sufficient to induce social avoidance (B), but has no effect on SPT (C) or TST (D). Mann-Whitney test; U = 162, * p < .05; n = 15, 14 for eGFP and ChR2 groups, respectively for SI and TST test, n = 7 for SPT).
(E–G) No effect of repeated stimulation of VP PV terminals in LHb in SI (E), SPT (F), or TST (G); n = 5 each condition.
(H–J) No change in depressive like phenotypes after subthreshold defeat stress with VP PV terminal stimulation in LHb (n = 5 each SI, SPT, TST). All data tested on Mann-Whitney U-test and presented as mean ± SEM. Red dashed lines indicate levels separating resilient and susceptible animals (SI ratio of 1 and 50% sucrose preference for SI, SPT, respectively).
We then reasoned that mice may require acute subthreshold environmental exposure to CD1 aggressors as a primer in order for PVVP→LHb stimulation to affect changes (Christoffel et al., 2015; Walsh et al., 2013). Again, PVVP→LHb terminal stimulation in conjunction with subthreshold exposure to aggressive CD1 mice was not sufficient to induce differences in SI, SPT, or TST, suggesting that PVVP→LHb terminal activation is necessary but not sufficient to elicit depressive-like behaviors (Figure 7H–7J).
Discussion
Uncovering neural circuitries for specific depressive symptoms
The DSM-V lists several core symptoms present in patients with MDD, and requires that at least five be present for official diagnosis. Inherent in this diagnostic guideline is the wide patient to patient variability of behavioral symptoms, yet few studies have taken a systematic approach to understand how different circuits may underlie the behavioral heterogeneity seen in MDD. We find that silencing distinct VP PV neuronal outputs ameliorates separate depressive-like behaviors. In particular, silencing PVVP→LHb neurons attenuates helplessness on the TST, while reduction of PVVP→VTA neuronal activity increases social interaction in animals susceptible to SDS. Furthermore, chronically reducing VP PV neuronal activity before animals undergo SDS promotes a pro-resilient phenotype in a subset of depressive-like behaviors: social withdrawal and behavioral despair/helplessness but not anhedonia.
In addition to increased activity of VP PV neurons being necessary for the development and expression of SDS-induced helplessness and social withdrawal behaviors, we also show that prolonged artificial stimulation of PVVP→VTA neuronal terminals in VTA is sufficient to induce social withdrawal, but not behavioral despair phenotypes (Figure 7). Surprisingly, this suggests that the PVVP→VTA circuit specifically encodes social withdrawal phenotypes independent of environmental primers such as acute subthreshold exposure to aggressive CD1 mice.
On the other hand, artificially driving PVVP→LHb terminal activity in an animal’s home cage or during subthreshold SDS is insufficient to induce any behavioral abnormalities, suggesting that this circuit may either not be sufficient to induce depressive behaviors or that it partakes in different forms of neural adaptations that may require more long term exposure to adverse environmental stimuli.
Enhanced PVVP→LHb and PVVP→VTA activity defines susceptibility to SDS
Previous studies report increased cellular activity in the LHb and dopaminergic neurons in the VTA in animal models of depression (Chaudhury et al., 2013; Li et al., 2011). Using ex vivo whole cell patch clamp electrophysiology we find that susceptible, but not resilient animals, are characterized by increased activity in both PVVP→LHb and PVVP→VTA populations via altered synaptic input, but by different mechanisms. Increased activity of PVVP→LHb and PVVP→VTA neurons observed in susceptible animals would thus have a net effect of enhanced postsynaptic activity in the LHb and VTA, in line with previous reports. It may be the case that aberrant ventral pallidal activity is upstream of the stress-induced increases in firing previously observed in the LHb and VTA.
The different synaptic adaptations between PVVP→LHb and PVVP→VTA populations may in part be explained by the distinct input patterns observed on to each population (Figure 2).
Our mapping study of the input-output relationship of VP PV neurons demonstrate that PVVP→VTA and PVVP→LHb neurons receive excitatory or inhibitory inputs from distinct brain areas. Thus, it is possible that projections from different areas preferentially targeting specific VP PV neuronal populations, such as the CeA which preferentially targets PVVP→VTA, may induce selective synaptic changes of individual populations in susceptible animals, which in turn elicits specific depressive behaviors. Future studies will need to parse the contributions of area-specific synaptic inputs onto VP PV neurons to understand which precise upstream pathways drive SDS-induced changes within the VP.
We also show that chronic fluoxetine (FLX), an FDA-approved antidepressant prescribed in humans, reverses the SDS-induced cellular adaptations in PVVP→LHb and PVVP→VTA neurons to healthy levels by modulating intrinsic properties and synaptic balance (Figure 3).
However, it is important to emphasize that while chronic FLX treatment induces changes in VP PV neurons, it remains unclear if this is a direct effect or whether FLX engages a larger circuit by which VP PV neurons are a part of. Many studies have underscored serotonergic neurons in the DRN as a critical mediator of FLX actions (Adachi et al., 2016; Neumaier et al., 1996). Our tracing data reveals that VP PV neurons receive input from the DRN (Figure 2), which could alternatively explain how FLX treatment affects VP PV activity rather than direct effects on VP PV neurons themselves. Further studies are required to parse this possibility, yet for now it is sufficient to say that VP PV neuronal activity is modulated – directly or indirectly – by chronic FLX treatment and correlates with the presentation of SDS-induced depressive-like symptoms.
Engagement of distinct downstream midbrain circuitry
How might different components of a related circuit mediate separate behaviors? One manner in which this may be possible is the engagement of separate downstream midbrain circuitries by PVVP→LHb and PVVP→VTA neurons. We show that PVVP→LHb neurons preferentially synapse onto LHb neurons projecting to the RMTg and cortex-projecting medial VTA neurons (Figure S5D; Lammel et al., 2012). Both the LHb→RMTg→VTA and LHb→medial VTA→cortex connections have been reported to encode aversive signals (Lammel et al., 2012b), consistent with our results showing that the activation of glutamatergic PVVP→LHb fibers induces aversion (Figure S6A–S6H).
It also follows that increased activity of PVVP→LHb neurons in susceptible animals would increase LHb excitatory inputs to the cortex-projecting medial VTA neurons and to RMTg neurons, which are mainly GABAergic, that have been reported to inhibit VTA DA neurons (Beier et al., 2015; Lammel et al., 2012a). Consequently, it may be the combined downstream effect of increased medial cortex-projecting VTA DA neuronal activity (via excitatory LHb projections) with decreased other VTA DA neuronal activity (via inhibitory RMTg projections) that engages changes in helplessness/behavioral despair in susceptible animals.
PVVP→VTA neurons preferentially project to the lateral portion of the VTA which contains largely non-overlapping populations of GABAergic and DAergic neurons. Previous studies indicate that lateral VTA neurons preferentially project to the lateral shell of the NAc, a finding supported by our own tracing data (Lammel et al., 2008; Figure S5). VP PV neurons synapse on both cell types, yet to separate degrees and strength. VP PV neurons send strong inhibitory projections to GABA neurons, while DA neurons receive mixed, but stronger excitatory projections.
It follows then that increased PVVP→VTA neuronal activity seen in susceptible animals would drive a reduction in GABA VTA activity and net increase in monosynaptic DA VTA activity. Previous studies have shown that GABA VTA neurons send local inhibitory projections to DA neurons (Van Zessen et al., 2012). Thus, in addition to direct DA excitation, the net reduction in GABAergic VTA activity would relieve its inhibition onto DAergic neurons and further drive increased VTA DA activity – a hallmark previously described in susceptible animals (Cao et al., 2010; Chaudhury et al., 2013).
Together, we see that the same manipulation at VP PV neuronal projections in different target structures may have distinct changes in neuronal activity within different populations of VTA neurons. Ultimately, the separate effects on specific downstream midbrain areas and different combinations of activity across regions may account for the discrete effects of PVVP→LHb and PVVP→VTA pathway in different depressive behaviors.
Of note, we surprisingly find no effect on social interaction silencing the PVVP→VTA pathway in healthy animals, yet after stress the same manipulation induces a robust attenuation of social withdrawal. This suggests that the synaptic connection between VP PV neurons and VTA neurons is weak in non-stressed animals, but may get selectively stronger in susceptible animals which is required to induce behavioral effects on social interaction. One possible explanation for the post-stress synaptic enhancement of this pathway is increased activity of PVVP→VTA neurons. In line with this reasoning, we show that PVVP→VTA neurons receive increased excitatory synaptic input in susceptible, but not resilient mice that persists up to 2 weeks after SDS (Figures 3J–3O). In effect, it is this increase in activity of PVVP→VTA neurons that is required for the expression of social avoidance behaviors, and explains why we only see effects of silencing PVVP→VTA neurons in susceptible, but not healthy animals. This also explains why silencing VP PV neuronal activity before SDS with Kir2.1 would reduce the development of social withdrawal symptoms in response to stress, and why reduction of PVVP→VTA neuronal activity with FLX ameliorates social avoidance in susceptible animals as well.
However, since PVVP→VTA neurons synapse onto two distinct cell types, there may be either: 1) a selective enhancement of one pathway over the other (GABA vs. DA) or, 2) an altered balance of synaptic drive onto DA neurons, in particular an increase in excitatory drive. Future interrogation into the downstream circuitries and midbrain cell types engaged by VP PV neurons and their neural adaptations in response to SDS will be required.
In sum, we delineate and describe two distinct PV-positive VP circuits that underlie separate depressive-like behaviors. We believe these results highlight the need for more circuit-based studies in neuropsychiatric disorders to parse the neural underpinnings of distinct behavioral symptoms in patients to move towards symptom-based treatments in the future.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Byungkook Lim (bklim@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All procedures to maintain and use mice were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California, San Diego. Mice were maintained on a 12-h:12-h light: dark cycle with regular mouse chow and water ad libitum. CD-1 male retired breeders and C57BL6 mice were purchased from Charles River Laboratories and Jackson Laboratories, respectively. Pvalb-ires-Cre, Rosa-CAG-LSL-tdTomato (Ai14), and PV-Flp mice were obtained from the Jackson Laboratory (00869, 007914, 022730, respectively). TH-GFP and GAD67-GFP mice were gifts from Dr. Davide Dulcis and Dr. Yuchio Yanagawa, respectively. The social defeat stress protocol used requires male C57BL6 mice (Golden et al., 2011), thus all subjects for behavior and electrophysiological experiments were 8–12 week adult hemizygous transgenic male mice and heterozygous knock-in males on a C57BL6/JL background. For behavioral experiments, all animals were singly housed after undergoing SDS. In all other cases, animals were group-housed. For tracing experiments, 8–12 week old male and female mice were used. No difference between sexes were observed so data was combined.
