

Abstract
Key points
Pharmacological, molecular and genetic data indicate a prominent role of low‐voltage‐activated T‐type calcium channels (T‐channels) in the firing activity of both pyramidal and inhibitory interneurons in the subiculum.
Pharmacological inhibition of T‐channels switched burst firing with lower depolarizing stimuli to regular spiking, and fully abolished hyperpolarization‐induced burst firing.
Our molecular studies showed that CaV3.1 is the most abundantly expressed isoform of T‐channels in the rat subiculum.
Consistent with this finding, both regular‐spiking and burst firing patterns were profoundly depressed in the mouse with global deletion of CaV3.1 isoform of T‐channels.
Selective inhibition of T‐channels and global deletion of CaV3.1 channels completely suppressed development of long‐term potentiation (LTP) in the CA1–subiculum, but not in the CA3–CA1 pathway.
Abstract
Several studies suggest that voltage‐gated calcium currents are involved in generating high frequency burst firing in the subiculum, but the exact nature of these currents remains unknown. Here, we used selective pharmacology, molecular and genetic approaches to implicate Cav3.1‐containing T‐channels in subicular burst firing, in contrast to several previous reports discounting T‐channels as major contributors to subicular neuron physiology. Furthermore, pharmacological antagonism of T‐channels, as well as global deletion of CaV3.1 isoform, completely suppressed development of long‐term potentiation (LTP) in the CA1–subiculum, but not in the CA3–CA1 pathway. Our results indicate that excitability and synaptic plasticity of subicular neurons relies heavily on T‐channels. Hence, T‐channels may be a promising new drug target for different cognitive deficits.
Keywords: calcium, hippocampus, low‐threshold‐activated
Key points
Pharmacological, molecular and genetic data indicate a prominent role of low‐voltage‐activated T‐type calcium channels (T‐channels) in the firing activity of both pyramidal and inhibitory interneurons in the subiculum.
Pharmacological inhibition of T‐channels switched burst firing with lower depolarizing stimuli to regular spiking, and fully abolished hyperpolarization‐induced burst firing.
Our molecular studies showed that CaV3.1 is the most abundantly expressed isoform of T‐channels in the rat subiculum.
Consistent with this finding, both regular‐spiking and burst firing patterns were profoundly depressed in the mouse with global deletion of CaV3.1 isoform of T‐channels.
Selective inhibition of T‐channels and global deletion of CaV3.1 channels completely suppressed development of long‐term potentiation (LTP) in the CA1–subiculum, but not in the CA3–CA1 pathway.
Abbreviations
- ADP
afterdepolarization potential
- AP
action potential
- fEPSP
field excitatory postsynaptic potential
- HF
hydrofluoric acid
- HFS
high frequency stimulation
- HVA
high‐voltage ‐activated
- I–V
current–voltage
- KO
knockout
- LTP
long‐term potentiation
- LVA
low‐voltage activated
- RMP
resting membrane potential
- TTA‐P2
3,5‐dichloro‐N‐[1‐(2,2‐dimethyl‐tetrahydro‐pyran‐4‐ylmethyl)‐4‐fluoropiperidin‐4‐ylmethyl]‐benzamide
- WT
wild type
- YFP
yellow fluorescent protein
Introduction
High‐frequency (burst) firing may increase the fidelity of synaptic transmission and thereby modulate memory processing (Lisman, 1997; Izhikevich et al. 2003). In thalamocortical neurons the switch from tonic to burst firing is mediated by T‐type calcium channels (T‐channels). Specifically, hyperpolarization of thalamocortical neurons de‐inactivates T‐channels, causing the switch to burst firing that allows the generation of low‐frequency thalamic oscillations (Llinás & Steriade, 2006). The subiculum, the main output of the hippocampal formation, is another structure where burst firing plays a prominent role in oscillatory activity, most notably during fast gamma rhythms (Stanford et al. 1998; Eller et al. 2015). Unlike thalamocortical neurons, the current responsible for burst firing in subicular neurons remains unknown. Dendritic calcium currents were initially implicated, as bursting can modulate synaptic plasticity through active backpropagation of somatic spikes along the proximal apical dendrite (Stewart & Wong, 1993; Taube, 1993). The importance of T‐currents for the Ca2+ influx in dendrites has been well established in pyramidal neurons of the hippocampal CA1 region (Christie et al. 1995; Magee & Carruth, 1999). However, Jung et al. (2001) proposed that the Ca2+ tail current mediated mainly by high‐voltage activated (HVA) Ca2+ channels (presumably R‐type), and not T‐channels, is the necessary component of the afterdepolarization potential (ADP) that drives bursting in the rat subiculum. More recently, the same group showed that slow inactivation of voltage‐gated sodium channels participates in the switch from a bursting to a regular‐spiking pattern, but without affecting the peak of ADP (Cooper et al. 2005). Therefore, it appears that multiple ionic currents contribute to the mechanisms underlying bursting in subiculum.
The role of T‐channels in subicular burst firing remains unclear, largely due to the lack of selective pharmacological tools. However, new selective T‐channel blockers have been synthesized and tested recently, both in preclinical (Shipe et al. 2008; Kraus et al. 2010) and clinical settings (Egan et al. 2013; Ziegler et al. 2015). Here, we conducted a comprehensive characterization of T‐channel function in the mouse and rat subiculum by combining whole‐cell patch‐clamp electrophysiology with pharmacological, molecular and genetic tools that selectively target T‐channels. Our results show that low‐threshold burst firing in subicular neurons is abolished by the pan‐selective T‐channel blocker TTA‐P2 (3,5‐dichloro‐N‐[1‐(2,2‐dimethyl‐tetrahydro‐pyran‐4‐ylmethyl)‐4‐fluoropiperidin‐4‐ylmethyl]‐benzamide). Importantly, inhibition of T‐channel‐mediated burst firing significantly reduced CA1–subiculum long‐term potentiation (LTP). Consistent with our pharmacology data, Cav3.1 knockout mice have reduced T‐current densities when compared to wild‐type (WT) mice. In addition, both depolarization‐ and hyperpolarization‐induced burst firing, as well as LTP are attenuated in neurons lacking CaV3.1 T‐channels. Our data provide the first direct evidence for the role of Cav3.1 isoform of T‐channels in burst firing of subicular neurons. Furthermore, subiculum serves as a relay centre between the hippocampal complex and numerous cortical and subcortical structures (O'Mara, 2005). Thus, our data strongly suggest a critical role of CaV3.1 channels in various hippocampal‐dependent cognitive processes.
