

Abstract
Key points
While autologous stem cell‐based therapies are currently being tested on elderly patients, there are limited data on the function of aged stem cells and in particular c‐kit+ cardiac progenitor cells (CPCs). We isolated c‐kit+ cells from young (3 months) and aged (24 months) C57BL/6 mice to compare their biological properties.
Aged CPCs have increased senescence, decreased stemness and reduced capacity to proliferate or to differentiate following dexamethasone (Dex) treatment in vitro, as evidenced by lack of cardiac lineage gene upregulation.
Aged CPCs fail to activate mitochondrial biogenesis and increase proteins involved in mitochondrial oxidative phosphorylation in response to Dex.
Aged CPCs fail to upregulate paracrine factors that are potentially important for proliferation, survival and angiogenesis in response to Dex.
The results highlight marked differences between young and aged CPCs, which may impact future design of autologous stem cell‐based therapies.
Abstract
Therapeutic use of c‐kit+ cardiac progenitor cells (CPCs) is being evaluated for regenerative therapy in older patients with ischaemic heart failure. Our understanding of the biology of these CPCs has, however, largely come from studies of young cells and animal models. In the present study we examined characteristics of CPCs isolated from young (3 months) and aged (24 months) mice that could underlie the diverse outcomes reported for CPC‐based therapeutics. We observed morphological differences and altered senescence indicated by increased senescence‐associated markers β‐galactosidase and p16 mRNA in aged CPCs. The aged CPCs also proliferated more slowly than their young counterparts and expressed lower levels of the stemness marker LIN28. We subsequently treated the cells with dexamethasone (Dex), routinely used to induce commitment in CPCs, for 7 days and analysed expression of cardiac lineage marker genes. While MEF2C, GATA4, GATA6 and PECAM mRNAs were significantly upregulated in response to Dex treatment in young CPCs, their expression was not increased in aged CPCs. Interestingly, Dex treatment of aged CPCs also failed to increase mitochondrial biogenesis and expression of the mitochondrial proteins Complex III and IV, consistent with a defect in mitochondria complex assembly in the aged CPCs. Dex‐treated aged CPCs also had impaired ability to upregulate expression of paracrine factor genes and the conditioned media from these cells had reduced ability to induce angiogenesis in vitro. These findings could impact the design of future CPC‐based therapeutic approaches for the treatment of older patients suffering from cardiac injury.
Keywords: aging, cardiac progenitor cells, c‐kit, stem cell
Key points
While autologous stem cell‐based therapies are currently being tested on elderly patients, there are limited data on the function of aged stem cells and in particular c‐kit+ cardiac progenitor cells (CPCs). We isolated c‐kit+ cells from young (3 months) and aged (24 months) C57BL/6 mice to compare their biological properties.
Aged CPCs have increased senescence, decreased stemness and reduced capacity to proliferate or to differentiate following dexamethasone (Dex) treatment in vitro, as evidenced by lack of cardiac lineage gene upregulation.
Aged CPCs fail to activate mitochondrial biogenesis and increase proteins involved in mitochondrial oxidative phosphorylation in response to Dex.
Aged CPCs fail to upregulate paracrine factors that are potentially important for proliferation, survival and angiogenesis in response to Dex.
The results highlight marked differences between young and aged CPCs, which may impact future design of autologous stem cell‐based therapies.
Abbreviations
- CPC
cardiac progenitor cell
- Dex
dexamethasone
- GAPDH
glyceraldehyde 3‐phosphate dehydrogenase
- GATA4
GATA binding protein 4
- GATA6
GATA binding protein 6
- MEF2C
myocyte enhancer factor 2c
- NDUFAF1
NADH:ubiquinone oxidoreductase complex assembly factor 1
- NRF‐1
nuclear respiratory factor 1
- NS
nucleostemin
- OXPHOS
mitochondrial oxidative phosphorylation
- PECAM
platelet endothelial cell adhesion molecule
- PGC1α
peroxisome proliferator‐activated receptor gamma coactivator 1 alpha
- PGC1β
peroxisome proliferator‐activated receptor gamma coactivator 1 beta
- POLG
proofreading‐deficient mitochondrial DNA polymerase γ
- SAβ‐Gal
senescence associated beta‐galactosidase
- SDF‐1
stromal cell‐derived factor‐1
- TGF‐β
transforming growth factor‐beta
- X‐gal
5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐galactopyranoside
Introduction
Average life expectancy has increased dramatically over the past century as a consequence of advances in the biomedical field. Currently 11% of the entire human population is composed of elderly individuals (>60 years old) and epidemiological studies project an increase to 22% by 2050 (Kanasi et al. 2016). The unprecedented increase in human lifespan over the past years has driven scientific interest in the study of human diseases that arise with ageing. Ageing can be defined as the process by which the physiological integrity of an organism decreases over time, ultimately leading to death. At the cellular level senescence is the degenerative process that takes place due to DNA damage during ageing (Terzi et al. 2016). This phenomenon is mainly, but not only, dependent on the organism age and lifespan.
The heart is one of the organs of the body that is most affected by ageing, undergoing more than three billion contraction cycles during the average human lifespan. In addition, the decline in cardiac function with ageing is often accompanied by other age‐related risk factors such as diabetes or hypertension that accelerate cardiovascular senescence (Brodsky et al. 2004; Kosugi et al. 2006). The loss of myocyte turnover (Bergmann et al. 2015) and decreased contraction due to impairment in calcium handling are some of the causes underlying the development of heart failure in the aged heart (Lakatta & Levy, 2003; Bergmann et al. 2015). Thus, it is not surprising that cardiovascular disease occurs more frequently in the elderly population (Writing Group Members et al. 2016).
Strategies to ‘rejuvenate’ the ageing heart are currently being investigated. Recent studies have identified follistatin like 1 (Masters & Riley, 2014; van Rooij, 2016), Pim‐1 (Cottage et al. 2010), growth differentiation factor 11 (Olson et al. 2015; Rochette et al. 2015; van Rooij, 2016) and insulin like growth factor 1 (Leifke et al. 2000; Fontana et al. 2012) as possible anti‐ageing factors capable of inducing endogenous regeneration in the ageing heart. Although it is unclear whether the heart can be made to regenerate, cardiac progenitor cells identified in the adult heart are multipotent and have the ability to differentiate (Beltrami et al. 2003; Yellamilli & van Berlo, 2016). A randomized control phase I clinical trial (SCIPIO) investigated whether isolating c‐kit+ cardiac progenitor cell (CPCs) from patients and re‐introducing them back into the injured heart would be beneficial. The study has produced encouraging results and paved the way for future phase II clinical trials (Bolli et al. 2011; Hong & Bolli, 2014). It is clear, however, that a major hindrance for cell therapy is the poor stem cell survival and engraftment in the injured heart. Although studies have suggested that CPCs are capable of differentiating into cardiomyocytes (Beltrami et al. 2003; Dawn et al. 2005; Hsieh et al. 2007; Bolli et al. 2013; Ellison et al. 2013), the prevailing theory is that the reparative capacity of CPCs may be mediated through paracrine mechanisms that modulate immune responses and promote cell survival and angiogenesis (Stastna et al. 2010; Khanabdali et al. 2016; Der Sarkissian et al. 2017; Sharma et al. 2017).