METHOD DETAILS
Virus generation
AAV vector plasmids were constructed using standard molecular cloning methods. Synaptophysin-eGFP, oPBG, and hM4D(Gi)-mCherry DNA fragments were obtained from pAAV-phSyn1(S)-FLEX-tdTomato-T2A-SypEGFP-WPRE (a gift from Hongkui Zeng; Addgene plasmid #51509) and pAAV-EF1α-DIO-oPBG (a gift from Edward M. Callaway), respectively. All ‘spaghetti monster’ fluorescent protein (smFP) DNA fragments were obtained from pCAG-smFP-FLAG, pCAG-smFP-HA, pCAG-smFP-Myc, and pCAG-smFP-V5 (gifts from Loren Looger; Addgene #59756–59759). A Flp-dependent, double-floxed inverted open reading frame (fDIO) was constructed with two heterospecific pairs of FRT and FRT5 sequences based on pAAV-EF1a-fDIO-hChR2(H134R)-eYFP (a gift from Karl Deisseroth; Addgene #55639). AAV vector plasmids encoding ChR2 (H134R variant) and eNPHR3.0 were gifts from Karl Deisseroth. We used EF1α promoter to drive the expression of target constructs for all AAV vectors except for AAV-DIO-mRuby2-T2A-Synaptophysion-eGFP which is driven by the human synapsin promoter. pAAV-hSyn-DIO-hM4D(Gi)-mCherry was a gift from Bryan Roth (Addgene plasmid # 44362)
All AAV vectors used in this study were packaged as serotype DJ and generated as previously described (Lim et al., 2012). In brief, AAV vectors were produced by transfection of AAV293 cells (Agilent) with three plasmids: an AAV vector plasmid carrying target constructs (DIO-mRuby2-T2A-Synaptophysin-eGFP, DIO-Synaptophysin-eGFP, DIO-Synaptophysin-smFP-FLAG, fDIO-eGFP, fDIO-oPBG, fDIO-mRuby2-P2A-TVA, DIO-ChR2(H134R)-eYFP, DIO-eNpHR3.0-eYFP, DIO-eYFP, fDIO-hM4D(Gi)-mCherry, or DIO-Kir2.1-T2A-ZsGreen), AAV helper plasmid (pHELPER; Agilent), and AAV rep-cap helper plasmid (pRC-DJ, gift from M. Kay). At 72 h post-transfection, the cells were collected and lysed by a repeated freeze-thaw procedure. Viral particles were then purified by an iodixanol step-gradient ultracentrifugation and subsequently concentrated using a 100-kDa molecular cutoff ultrafiltration device (Millipore). The genomic titer was determined by quantitative PCR. The AAV vectors were diluted in PBS to a working concentration of approximately 1013 viral particles/mL.
EIAV genomic vector plasmids were constructed from the initial pEIAV-SIN6.1-CBGFPW (a gift from John Olsen; Addgene #44173) by replacing eGFP coding sequence with DNA fragments containing DIO-Flp and fDIO-Cre, respectively. RG-EIAV vectors were generated by a modified version of a published protocol (Cetin and Callaway, 2014). Briefly, HEK293-T cells were transfected with three plasmids: an EIAV genomic vector (pEIAV-DIO-Flp or pEIAV-fDIO-Cre), a helper packaging plasmid (pEV53B; a gift from Edward M. Callaway), and a pseudotyping plasmid encoding fusion protein FuG-B2 (a gift from Kazuto Kobayashi). At 72 h post-transfection, viral particles were harvested from the media by centrifugation using SureSpin630 swinging bucket rotor (Thermo Scientific) at 5,700 rpm and 16,200 rpm for 16 h and 2 h, respectively. EIAV viral particles were reconstituted from the pellets with PBS and immediately stored at −80 °C.
Rabies viruses were designed and generated from a full-length cDNA plasmid containing all components of the virus (a gift from Karl-Klaus Conzelmann) as previously described (Osakada and Callaway, 2013). We replaced the coding sequence for viral glycoprotein with tdTomato, mTagBFP2, iRFP670, smFP-HA, smFP-Myc, or smFP-V5 to produce rabies viruses expressing three different fluorescent markers. In brief, B7GG cells were transfected with a total of five plasmids: four plasmids expressing the viral components pcDNA-SADB16N, pcDNA-SADB16P, pcDNA-SADB16L, pcDNA-SADB16G and the aforementioned rabies virus genomic vector. The virus-containing media was collected 3–4 days post-transfection and used for further amplification. Viral particles were harvested from the media by centrifugation using SureSpin630 rotor at 20,000 rpm for 2 h. Rabies viral particles were reconstituted from the pellets with PBS and immediately stored at −80 °C. To generate EnvA-pseudotyped, glycoprotein-deleted rabies virus expressing eGFP (EnvA-SADΔG-eGFP), we used a modified version of a published protocol (Osakada and Callaway, 2013). Plasmids expressing the rabies viral components, B7GG, BHK-EnvA and HEK-TVA cells were gifts from Edward M. Callaway.
Stereotaxic injections and optic fiber/cannula implantations
Animals were group housed in a 12 hour dark/light cycle (lights on 07:00) prior to surgery. Mice were anesthetized with a mixture of ketamine (100 mg/kg) and dexmedetomidine (1 mg/kg) and placed in a stereotaxic apparatus (David Kopf Instruments) as previously described. All animals were kept on a heating pad while recovering from anesthesia. All tracing experiments were conducted in either male or female PV-Cre adult mice (8–12 weeks) and infused unilaterally. Stereotactic coordinates were derived from Paxinos and Franklin mouse brain atlas and empirically adjusted. Empirically determined coordinates were on average shifted 0.5 mm anterior relative to atlas. For experiments examining outputs of VP PV neurons, 250 nL AAV-DIO-mRuby-T2A-Synaptophysin-eGFP was infused into VP (anteroposterior, 0.65 mm; mediolateral, +/− 1.45 mm; dorsoventral, −4.85 mm from top of skull).
For input-output mapping experiments with EnvA-pseudotyped rabies virus (SADΔG-eGFP(EnvA)), 250 nL of RG-EIAV-DIO-Flp was unilaterally infused into either VTA (AP, −2.9 mm; ML, +/− 0.6 mm; DV, −4.4 mm skull) or LHb (AP, −1.45 mm; ML, +/− 0.41 mm; DV, −3.0 mm skull) along with a 1:1 mixture of AAV-fDIO-oPBG and AAV-fDIO-TVA-tdTomato into ipsilateral VP at a rate of 100 nL/min. In order to control for leakage in the relatively small LHb, 175 nL was infused at a rate of 75 nL/min. After allowing for 3 weeks of expression, animals were again anesthetized as previously described and SADΔG-eGFP(EnvA) was infused into ipsilateral VP using scar on skull as guide validation. Six day post SADΔG-eGFP(EnvA) infusion animals were sacrificed for circuit mapping analysis.
For downstream targets of neurons in LHb or VTA, 250 nL of AAV-DIO-Synaptophysin-eGFP or AAV-DIO-Synaptophysin-smFP-FLAG was infused into unilateral VP of PV-Cre mice. After 2 weeks of expression, animals were injected with 300 nL RVΔG-tdTomato, RVΔG-mTag2-BFP, and RVΔG-iRFP670; or, RVΔG-smFP_myc, RVΔG-smFP_V5, and RVΔG-smFP_HA into either DRN (AP, −4.3 mm; ML, 0.0 mm; DV, −2.8 mm skull), NAc medial shell (AP, 1.9 mm; ML, 0.5 mm; DV, −4.4 mm skull), NAc lateral shell (AP, 1.65 mm; ML, 1.8 mm; DV, −4.4 mm skull), RMTg (AP, −3.65 mm; ML, −0.4 mm; DV, −4.0 mm skull), VTA, LHb, or VP. Different rabies and smFPs were randomly assigned to brain areas and interleaved between animals.
For electrophysiology experiments, 8 week old PV-Flp×Ai14 (Ai14-LSL-tdTomato) double transgenic males were injected bilaterally with RV-EIAV-fDIO-Cre into VTA or LHb using same volumes and coordinates as previously stated. Two weeks were given before animals were sacrificed for electrophysiology. For VTA or LHb target recordings, AAV-DIO-ChR2-eYFP was infused bilaterally into VP of PV-Cre animals.
For optogenetic behavioral experiments, adult (8–12 weeks) male PV-Cre animals were bilaterally injected with 250nL AAV-DIO-ChR2-eYFP into VP. Bilateral chronic optic fiber cannulae (200 um, 0.22 NA; Doriclenses, Canada) were implanted during same surgery session above either lateral habenula (LHb) or ventral tegmental area (VTA). One initial layer of adhesive cement (C&B metabond; Parkell) was used to cement fiber in place. Once dried, a second layer of dental cement was used to secure implant to skull. Finally, sutures and sterile tissue adhesive (Vetbond; 3M) was used to close up head incision. Four weeks were given for viral expression in terminals before behavioral testing commenced. Upon completion of behavioral experiments, viral injections and fiber placement were confirmed.
Ex vivo electrophysiology
Mice were deeply anesthetized with isoflurane inhalation. Acute 250 µm coronal brain sections were prepared after intracardial perfusion of ice-cold choline-based slicing solution containing (in mM): 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7 MgCl2, 25 glucose, 0.5 CaCl2, 110 choline chloride, 11.6 ascorbic acid, 3.1 pyruvic acid). Brain was quickly transferred and sliced in the same solution with vibratome (LeicaVT1200). Sections were transferred to a recovery chamber and incubated for 15–20 minutes at 28–31°C in recovery solution consisting of (in mM): 118 NaCl, 2.6 NaHCO3, 11 gluocse, 15 HEPES, 2.5 KCl, 1.25 NaH2PO4, 2 sodium pyruvate, 0.4 sodium ascorabate, 2 CaCl2, 1 MgCl2. Slices were maintained at room temperature for at least one hour until transferred to bath for recording. Cutting solution, recovery solution, and ACSF were constantly bubbled with 95% O2/5% CO2.
For recordings from VP neurons, slices were transferred to a recording chamber on an upright fluorescent microscope continuously perfused with oxygenated ACSF (in mM): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 11 glucose, 1.3 MgCl2 and 2.5 CaCl2 at 28–31°C using a feedback temperature controller. Neurons labeled by fluorescent markers were visualized with a 40X water-immersion objective (Olympus) with epifluorescence and infrared differential interference contrast video microscopy.