Methods
Drugs
TTA‐P2 (Alomone Labs, Israel) was prepared as a 3 mm stock solution in dimethyl sulfoxide (DMSO); aliquots were kept at –20°C and diluted for use at final concentrations of 5 μm and 10 μm, known to selectively block T‐type calcium channels (Shipe et al. 2008; Dreyfus et al. 2010; Choe et al. 2011). In addition, we have recently shown that 10 μm TTA‐P2 does not affect the shape or amplitude of baseline excitatory synaptic currents in thalamic neurons, suggesting a lack of effect on NMDA and AMPA receptors (DiGruccio et al. 2015). Thus, it is unlikely that possible alternative molecular targets, except for T‐channels, can account for the effects of TTA‐P2 on neuronal excitability in the subiculum.
Animals
Young and adult C57BL/6J, CaV3.1 knockout (KO) and CaV3.2 KO mice (P7–P208), and young Sprague‐Dawley (P7–P23) and Wistar rats (P11–P28) of both genders were used for this study. Sprague‐Dawley and Wistar rats were obtained from Envigo (Indianapolis, IN, USA). C57BL/6J mice were obtained from The Jackson Laboratory (USA). CaV3.1 (α1G) (Ricken BioResources Centre, Japan) and CaV3.2 (α1H) (The Jackson Laboratory) null mice were generated as previously described (see Petrenko et al. 2007 and Chen et al. 2003, respectively). For the characterization of T‐channels in GABA interneurons, we used young (P11–P28) transgenic rats of both genders that co‐express Venus, a derivative of yellow fluorescent protein, with the vesicular GABA transporter (VGAT‐Venus rat) (Ricken BioResources Centre; as described in Uematsu et al. 2008). All animals were maintained on a 12 h light–dark cycle with food and water ad libitum. Animals were housed within accredited animal facilities according to protocols approved by the University of Virginia Animal Care and Use Committee, University of Colorado Anschutz Medical Campus or the Washington University Animal Studies Committee. Treatments of rats adhered to guidelines set forth in the NIH Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize animal suffering and to use only the number of animals necessary to produce reliable scientific data.
Quantitative real‐time reverse transcription‐PCR
mRNA expression of T‐type calcium channel isoforms was performed on subicular tissue of P9–P10 Sprague‐Dawley rat pups (n = 12). After anesthetizing animals with isoflurane and decapitation, each brain was sliced on a vibratome (Leica VT 1200S) in ice‐cold PBS, creating 500–700 μm thick horizontal slices. Under a dissecting microscope, the subiculum from each slice was collected and placed on dry ice. RNA was isolated using RNeasy Microarray Tissue Mini Kit with QiAzol (Qiagen), and quantitative real‐time reverse transcription (qRT)‐PCR was performed on BioRad Icycler, with RT2 First Strand Kit and RT2 SYBR Green qPCR Mastermix (Qiagen) according to manufacturer's protocols. Primers for CaV3.1 and CaV3.3 T‐type calcium channel isoforms were purchased from Qiagen (CACNA1G NM_031601.4; CACNA1I NM_020084.3). For CaV3.2 channel, primer was constructed by Eurofins according to the primer sequence from Bourinet et al. (2005) (forward primer 5′‐TCATCACTCACACGGCAGCTA‐3′; reversed primer 5′‐TGGGCCACGTCCAGGTT‐3′). Cyclophilin was used as an internal standard (Qiagen, Ppid NM_001004279.1). Obtained qRT‐PCR data were analysed as previously described (Jagodic et al. 2007). In brief, the cycle threshold (Ct) for cyclophilin was subtracted from each corresponding channel Ct value from each sample, and relative mRNA levels were expressed as 2−∆Ct.
Brain slice preparation for patch‐clamp experiments
Animals were anesthetized briefly with isoflurane and decapitated, and their brains rapidly removed. Live 250–300 μm thick horizontal or sagittal brain slices were sectioned at 4°C in pre‐chilled solution containing (in mm): sucrose 260, d‐glucose 10, NaHCO3 26, NaH2PO4 1.25, KCl 3, CaCl2 2, MgCl2 2, using a vibrating micro‐slicer (Ted Pella Instruments DTK 1000). Brain slices were then immediately incubated for 45 min in solution containing (in mm): NaCl 124, d‐glucose 10, NaHCO3 26, NaH2PO4 1.25, KCl 4, CaCl2 2, MgCl2 2 at 37°C prior to use in electrophysiology recordings, which were done at room temperature except for some current‐clamp experiments that were done at 33°C. During incubation, slices were constantly perfused with a gas mixture of 95 vol% O2 and 5 vol% CO2.
Electrophysiological recordings were made randomly from subicular neurons throughout the pyramidal and polymorphic layers between CA1 and presubiculum. This was of particular importance for the comparison of different firing patterns in mice (WT vs. CaV3.1 KO vs. CaV3.2 KO) since it has been reported that the distribution of bursting and regular‐spiking neurons might differ along the proximal–distal axis (Staff et al. 2000; Kim & Spruston, 2012). To exclude possible differences along the dorsal–ventral axis, these experiments were done using both horizontal and sagittal slices.