It has been suggested that the decline in regenerative capacity of stem cells contributes to the loss of organ function and tissue homeostasis (Hariharan & Sussman, 2015). Taking advantage of the relatively short mouse lifespan, genetically modified mouse models have been used to study CPC activity during ageing (Cottage et al. 2010; Goichberg et al. 2011; Toko et al. 2014). For example, Hariharan et al. (2015) demonstrated that nucleostemin (NS) expression is lower in CPCs isolated from aged compared to young mice. Aged CPCs engineered with NS had preserved ‘stemness’ properties while deficiency of NS led to myocardial ageing due to telomere shortening. Ageing is also associated with accumulation of mitochondrial DNA mutations that we previously reported to lead to impaired function of CPCs (Orogo et al. 2015). Recent proteomics analysis of human CPCs revealed basal age‐based differences, which could account for variability in the capacity of stem cells to promote myocardial recovery (Sharma et al. 2017). However, there is a lack of information in the literature regarding the cell biology of aged CPCs under conditions that promote differentiation.
In the present study we compared CPCs isolated from 3‐ and 24‐month‐old C57Bl/6 mice, ages that roughly correspond to <20 years and >60 years of age in humans (Ferando et al. 2016). We have recently demonstrated that young CPCs have the potential to upregulate markers for multiple cardiac cell types in response to dexamethasone (Dex) treatment (Castaldi et al. 2016). Here we demonstrate that aged CPCs not only have decreased proliferation and increased senescence compared to young CPCs, but that their ability to respond to Dex with increased expression of cardiac lineage markers and OXPHOS mitochondrial proteins as well as mitochondrial biogenesis was repressed. In addition, aged CPCs failed to show Dex‐induced upregulation of factors involved in survival and proliferation and to secrete factors important for angiogenesis. Thus, our findings demonstrate marked differences in the young and aged CPCs, basally and under conditions of differentiation, that could impact design of future CPC‐based therapeutic approaches for the treatment of elderly patients suffering from cardiac injury.
Methods
Ethical approval
All procedures were performed in accordance with NIH Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at the University of California San Diego. Male 3‐ and 24‐month‐old C57BL/6J mice were used for the isolation of c‐kit+ CPCs. Young mice were bred at the University of California San Diego and aged mice were obtained from the National Institute on Aging aged rodent colony (Bethesda, MD, USA).
Cardiac c‐kit+ cell isolation and culture
Adult c‐kit+ CPCs were isolated from 3 (young)‐ and 24 (aged)‐month‐old C57BL/6J mice. Three young and three aged mice were killed with CO2 followed by cervical dislocation and CPCs were isolated and cultured in growth medium as previously described (Castaldi et al. 2016). For isolation, the purity of the CPC population was assessed by fluorescence‐activated cell sorting analysis for c‐kit+ and being haematopoietic negative at early passages (∼60–80% c‐kit+ at early passage and 100% by passage 15). Cells were used for experiments between passages 15 and 20. The data reported here are from independent experiments, each performed in triplicate on three young or three aged cell lines.
Immunofluorescence
CPCs were seeded on six‐well tissue culture plates (10 000 cells per well) in growth medium and fixed the following day in paraformaldehyde solution 4% in PBS, permeabilized with 10% Triton X‐100, and incubated overnight with LIN28 antibody 1:200 (LSBio, Seattle, WA, USA). The following day, cells were washed and incubated with secondary antibody, Alexa‐fluor 488 dye 1:200 (Thermo Fisher Scientific, Waltham, MA, USA) and rhodamine phalloidin 1:200 (Invitrogen, Carlsbad, CA, USA) for 1 h. After washing, the slides were mounted using Vectashield with DAPI (Vector Laboratories, Burlingame, CA, USA) and images were acquired using a Leica SP5 confocal microscope (40× oil immersion objective). Images were acquired at a focal distance of 0.49 μm and z‐stacks were compressed to provide a composite image (ImageJ software analysis, plugin Bio‐format). For morphological evaluation, permeabilized CPCs were stained with Phalloidin‐488 (Invitrogen) for 1 h. After washing, the slides were mounted using Vectashield with DAPI (Vector Laboratories) and images were acquired using a Leica DMi8 fluorescence microscope (20× objective).
β‐Galactosidase staining
CPCs were seeded on six‐well tissue culture plates (10 000 cells per well) in growth medium and fixed with a paraformaldehyde solution 4% in PBS for 15 min at room temperature. Senescence‐associated β‐galactosidase (SA‐β‐Gal) was detected using the Senescence Detection Kit (Abcam, Cambridge, MA, USA) following the manufacturer's protocol, as previously described (Hariharan et al. 2015). Images were acquired using a Leica DMi8 microscope (20× magnification objective). Images from four independent experiments were analysed and the percentage of cells positive for SA‐β‐Gal was calculated for each (eight fields per experiment).
Glycolysis assay
CPCs were seeded on 12‐well tissue culture plates (5000 cells per well) and serum removed the following day. Growth medium complete with 10% embryonic stem cell fetal bovine serum was added back to the cells 24 h after starvation. l‐Lactate concentration in the growth medium was measured after 4 days using a glycolysis cell‐based assay kit (Cayman Chemical, Ann Harbor, MI, USA) according to the manufacturer's protocol, as previously described (Orogo et al. 2015). CPCs were lysed in RIPA buffer and protein concentration was evaluated with Micro BCA (Thermo Fischer Scientific). l‐Lactate concentration was normalized to total protein. Colorimetric measurements were made using an INFINTE M200 microplate reader (TECAN, Männedorf, Switzerland) and I‐Control 1.6 Software.
ATP assay
CPCs were seeded on 10 cm culture dishes (150 000 cells per dish) and harvested the following day. ATP levels were measured using a CellTiter‐Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA) according to the manufacturer's protocol, as previously described (Orogo et al. 2015). CPCs (40 000 cells/100 μl) were added to a 96‐well black plate. Luminescence measurements were made using an INFINTE M200 microplate reader (TECAN) and I‐Control 1.6 Software.
Proliferation assay
CPCs were seeded on a transparent 48‐well tissue culture plate (8000 cells per well) cultured in growth medium. Cell growth was measured on day 0 and after 1, 2 and 3 days using a CyQUANT NF Cell Proliferation Assay (Invitrogen). Plates were assayed according to the manufacturer's protocol and as previously described (Castaldi et al. 2016). Fluorescence intensity was determined using an INFINTE M200 microplate reader (TECAN) and I‐Control 1.6 Software.
Dexamethasone treatment
CPCs were seeded on 10 cm culture dishes (30 000 cells per dish) in growth medium. The following day medium was changed to minimum essential medium Eagle‐alpha modification supplemented with 10% fetal bovine serum and Dex (10 nM). Dex was added fresh every 3 days, as previously described (Fischer et al. 2011). Cells were harvested on day 7 and gene/protein expression was compared to Time 0 (no Dex).