For current clamp recording, patch pipettes (3–6 MΩ) were pulled from borosilicate glass (G150TF-4; Warner Instruments) and filled with internal solution containing (in µM): 135 K-gluconate, 5 KCl, 10 HEPES, 0.1 EGTA, 2 MgCl2, 2 Mg2+-ATP, and 0.2 Na+-GTP, pH 7.35 (290–300 mOsm, pH 7.4). Electrophysiological recordings were made using a MultiClamp700B amplifier and PClamp software (Molecular Devices). Data was low-pass filtered at 1 kHz and digitized at 10 kHz with Digidata 1440 (Molecular Devices). For measuring firing frequency, steady state current was injected in +20 pA increments from −100 pA to 200 pA. All action potential properties and excitability recordings in Figure S8 were performed in the presence of 10 µM NBQX and 100 µM picrotoxin.
Action potential threshold was measured as the change in voltage from rest at which the slope = 20 V/s. Peak amplitude measured as change in voltage from threshold to peak, after hyperpolarization (AHP) the change in voltage and time from threshold to minimum after peak (see Figure S8A). Half-width calculated in Clampfit software (Molecular Devices) as full-width at half max amplitude. All measures quantified while eliciting a single action potential. Capacitance was calculated by Clampfit during the first minute after breaking in to cell. Membrane resistance was calculated from the change in voltage elicited after a 50 ms 5 mV hyperpolarizing step from −70 mV (the last 10 ms of the step from baseline was taken as ΔV).
For measuring the ratio of excitatory and inhibitory inputs (E/I ratio), a Cs-based internal solution was used containing (in mM): 115 Cs+-methanesulphonate, 10 HEPES, 1 EGTA, 1.5 MgCl2, 4 Mg+-ATP, 0.3 Na+-GTP, 10 Na+-phosphocreatine, 2 QX 314-Cl, 10 BAPTA-tetracesium (290–300 mOsm, pH 7.4). EPSCs and IPSCs were recorded at the reversal potential for IPSCs (−70 mV) and EPSCs (0 mV), respectively. E/I ratio was calculated by dividing amplitude of AMPA-mediated EPSC while voltage-clamping at −70 mV by amplitude of GABA-mediated IPSC at 0 mV. Spontaneous EPSC and IPSC were also recorded at −70mV and 0mV, respectively. Liquid junction potential (5–8 mV) was not corrected for any experiments.
For target recordings in the LHb or VTA, AAV-DIO-ChR2-eYFP was injected into the VP of PV-Cre animals and cells in the LHb or VTA near eYFP+ fibers were patched. For photostimulation of ChR2-expressing axon terminals, 5 ms blue light pulse was emitted from a collimated light-emitting diode (473 nm; Thorlabs) driven by a T-Cube LED Driver (Thorlabs) under the control of an Axon Digidata 1440A Data Acquisition System and pClamp software. Light was delivered through the reflected light fluorescence illuminator port and the 40X objective (light power at max setting measured at 10.68 mW). AMPA-mediated EPSCs were blocked by bath-application of 15 µM of NBQX, GABAA-mediated IPSCs with 100 µM picrotoxin. (Tocris). All recordings were excluded if holding current exceeded −200 pA or if series resistance was greater than 30 MΩ. Quantitation for electrophysiological records was performed in either Clampfit (Molecular Devices) or OriginPro 2016 (Origin Lab).
Immunohistochemistry
Immunohistochemistry was performed as previously described (Lammel et al., 2012). Briefly, animals were intracardially perfused with 4% paraformaldehyde in PBS and post-fixed at 4 °C overnight. 50 µm coronal sections were sliced on a Leica VT1000 vibratome. Primary antibodies used were: guinea pig anti-Vglut1 (1:250; Millipore), guinea pig anti-Vglut2 (1:1000; Millipore), rabbit anti-TH (1:1000; Millipore), rabbit anti-NeuN (1:2000; Abcam), mouse anti-GAD65 (1:250; DSHB U. of Iowa), guinea pig anti-substance P (1:500; Abcam). For spaghetti monster experiments, epitope antibodies used were: rabbit anti-FLAG (1:1000; Sigma), goat anti-myc (1:1000; Abcam), mouse anti-V5 (1:1000; Life Technologies), rat anti-HA (1:1000; Roche). Alexa Fluor dyes conjugated to either 405, 488, 568, or 647 were used for secondary antibodies (Life Technologies, Abcam). All dilutions used were 1:1000 except for Alexa Fluor 405 at 1:500. All images were acquired with an Olympus FluoView FV1200 confocal microscope.
Circuit mapping quantitation
For whole-brain circuit mapping, cell counting was quantified as previously described with slight alterations (Beier et al., 2015). Briefly, 60 µm coronal sections were obtained across the antero-posterior axis of the brain and imaged on an Olympus VS120 slide scanner. Every third section was quantitated and all GFP-positive input neurons were counted excluding the site of SADΔG-eGFP(EnvA) injection. Data is presented as a percentage of total brain-wide inputs. That is, (total cells quantified in region / all cells quantified across brain). Fourteen total animals were injected, however only those that had the majority of starter cells located in VP were quantified (final n = 7). Brain regions were determined by anatomical landmarks and based on Mouse Brain Atlas in Stereotaxic Coordinates, Franklin and Paxinos, 2nd edition. Olfactory cortex (Olfactory ctx) included anterior olfactory areas, piriform cortex, and olfactory tubercle. Preoptic – lateral preoptic area, medial preoptic area, medial preoptic nucleus. STN – subthalamic nucleus and parasubthalamic nucleus. Areas that exhibited < 1% of all inputs for both LHb-PV and VTA-PV populations were not included in graph. Virus titer and signal of SADΔG-eGFP(EnvA) was sufficiently high, so GFP signal was not amplified with immunofluorescence.
For VP PV neuronal fiber quantitation, images were taken on Olympus FluoView FV1200 confocal microscope. All images were taken in roughly the same imaging area, as denoted in Figure S2A. Images were acquired at 20X with 2× zoom, 570 HSV (PMT sensitivity), 5.8% laser power, using a high-sensitivity Gallium arsenide phosphide (GaAsP) detector. Analysis was conducted in Fiji (Image J) and quantified as a percentage of area of thresholded pixels normalized to the RG-EIAV injection site. Qualitatively determined threshold values were maintained consistent throughout animals and obtained by determining the level that best mirrored the original image without introducing background.
For spaghetti monster downstream connectivity quantitation, images of RV-smFP+ (myc, V5, or HA) labeled neurons were imaged at 60X zoom 2 magnification. Z-stacks, step size 2 µm were taken to fully capture labeled dendritic tree (60 µm slices). Number of synaptophysin-smFP_FLAG+ puncta on cell body and dendrites were manually quantitated using FV10 Viewer (Olympus) to ensure puncta were in same z-plane as soma and neurites.
FISH assay
PV-Flp×Ai14 animals were injected with RG-EIAV-fDIO-Cre into LHb or VTA to label LHb-PV or VTA-PV subpopulations, respectively. Fresh-frozen brains were collected from mice and frozen down with isopentane (Sigma) chilled with dry ice in 70% ethanol. 20 µm coronal brain slices were sectioned on a cryostat (Thermo-Fisher). Sample preparation and mRNA labeling was performed exactly as described in RNAscope online protocol (ACD; Advanced Cell Diagnostics). All probes were purchased from ACD.
Behavioral Assays
For ChR2-mediated stimulation experiments optical fibers (Doric) were connected to a 473 nm blue laser diode (OEM Laser Systems). For LHb stimulation, 5 ms pulses of 20 Hz light were delivered as outlined in previous studies that optogenetically manipulated LHb (Shabel et al., 2012). For VTA, phasic firing has been shown to induce depressive-like symptoms in mice (Chaudhury et al., 2013). As such, phasic stimulation (5 pulses of 40ms light at 20Hz, once every 10 seconds) was used in SI and subthreshold/sufficiency experiments. For all others, 20 Hz, 5 ms pulse was used as previously described (Beier et al., 2015). Laser power was measured (Thorlabs) before each experiment and measured to be 10–15 mW (measured from bilateral fibers, assumed that roughly half power would be delivered to each hemisphere). For NpHR-mediated inhibition, fibers were connected to a 593 nm yellow laser diode. For both LHb and VTA inhibition, 0.1 Hz 9000 ms duration (9 s on, 1 s off) was delivered.
Chronic social defeat stress
Chronic social defeat stress (SDS) was carried out as previously described (Golden et al., 2011). Briefly, prior to experiment retired male breeder CD1 mice were screened on three consecutive days for aggressive characteristics. For ten consecutive days, experimental male C57BL6/JL mice (intruder) were subjected to physical contact and defeat by the aggressive CD1 (resident) in resident home cage for ten minutes (one defeat / day). After physical defeat a clear, perforated divider was placed in the cage preventing physical, yet allowing for sensory contact between intruder and resident. Sensory defeat stage of SDS continued for 24h until next bout of physical defeat. Intruder mice always faced novel residents. After 10 days of SDS, animals were singly housed and tested 24h later for social avoidance behavior. Control animals were housed together separated by the perforated barrier, and switched each day. They were never in physical or sensory contact with CD1s.
Fluoxetine injections were performed as previously described (Cao et al., 2010). Briefly, animals were injected intraperitoneally with fluoxetine (20 mg/kg; Spectrum Chemical) from 9:00 A.M. – 10:00 A.M. once per day for 14–28 days. The day prior to sacrifice, animals underwent one more round of SI test to ascertain whether behavioral deficits were reversed. If animals did not exhibit reversal (judged to be SI ratio > 1), then animals were not used for FLX recordings. Control animals were treated identically except with 100 µL i.p. saline injection.
Social interaction and avoidance
Social avoidance behavior was measured using the two-stage social interaction (SI) test. In the first stage, animals were placed in an open field arena (44 cm × 44 cm × 44 cm) with an empty metal cage (9.5 cm × 9.5 cm × 8 cm). In all behavioral experiments, animals were monitored with the BIOBSERVE video-tracking software. Time spent in an area surrounding the cage (‘interaction zone,’ 8 cm region flanking cage), and the area along the wall opposite to the cage (‘opposing zone’ 9 cm region along wall opposing cage) was measured. The time spent in the interaction zone in this first stage was termed ‘NO CD1.’ Animals were then returned to home cage for one minute. In the second stage, a novel, aggressive CD1 mouse was placed in the cage and the same metrics were measured. From these two stages, an SI index was calculated (SI = time spent interaction zone CD1 / time spent interaction zone NO CD1). Animals were deemed susceptible if SI < 1, and resilient if SI > 1.