Electrophysiology experiments
The external solution for all whole‐cell electrophysiology experiments consisted of (in mm): NaCl 125, d‐glucose 25, NaHCO3 25, NaH2PO4 1.25, KCl 2.5, MgCl2 1, CaCl2 2. This solution was equilibrated with a mixture of 95 vol% O2 and 5 vol% CO2 for at least 30 min with a resulting pH of about 7.4. For current‐clamp experiments, the internal solution consisted of (in mm): potassium‐d‐gluconate 130, EGTA 5, NaCl 4, CaCl2 0.5, Hepes 10, Mg ATP 2, Tris GTP 0.5, pH 7.2. The internal solution for experiments examining well isolated T‐currents consisted of (in mm): tetramethyl ammonium (TMA)‐OH 135, EGTA 10, MgCl2 2, and Hepes 40, titrated to pH 7.2 with hydrofluoric acid (HF) (Todorovic & Lingle, 1998). Glass micropipettes (Sutter Instruments O.D. 1.5 mm) were pulled using a Sutter Instruments model P‐97 or P‐1000 and fabricated to maintain an initial resistance of 3–5 MΩ. Neuronal membrane responses were recorded using an Axopatch 200B or Multiclamp 700B amplifier (Molecular Devices, Foster City, CA, USA). Voltage current commands and digitization of the resulting voltages and currents were performed with Clampex 8.3 software (Molecular Devices) running on an IBM‐compatible computer. Resulting current and voltage traces were analysed using Clampfit 10.5 (Molecular Devices).
Current‐clamp experiments
Regular‐spiking and depolarization‐induced burst firing properties of subicular neurons were characterized by injecting a family of depolarizing (5–190 pA) current pulses of 500 ms duration in 10 pA incremental steps through the recording pipette, in the presence of synaptic blockers ((20 μm picrotoxin, 50 μm d‐2‐amino‐5‐phosphonovalerate (d‐APV) and 5 μm 2,3‐dihydroxy‐6‐nitro‐7‐sulfamoyl‐benzo[f]quinoxaline‐2,3‐dione (NBQX)) in the external solution. To assess hyperpolarization‐induced (rebound) firing, a multi‐step protocol was used by injecting a depolarizing current of 200 pA followed by a series of hyperpolarizing currents in 50 pA increments stepping from −200 to −400 pA. Subsequent resting membrane potentials, tonic action potential frequencies, rebound action potentials and input resistances were determined. Resting membrane potential was measured at the beginning of each recording and was not corrected for the liquid junction potential, which was between 5 and 10 mV in our experiments. The membrane input resistance was calculated by dividing the steady‐state hyperpolarizing voltage deflection by the injected current. In some experiments, pyramidal neurons were filled with 0.5% biocytin and processed for visualization by using an avidin–horseradish peroxidase (HRP) 3,3′‐diaminobenzidine reaction. Current‐clamp experiments designed to determine whether T‐currents contribute to the excitability of subicular neurons were done using wild‐type Sprague‐Dawley and Wistar rat strains. These data were initially analysed separately but we found no apparent difference between the two strains and thus we combined the data for final analysis.
T‐current properties
T‐channel activation was measured by stepping the membrane potential from an initial holding potential (V h) of −90 mV to test potentials (V t) of −80 mV to +5 mV in 5 mV increments over a period of 320 ms. Current–voltage (I–V) curves were generated, and peak current amplitudes and inactivation properties of current waveforms were established and compared in both mice and rats. Steady‐state inactivation curves were generated by using a standard double‐pulse protocol with 3.6 s‐long pre‐pulses to variable voltages (from −120 to −50 mV in 5 mV increments) and test potentials to −50 mV. The voltage dependencies of activation and steady‐state inactivation were described with single Boltzmann distributions of the following forms:
In these forms, I max is the maximal amplitude of current; G max is the maximal conductance (calculated by dividing current amplitude by estimated reversal potential); V 50 is the voltage at which half of the current is activated or inactivated; and k represents the voltage dependence (slope) of the distribution. The time constant of T‐current inactivation in subicular neurons was assessed by fitting the decaying portions of the current waveforms at the peak potential of the I–V relationships (−40 mV) with a single exponential function.
Recordings from intact brain slices offer great advantages for studying neurons in an intact setting in vitro. However, the presence of extensive neuron processes compromises voltage control in whole‐cell recordings from slices so that all biophysical measurements must be interpreted with caution. Accordingly, we paid close attention to signs of good voltage control. Specifically, we analysed cells in which there was no extensive delay in the onset of current; also, the onset and offset kinetics depended on voltage, not on the amplitude of current. In whole‐cell experiments, because intact subicular neurons have long processes, rapid components of recorded current, such as tail currents, are unlikely to reflect the true amplitude and time course of calcium current behaviour. The use of internal solution with with TMA and HF allowed us to isolate T‐currents from high‐voltage‐activated (HVA) calcium currents (Todorovic & Lingle, 1998). The amplitude of T‐currents was measured from the peak, which was subtracted from the current at the end of a depolarizing test potential to avoid contamination with a small HVA current component. The inactivation time constant of T‐currents was assessed only in neurons where the amplitude of residual HVA current amplitude was less than 10 pA.
All drugs were applied during the time period ranging from 5 to 15 min until an apparent steady‐state effect was achieved. TTA‐P2 was pre‐incubated with slices for 15 min for the LTP experiments in rats. The quantitative assessment of drug effects is limited since delivery of drug‐containing solutions in vitro may be compromised due to diffusion through the sliced tissue. Therefore, the actual concentrations of all compounds at their sites of action are likely to be lower than those reported.
Long‐term potentiation study in rats
The hippocampus was dissected from postnatal day 28–32 Sprague‐Dawley rats under isoflurane anesthesia. Slices (500 μm thick) were cut from the middle part of hippocampus with a rotary slicer in artificial cerebrospinal fluid (ACSF) containing (in mm): NaCl 124, KCl 5, MgSO4 2, CaCl2 2, NaH2PO4 1.25, NaHCO3 22, and glucose 10, bubbled with 95% O2–5% CO2 at 4–6°C. After recovering for at least one hour at 30°C, hippocampal slices were transferred to a submerged recording chamber with continuous bath perfusion of ACSF bubbled with 95% O2–5% CO2 at 2 ml min−1 at 30°C (Tokuda et al. 2010). CA1 extracellular recordings were obtained from the apical dendritic layer of the CA1 region elicited with 0.1 ms constant current pulses through a bipolar stimulating electrode placed in stratum radiatum. For the subiculum extracellular recordings, a bipolar electrode was placed in stratum oriens at the very end of the CA1 region. Field excitatory postsynaptic potentials (fEPSPs) were monitored using a half‐maximal stimulus based on a baseline input–output curve. For LTP induction in the CA1 region a 100 Hz × 1 s high frequency stimulation (HFS) was delivered once, whereas in the subiculum a 200 Hz × 1 s HFS was delivered twice at an interval of 30 s. Another input–output curve was obtained 60 min after HFS for analysis of changes in fEPSPs.