Western blot analysis
For LIN28 protein expression analysis, CPCs were lysed in RIPA buffer (150 mm NaCl, 50 mm Tris‐HCl pH 7.4, 1% NP40, 1% sodium deoxycholate (NaDoc), 0.1% SDS, 2 mm EDTA, 50 mm NaF) with freshly added leupeptin (10 μg), phenylmethylsulfonyl fluoride (PMSF; 1 mm), p‐nitrophenylphosphate (PNPP; 1 mm), Na3VO4 (0.2 mm) and aprotinin (0.6%; Fischer BioReagents). For mitochondrial oxidative phosphorylation proteins (OXPHOS) analysis, CPCs were lysed in lysis buffer as previously described (Orogo et al. 2015). In total, 30 μg of lysate was loaded on NuPAGE Bis‐Tris Gels (12% for LIN28 and 10% for OXPHOS; Thermo Fisher). The membranes were probed with the following antibodies: MitoProfile Total OXPHOS Rodent WB antibody mixture 1:500 (MitoSciences, Eugene, OR, USA), LIN28 1:1000 (LSBio), Actin and α‐Tubulin 1:1000 (Cell Signaling Technologies, Danvers, MA, USA). Membranes were imaged using a MyECL Imager (Thermo Fischer Scientific) and signal quantified using AlphaView SA Software.
Gene expression analysis
RNA was isolated from CPCs using Trizol (Invitrogen) following the manufacturer's protocol, cDNA synthesized with a High‐Capacity cDNA Reverse Transcription Kit (Applied Biosystems ABI, Foster City, CA, USA) and real‐time quantitative PCR performed with TaqMan Universal Master Mix II, with UNG (Applied Biosystems ABI). IDT TaqMan probes for mouse myocyte enhancer factor 2C (MEF2C), GATA binding protein 4 (GATA4), GATA binding protein 6 (GATA6), platelet endothelial cell adhesion molecule 1 (PECAM), p16, complex IV subunit 4, NADH:ubiquinone oxidoreductase complex assembly factor 1 (NDUFAF1), NADH:ubiquinone oxidoreductase complex assembly factor 1 (NRF‐1), peroxisome proliferator‐activated receptor gamma coactivator 1‐alpha (PGC1α), peroxisome proliferator‐activated receptor gamma coactivator 1‐beta (PGC1β), stromal cell derived factor‐1 (SDF‐1), transforming growth factor‐beta (TGF‐β), beta‐catenin (β‐catenin) and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) were used. Relative quantification was calculated using the ΔΔCt method and expressed as fold change.
Endothelial tube formation assay
CPCs (young and aged) were seeded on 24‐well plates (8000 cells per well) and treated with Dex as described above for 7 days to generate conditioned medium. Bovine aortic endothelial cells (75 000 cells per well) (Lonza, Walkersville, MD, USA) were seeded on Geltrex (Invitrogen)‐coated 24‐well dishes with conditioned media from young or aged CPCs. After 6 h, tube formation was visualized and images from six fields per well were captured using a Leica DMi8 microscope (10× magnification). The number of tubes/field was calculated, as previously described (Moc et al. 2015).
Statistical analyses
Researchers were blinded to the treatment group during analyses. Data are represented as means ± SEM. Differences at P < 0.05 were considered statistically significant and were assessed using unpaired Student's t test (for two groups), one‐way (for multiple comparisons) or two‐way (for multiple comparisons with more than one variable) ANOVA with post hoc Tukey analysis using GraphPad Prism software (GraphPad, La Jolla, CA, USA).
Results
c‐kit+ CPCs become senescent with ageing
To determine how ageing affects the functional properties of c‐kit+ CPCs, CPCs were isolated from 3‐ and 24‐month‐old C57BL/6 mice. These are referred to in the present work as young and aged mouse CPCs. Three separate CPC preparations, isolated from animals at either 3 or 24 months, were passaged and analysed.
There were evident morphological differences between young and aged CPCs, the former being elongated and the latter more flat (Fig. 1 A). Flattened cell morphology can be indicative of senescence (Goichberg et al. 2013; Hariharan et al. 2015), so we investigated whether aged CPCs presented a more senescent phenotype. Young and aged mouse CPCs were stained with X‐gal (5‐bromo‐4‐chloro‐3‐indolyl‐β‐d‐galactopyranoside) and the SA‐β‐Gal activity was detected as a blue/dark signal (Fig. 1 B). As hypothesized, there was a significantly higher percentage of SA‐β‐Gal‐positive cells in the aged mouse CPCs (∼60%) compared to young mouse CPCs (∼35%; Fig. 1 C). In addition, aged mouse CPCs expressed higher levels of the senescence marker p16 (Fig. 1 D). It has been shown that senescence is associated with a metabolic switch from oxidative phosphorylation to glycolysis (Feng et al. 2016). Therefore we analysed levels of lactate, a byproduct of glycolysis, in young and aged mouse CPCs cultured for 4 days in growth medium. Lactate content was significantly higher in the aged versus young CPCs (Fig. 1 E), suggesting that cells from older animals at baseline are more glycolytic then their younger counterparts. ATP concentration was modestly but not significantly lower in aged compared to young mouse CPCs (Fig. 1 F). These data suggest that aged CPCs are more senescent and have altered function, presenting an overall phenotype distinct from that of young mouse CPCs.
Figure 1. Aged mouse c‐kit+ CPCs present a flat, senescent and glycolytic phenotype compared to young mouse CPCs.
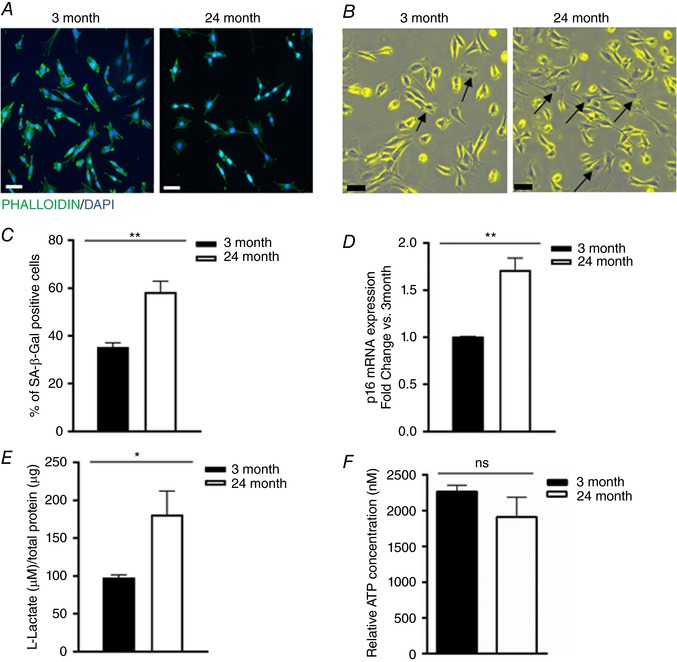
A, morphological evaluation of young (3 months) and aged (24 months) mouse CPCs: representative immunofluorescence. Cells were stained with Phalloidin (green) and DAPI (blue). Images were acquired on a Leica DMi8 microscope (20× magnification). Scale bar = 50 μm. B, senescence‐associated expression of β‐galactosidase (SA‐β‐Gal staining): young and aged mouse CPCs were stained with X‐gal to detect endogenous β‐galactosidase activity. Brightfield images were acquired on a Leica DMi8 microscope (20× magnification), Scale bar = 50 μm. C, quantification of the SA‐β‐Gal staining: eight fields per slide were counted, from three independent experiments; t test ** P < 0.01. D, mRNA expression of the senescence marker p16 in aged vs. young mouse CPCs; three independent experiments, t test * P < 0.05. E, l‐lactate concentration in 4 day conditioned media from young and aged mouse CPCs; three independent experiments, t test * P < 0.05. F, ATP concentration in young and aged CPCs; three independent experiments, t test ns = not significant. [Color figure can be viewed at wileyonlinelibrary.com]
Ageing impairs proliferative capacity and ‘stemness’ of c‐kit+ CPCs
To further investigate whether the biological function of CPCs is altered with ageing, we compared the proliferative capacity of young and aged CPCs in serum‐containing medium. Aged CPCs proliferated at a significantly lower rate, showing a 40% decrease compared to young mouse CPCs after 2 days in serum (Fig. 2 A). The combined decrease in proliferation and increase in markers of senescence suggested that ‘stemness’ might be diminished in the aged mouse CPCs. To address this we examined the expression of LIN28, an RNA binding protein involved in pluripotency and tissue repair (Zhong et al. 2010; Oshima et al. 2016) by both immunofluorescence and western blotting. Immunofluorescence was detectable but considerably lower in the aged compared to the young mouse CPCs (Fig. 2 B, 3 month left panel, 24 month right panel). Western blotting also revealed a considerably lower level of LIN28 protein expression in aged compared to young mouse CPCs (Fig. 2 C).