For initial optogenetic manipulation experiments, animals were subjected to light epochs of 3 min OFF / 3 min ON / 3 min OFF in NO CD1 stage to determine whether stimulation or inhibition changed interaction with an empty cage. Since no change was seen, most experiments omitted the 3 min OFF / 3 min ON / 3 min OFF light epochs for NO CD1 stage, and restricted light stimulation or inhibition to only CD1 stage. Both conditions were pooled together. To represent light-mediated increases or decreases in social interaction, data is presented as a ratio: time in interaction zone CD1 light ON / time in interaction zone CD1 light OFF. Thus, a ratio of 1 indicates no change, while a ratio > 1 indicates increased time in interaction zone and vice versa. For post-stress optogenetic social interaction experiments, data from only susceptible mice was reported as resilient animals would readily approach CD1 interaction zone. Previous reports indicate that SDS is not a simple fear memory of the CD1, as avoidance behavior is present with novel C57 mice as well so these were not carried out in the present study (Chaudhury et al., 2013).
For DREADD experiments, to account for difference in time course of CNO-mediated inhibition relative to optogenetics, social interaction was increased to 10 minutes. 24 hours after SDS completion, mice underwent 10 minutes NO CD1 SI. Animals were then injected with saline and 30 minutes later were tested for 10 minutes in the SI test with CD1. From this ratio, it was determined whether an animal was resilient or susceptible (only susceptible animals were tested with CNO). 24 hours later, animals were injected with CNO, and 30 minutes after CNO injection were again subjected to SI test with CD1.
Conditioned-place preference/aversion
For context-dependent CPP/CPA (Figure S6), a three chambered rectangular arena was used with two chambers on each side (28 × 24 cm) separated by a neutral central chamber (11.5 cm × 24 cm)(Lammel et al., 2012). In one side chamber, the floor had punched, checker-like flooring with black and white checkered pattern on the walls. The opposite chamber contained a metal grill floor with black and white stripe patterns on the walls. The central, neutral chambers had solid, clear flooring with white walls. The CPP/CPA experiments took place over 3 days as habituation, training, and testing phases. During habituation, animals were placed in the center chamber and allowed free access to explore all chambers for fifteen minutes while time spent in each chamber was recorded with BIOBSERVE. On day 2, animals were placed and restricted to one context and received no stimulation for 15 min. After, animals were restricted to the opposite context and received either blue light stimulation or yellow light inhibition for 15 min. Animals were randomly assigned which context would receive light treatment and alternated between subjects. On day 3, animals were again allowed free access to all chambers. To represent acquired preference or aversion, two metrics were calculated. CPP index represents the time spent in the stimulated side on day 3 (testing) divided by time spent in stimulated side on day 1 (habituation). 1 would indicate no change, while >1 and < 1 indicates preference and aversion, respectively. To more clearly represent aversion and preference, the data was also presented as a difference score. Instead of dividing, timehabituation was subtracted from timetesting. In this manner, negative values denoted acquired aversion, and positive values preference. After trends were established after first rounds of experiments, a biased CPP procedure was used. In order to account for potential chamber preferences between animals, NpHR inhibition was restricted to the non-preferred side while ChR2 stimulation was restricted to the preferred side. Thus, optogenetic experiments worked against the animals’ innate preference. Since no significant differences were seen between these two approaches, results were pooled together. CPP with cocaine experiments were carried out in identical fashion with the exception that on day 2 animals received an intraperitoneal saline injection with no light stimulation in one context, and light delivery was paired with a 20 mg/kg i.p. injection of cocaine in the other.
Open field test
A plain, 44 × 44 cm open field arena was used while total locomotor activity was measured. After a 3 minute habituation period, locomotion was measured in 3 minute light epochs (3 min OFF / 3 min ON / 3 min OFF). Time spent in center (20 × 20 cm) and surround of arena was also measured. For non-optogenetic experiments, locomotion was recorded in one 10 minute session.
Elevated plus maze
Mice were placed in the center of a plus-shaped apparatus (each arm protruding 30 cm from center, 6.35 cm wide) containing two closed arms facing each other, and two open arms. Closed arms had walls flanking the platform (15.24cm high), while closed arms had small (2 cm) ‘railings’ to prevent animals falling. Apparatus was elevated 30.5 cm off the ground. Time spent in closed and open arms, excluding the middle neutral area, was recorded in 5 minute light OFF / 5 minute light ON / 5 minute light OFF epochs.
Sucrose preference test
In home cage, animals were presented with choice of two identical bottles with ball-bearing sipper tubes, one containing water and the other containing a 1% sucrose solution (bottle locations were randomly assigned and flipped at 12h to control for preference in side). The amount of liquid consumed from each bottle was measured at 0, 12, and 24h time points. No difference between 12h and 24h time points were observed, so data is presented at 12h.
Tail suspension test
TST was performed as previously described (Steru et al., 1985). Briefly, mice were suspended by tails with adhesive tape 1 cm from tip of tail and roughly 50 cm above ground so no contact could be made. Plastic tubes were placed over mouse tails to ensure mice could not climb or hang on to their tail. Animals were video recorded for 5 minutes and time spent immobile was quantitated over the full 5 minute interval post hoc. For optogenetic experiments, to control for fatigue over a 10 minute interval, animals were subjected to two separate 5 minute bouts (light OFF, then light ON) separated by 5 minutes of rest.
For DREADD experiments, animals were tested on consecutive days, 24 hours apart. First day animals were always injected with saline, and the second day with CNO. Animals were tested 30 minutes post injection.
Stimulation sufficiency
All light delivery was performed in the home cage of single-housed animals and stimulation protocol was performed as previously indicated. Two times a day (08:00 and 20:00) animals received blue light stimulation for ten minutes. 24h after day 10, animals underwent SI test and later, other behavior tests (Figure S7I and S7J). After behavior, animals were sacrificed to check for injection site and fiber placement.
Subthreshold defeat protocol was performed as previously described (Chaudhury et al., 2013). Briefly, experimental C57BL6/JL mouse was subjected to 3 consecutive bouts of 3 minutes physical defeat and 10 minutes sensory defeat on a single day. Light stimulation was delivered during sensory portion of defeat. 5 minutes of rest in home cage was given between sensory defeat and subsequent bout of physical defeat.
QUANTIFICATION AND STASTISTICAL ANALYSIS
Student’s t-tests, Mann-Whitney U-tests, and one- or two-way ANOVA tests were used as appropriate to determine statistical differences using OriginPro 2016 software (Origin Labs). Shapiro-Wilk test was used to assess normality. Tukey post test was applied in ANOVAs to ascertain differences within groups and to correct for multiple comparisons. Clampfit (Molecular Devices) was used for event detection in electrophysiological experiments. All statistical data can be found in figure legends with corresponding sample sizes. Statistical significance was set at * P < 0.05, ** P < 0.001, *** P < 0.0001. All data is presented as mean ± SEM.
Supplementary Material
(A) Immunofluorescent images showing staining for Substance P (to delineate VP border), PV (from PV-tdTomato mouse), and NeuN (neuronal marker). Scale: 100 µm.
(B) Quantitation of PV neurons as a percentage of the total population of ventral pallidal neurons. PV neurons comprise 14% (# PV+ neurons / total # NeuN+ neurons within borders of Substance P staining). > 25,000 neurons across 4 animals quantified.
(C) Validation showing virus expression can be precisely localized to the VP without leakage into surrounding brain areas. 250 nL of AAV-DIO-eGFP injected into a PV-Cre mouse. Scale: 400 µm.
(D) Other outputs of VP PV neurons. Yellow boxes indicate zoomed areas in bottom row. Scale bars: 50 µm top, 150 µm bottom. Related to Figures 1C–1E.
Abbreviations: NAc, nucleus accumbens; mSh, medial shell; lSh, lateral shell; LH, lateral hypothalamus; MD, mediodorsal nucleus of the thalamus; BLA, basolateral amygdala.
(E) Sagittal view showing areas along antero-posterior axis images are taken for fiber quantitation analysis. Coronal views of the areas in which confocal images in (B) are taken (red squares).
(F) Representative images of fibers in target areas after RG-EIAV-DIO-Flp injection into either LHb (top) or VTA (bottom) and AAV-fDIO-eGFP into VP. Scale: 80 µm.
(G) Fiber quantitation of PVVP→LHb and PVVP→VTA neurons. Dashed line indicates normalized level of injection site. For analysis, 3-4 images were taken for each brain area within an individual animal (n=4) and averaged. Data presented as mean ± SEM.
(H) Experimental schematic. Cholera toxin subunit B conjugated to Alexa Fluor 488 and 647 injected into LHb or VTA in a PV-Cre×Ai14-tdTomato animal to label PVVP→LHb and PVVP→VTA neurons.
(I) Representative images of bead injection sites in LHb (left) and VTA (middle) and retrograde labelling in VP (right). Green arrows depict colocalization of CTbVTA beads and PV-tdT, blue arrow CTbLHb and PV-tdT, and red arrow PV neuron with no bead expression that likely projects to a separate target. Scales: 100 µm.
(J) Quantitation of VP PV neurons that colocalized with CTb beads injected into the VTA, LHb (CTbVTA and CTbLHb, respectively), or both. Only 2.9% of bead-labeled PV neurons projected to both LHb and VTA indicating minimal overlap between these two populations (n = 1972 neurons).
Abbreviations: NAc, nucleus accumbens; mSh, medial shell; lSh, lateral shell; ac, anterior commissure LH, lateral hypothalamus; MD, mediodorsal nucleus of the thalamus; BLA, basolateral amygdala; EP, entopeduncular nucleus; SNr, substantia nigra reticulata.
Localization of PVVP→LHb (red) and PVVP→VTA (blue) neurons across the A–P axis in VP. Only cells in VP are shown. Each dot represents a single neuron. All neurons presented are cumulative across 4 animals for each.
NAc, nucleus accumbens; ac, anterior commissure; CPu, caudate putamen; VP, ventral pallidum; HDB, horizontal diagonal band; MCPO, magnocellular preoptic nucleus; SI, substantia inominata; BNST, bed nucleus of stria terminalis; LPO, lateral preoptic area; IP, interstitial nucleus; Pir, piriform cortex; Tu, olfactory tubercle; LGP, lateral globus pallidus.
(A and E) Absolute amplitude of eEPSCs and eIPSCs of LHb (n = 40; (A)) and VTA (green, GABAergic neurons, n = 25; blue, dopaminergic (DA) neurons, n=40; (E)) neurons evoked by light-stimulation of axonal terminals of VP PV neurons. Individual responses depicted in Figure 1J quantitation.