Long‐term potentiation study in mice
Hippocampal slices (300 μm) were dissected from postnatal day 28–42 CaV3.1 KO mice or control age‐matched wild‐type C57BL/6J mice. fEPSPs were monitored as above. LTP for both the CA3–CA1 and CA1–subiculum pathway was induced by delivering a theta burst stimulation (TBS) train (four pulses delivered at 100 Hz in 30 ms bursts, repeated 10 times with 200 ms interburst intervals) as described previously (Orfila et al. 2014; Dietz et al. 2016; Deng et al. 2017). The amount of potentiation was calculated as the percentage change from baseline (the averaged 10 min slope value from 50 to 60 min post‐TBS divided by the averaged slope value at 10 min prior to TBS).
Data analysis
In all electrophysiology experiments, we attempted to obtain as many neurons as possible from each animal in order to minimize number of animals used. Results are presented as means ± SEM. Statistical analysis was performed using Student's two‐tailed t test, or paired t test where appropriate, as well as two‐way ANOVA with repeated measures; significance was accepted as P < 0.05. Where applicable, Tukey's test for post hoc comparisons was also used. For continuous data sets (repeated measures experiments), we applied linear mixed models (LMM) in order to account for intracluster correlation effects, and to avoid reducing data to means per animal (Galbraith et al. 2010; DiGruccio et al. 2015). We did not present the outputs of this statistical approach since it yielded very similar results to two‐way ANOVA (per neuron analysis). For comparison of proportions, Fisher's exact test was used. Issues of normality and heterogeneity of variation were evaluated to determine the adequacy of the ANOVA models and whether additional manipulations were warranted. Statistical and graphical analyses were performed using GraphPad Prism 5.01 software (GraphPad Software, La Jolla, CA, USA), Origin 7.0 (OriginLab, Northhampton, MA, USA) or SigmaPlot 12.0 (Systat Software Inc., Chicago, IL, USA). Linear mixed model statistical analysis was conducted using PASW Statistics 18 (SPSS Inc., Chicago, IL, USA).
Results
Biophysical properties of T‐currents and effects of T‐channel antagonism on spike firing patterns in the rat subiculum
The majority of studies investigating the ionic currents that underlie burst firing in the subiculum were conducted in rats (Jung et al. 2001; Menendez de al Prida et al. 2003; Cooper et al. 2005). However, the isolated T‐currents in the rat subiculum are yet to be reported. Therefore, we first examined the biophysical properties of T‐currents using standard voltage‐clamp protocols in rat brain slices. As expected, extensive neuron processes could hamper voltage control in whole‐cell recordings from brain slices. To avoid this potential ‘space‐clamp’ problem, we investigated biophysical properties of T‐currents from younger rats (P7–P14). Representative traces from our double‐pulse experiments that are used to assess voltage‐dependent inactivation are presented in Fig. 1 A. Plots in Fig. 1 B and C show that average maximal peak T‐current amplitude was about 250 pA with a corresponding current density of about 4 pA/pF. Figure 1 D shows that the average V 50 value for steady‐state inactivation was −88.9 ± 0.4 mV with a slope factor of 7.7 ± 0.3 (n = 22 neurons, 6 rats). Representative traces from our current–voltage (I–V) experiments that are used to assess voltage‐dependent activation of T‐currents are presented in Fig. 1 E. Plots in Fig. 1 F and G show that average maximal peak T‐current amplitude was about 200 pA with a corresponding current density of about 3.5 pA/pF. Figure 1 H shows that the average V 50 for current activation was −52.4 ± 0.4 with slope factor of 4.5 ± 0.3 (n = 14 neurons, 5 rats).
Figure 1. Biophysical properties of T‐currents in the rat subiculum.
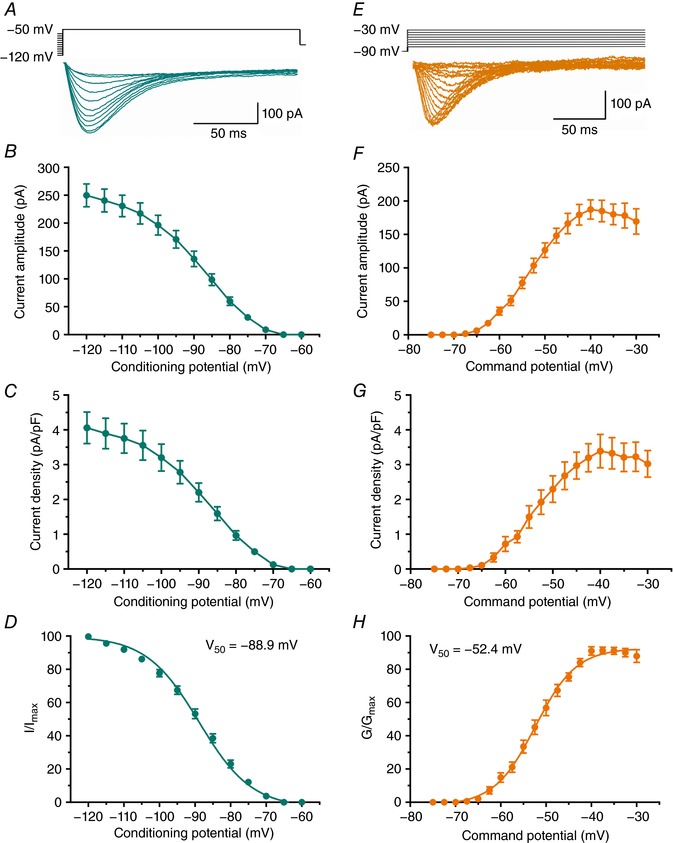
A, averaged T‐current traces from representative subicular neurons generated using a double‐pulse protocol with 3.6‐s‐long prepulses to variable voltages (from −120 to −60 mV in 5 mV increments) and test potential (V t) of −50 mV. B, average current amplitudes over the range of potentials obtained from the steady‐state inactivation protocol. C, average current density, as calculated from the steady‐state inactivation protocol. D, the average steady‐state inactivation (I/I max) curve with V 50 value noted on the graph. E, averaged T‐current I–V traces from representative subicular neurons in the voltage range of V t from −75 to −40 mV from V h of −90 mV in 2.5 mV increments. F, average current amplitudes from multiple I–V curves. G, average current density, as calculated from the I–V curves. H, the average voltage dependence of steady‐state activation (G/G max) curve with V 50 value noted on the graph.