Figure 2. Aged mouse c‐kit+ CPCs have slower proliferation and lower levels of the stemness marker LIN28.
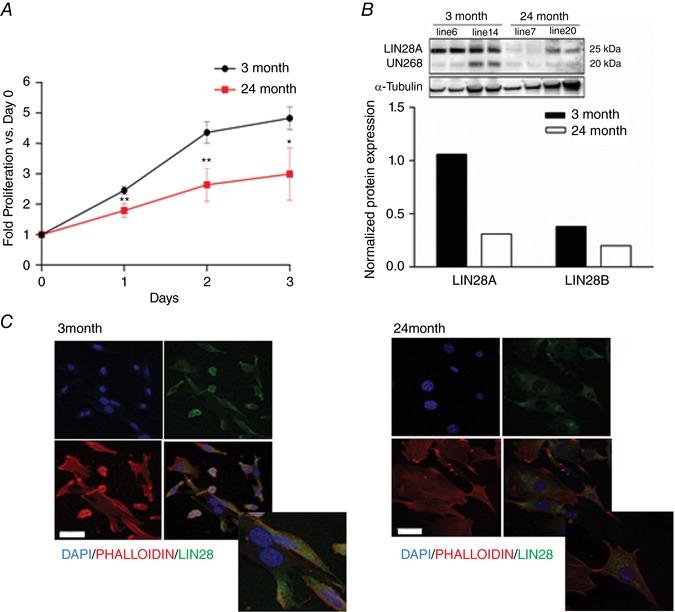
A, proliferation of young (3 months) and aged (24 months) mouse CPCs in growth media was measured using CyQuant. Three independent experiments, t test vs. 3 months * P < 0.05 ** P < 0.01. B, western blot analysis of LIN28A and LIN28B protein expression and quantification in young (3 months) and aged (24 months) mice; two independent lines of each were analysed in duplicate. C, representative immunofluorescence of young and aged mouse CPCs. Cells were stained with DAPI (blue), Phalloidin (red) and LIN28 antibody (green) and images were acquired using a Leica SP5 confocal microscope. Scale bar = 50 μm. [Color figure can be viewed at wileyonlinelibrary.com]
Aged CPCs fail to upregulate lineage markers in response to Dex treatment
Protocols to induce the expression of cardiac lineage markers in c‐kit+ CPCs in culture are well established, the most routinely used being Dex treatment (Fischer et al. 2011; Castaldi et al. 2016). We subjected young and aged CPCs to 7 day treatment with Dex and evaluated mRNA expression of cardiac lineage markers MEF2C and GATA4, as well as the vascular smooth muscle marker GATA6 and endothelial marker PECAM by quantitative PCR analysis. As demonstrated in Fig. 3, Dex increased mRNA levels for MEF2C, GATA4, GATA6 and PECAM in young CPCs by ∼3‐, ∼4‐, ∼7‐ and ∼8‐fold, respectively. In contrast, aged CPCs failed to upregulate any of these lineage markers in response to Dex. There did not appear to be a generalized defect in transcriptional activity in the aged mouse CPCs, however, as tunicamycin induced the endoplasmic reticulum stress marker Grp78 to the same extent as in young mouse CPCs (data not shown).
Figure 3. Cardiac lineage markers are not induced by dexamethasone (Dex) in aged mouse CPCs.
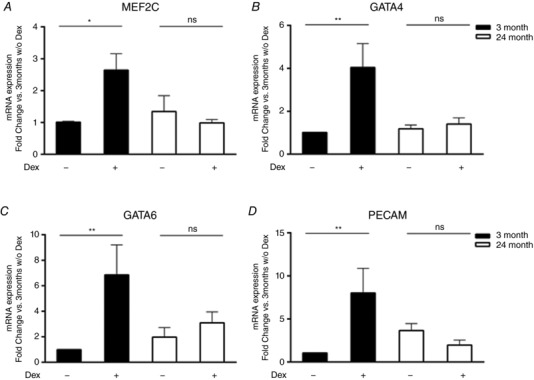
Quantitative PCR analysis of cardiac lineage genes MEF2C (A) and GATA4 (B), smooth muscle lineage gene GATA6 (C) and endothelial lineage gene PECAM (D) in young and aged mouse CPCs treated with Dex (10 nm) for 7 days. Six independent experiments, two‐way ANOVA * P < 0.05, ** P < 0.01, ns = not significant.
Aged CPCs fail to increase oxidative phosphorylation and mitochondrial biogenesis in response to Dex treatment
We have previously shown that Dex treatment increases OXPHOS and mitochondrial biogenesis in CPCs (Orogo et al. 2015). Here, using the young mouse CPCs isolated from C57Bl/6 mice, we confirmed that OXPHOS protein levels increased following 3 and 7 days of Dex treatment (Fig. 4 A, left panel and Fig. 4 B, black bars). Aged mouse CPCs, by contrast, completely failed to increase expression of subunits in respiratory Complex III and Complex IV in response to Dex treatment (Fig. 4 A, right panel and Fig. 4 B, white bars). We also observed upregulation of mRNA for Complex IV Subunit 4 and of the mRNA for NDUFAF1 following Dex in young CPCs, responses that were markedly attenuated in aged CPCs (Fig. 4 C). These data suggest that the aged CPCs are impaired in their capacity for transcriptional activation of genes involved in OXPHOS. Consistent with these observations, aged mouse CPCs displayed impairment in mitochondrial biogenesis, as indicated by failure to upregulate NRF‐1, PGC1α and PGC1β mRNAs in response to Dex treatment (Fig. 4 D). Taken together, our findings demonstrate that aged CPCs exhibit an overall impairment in the induction of cardiac lineage commitment and a failure to induce mitochondrial biogenesis and upregulation of OXPHOS proteins following Dex.
Figure 4. Aged CPCs do not increase mitochondrial OXPHOS and mitochondrial biogenesis in response to dexamethasone (Dex) treatment.