(B and F) Representative traces of LHb (B) and VTA (F) neurons. Colored traces represent baseline responses, and black traces after bath-application of indicated drug. Scale: (B) 50 pA, 100 ms; (F, blue) 25 pA, 50 ms; (D, green) 100 pA, 100 ms.
(C and G) Average EPSC and IPSC amplitude after bath application of NBQX and PTX. Data presented as normalized % of PSC amplitude after drug application. For (C): n = 5 each condition. For (G, DA): n = 5, 3 for NBQX, PTX respectively. (G, GABA): n = 5.
(D and H) Latency of beginning of synaptic response to initiation of light delivery on slice.
(I) Quantitation of net post-synaptic potential (PSP) responses using a potassium gluconatebased internal in DAergic and GABAergic VTA neurons in current-clamp mode. Example traces of evoked IPSPs and EPSPs on right. Scale: 2 mV, 100 ms.
(J) Latencies of EPSP and IPSP responses in GABAergic and DAergic VTA neurons to initiation of light delivery on slice.
(A) mRNA labeling of PV and tdTomato (from PV-Flp×Ai14 animal injected with RG-EIAV-fDIO-Cre, related to Figures 1K–1L) depicting specificity of RG-EIAV virus. tdTomato mRNA localizes predominantly to PV+ neurons. Scale: 10 µm.
(B) mRNA viral validation quantitation. Roughly 75% of tdTomato+ neurons colocalize with PV mRNA.
(C) Image shows labeled PV-tdTomato+ neuron in prepared ex vivo slice with same viral labeling strategy used in (A).
(D) The RT-PCR of picked tdTomato+ neurons in slice to further validate RG-EIAV virus specificity. Example gel showing bands for GAPDH (control), PV, and GAD (glutamate decarboxylase, marker for GABAergic neurons).
(E) 100% of tdTomato+ neurons patched and picked (n = 7) expressed mRNA for PV.
(F) Fluorescent mRNA labeling showing triple co-expression of probes to tdTomato (from PV-Flp×Ai14 mouse injected with RG-EIAV-fDIO-Cre), PV, and VGluT2. Validation that virally labeled cells (tdT+) are also PV+ and VGluT2+. Scale: 20 µm.
(G) mRNA labeling for PV and VGluT2 in the VP. Scale: 200 µm, inset 50 µm. White arrowhead indicates PV/VGluT2 co-localization, blue arrowhead indicates VGluT2− PV+ neuron.
(H) Quantitation of all VP PV neurons imaged (non-projection specific) that either co-localized (VGluT2+ | PV+, 19.5%) or did not co-localize (VGluT2− | PV+, 80.5%) with VGluT2 probe. n = 67 / 343 neurons VGluT2+ | PV+, from 2 animals.
(I) Viral labeling of PV+ neurons (red) and VGluT2+ neurons (green) by viral injections in double transgenic (PV-Flp×VGluT2-Cre) animals. Colocalized VGluT2/PV-positive neurons denoted by yellow arrows. Scale: 100 µm.
(J) Immunohistochemistry showing co-expression of synaptophysin-eGFP puncta with VGluT1 and VGluT2, but not GAD65 in LHb (left), and VTA (middle). AAV-DIO-Synaptophysin-eGFP was injected in VP of PV-Cre mice. Dense colocalization of synaptophysin to GAD65 was observed in the MD thalamus. Arrowheads indicate synaptophysin-eGFP puncta co-localized with respective marker. Scale: 2 µm.
(A) Schematic of viral injections labeling outputs of neuronal targets of PVVP→LHb and PVVP→VTA neurons. AAV-DIO-Synaptophysin-eGFP was injected into VP of PV-Cre mice. After 2 weeks of expression, same animals were injected with retrograde rabies viruses expressing various fluorophores including RVΔG-tdTomato, RVΔG-mTagBFP2, and RVΔG-iRFP670 into output structures of LHb and VTA (RMTg, DRN, and VTA for LHb; NAc medial shell (mSh), NAc lateral shell (lSh), VP, prefrontal cortex (PFC), and LHb for VTA). Colors and brain structures were alternated across animals. RV+ neurons were imaged and synaptophysin-eGFP puncta colocalizing with RV+ soma were quantitated.
(B) Quantitation of puncta/rabies+ cell body, data presented as mean ± SEM. n = 8 total animals. Cells: LHb outputs (VTA, n = 18; RMTg, n = 13; DRN, n = 7), VTA outputs (NAc mSh, n = 8; NAc lSh, n = 17; PFC, n = 3; VP, n = 5; LHb, n = 8).
(C) Representative confocal images. Scale: 10 µm, inset 4 µm. Arrow denotes zoomed in area. NAc mSh and mPFC projecting VTA neurons were located predominantly in medial VTA, as such very few if any puncta were located nearby.
(D) Overall inputs, outputs, and targets of outputs circuit diagram of LHb-PV and VTA-PV neurons. NAc, nucleus accumbens; mSh, medial shell; lSh, lateral shell; RMTg, rostromedial tegmental nucleus; DRN, dorsal raphe nucleus; PFC, prefrontal cortex.
(E) Schematic of spaghetti monster fluorescent proteins (smFPs) tracing experiments. AAV-DIO-synaptophysin-smFP_FLAG injected into VP of PV-Cre mouse while. RV-smFP_HA, RV-smFP_myc, and RV-smFP_V5 injected into output targets of VTA or LHb (1). After viral expression, IHC to epitope tags performed to enhance and amplify fluorescent signal (2). Animals are then perfused and imaged (3).
(F) Representative low magnification confocal images at 20X. Top: RMTg-projecting LHb neurons in green, VTA-projecting LHb neurons in red, DRN-projecting LHb neurons in blue. VP PV synaptophysin terminals in yellow. Bottom: NAc lSh-projecting neurons in red, NAc mSh-projecting neurons in green, VP-projecting neurons in blue. Scale: 100 µm.
(G) Representative high magnification images of LHb- or VTA-output neurons. Scale: 10 µm. Arrowheads indicate synaptic contact with cell body, arrows indicate synaptic contact with dendrite.
(H and I) Quantitation for number of synaptophysin+ puncta/cell body (H) or per 10 µm of dendrite (I). N = 12, 11, 8, 14, 12, 11 cell bodies and n = 6, 14, 9, 28, 14, 19 dendrites for VTA, RMTg, NAc lSh, NAc mSh, VP, LHb, respectively.
(A and B) Schematic of viral injections in the VP and optical fiber implantations in the LHb and VTA for optogenetic-mediated terminal stimulation (AAV-DIO-ChR2), inhibition (AAV-DIO-NpHR), or control (AAV-DIO-eYFP).
(C) Behavioral paradigm of conditioned place preference or aversion (CPP or CPA).
(D) Quantitation of CPP/CPA. Activation or silencing of axonal fibers in LHb, but not in VTA, produces aversion and preference, respectively. LHb: one-way ANOVA F2,25 = 13.95, P < .001. Tukey post-hoc test to determine significance between individual groups. * P < .05, eYFP-NpHR * P < .05, ChR2-NpHR ** P < .001. n = 9, 9, 10 for eYFP, ChR2, NpHR groups, respectively. n = 5, 8, 8 for each group in VTA-implant animals.
(E) Example behavior heat map of CPP/CPA on day 3. Shaded yellow area on left indicates side where yellow light was delivered on day 2. Warmer colors denote more time spent in designated area. Dashed lines indicate neutral center area.
(F) Same CPP/CPA behavior paradigm, except i.p. injection of cocaine (20 mg / kg) was paired with light delivery.
(G) Terminal stimulation of VP PV neurons in LHb, but not in VTA, is sufficient to block expression of cocaine-induced CPP. LHb: one-way ANOVA F2,18 = 4.804, P < .05, Tukey post test: eYFP-ChR2 * P < .05. n = 5, 8, 8 for eYFP, ChR2, and NpHR respectively in both LHb and VTA animals.
(H) Representative trace of animal track on day 3, blue shading and needle indicate side on day 2 where light delivery and cocaine was administered. All data presented as mean ± SEM.
(I and J) Optogenetic stimulation or inhibition of VP PV terminals in LHb (A) and VTA (B) does not induce changes in locomotion (OFT), anxiety (EPM), or basal social interaction (SI test). Data presented as fold change in light ON epoch divided by light OFF epoch. Dashed line indicates no change.
(A and B) Spikes elicited at all current injection levels. Supplemental to Figures 3E and 3K. For (A): two-way ANOVA with main effect on SDS group, F3,345 = 54.1, * P < .001. N = 9, 11, 10, 8 cells for healthy, resilient, susceptible, FLX, respectively. For (B): two-way ANOVA with main effect on SDS group, F3,358 = 32.58 * P < .001. n = 12, 10, 10, 8 cells from each group.
(C) SI ratio before and after 2 weeks FLX or saline injections in susceptible animals. FLX data point adapted from Fig 3C. No spontaneous recovery observed 2 weeks post SDS.
(D and E) E/I ratio of susceptible animals administered 2 weeks saline injections (grey bar). Other data points adapted from Figures 3H and 3N for comparison (dashed lines around bars). All data presented as mean ± SEM.
(F to I) Differential changes in spontaneous excitatory (F and G) and inhibitory (H and I) postsynaptic currents (sEPSC, sIPSC, respectively) in control, resilient, susceptible, and FLX mice (light tan, brown, light blue, navy, respectively) in PVVP→LHb neurons. Amplitude of sEPSCs and sIPSCs (right) and frequency (left). Scale in (F): 10 pA, 500 ms. For (H): control, 100 pA, 500 ms; resilient, susceptible, and FLX, 50 pA, 500ms. Significant decreases in sIPSC frequency (left, F3,37 = 8.468, P < .001) and amplitude (right, F3,37 = 3.916, P < .05) in susceptible animals in PVVP→LHb neurons, with recovery in amplitude in FLX animals.
(J to M) PVVP→VTA neurons in susceptible animals exhibit significant increases in frequency (left) and amplitude (right) of sEPSCs (J and K), but not sIPSCs (L and M) with recovery in FLX animals. (K) frequency, F3,38 = 3.606, P < .05; amplitude, F3,38 = 4.699, P < .01. (G) amplitude, F3,41 = 3.389, P < .05. Scale for (J): 10 pA, 500 ms. (L): 20 pA, 500 ms. PVVP→LHb neurons: n = 6, 8, 13, 14 from 3, 4, 4, 4 animals in control, resilient, susceptible, FLX groups, respectively. PVVP→VTA cells: n = 9, 14, 8, 14 from 3, 4, 4, 4 animals in control, resilient, susceptible, FLX groups, respectively.