A relatively fast time constant of T‐current inactivation at the peak current activation of V t = −40 mV (33.0 ± 2.1 ms, n = 12 neurons) in current–voltage relationships strongly suggests the dominant expression of CaV3.1 and/or 3.2 T‐channels in the rat subiculum. Similar properties of subicular T‐currents were observed in brain slices from older rats (P15–P28, data not shown).
Next, we performed current‐clamp experiments to investigate if T‐currents contribute to excitability of rat subicular neurons. We found that 10 μm TTA‐P2 strongly attenuated ADP, thus promoting the switch from bursting to regular‐spiking pattern (Fig. 2 A and B, n = 3), at least in response to smaller current injections. The same effect was observed using an even lower concentration of 5 μm TTA‐P2 (n = 5 neurons, data not shown). In addition, we found that 10 μm TTA‐P2 alone may completely abolish rebound burst firing (Fig. 2 C; P = 0.002, two‐tailed paired t test, n = 5 neurons, 4 rats). These findings were confirmed using 5 μm TTA‐P2 (n = 5 neurons).
Figure 2. Pharmacological characterization of burst‐firing pattern in the rat subiculum.
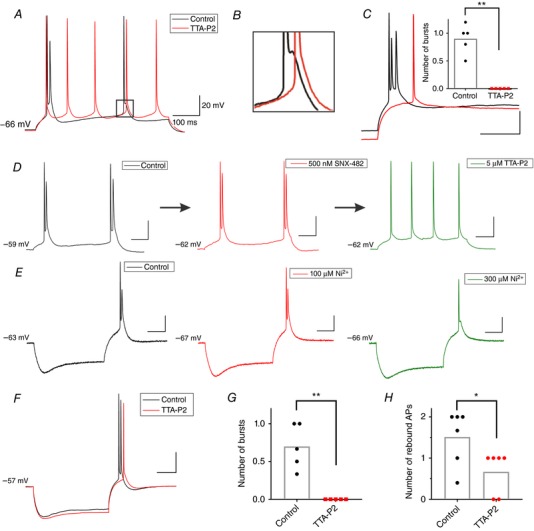
A, original traces from a representative bursting neuron portraying the change in the firing pattern (from burst to regular‐spiking) as a response to a depolarizing 20 pA current injection, before (black) and after the addition of 10 μm TTA‐P2 (red). B, zoomed in trace from A, showing the elimination of ADP hump due to the application of TTA‐P2. C, original trace of rebound bursting (black) which was eliminated by 10 μm TTA‐P2 (red), and instead displaying a single rebound spike after 200 pA hyperpolarizing current injection. The inset graph represents the average number of rebound bursts before and after TTA‐P2 (* P < 0.05, ** P < 0.01, two‐tailed paired t test). D, original traces from a representative bursting neuron (Control; black trace) depicting the lack of effect of SNX‐482 (500 nm) on the burst‐firing pattern as a response to a depolarizing 30 pA current injection (red trace). When the low concentration of TTA‐P2 (5 μm) was added to the bath, the same neuron fired a regular train of action potentials (green trace). E, original traces of hyperpolarization‐induced bursting before (Control; black trace) and after 100 μm Ni2+ (red) that failed to attenuate bursting, followed by the same neuron with 300 μm Ni2+ (green), which successfully eliminated bursting, but without fully removing ADP. F, representative traces from a current‐clamp experiment performed at 33°C showing rebound burst firing in a rat neuron in control conditions (black trace) and after application of 5 μm TTA‐P2 (red trace). G, TTA‐P2 abolished rebound bursts in the experiment performed at 33°C. H, TTA‐P2 decreased the number of rebound APs in the experiments at 33°C.
The lower concentration of TTA‐P2 (5 μm) was used to exclude possible off‐target effects of this selective T‐channel antagonist, especially those concerning CaV2.3 R‐type calcium channels (Choe et al. 2011). To further examine the possible contribution of these HVA calcium channels to bursting, we used SNX‐482, a high‐affinity CaV2.3 R‐type blocker (Newcomb et al. 1998). Interestingly, even at a relatively high concentration of 500 nm (n = 6 neurons), this R‐type blocker had no apparent effect on the bursting pattern (Fig. 2 D, red trace). On the other hand, when TTA‐P2 (5 μm) was added to the external solution, the same bursting neuron was converted to a regular‐spiking train of action potentials (Fig. 2 D, green trace), thus confirming our findings that T‐type, but not R‐type channels are crucially involved in depolarization‐induced burst firing.