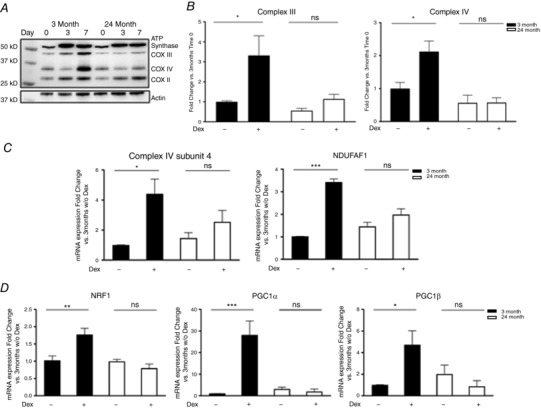
A, OXPHOS protein expression in young and aged mouse CPCs treated with Dex for 3 and 7 days. Representative western blot analysis. B, quantification of Complex III and Complex IV protein level at time 0 and 7 days after Dex. C and D, mRNA expression levels of the complex IV subunit 4 and complex assembly factor NDUFAF1 (C) and of the mitochondria biogenesis markers NRF‐1, PGC1α and β (D) by quantitative PCR in aged (24 months) vs. young cells (3 months), 7 days after Dex treatment. Three independent experiments; two‐way ANOVA * P < 0.05, ** P < 0.01, *** P < 0.005.
Aged CPCs fail to upregulate paracrine factors and generate angiogenic factors in response to Dex treatment
While the cardiogenic potential of CPCs is highly debated there is a growing consensus that CPCs have the potential to secrete trophic factors involved in survival, repair or angiogenesis (van Berlo et al. 2014; Khanabdali et al. 2016; Cai & Molkentin, 2017; Der Sarkissian et al. 2017; Sharma et al. 2017). Age‐dependent changes in secreted proteins from human CPCs were recently reported (Sharma et al. 2017), but the ability of young and aged CPCs to upregulate paracrine signalling in response to Dex‐induced differentiation has not been explored. We observed a significant increase in β‐catenin, TGF‐β and SDF‐1 mRNA expression following Dex treatment in young CPCs, which was not recapitulated in aged CPCs (Fig. 5). To determine if functionally different factors were secreted by young and aged CPCs we tested conditioned medium in an endothelial tube formation assay. Conditioned media from Dex‐treated aged CPCs elicited significantly less tube formation than that from young CPCs (Fig. 6). Thus, treatment of CPCs with Dex induces factors that are potentially important for survival and proliferation and that promote angiogenesis in vitro, and these responses become impaired with ageing.
Figure 5. Paracrine factors are not induced by dexamethasone (Dex) in aged mouse CPCs.
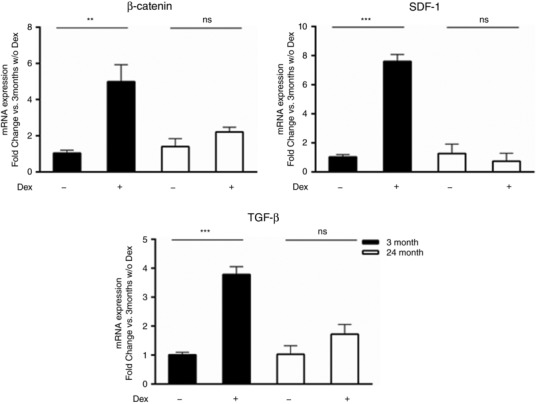
Quantitative PCR analysis of paracrine factors (β‐catenin, SDF‐1 and TGF‐β) in young and aged mouse CPCs treated with Dex (10 nm) for 7 days. Four independent experiments, two‐way ANOVA ** P < 0.01, *** P < 0.005, ns = not significant.
Figure 6. Conditioned medium from aged CPCs has impaired ability to induce endothelial tube formation.
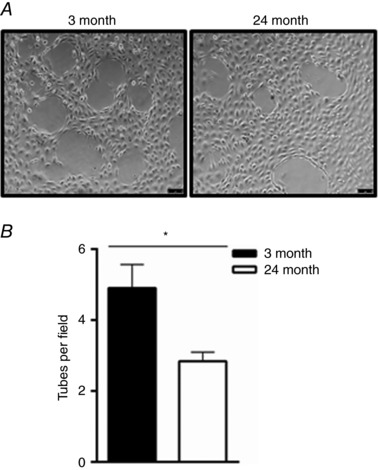
A, bovine aortic endothelial cells were plated in conditioned media from young (3 months) or aged (24 months) CPCs for 6 h and tube formation was visualized using a Leica DMi8 microscope (10× magnification). Scale bar = 75 μm. B, quantification of the number of tubes per field. Three independent experiments, t test * P < 0.05.
Discussion
Perhaps the greatest controversy in the field of cardiac research over the past decade has been the existence and the functional role of adult resident c‐kit+ CPCs. Since their discovery more than 10 years ago (Beltrami et al. 2003), multiple research studies have been carried out to understand the potential role of these cells in cardiac regeneration following injury. CPCs isolated from rodents and porcine models as well as humans have been expanded in vitro and re‐introduced into the injured heart with beneficial outcomes (Tang et al. 2010; Bolli et al. 2011, 2013; Li et al. 2011; McCall et al. 2012). In addition, mouse models have been used to surgically impose cardiac injury and demonstrate recruitment of endogenous stem cells along the border zone area (Williams et al. 2013; Quijada et al. 2015; Zhang et al. 2016). A major bias in these studies, both in vitro and in vivo, has been the use of relatively young animals. In contrast, the portion of the human population that suffers from cardiac disease and would be most likely candidates for stem cell‐based therapy are mainly the elderly. Studies have been carried out using CPCs isolated from human heart biopsies from heart failure patients (Samse et al. 2015), but the heterogeneity between samples and the lack of a direct younger control make these less than ideal for clarifying the effect of ageing on the properties of CPCs.
The findings presented here demonstrate marked differences in CPCs isolated from hearts of young (3 months) and aged (24 months) mice. Based on a variety of factors such as sexual maturity, deterioration of locomotion and endurance, social behaviour and cognitive development, 24‐month‐old mice are considered the equivalent of an elderly human (Ferando et al. 2016). The present study is, to our knowledge, the first to explore differences between young and aged CPCs isolated from the same strain of mice and grown under identical conditions. Our data demonstrate that CPCs isolated from aged mice differ from those of young mice in (a) morphology and expression of molecular markers of senescence, (b) diminished proliferation rate and ‘stemness’, (c) failure to respond to Dex by induction of cardiac lineage genes or increased oxidative metabolism and (d) diminished induction of paracrine and release of angiogenic factors following Dex.
Aged mouse CPCs present a flattened morphology accompanied by increased SA‐β‐Gal and expression of p16 mRNA. This is in line with a previous study in which it was reported that CPCs isolated from 13‐month FVBN mice presented flattened morphology and greater SA‐β‐Gal expression than a younger FVBN cohort (Hariharan et al. 2015). It has also been shown that p16 accumulates in various stem/progenitor cells including the brain, bone marrow, pancreas and heart in aged rodents (Krishnamurthy et al. 2004; Torella et al. 2004; Janzen et al. 2006; Molofsky et al. 2006; Rota et al. 2015). Skeletal muscle stem cells in aged mice also have elevated expression of p16; this contributes to a switch from quiescent to senescent and their lack of activation upon injury (Sousa‐Victor et al. 2014). In human CPCs, p16 expression has been observed to increase during culture expansion (Goichberg et al. 2013).
From a metabolic point of view, mammalian organs such as liver, skeletal muscle and brain shift toward increased glycolysis with ageing (Feng et al. 2016). We report here that aged CPCs also appear to rely more on glycolysis than oxidative phosphorylation for energy production, as indicated by the increased release of lactate into the medium. Stem cells have been shown to decrease their replicative capacity during in vitro expansion (Wong et al. 2015). Here, we report a reduction in proliferative capacity that could extrapolate to donor age, as CPCs from aged mice proliferate at a rate that is 40% slower than that of young CPCs. Similar observations have been made with human mesenchymal stem cells and CPCs isolated from elderly compared with paediatric donors (Baxter et al. 2004; Sharma et al. 2017). Interestingly, we also observed a concomitant and dramatic downregulation of the ‘stemness’ marker LIN28 in aged CPCs, a change that may influence their overall function.