*** P < .001, ** P < .01, * P < .05. All tests conducted one-way ANOVA with post hoc Tukey test for multiple comparisons.
(A) Schematic defining how certain action potential (a.p.) properties were quantified (for further information see STAR methods). All measurements were recorded in the presence of the synaptic blockers NBQX and picrotoxin. AHP: after-hyperpolarization.
(B) Spikes elicited in PVVP→LHb (top) or PVVP→VTA (bottom) neurons in response to current injection. Same as Figures S7A and S7B, but with synaptic blockers. For PVVP→LHb neurons, F2,359 = 106.72, P < .001; n = 11, 13, 12 cells from 3, 4, 4 animals for control, resilient, susceptible, respectively. P < .01 for all individual group interactions. For PVVP→VTA neurons, F2, 387 = 49.33, P < .001; n = 12, 10, 14 cells from 4, 4, 4 animals for control, resilient, susceptible, respectively. Susceptible and control significantly different from resilient. P < .001 for healthy-resilient and susceptible-resilient comparisons. Two-way ANOVA with post hoc Tukey test for multiple comparisons, main effect on stress condition for both.
(C and M) Sample action potential traces of PVVP→LHb and PVVP→VTA neurons in healthy, resilient, and susceptible animals. Scale: 2 ms, 10 mV.
(D–L) Rheobase (D, minimum current required to elicit action potential. F2,32 = 4.19, P < .05), resting membrane potential (E, Vmr), peak a.p. amplitude (F), a.p. half width (G, full width at half max. F2,31 = 11.83, P < .001), a.p. threshold (H, F2,31 = 6.48, P < .01), AHP latency (I), AHP amplitude (J), membrane capacitance (K), and membrane resistance (L) for PVVP→LHb neurons.
(N–V) Rheobase (N, F2,35 = 4.68, P < .05), Vmr (O), peak a.p. amplitude (P), a.p. half width (Q; F2,36 = 5.86, P < .01), a.p. threshold (R), AHP latency (S), AHP amplitude (T), membrane capacitance (U; F2,36 = 6.99, P < .01), and membrane resistance (V; F2,36 = 5.12, P < .05) for PVVP→VTA neurons.
All data presented as mean ± SEM. * P < .05, ** P < .01, *** P < .001. All data except for (B) tested on one-way ANOVA with Tukey post-hoc test to assess inter-group differences. Two-way ANOVA used for (B).
Highlights.
-
-
VP PV neurons project to the LHb and VTA and release different transmitters
-
-
Elevated VP PV neuronal activity is a hallmark of depressed animals
-
-
VP PV neurons mediate distinct depressive-like symptoms based on projection target
Acknowledgments
X. Wang for technical assistance, H. Pribiag and Lim laboratory for comments on manuscript. This work was supported by Klingenstein foundation, Searle scholar program (Kinship foundation), Whitehall foundation, NARSAD young investigator grant and grants from NIH (MH107742 and MH108594). V.L. is also supported by Anandamahidol Foundation Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions Conceptualization, D.K. and B.K.L.; Investigation, D.K., V.L., C.P.P.; Resources, V.L., S.S.; Software, E.H.W.; Writing – original draft, D.K.; Writing – Review & Editing, D.K., V.L., E.H.W., B.K.L.; Funding Acquisition, B.K.L.; Supervision, B.K.L.
References
- Adachi M, Autry AE, Mahgoub M, Suzuki K, Monteggia LM. TrkB Signaling in Dorsal Raphe Nucleus is Essential for Antidepressant Efficacy and Normal Aggression Behavior. Neuropsychopharmacology. 2016;42:1–9. doi: 10.1038/npp.2016.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. U. S. A. 2007;104:5163–5168. doi: 10.1073/pnas.0700293104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagot RC, Parise EM, Peña CJ, Zhang H-X, Maze I, Chaudhury D, Persaud B, Cachope R, Bolaños-Guzmán Ca, Cheer J, et al. Ventral hippocampal afferents to the nucleus accumbens regulate susceptibility to depression. Nat. Commun. 2015;6:7062. doi: 10.1038/ncomms8062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, Gao XJ, Kremer EJ, Malenka RC, Luo L. Circuit Architecture of VTA Dopamine Neurons Revealed by Systematic Input-Output Mapping. Cell. 2015;162:622–634. doi: 10.1016/j.cell.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton O, McClung Ca, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos Ca, Rios M, et al. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Brauer K, Härtig W, Bigl V, Brückner G. Distribution of parvalbumin-containing neurons and lectin-binding perineuronal nets in the rat basal forebrain. Brain Res. 1993;631:167–170. doi: 10.1016/0006-8993(93)91205-7. [DOI] [PubMed] [Google Scholar]
- Cao J-L, Covington HE, Friedman aK, Wilkinson MB, Walsh JJ, Cooper DC, Nestler EJ, Han M-H. Mesolimbic Dopamine Neurons in the Brain Reward Circuit Mediate Susceptibility to Social Defeat and Antidepressant Action. J. Neurosci. 2010;30:16453–16458. doi: 10.1523/JNEUROSCI.3177-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cetin a, Callaway EM. Optical control of retrogradely infected neurons using drug-regulated “TLoop” lentiviral vectors. J. Neurophysiol. 2014;111:2150–2159. doi: 10.1152/jn.00495.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, Ferguson D, Tsai H-C, Pomeranz L, Christoffel DJ, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–536. doi: 10.1038/nature11713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffel DJ, Golden Sa, Walsh JJ, Guise KG, Heshmati M, Friedman AK, Dey A, Smith M, Rebusi N, Pfau M, et al. Excitatory transmission at thalamo-striatal synapses mediates susceptibility to social stress. Nat. Neurosci. 2015;18:6–11. doi: 10.1038/nn.4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creed M, Ntamati NR, Chandra R, Lobo MK, Lüscher C. Convergence of Reinforcing and Anhedonic Cocaine Effects in the Ventral Pallidum. Neuron. 2016:214–226. doi: 10.1016/j.neuron.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donato F, Rompani SB, Caroni P. Parvalbumin-expressing basket-cell network plasticity induced by experience regulates adult learning. Nature. 2013;504:272–276. doi: 10.1038/nature12866. [DOI] [PubMed] [Google Scholar]
- Franklin TB, Saab BJ, Mansuy IM. Neural Mechanisms of Stress Resilience and Vulnerability. Neuron. 2012;75:747–761. doi: 10.1016/j.neuron.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Friedman AK, Walsh JJ, Juarez B, Ku SM, Chaudhury D, Wang J, Li X, Dietz DM, Pan N, Vialou VF, et al. Enhancing Depression Mechanisms in Midbrain Dopamine Neurons Achieves Homeostatic Resilience. Science. 2014;344:313–319. doi: 10.1126/science.1249240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaykema RPA, Zaborszky L. Parvalbumin-containing neurons in the basal forebrain receive direct input from the substantia nigra-ventral tegmental area. Brain Res. 1997:173–179. doi: 10.1016/s0006-8993(96)01309-1. [DOI] [PubMed] [Google Scholar]
- Golden Sa, Covington HE, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nat. Protoc. 2011;6:1183–1191. doi: 10.1038/nprot.2011.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Groenewegen HJ, Grove Ea, Nauta WJ. Efferent connections of the ventral pallidum: evidence of a dual striato pallidofugal pathway. J. Comp. Neurol. 1985;235:322–335. doi: 10.1002/cne.902350304. [DOI] [PubMed] [Google Scholar]
- Haubensak W, Kunwar PS, Cai H, Ciocchi S, Wall NR, Ponnusamy R, Biag J, Dong H-W, Deisseroth K, Callaway EM, et al. Genetic dissection of an amygdala microcircuit that gates conditioned fear. Nature. 2010:468. doi: 10.1038/nature09553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimer L, Switzer RD, Van Hoesen GW. Ventral striatum and ventral pallidum. Components of the motor system? Trends Neurosci. 1982;5:83–87. [Google Scholar]
- Kato S, Kobayashi K, Inoue K, Kuramochi M, Okada T, Yaginuma H, Morimoto K, Shimada T, Takada M, Kobayashi K. A lentiviral strategy for highly efficient retrograde gene transfer by pseudotyping with fusion envelope glycoprotein. Hum. Gene Ther. 2011;22:197–206. doi: 10.1089/hum.2009.179. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Jacobs MW, Ito-Cole T, Callaway EM. Improved Monosynaptic Neural Circuit Tracing Using Engineered Rabies Virus Glycoproteins. Cell Rep. 2016;15:692–699. doi: 10.1016/j.celrep.2016.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Han MH, Graham DL, Berton O, Renthal W, Russo SJ, LaPlant Q, Graham A, Lutter M, Lagace DC, et al. Molecular Adaptations Underlying Susceptibility and Resistance to Social Defeat in Brain Reward Regions. Cell. 2007;131:391–404. doi: 10.1016/j.cell.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Kuo H, Chang HT. Ventral pallido-striatal pathway in the rat brain: a light and electron microscopic study. J. Comp. Neurol. 1992;321:626–636. doi: 10.1002/cne.903210409. [DOI] [PubMed] [Google Scholar]
- Kupchik YM, Brown RM, Heinsbroek Ja, Lobo MK, Schwartz DJ, Kalivas PW. Coding the direct/indirect pathways by D1 and D2 receptors is not valid for accumbens projections. Nat. Neurosci. 2015;18:1230–1232. doi: 10.1038/nn.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique Properties of Mesoprefrontal Neurons within a Dual Mesocorticolimbic Dopamine System. Neuron. 2008;57:760–773. doi: 10.1016/j.neuron.2008.01.022. [DOI] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012a;491:212–217. doi: 10.1038/nature11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012b;491:212–217. doi: 10.1038/nature11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Piriz J, Mirrione M, Chung C, Proulx CD, Schulz D, Henn F, Malinow R. Synaptic potentiation onto habenula neurons in the learned helplessness model of depression. Nature. 2011;470:535–539. doi: 10.1038/nature09742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim BK, Huang KW, Grueter Ba, Rothwell PE, Malenka RC. Anhedonia requires MC4R–mediated synaptic adaptations in nucleus accumbens. Nature. 2012;487:183–189. doi: 10.1038/nature11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahn M, Prigge M, Ron S, Levy R, Yizhar O. Biophysical constraints of optogenetic inhibition at presynaptic terminals. Nat. Neurosci. 2016;19:554–558. doi: 10.1038/nn.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Henry S, Hu J, Gallezot JD, Planeta-Wilson B, Neumaier JF, Neumeister A. Reduced ventral striatal/ventral pallidal serotonin1B receptor binding potential in major depressive disorder. Psychopharmacology (Berl) 2011;213:547–553. doi: 10.1007/s00213-010-1881-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumaier JF, Root DC, Hamblin MW. Chronic fluoxetine reduces serotonin transporter mRNA and 5-HT(1B) mRNA in a sequential manner in the rat dorsal raphe nucleus. Neuropsychopharmacology. 1996;15:515–522. doi: 10.1016/S0893-133X(96)00095-4. [DOI] [PubMed] [Google Scholar]