There are three known subtypes of T‐type calcium channels based on the cloned pore‐forming α1 subunit: CaV3.1 (α1G), CaV3.2 (α1H) and CaV3.3 (α1I) that are encoded by CACNA1G, CACNA1H and CACNA1I genes, respectively (reviewed by Perez‐Reyes, 2003). Since TTA‐P2 possesses a comparable affinity for all three T‐channel subtypes (Schipe et al. 2008), we investigated the individual contribution of different T‐channels to rebound bursting in the rat subiculum using a concentration of Ni2+ (100 μm) known to preferentially block CaV3.2 vs. CaV3.1 and CaV3.3 channels (Lee et al. 1999). We found that rebound burst firing was not affected by 100 μm Ni2+, suggesting that blocking CaV3.2 channels alone could not prevent the neurons from bursting upon hyperpolarization (red trace in Fig. 2 E, n = 4 neurons), as assessed with insignificant changes in the current injection required to induce burst firing (−250 ± 50 pA vs. −275 ± 75 pA), and the burst threshold (−47.5 ± 1.4 mV vs. −47.1 ± 0.9 mV). When the concentration of Ni2+ was increased to 300 μm (n = 6), both rebound bursting and ADP were completely abolished in four out of six neurons, while in two tested neurons only bursting was blocked but ADP still persisted. The green trace in Fig. 2 E represents one of the neurons where 300 μm Ni2+ abolished bursting but ADP was spared. Taken together, our results with TTA‐P2 and Ni2+ imply that neither CaV3.2 nor CaV2.3 channels play a major role in the rebound burst firing in the rat subiculum.
We next performed additional current‐clamp experiments with TTA‐P2 at a more physiological temperature of 33°C, as this could potentially influence rebound properties of hippocampal neurons in CA1 region (Magee, 1998). Representative traces from these experiments are depicted in Fig. 2 F. Summary data from these experiments show that the inhibitory effect of 5 μm TTA‐P2 at higher temperature in terms of number of rebound bursts (P = 0.006, paired t test; n = 5 neurons, 2 rats; Fig. 2 G) and the number of rebound APs (P = 0.016, paired t test; n = 6 neurons, 3 rats; Fig. 2 H) were similar to its effect at room temperature (Fig. 2 C). Likewise, we found that at 33°C the average number of depolarization‐induced bursts from the resting membrane potentials (as depicted on Fig. 2 A) was greatly decreased after application of 5 μm TTA‐P2 (0.4 ± 0.2) when compared to the control pre‐drug values (1.2 ± 0.3) in these neurons (P = 0.003, paired t test, n = 6 neurons, 3 rats; data not shown).
Based on our findings of insensitivity of burst and ADP to low concentrations of Ni2+, fast T‐current inactivation kinetics, short duration of bursts, and available in situ hybridization data (Talley et al. 1999), it appears that CaV3.1 isoform may constitute the largest portion of T‐channels in the subiculum. To investigate this idea further, we used qRT‐PCR to assess mRNA expression of all three genes encoding for different T‐channel isoforms in the rat subiculum (n = 12 rats). This experiment revealed significantly higher mRNA expression of CaV3.1 than CaV3.2 or CaV3.3 isoforms (P < 0.001 vs. both isoforms, one‐way ANOVA followed by Tukey's post hoc test; Fig. 3), whereas the expression levels of CaV3.2 were higher than the CaV3.3 isoform (P = 0.042). Furthermore, CaV3.1 mRNA expression levels were around twofold higher than CaV3.2, and even threefold higher than the least expressed CaV3.3 isoform, confirming CaV3.1 channels as the dominant T‐channel isoform in the rat subiculum.
Figure 3. Dominant expression of CaV3.1 isoform of T‐channel in the rat subiculum.
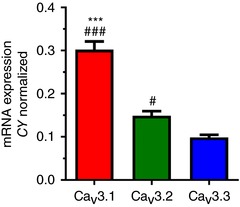
Bar graph with qRT–PCR analysis on excised subicular tissue revealed the dominant expression of CaV3.1 isoform of T‐channels (*** P < 0.001 vs. CaV3.2, # P < 0.05 and ### P < 0.001 vs. CaV3.3 isoform, Tukey's post hoc test).
Rebound firing of certain subicular fast‐spiking GABA interneurons is also T‐channel dependent
In the subiculum, among typical larger pyramidal neurons there are also smaller neurons – putative GABAergic inhibitory interneurons that comprise less than 20% of all cells (Amaral & Witter, 1995; Menendez de la Prida et al. 2003). Little is known, though, about the characteristics and firing patterns of GABAergic interneurons. To characterize putative T‐currents and examine their possible contribution to the rebound spiking in non‐pyramidal (GABAergic), as well as in pyramidal (glutamatergic) neurons, we used Wistar rats that express yellow fluorescent protein (YFP) in VGAT‐containing GABAergic cells. This type of labelling facilitated the identification of GABA interneurons, especially those with morphological features resembling that of pyramidal neurons (Uematsu et al. 2008).
The original traces of T‐currents from GABA interneurons (VGAT+) and pyramidal neurons (VGAT–) are presented in Fig. 4 A. Bar graphs on Fig. 4 B revealed that T‐current amplitudes and densities were almost twofold smaller in VGAT+ neurons (n = 15 neurons, 7 rats) compared to VGAT– neurons (n = 14 neurons, 5 rats) (P < 0.001 and P = 0.004, respectively). In addition, the percentage of neurons that had detectable T‐currents was only about 47% of VGAT‐labelled neurons whereas almost every pyramidal neuron had prominent T‐currents (P < 0.001, Fisher's exact test; Fig. 4 C). It is also noteworthy that pyramidal neurons and GABA interneurons had very similar and relatively fast time constants of macroscopic T‐current inactivation (26.6 ± 1.7 ms, n = 11 and 28.8 ± 3.9 ms, n = 6, respectively), which suggests a negligible contribution of slowly inactivating CaV3.3 isoform, which has an average inactivation time constant ranging from 73 to 83 ms (see Joksovic et al. 2005).
Figure 4. Properties of T‐currents in subicular GABAergic interneurons and their classification.