Multipotent stem cells have the potential to differentiate into various tissue‐specific cell lineages. At present there is no evidence that CPCs are capable of differentiating into mature cardiac myocytes in vitro. They have, however, been shown to upregulate cardiac lineage markers in response to Dex treatment (Fischer et al. 2011; Castaldi et al. 2016). In this study comparing the response of young and aged CPCs to Dex treatment we show that the aged CPCs failed to upregulate cardiac lineage markers. We also discovered that aged CPCs failed to activate mitochondrial biogenesis and increase proteins involved in OXPHOS in response to Dex treatment. We have previously found that mitochondrial biogenesis and OXPHOS proteins are upregulated in young CPCs during differentiation when the differentiating cells are switching to mitochondrial respiration for more efficient ATP production (Orogo et al. 2015). Also, pluripotent stem cells typically switch their metabolism from glycolytic to oxidative in the transition from a quiescent to activated state (Xu et al. 2013; Ito & Suda, 2014). Thus, our findings in the present study suggest that the mitochondrial biogenesis response is impaired in aged CPCs. This is in agreement with our previous study on CPCs isolated from mice expressing a proofreading‐deficient mitochondrial DNA polymerase γ (POLG) (Orogo et al. 2015). Ageing is associated with accumulation of mitochondrial DNA mutations, which may explain in part why aged CPCs resemble those from POLG mice in failing to activate mitochondrial biogenesis and increase OXPHOS proteins in response to Dex treatment.
Recently accumulated evidence suggests that the limited but documented cardiac repair observed with autologous therapy results from release of cytokines and growth factors from stem cells including CPCs (Torella et al. 2004; Rota et al. 2015; Khanabdali et al. 2016). SDF‐1, otherwise known as CXCL12, is a chemokine that mediates recruitment and trafficking of both haematopoietic and non‐haematopoietic stem cells (Abbott et al. 2004; Bromage et al. 2014). SDF‐1 has been found to be secreted at high levels from patient‐derived cardiosphere‐derived stem cells in vitro (Cheng et al. 2014), although adult human CPCs secrete much lower levels than do neonatal cells (Sharma et al. 2017). TGF‐β is another factor examined in various stem cell cultures in which upregulation of TGF‐β has been demonstrated to promote differentiation and the Wnt/β‐catenin pathway to promote expansion and survival (Goumans et al. 2007; Cohen et al. 2008; Khanabdali et al. 2016; Der Sarkissian et al. 2017). We show here that β‐catenin, SDF‐1 and TGF‐β gene expression can be upregulated in young CPCs in response to Dex treatment whereas these responses are completely absent in aged cells. While in vitro observations such as those made in our studies might not mimic the physiological environment, it is important to consider that protocols for use of CPCs in clinical trials require cell isolation and expansion in vitro prior to re‐injection into patients at the site of injury. The inability of aged CPCs to respond to proliferative and differentiation cues, or generate and release trophic factors could account for the limited regenerative potential of aged cells.
While CPCs have limited capacity to differentiate into cardiomyocytes, it has been recently theorized that these cells may be more endothelial‐like (van Berlo et al. 2014; Cai & Molkentin, 2017) and capable of producing paracrine factors (Cai & Molkentin, 2017; Sharma et al. 2017). Recent studies have shown that media from human CPCs (Sharma et al. 2017) as well as human cardiospheres (Chimenti et al. 2010) can promote tube formation in vitro. Our study revealed that in response to differentiation signalling, young CPCs generated factors that were able to stimulate endothelial tube formation, a response that was significantly impaired in the aged CPCs. While the ability of aged CPC conditioned media to promote endothelial tube formation is clearly impaired, the limited angiogenic effect provided by aged CPCs could nonetheless afford partial benefit following ischaemic damage to the heart.
In conclusion, there are multiple previously undescribed and unanticipated differences in c‐kit+ CPCs isolated from young and aged mouse hearts. The more accommodating behaviour of the commonly studied young CPCs belies the more limited proliferative capacity, increased senescence and refractoriness to ‘differentiation’ and paracrine signalling of the aged mouse CPCs. Attempts to understand and perhaps shift the deficits seen in the aged cells may at least enhance their ability to expand in vitro and potentially improve their ability to express unknown factors that contribute to their reported ability to improve the failing heart.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
Experiments were performed in the Department of Pharmacology and in the Skaggs School of Pharmacy and Pharmaceutical Sciences at the University of California San Diego. JHB, NHP and ÅBG contributed to the experimental design of the work. JHB, NHP, ÅBG, AC, RMD, AMO, CMZ and RHN contributed to the acquisition, analysis or interpretation of the data. JHB, NHP and AC wrote the paper. All authors approved the final version of the manuscript and agreed to be accountable for all aspects of the work and for ensuring that questions related to the accuracy or integrity of any part of the work were appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by grants from the National Heart, Lung and Blood Institute Grants: HL085577 (JHB, NHP, ÅBG and AC), HL028143 (JHB), HL114949 (NHP) and HL087023 (ÅBG), and by the National Institutes of Health NRSA Predoctoral Fellowship HL123309 (AMO). ÅBG is supported by an AHA Established Investigator Award.
Acknowledgements
We would like to thank Melissa S. Barlow for her technical assistance in isolating the mouse cardiac progenitor cell lines.
Linked articles This article is highlighted by a Perspective by Garikipati & Kishore. To read this Perspective, visit https://doi.org/10.1113/JP274989.
This is an Editor's Choice article from the 1 October 2017 issue.
Contributor Information
Joan Heller Brown, Email: jhbrown@ucsd.edu.