- Osakada F, Callaway EM. Design and generation of recombinant rabies virus vectors. Nat Protoc. 2013;8:1583–1601. doi: 10.1038/nprot.2013.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoli V, Terrier J, Hiver A, Lüscher C. Sufficiency of Mesolimbic Dopamine Neuron Stimulation for the Progression to Addiction. Neuron. 2015;88:1054–1066. doi: 10.1016/j.neuron.2015.10.017. [DOI] [PubMed] [Google Scholar]
- Proulx CD, Hikosaka O, Malinow R. Reward processing by the lateral habenula in normal and depressive behaviors. Nat. Neurosci. 2014;17:1146–1152. doi: 10.1038/nn.3779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root DH, Melendez RI, Zaborszky L, Napier TC. The ventral pallidum: Subregion-specific functional anatomy and roles in motivated behaviors. Prog. Neurobiol. 2015;1369:1–43. doi: 10.1016/j.pneurobio.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell. 2014;158:198–212. doi: 10.1016/j.cell.2014.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat. Rev. Neurosci. 2013;14:609–625. doi: 10.1038/nrn3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz La, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, et al. Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature. 2015;524:88–92. doi: 10.1038/nature14600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabel SJ, Proulx CD, Trias A, Murphy RT, Malinow R. Input to the lateral habenula from the basal ganglia is excitatory, aversive, and suppressed by serotonin. Neuron. 2012;74:475–481. doi: 10.1016/j.neuron.2012.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Tindell AJ, Aldridge JW, Berridge KC. Ventral pallidum roles in reward and motivation. Behav. Brain Res. 2009;196:155–167. doi: 10.1016/j.bbr.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steru L, Chermat R, Thierry B, Simon P. The tail suspension test: A new method for screening antidepressants in mice. Psychopharmacology (Berl) 1985;85:367–370. doi: 10.1007/BF00428203. [DOI] [PubMed] [Google Scholar]
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C. GABA Neurons of the VTA Drive Conditioned Place Aversion. Neuron. 2012;73:1173–1183. doi: 10.1016/j.neuron.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA Neurons Disrupts Reward Consumption. Neuron. 2012;73:1184–1194. doi: 10.1016/j.neuron.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswanathan S, Williams ME, Bloss EB, Stasevich TJ, Speer CM, Nern A, Pfeiffer BD, Hooks BM, Li W-P, English BP, et al. High-performance probes for light and electron microscopy. Nat. Methods. 2015;12:568–576. doi: 10.1038/nmeth.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JJ, Friedman AK, Sun H, Heller EA, Ku SM, Juarez B, Burnham VL, Mazei-robison MS, Ferguson D, Golden SA, et al. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat. Publ. Gr. 2013;17:27–29. doi: 10.1038/nn.3591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbourd B, Ren J, DeLoach KE, Guenthner CJ, Miyamichi K, Luo L. Presynaptic Partners of Dorsal Raphe Serotonergic and GABAergic Neurons. Neuron. 2014;83:645– 662. doi: 10.1016/j.neuron.2014.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Lyon DC, Barnard RJO, Mori T, Finke S, Conzelmann K-K, Young Ja T, Callaway EM. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H, Yokoyama C, Mizuma H, Kurai S, Finnema SJ, Halldin C, Doi H, Onoe H. A possible mechanism of the nucleus accumbens and ventral pallidum 5-HT1B receptors underlying the antidepressant action of ketamine: a PET study with macaques. Transl. Psychiatry. 2014;4:e342. doi: 10.1038/tp.2013.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahm DS, Williams E, Wohltmann C. Ventral striatopallidothalamic projection: Relative involvements of neurochemically distinct subterritories in the ventral pallidum and adjacent parts of the rostroventral forebrain. J. Comp. Neurol. 1996;364:340–362. doi: 10.1002/(SICI)1096-9861(19960108)364:2<340::AID-CNE11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Immunofluorescent images showing staining for Substance P (to delineate VP border), PV (from PV-tdTomato mouse), and NeuN (neuronal marker). Scale: 100 µm.
(B) Quantitation of PV neurons as a percentage of the total population of ventral pallidal neurons. PV neurons comprise 14% (# PV+ neurons / total # NeuN+ neurons within borders of Substance P staining). > 25,000 neurons across 4 animals quantified.
(C) Validation showing virus expression can be precisely localized to the VP without leakage into surrounding brain areas. 250 nL of AAV-DIO-eGFP injected into a PV-Cre mouse. Scale: 400 µm.
(D) Other outputs of VP PV neurons. Yellow boxes indicate zoomed areas in bottom row. Scale bars: 50 µm top, 150 µm bottom. Related to Figures 1C–1E.
Abbreviations: NAc, nucleus accumbens; mSh, medial shell; lSh, lateral shell; LH, lateral hypothalamus; MD, mediodorsal nucleus of the thalamus; BLA, basolateral amygdala.
(E) Sagittal view showing areas along antero-posterior axis images are taken for fiber quantitation analysis. Coronal views of the areas in which confocal images in (B) are taken (red squares).
(F) Representative images of fibers in target areas after RG-EIAV-DIO-Flp injection into either LHb (top) or VTA (bottom) and AAV-fDIO-eGFP into VP. Scale: 80 µm.
(G) Fiber quantitation of PVVP→LHb and PVVP→VTA neurons. Dashed line indicates normalized level of injection site. For analysis, 3-4 images were taken for each brain area within an individual animal (n=4) and averaged. Data presented as mean ± SEM.
(H) Experimental schematic. Cholera toxin subunit B conjugated to Alexa Fluor 488 and 647 injected into LHb or VTA in a PV-Cre×Ai14-tdTomato animal to label PVVP→LHb and PVVP→VTA neurons.
(I) Representative images of bead injection sites in LHb (left) and VTA (middle) and retrograde labelling in VP (right). Green arrows depict colocalization of CTbVTA beads and PV-tdT, blue arrow CTbLHb and PV-tdT, and red arrow PV neuron with no bead expression that likely projects to a separate target. Scales: 100 µm.
(J) Quantitation of VP PV neurons that colocalized with CTb beads injected into the VTA, LHb (CTbVTA and CTbLHb, respectively), or both. Only 2.9% of bead-labeled PV neurons projected to both LHb and VTA indicating minimal overlap between these two populations (n = 1972 neurons).
Abbreviations: NAc, nucleus accumbens; mSh, medial shell; lSh, lateral shell; ac, anterior commissure LH, lateral hypothalamus; MD, mediodorsal nucleus of the thalamus; BLA, basolateral amygdala; EP, entopeduncular nucleus; SNr, substantia nigra reticulata.
Localization of PVVP→LHb (red) and PVVP→VTA (blue) neurons across the A–P axis in VP. Only cells in VP are shown. Each dot represents a single neuron. All neurons presented are cumulative across 4 animals for each.
NAc, nucleus accumbens; ac, anterior commissure; CPu, caudate putamen; VP, ventral pallidum; HDB, horizontal diagonal band; MCPO, magnocellular preoptic nucleus; SI, substantia inominata; BNST, bed nucleus of stria terminalis; LPO, lateral preoptic area; IP, interstitial nucleus; Pir, piriform cortex; Tu, olfactory tubercle; LGP, lateral globus pallidus.
(A and E) Absolute amplitude of eEPSCs and eIPSCs of LHb (n = 40; (A)) and VTA (green, GABAergic neurons, n = 25; blue, dopaminergic (DA) neurons, n=40; (E)) neurons evoked by light-stimulation of axonal terminals of VP PV neurons. Individual responses depicted in Figure 1J quantitation.
(B and F) Representative traces of LHb (B) and VTA (F) neurons. Colored traces represent baseline responses, and black traces after bath-application of indicated drug. Scale: (B) 50 pA, 100 ms; (F, blue) 25 pA, 50 ms; (D, green) 100 pA, 100 ms.
(C and G) Average EPSC and IPSC amplitude after bath application of NBQX and PTX. Data presented as normalized % of PSC amplitude after drug application. For (C): n = 5 each condition. For (G, DA): n = 5, 3 for NBQX, PTX respectively. (G, GABA): n = 5.
(D and H) Latency of beginning of synaptic response to initiation of light delivery on slice.
(I) Quantitation of net post-synaptic potential (PSP) responses using a potassium gluconatebased internal in DAergic and GABAergic VTA neurons in current-clamp mode. Example traces of evoked IPSPs and EPSPs on right. Scale: 2 mV, 100 ms.
(J) Latencies of EPSP and IPSP responses in GABAergic and DAergic VTA neurons to initiation of light delivery on slice.
(A) mRNA labeling of PV and tdTomato (from PV-Flp×Ai14 animal injected with RG-EIAV-fDIO-Cre, related to Figures 1K–1L) depicting specificity of RG-EIAV virus. tdTomato mRNA localizes predominantly to PV+ neurons. Scale: 10 µm.
(B) mRNA viral validation quantitation. Roughly 75% of tdTomato+ neurons colocalize with PV mRNA.
(C) Image shows labeled PV-tdTomato+ neuron in prepared ex vivo slice with same viral labeling strategy used in (A).
(D) The RT-PCR of picked tdTomato+ neurons in slice to further validate RG-EIAV virus specificity. Example gel showing bands for GAPDH (control), PV, and GAD (glutamate decarboxylase, marker for GABAergic neurons).
(E) 100% of tdTomato+ neurons patched and picked (n = 7) expressed mRNA for PV.
(F) Fluorescent mRNA labeling showing triple co-expression of probes to tdTomato (from PV-Flp×Ai14 mouse injected with RG-EIAV-fDIO-Cre), PV, and VGluT2. Validation that virally labeled cells (tdT+) are also PV+ and VGluT2+. Scale: 20 µm.
(G) mRNA labeling for PV and VGluT2 in the VP. Scale: 200 µm, inset 50 µm. White arrowhead indicates PV/VGluT2 co-localization, blue arrowhead indicates VGluT2− PV+ neuron.