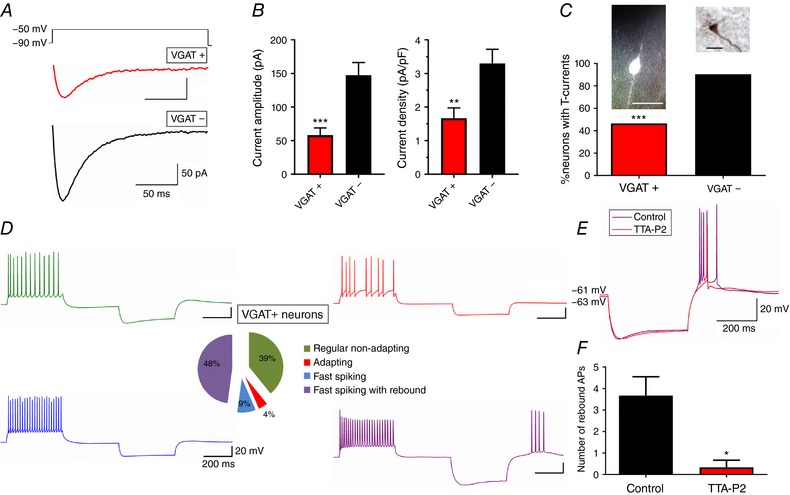
A, average original T‐current traces from representative VGAT+ (red) and VGAT– (black) neurons, generated using a double‐pulse protocol with 3.6‐s‐long prepulses to −90 mV and test potential of −50 mV. B, GABA interneurons (VGAT+) have smaller T‐current amplitudes and density compared to pyramidal neurons (VGAT–). (* P < 0.05, ** P < 0.01 and *** P < 0.001, Tukey's post hoc test) C, relative percentages of VGAT+ and VGAT– neurons with detectable T‐currents accompanied by fluorescently labelled VGAT+ interneuron (left), and biocytin labelled VGAT– pyramidal neuron (right) determined morphologically. Scale bars: 10 μm. (*** P < 0.001, Fisher's exact test) D, classification of identified VGAT‐labelled neurons according to their firing patterns, with largest constituent being fast‐spiking with rebound (48%, purple trace). E, original traces of VGAT+ neuron characterized as fast‐spiking with rebound before (purple) and after the application of 10 μm TTA‐P2 (red). F, number of rebound action potentials before (Control) and after TTA‐P2. (* P < 0.05, two‐tailed paired t test).
Next, we wanted to identify and classify all VGAT‐labelled neurons according to their firing patterns using the Petilla Interneuron Nomenclature Group terminology (2008). Out of twenty‐three neurons recorded in six VGAT‐Venus rats, eleven were fast‐spiking with rebound firing (48%), nine were regular non‐adapting (39%), two were fast‐spiking without rebound firing (9%), and only one neuron had adapting properties (4%) (Fig. 4 D). Since the fraction of fast‐spiking interneurons with rebound firing remarkably coincided with the percentage of VGAT‐labelled neurons that contain prominent T‐currents, we speculated that the hyperpolarization‐induced spiking in at least some interneurons depends on T‐channels. Indeed, the application of 10 μm TTA‐P2 strongly attenuated the rebound firing (Fig. 4 E), observed through the decrease in the number of rebound APs (Fig. 4 F; P = 0.038, two‐tailed paired t test, n = 3 neurons). The sag ratio, as well as input resistance, was not significantly affected after the addition of TTA‐P2, implying that I h alone could not elicit more than one rebound spike, despite a prominent sag voltage deflection in these neurons.
Selective inhibition of T‐currents abolished burst firing in the subiculum of wild‐type (WT) C57BL/6J mice
Next, we began to characterize T‐channel function in the subiculum of C57BL/6J mice. First, we characterized biophysical properties of subicular T‐currents in young (P7–P14) WT mice using the same protocols as we did for studies in the rat subiculum (Fig. 1). Representative traces from our double‐pulse experiments used to assess voltage‐dependent inactivation are presented in Fig. 5 A. Plots in Fig. 5 B and C show that average maximal peak T‐current amplitude was about 400 pA with a corresponding current density of about 6 pA/pF. Furthermore, Fig. 5 D shows that similar to young rats, the average V 50 value for steady‐state inactivation was −86.8 ± 0.6 mV with a slope factor of 7.7 ± 0.6 (n = 11 neurons, 3 mice). Representative traces from our I–V experiments used to assess voltage‐dependent activation of subicular T‐currents in young mice are presented in Fig. 5 E. Plots in Fig. 5 F and G show that average maximal peak T‐current amplitude was about 250 pA with a corresponding current density of about 4 pA/pF. Figure 5 H shows that the average V 50 for steady‐state activation was −51.8 ± 0.4 with a slope factor of 5.4 ± 0.4 (n = 11 neurons, 3 mice). It is interesting to note that slopes for activation curves in both rats and mice correspond reasonably well to the value of the slope for activation curves of 5.0 that we reported in HEK293 cells expressing CaV3.1 channels (Eckle & Todorovic, 2010). These data indicate that we had reasonably well‐clamped recordings of native T‐currents in brain slices from young animals in our experimental conditions.
Figure 5. Biophysical properties of T‐type currents in subiculum of C57BL/6J WT mice.