Nicole H. Purcell, Email: npurcell@ucsd.edu
References
- Abbott JD, Huang Y, Liu D, Hickey R, Krause DS & Giordano FJ (2004). Stromal cell‐derived factor‐1α plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation 110, 3300–3305. [DOI] [PubMed] [Google Scholar]
- Baxter MA, Wynn RF, Jowitt SN, Wraith JE, Fairbairn LJ & Bellantuono I (2004). Study of telomere length reveals rapid aging of human marrow stromal cells following in vitro expansion. Stem Cells 22, 675–682. [DOI] [PubMed] [Google Scholar]
- Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal‐Ginard B & Anversa P (2003). Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 114, 763–776. [DOI] [PubMed] [Google Scholar]
- Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, Malm T, Andra M, Jashari R, Nyengaard JR, Possnert G, Jovinge S, Druid H & Frisen J (2015). Dynamics of cell generation and turnover in the human heart. Cell 161, 1566–1575. [DOI] [PubMed] [Google Scholar]
- Bolli R, Chugh AR, D'Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J & Anversa P (2011). Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet 378, 1847–1857. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bolli R, Tang XL, Sanganalmath SK, Rimoldi O, Mosna F, Abdel‐Latif A, Jneid H, Rota M, Leri A & Kajstura J (2013). Intracoronary delivery of autologous cardiac stem cells improves cardiac function in a porcine model of chronic ischemic cardiomyopathy. Circulation 128, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky SV, Gealekman O, Chen J, Zhang F, Togashi N, Crabtree M, Gross SS, Nasjletti A & Goligorsky MS (2004). Prevention and reversal of premature endothelial cell senescence and vasculopathy in obesity‐induced diabetes by ebselen. Circ Res 94, 377–384. [DOI] [PubMed] [Google Scholar]
- Bromage DI, Davidson SM & Yellon DM (2014). Stromal derived factor 1α: a chemokine that delivers a two‐pronged defence of the myocardium. Pharmacol Ther 143, 305–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai CL & Molkentin JD (2017). The elusive progenitor cell in cardiac regeneration: slip slidin’ away. Circ Res 120, 400–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaldi A, Chesini GP, Taylor AE, Sussman MA, Brown JH & Purcell NH (2016). Sphingosine 1‐phosphate elicits RhoA‐dependent proliferation and MRTF‐A mediated gene induction in CPCs. Cell Signal 28, 871–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Malliaras K, Smith RR, Shen D, Sun B, Blusztajn A, Xie Y, Ibrahim A, Aminzadeh MA, Liu W, Li TS, De Robertis MA, Marban L, Czer LS, Trento A & Marban E (2014). Human cardiosphere‐derived cells from advanced heart failure patients exhibit augmented functional potency in myocardial repair. JACC Heart Fail 2, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimenti I, Smith RR, Li TS, Gerstenblith G, Messina E, Giacomello A & Marban E (2010). Relative roles of direct regeneration versus paracrine effects of human cardiosphere‐derived cells transplanted into infarcted mice. Circ Res 106, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen ED, Tian Y & Morrisey EE (2008). Wnt signaling: an essential regulator of cardiovascular differentiation, morphogenesis and progenitor self‐renewal. Development 135, 789–798. [DOI] [PubMed] [Google Scholar]
- Cottage CT, Bailey B, Fischer KM, Avitabile D, Collins B, Tuck S, Quijada P, Gude N, Alvarez R, Muraski J & Sussman MA (2010). Cardiac progenitor cell cycling stimulated by pim‐1 kinase. Circ Res 106, 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawn B, Stein AB, Urbanek K, Rota M, Whang B, Rastaldo R, Torella D, Tang XL, Rezazadeh A, Kajstura J, Leri A, Hunt G, Varma J, Prabhu SD, Anversa P & Bolli R (2005). Cardiac stem cells delivered intravascularly traverse the vessel barrier, regenerate infarcted myocardium, and improve cardiac function. Proc Natl Acad Sci USA 102, 3766–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Der Sarkissian S, Levesque T & Noiseux N (2017). Optimizing stem cells for cardiac repair: current status and new frontiers in regenerative cardiology. World J Stem Cells 9, 9–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellison GM, Vicinanza C, Smith AJ, Aquila I, Leone A, Waring CD, Henning BJ, Stirparo GG, Papait R, Scarfo M, Agosti V, Viglietto G, Condorelli G, Indolfi C, Ottolenghi S, Torella D & Nadal‐Ginard B (2013). Adult c‐kit(pos) cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 154, 827–842. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hanson RW, Berger NA & Trubitsyn A (2016). Reprogramming of energy metabolism as a driver of aging. Oncotarget 7, 15410–15420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferando I, Faas GC & Mody I (2016). Diminished KCC2 confounds synapse specificity of LTP during senescence. Nat Neurosci 19, 1197–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer KM, Cottage CT, Konstandin MH, Volkers M, Khan M & Sussman MA (2011). Pim‐1 kinase inhibits pathological injury by promoting cardioprotective signaling. J Mol Cell Cardiol 51, 554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana L, Vinciguerra M & Longo VD (2012). Growth factors, nutrient signaling, and cardiovascular aging. Circ Res 110, 1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goichberg P, Bai Y, D'Amario D, Ferreira‐Martins J, Fiorini C, Zheng H, Signore S, del Monte F, Ottolenghi S, D'Alessandro DA, Michler RE, Hosoda T, Anversa P, Kajstura J, Rota M & Leri A (2011). The ephrin A1‐EphA2 system promotes cardiac stem cell migration after infarction. Circ Res 108, 1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Goichberg P, Kannappan R, Cimini M, Bai Y, Sanada F, Sorrentino A, Signore S, Kajstura J, Rota M, Anversa P & Leri A (2013). Age‐associated defects in EphA2 signaling impair the migration of human cardiac progenitor cells. Circulation 128, 2211–2223. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Goumans MJ, de Boer TP, Smits AM, van Laake LW, van Vliet P, Metz CH, Korfage TH, Kats KP, Hochstenbach R, Pasterkamp G, Verhaar MC, van der Heyden MA, de Kleijn D, Mummery CL, van Veen TA, Sluijter JP & Doevendans PA (2007). TGF‐β1 induces efficient differentiation of human cardiomyocyte progenitor cells into functional cardiomyocytes in vitro . Stem Cell Res 1, 138–149. [DOI] [PubMed] [Google Scholar]
- Hariharan N, Quijada P, Mohsin S, Joyo A, Samse K, Monsanto M, De La Torre A, Avitabile D, Ormachea L, McGregor MJ, Tsai EJ & Sussman MA (2015). Nucleostemin rejuvenates cardiac progenitor cells and antagonizes myocardial aging. J Am Coll Cardiol 65, 133–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan N & Sussman MA (2015). Cardiac aging – Getting to the stem of the problem. J Mol Cell Cardiol 83, 32–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong KU & Bolli R (2014). Cardiac stem cell therapy for cardiac repair. Curr Treat Options Cardiovasc Med 16, 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J & Lee RT (2007). Evidence from a genetic fate‐mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med 13, 970–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K & Suda T (2014). Metabolic requirements for the maintenance of self‐renewing stem cells. Nat Rev Mol Cell Biol 15, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, DePinho RA, Sharpless NE & Scadden DT (2006). Stem‐cell ageing modified by the cyclin‐dependent kinase inhibitor p16INK4a. Nature 443, 421–426. [DOI] [PubMed] [Google Scholar]
- Kanasi E, Ayilavarapu S & Jones J (2016). The aging population: demographics and the biology of aging. Periodontol 2000 72, 13–18. [DOI] [PubMed] [Google Scholar]
- Khanabdali R, Rosdah AA, Dusting GJ & Lim SY (2016). Harnessing the secretome of cardiac stem cells as therapy for ischemic heart disease. Biochem Pharmacol 113, 1–11. [DOI] [PubMed] [Google Scholar]