(H) Quantitation of all VP PV neurons imaged (non-projection specific) that either co-localized (VGluT2+ | PV+, 19.5%) or did not co-localize (VGluT2− | PV+, 80.5%) with VGluT2 probe. n = 67 / 343 neurons VGluT2+ | PV+, from 2 animals.
(I) Viral labeling of PV+ neurons (red) and VGluT2+ neurons (green) by viral injections in double transgenic (PV-Flp×VGluT2-Cre) animals. Colocalized VGluT2/PV-positive neurons denoted by yellow arrows. Scale: 100 µm.
(J) Immunohistochemistry showing co-expression of synaptophysin-eGFP puncta with VGluT1 and VGluT2, but not GAD65 in LHb (left), and VTA (middle). AAV-DIO-Synaptophysin-eGFP was injected in VP of PV-Cre mice. Dense colocalization of synaptophysin to GAD65 was observed in the MD thalamus. Arrowheads indicate synaptophysin-eGFP puncta co-localized with respective marker. Scale: 2 µm.
(A) Schematic of viral injections labeling outputs of neuronal targets of PVVP→LHb and PVVP→VTA neurons. AAV-DIO-Synaptophysin-eGFP was injected into VP of PV-Cre mice. After 2 weeks of expression, same animals were injected with retrograde rabies viruses expressing various fluorophores including RVΔG-tdTomato, RVΔG-mTagBFP2, and RVΔG-iRFP670 into output structures of LHb and VTA (RMTg, DRN, and VTA for LHb; NAc medial shell (mSh), NAc lateral shell (lSh), VP, prefrontal cortex (PFC), and LHb for VTA). Colors and brain structures were alternated across animals. RV+ neurons were imaged and synaptophysin-eGFP puncta colocalizing with RV+ soma were quantitated.
(B) Quantitation of puncta/rabies+ cell body, data presented as mean ± SEM. n = 8 total animals. Cells: LHb outputs (VTA, n = 18; RMTg, n = 13; DRN, n = 7), VTA outputs (NAc mSh, n = 8; NAc lSh, n = 17; PFC, n = 3; VP, n = 5; LHb, n = 8).
(C) Representative confocal images. Scale: 10 µm, inset 4 µm. Arrow denotes zoomed in area. NAc mSh and mPFC projecting VTA neurons were located predominantly in medial VTA, as such very few if any puncta were located nearby.
(D) Overall inputs, outputs, and targets of outputs circuit diagram of LHb-PV and VTA-PV neurons. NAc, nucleus accumbens; mSh, medial shell; lSh, lateral shell; RMTg, rostromedial tegmental nucleus; DRN, dorsal raphe nucleus; PFC, prefrontal cortex.
(E) Schematic of spaghetti monster fluorescent proteins (smFPs) tracing experiments. AAV-DIO-synaptophysin-smFP_FLAG injected into VP of PV-Cre mouse while. RV-smFP_HA, RV-smFP_myc, and RV-smFP_V5 injected into output targets of VTA or LHb (1). After viral expression, IHC to epitope tags performed to enhance and amplify fluorescent signal (2). Animals are then perfused and imaged (3).
(F) Representative low magnification confocal images at 20X. Top: RMTg-projecting LHb neurons in green, VTA-projecting LHb neurons in red, DRN-projecting LHb neurons in blue. VP PV synaptophysin terminals in yellow. Bottom: NAc lSh-projecting neurons in red, NAc mSh-projecting neurons in green, VP-projecting neurons in blue. Scale: 100 µm.
(G) Representative high magnification images of LHb- or VTA-output neurons. Scale: 10 µm. Arrowheads indicate synaptic contact with cell body, arrows indicate synaptic contact with dendrite.
(H and I) Quantitation for number of synaptophysin+ puncta/cell body (H) or per 10 µm of dendrite (I). N = 12, 11, 8, 14, 12, 11 cell bodies and n = 6, 14, 9, 28, 14, 19 dendrites for VTA, RMTg, NAc lSh, NAc mSh, VP, LHb, respectively.
(A and B) Schematic of viral injections in the VP and optical fiber implantations in the LHb and VTA for optogenetic-mediated terminal stimulation (AAV-DIO-ChR2), inhibition (AAV-DIO-NpHR), or control (AAV-DIO-eYFP).
(C) Behavioral paradigm of conditioned place preference or aversion (CPP or CPA).
(D) Quantitation of CPP/CPA. Activation or silencing of axonal fibers in LHb, but not in VTA, produces aversion and preference, respectively. LHb: one-way ANOVA F2,25 = 13.95, P < .001. Tukey post-hoc test to determine significance between individual groups. * P < .05, eYFP-NpHR * P < .05, ChR2-NpHR ** P < .001. n = 9, 9, 10 for eYFP, ChR2, NpHR groups, respectively. n = 5, 8, 8 for each group in VTA-implant animals.
(E) Example behavior heat map of CPP/CPA on day 3. Shaded yellow area on left indicates side where yellow light was delivered on day 2. Warmer colors denote more time spent in designated area. Dashed lines indicate neutral center area.
(F) Same CPP/CPA behavior paradigm, except i.p. injection of cocaine (20 mg / kg) was paired with light delivery.
(G) Terminal stimulation of VP PV neurons in LHb, but not in VTA, is sufficient to block expression of cocaine-induced CPP. LHb: one-way ANOVA F2,18 = 4.804, P < .05, Tukey post test: eYFP-ChR2 * P < .05. n = 5, 8, 8 for eYFP, ChR2, and NpHR respectively in both LHb and VTA animals.
(H) Representative trace of animal track on day 3, blue shading and needle indicate side on day 2 where light delivery and cocaine was administered. All data presented as mean ± SEM.
(I and J) Optogenetic stimulation or inhibition of VP PV terminals in LHb (A) and VTA (B) does not induce changes in locomotion (OFT), anxiety (EPM), or basal social interaction (SI test). Data presented as fold change in light ON epoch divided by light OFF epoch. Dashed line indicates no change.
(A and B) Spikes elicited at all current injection levels. Supplemental to Figures 3E and 3K. For (A): two-way ANOVA with main effect on SDS group, F3,345 = 54.1, * P < .001. N = 9, 11, 10, 8 cells for healthy, resilient, susceptible, FLX, respectively. For (B): two-way ANOVA with main effect on SDS group, F3,358 = 32.58 * P < .001. n = 12, 10, 10, 8 cells from each group.
(C) SI ratio before and after 2 weeks FLX or saline injections in susceptible animals. FLX data point adapted from Fig 3C. No spontaneous recovery observed 2 weeks post SDS.
(D and E) E/I ratio of susceptible animals administered 2 weeks saline injections (grey bar). Other data points adapted from Figures 3H and 3N for comparison (dashed lines around bars). All data presented as mean ± SEM.
(F to I) Differential changes in spontaneous excitatory (F and G) and inhibitory (H and I) postsynaptic currents (sEPSC, sIPSC, respectively) in control, resilient, susceptible, and FLX mice (light tan, brown, light blue, navy, respectively) in PVVP→LHb neurons. Amplitude of sEPSCs and sIPSCs (right) and frequency (left). Scale in (F): 10 pA, 500 ms. For (H): control, 100 pA, 500 ms; resilient, susceptible, and FLX, 50 pA, 500ms. Significant decreases in sIPSC frequency (left, F3,37 = 8.468, P < .001) and amplitude (right, F3,37 = 3.916, P < .05) in susceptible animals in PVVP→LHb neurons, with recovery in amplitude in FLX animals.
(J to M) PVVP→VTA neurons in susceptible animals exhibit significant increases in frequency (left) and amplitude (right) of sEPSCs (J and K), but not sIPSCs (L and M) with recovery in FLX animals. (K) frequency, F3,38 = 3.606, P < .05; amplitude, F3,38 = 4.699, P < .01. (G) amplitude, F3,41 = 3.389, P < .05. Scale for (J): 10 pA, 500 ms. (L): 20 pA, 500 ms. PVVP→LHb neurons: n = 6, 8, 13, 14 from 3, 4, 4, 4 animals in control, resilient, susceptible, FLX groups, respectively. PVVP→VTA cells: n = 9, 14, 8, 14 from 3, 4, 4, 4 animals in control, resilient, susceptible, FLX groups, respectively.
*** P < .001, ** P < .01, * P < .05. All tests conducted one-way ANOVA with post hoc Tukey test for multiple comparisons.
(A) Schematic defining how certain action potential (a.p.) properties were quantified (for further information see STAR methods). All measurements were recorded in the presence of the synaptic blockers NBQX and picrotoxin. AHP: after-hyperpolarization.
(B) Spikes elicited in PVVP→LHb (top) or PVVP→VTA (bottom) neurons in response to current injection. Same as Figures S7A and S7B, but with synaptic blockers. For PVVP→LHb neurons, F2,359 = 106.72, P < .001; n = 11, 13, 12 cells from 3, 4, 4 animals for control, resilient, susceptible, respectively. P < .01 for all individual group interactions. For PVVP→VTA neurons, F2, 387 = 49.33, P < .001; n = 12, 10, 14 cells from 4, 4, 4 animals for control, resilient, susceptible, respectively. Susceptible and control significantly different from resilient. P < .001 for healthy-resilient and susceptible-resilient comparisons. Two-way ANOVA with post hoc Tukey test for multiple comparisons, main effect on stress condition for both.
(C and M) Sample action potential traces of PVVP→LHb and PVVP→VTA neurons in healthy, resilient, and susceptible animals. Scale: 2 ms, 10 mV.
(D–L) Rheobase (D, minimum current required to elicit action potential. F2,32 = 4.19, P < .05), resting membrane potential (E, Vmr), peak a.p. amplitude (F), a.p. half width (G, full width at half max. F2,31 = 11.83, P < .001), a.p. threshold (H, F2,31 = 6.48, P < .01), AHP latency (I), AHP amplitude (J), membrane capacitance (K), and membrane resistance (L) for PVVP→LHb neurons.
(N–V) Rheobase (N, F2,35 = 4.68, P < .05), Vmr (O), peak a.p. amplitude (P), a.p. half width (Q; F2,36 = 5.86, P < .01), a.p. threshold (R), AHP latency (S), AHP amplitude (T), membrane capacitance (U; F2,36 = 6.99, P < .01), and membrane resistance (V; F2,36 = 5.12, P < .05) for PVVP→VTA neurons.
All data presented as mean ± SEM. * P < .05, ** P < .01, *** P < .001. All data except for (B) tested on one-way ANOVA with Tukey post-hoc test to assess inter-group differences. Two-way ANOVA used for (B).