- Kosugi R, Shioi T, Watanabe‐Maeda K, Yoshida Y, Takahashi K, Machida Y & Izumi T (2006). Angiotensin II receptor antagonist attenuates expression of aging markers in diabetic mouse heart. Circ J 70, 482–488. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al‐Regaiey K, Su L & Sharpless NE (2004). Ink4a/Arf expression is a biomarker of aging. J Clin Invest 114, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakatta EG & Levy D (2003). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation 107, 346–354. [DOI] [PubMed] [Google Scholar]
- Leifke E, Gorenoi V, Wichers C, Von Zur Muhlen A, Von Buren E & Brabant G (2000). Age‐related changes of serum sex hormones, insulin‐like growth factor‐1 and sex‐hormone binding globulin levels in men: cross‐sectional data from a healthy male cohort. Clin Endocrinol (Oxf) 53, 689–695. [DOI] [PubMed] [Google Scholar]
- Li Q, Guo Y, Ou Q, Chen N, Wu WJ, Yuan F, O'Brien E, Wang T, Luo L, Hunt GN, Zhu X & Bolli R (2011). Intracoronary administration of cardiac stem cells in mice: a new, improved technique for cell therapy in murine models. Basic Res Cardiol 106, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters M & Riley PR (2014). The epicardium signals the way towards heart regeneration. Stem Cell Res 13, 683–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall FC, Telukuntla KS, Karantalis V, Suncion VY, Heldman AW, Mushtaq M, Williams AR & Hare JM (2012). Myocardial infarction and intramyocardial injection models in swine. Nat Protoc 7, 1479–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moc C, Taylor AE, Chesini GP, Zambrano CM, Barlow MS, Zhang X, Gustafsson AB & Purcell NH (2015). Physiological activation of Akt by PHLPP1 deletion protects against pathological hypertrophy. Cardiovasc Res 105, 160–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE & Morrison SJ (2006). Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KA, Beatty AL, Heidecker B, Regan MC, Brody EN, Foreman T, Kato S, Mehler RE, Singer BS, Hveem K, Dalen H, Sterling DG, Lawn RM, Schiller NB, Williams SA, Whooley MA & Ganz P (2015). Association of growth differentiation factor 11/8, putative anti‐ageing factor, with cardiovascular outcomes and overall mortality in humans: analysis of the Heart and Soul and HUNT3 cohorts. Eur Heart J 36, 3426–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orogo AM, Gonzalez ER, Kubli DA, Baptista IL, Ong SB, Prolla TA, Sussman MA, Murphy AN & Gustafsson AB (2015). Accumulation of mitochondrial DNA mutations disrupts cardiac progenitor cell function and reduces survival. J Biol Chem 290, 22061–22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M, Hasegawa N, Mochizuki‐Kashio M, Muto T, Miyagi S, Koide S, Yabata S, Wendt GR, Saraya A, Wang C, Shimoda K, Suzuki Y & Iwama A (2016). Ezh2 regulates the Lin28/let‐7 pathway to restrict activation of fetal gene signature in adult hematopoietic stem cells. Exp Hematol 44, 282–296.e283. [DOI] [PubMed] [Google Scholar]
- Quijada P, Salunga HT, Hariharan N, Cubillo JD, El‐Sayed FG, Moshref M, Bala KM, Emathinger JM, De La Torre A, Ormachea L, Alvarez R Jr, Gude NA & Sussman MA (2015). Cardiac stem cell hybrids enhance myocardial repair. Circ Res 117, 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rochette L, Zeller M, Cottin Y & Vergely C (2015). Growth and differentiation factor 11 (GDF11): functions in the regulation of erythropoiesis and cardiac regeneration. Pharmacol Ther 156, 26–33. [DOI] [PubMed] [Google Scholar]
- Rota M, Goichberg P, Anversa P & Leri A (2015). Aging effects on cardiac progenitor cell physiology. Compr Physiol 5, 1775–1814. [DOI] [PubMed] [Google Scholar]
- Samse K, Emathinger J, Hariharan N, Quijada P, Ilves K, Volkers M, Ormachea L, De La Torre A, Orogo AM, Alvarez R, Din S, Mohsin S, Monsanto M, Fischer KM, Dembitsky WP, Gustafsson AB & Sussman MA (2015). Functional effect of Pim1 depends upon intracellular localization in human cardiac progenitor cells. J Biol Chem 290, 13935–13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Mishra R, Bigham GE, Wehman BP, Khan MM, Datla SR, Saha P, Goo YA, Chen L, Goodlett DR & Kaushal S (2017). Deep proteome analysis identified complete secretome as the functional unit of human cardiac progenitor cells. Circulation 120, 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa‐Victor P, Gutarra S, Garcia‐Prat L, Rodriguez‐Ubreva J, Ortet L, Ruiz‐Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, Perdiguero E & Munoz‐Canoves P (2014). Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321. [DOI] [PubMed] [Google Scholar]
- Stastna M, Chimenti I, Marban E & Van Eyk JE (2010). Identification and functionality of proteomes secreted by rat cardiac stem cells and neonatal cardiomyocytes. Proteomics 10, 245–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XL, Rokosh G, Sanganalmath SK, Yuan F, Sato H, Mu J, Dai S, Li C, Chen N, Peng Y, Dawn B, Hunt G, Leri A, Kajstura J, Tiwari S, Shirk G, Anversa P & Bolli R (2010). Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30‐day‐old infarction. Circulation 121, 293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzi MY, Izmirli M & Gogebakan B (2016). The cell fate: senescence or quiescence. Mol Biol Rep 43, 1213–1220. [DOI] [PubMed] [Google Scholar]
- Toko H, Hariharan N, Konstandin MH, Ormachea L, McGregor M, Gude NA, Sundararaman B, Joyo E, Joyo AY, Collins B, Din S, Mohsin S, Uchida T & Sussman MA (2014). Differential regulation of cellular senescence and differentiation by prolyl isomerase Pin1 in cardiac progenitor cells. J Biol Chem 289, 5348–5356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, Urbanek K, Nadal‐Ginard B, Kajstura J, Anversa P & Leri A (2004). Cardiac stem cell and myocyte aging, heart failure, and insulin‐like growth factor‐1 overexpression. Circ Res 94, 514–524. [DOI] [PubMed] [Google Scholar]
- van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marban E & Molkentin JD (2014). c‐kit+ cells minimally contribute cardiomyocytes to the heart. Nature 509, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij E (2016). Cardiac repair after myocardial infarction. N Engl J Med 374, 85–87. [DOI] [PubMed] [Google Scholar]
- Williams AR, Hatzistergos KE, Addicott B, McCall F, Carvalho D, Suncion V, Morales AR, Da Silva J, Sussman MA, Heldman AW & Hare JM (2013). Enhanced effect of combining human cardiac stem cells and bone marrow mesenchymal stem cells to reduce infarct size and to restore cardiac function after myocardial infarction. Circulation 127, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong TY, Solis MA, Chen YH & Huang LL (2015). Molecular mechanism of extrinsic factors affecting anti‐aging of stem cells. World J Stem Cells 7, 512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Writing Group Members , Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics Committee & Stroke Statistics Subcommittee (2016). Heart disease and stroke statistics‐2016 update: a report from the American Heart Association. Circulation 133, e38–360. [DOI] [PubMed] [Google Scholar]
- Xu X, Duan S, Yi F, Ocampo A, Liu GH & Izpisua Belmonte JC (2013). Mitochondrial regulation in pluripotent stem cells. Cell Metab 18, 325–332. [DOI] [PubMed] [Google Scholar]
- Yellamilli A & van Berlo JH (2016). The role of cardiac side population cells in cardiac regeneration. Front Cell Dev Biol 4, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang NK, Cao Y, Zhu ZM, Zheng N, Wang L, Xu XH & Gao LR (2016). Activation of endogenous cardiac stem cells by apelin‐13 in infarcted rat heart. Cell Transplant 25, 1645–1652. [DOI] [PubMed] [Google Scholar]
- Zhong X, Li N, Liang S, Huang Q, Coukos G & Zhang L (2010). Identification of microRNAs regulating reprogramming factor LIN28 in embryonic stem cells and cancer cells. J Biol Chem 285, 41961–41971. [DOI] [PMC free article] [PubMed] [Google Scholar]