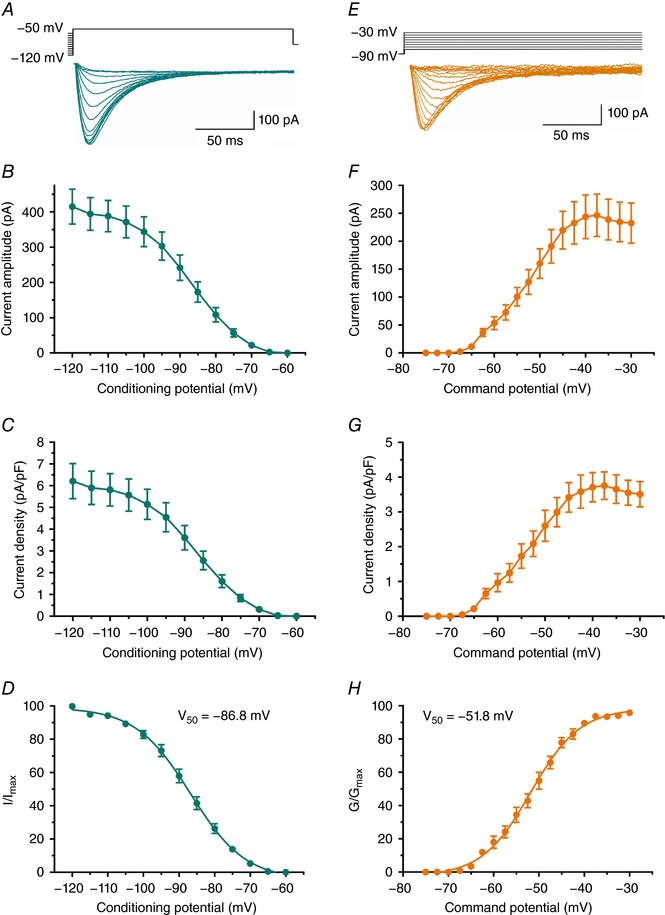
A, original averaged T‐current traces (green lines) from representative subicular neurons generated using a double‐pulse protocol with 3.6‐s‐long prepulses to variable voltages (from −120 to −60 mV in 5 mV increments) and test potential of −50 mV. B, averaged current amplitude from multiple neurons obtained by our steady‐state inactivation protocol similar to experiments depicted in A. C, average current density, as calculated from the steady‐state inactivation protocol. D, the steady‐state inactivation (I/I max) curve with V 50 value noted on the graph. E, original averaged I–V T‐current traces from representative subicular neurons in the voltage range of Vt from −75 to −40 mV from Vh of −90 mV in 2.5 mV increments. F, averaged current amplitudes obtained using the I–V protocols. G, averaged current density across different potentials was calculated from the I–V protocols. H, the voltage dependence of steady‐state activation (G/G max) curve with V 50 value noted on the graph.
Next, we used current‐clamp experiments to investigate the role of T‐currents in excitability of neurons in the subiculum of WT mice. The first protocol aimed to assess the depolarization‐induced burst firing by gradually increasing current injections. The original traces presented in Fig. 6 A show the effect of 10 μm TTA‐P2 on burst firing of a subicular neuron, where the same 30 pA current injection at nearly the same resting membrane potential resulted in two different firing patterns. Blocking T‐channels with TTA‐P2 effectively switched spike firing from bursting (black trace) to regular‐spiking (red trace) (n = 5), as evidenced by the significantly lower average number of bursts and action potentials within the burst (Fig. 6 B, P = 0.027 and P < 0.001, respectively, two‐tailed paired t test).
Figure 6. TTA‐P2 inhibits burst firing in C57BL/6J mice.
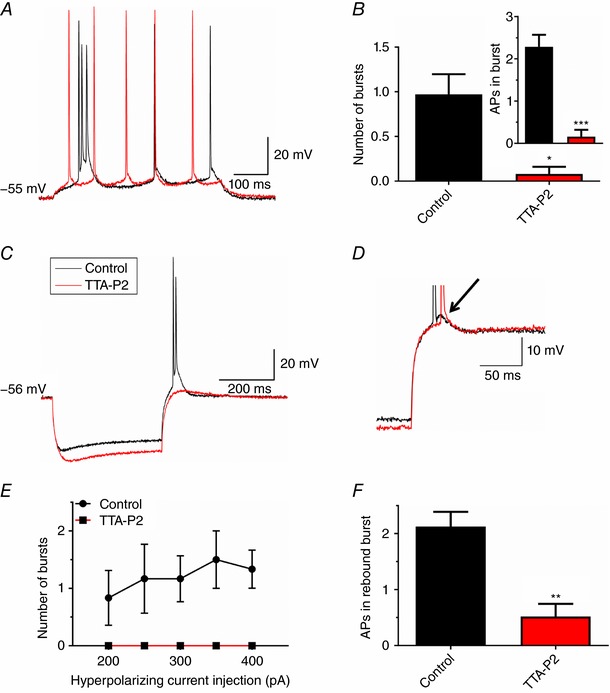
A, original traces from a representative subicular neuron before (black) and after the addition of10 μm TTA‐P2 (red) showing the change in firing pattern from bursting to regular spiking as a response to 30 pA depolarizing current injection via the recording electrode. B, TTA‐P2 (10 μm) decreased the average number of depolarization‐induced bursts. The inset shows lower number of action potentials during burst firing upon the addition of TTA‐P2. C, original traces from a representative subicular neuron before (black) and after the addition of 10 μm TTA‐P2 (red) depict the loss of hyperpolarization‐induced burst firing. D, the afterdepolarization potential (ADP) was clearly attenuated after the addition of TTA‐P2 (red), compared to the control trace (black). E, TTA‐P2 (10 μm) abolished rebound burst firing across different hyperpolarizing current injections. F, TTA‐P2 (10 μm) significantly decreased the number of rebound action potentials. * P < 0.05, ** P < 0.01 and *** P < 0.001, two‐tailed t test.
Figure 6C shows that TTA‐P2 (red trace) also abolished burst firing (black trace) as a response to a hyperpolarizing current injection. It was also evident that ADP, which underlies bursting, was completely abolished by TTA‐P2 (Fig. 6 D). The resting membrane potential was slightly more negative (−55.5 ± 0.7 vs. −58.5 ± 0.7 mV; P = 0.03, two‐tailed paired t test, n = 6), whereas input resistance and sag voltage deflection were higher, but non‐significantly, after the addition of TTA‐P2. These changes in passive neuronal properties could not account for the lower excitability, which was also evidenced by a significant decrease in the number of rebound bursts (Fig. 6 E; no significant interaction, factor treatment: F(1,5) = 7.11, P = 0.045, two‐way ANOVA with repeated measures, n = 5), as well as the decrease in average number of action potentials within the burst (Fig. 6 F; P = 0.009, two‐tailed paired t test, n = 5). Thus, these results give direct evidence that both depolarization‐ and hyperpolarization‐induced burst firing in the mouse subiculum are T‐channel dependent.
CaV3.1 KO mice have decreased T‐current amplitudes and a smaller fraction of burst firing neurons in the subiculum
Since our mRNA expression data in rats, fast current kinetics typical for CaV3.1/3.2 vs. CaV3.3 isoforms (Peres‐Reyes, 2003), as well as previous in situ hybridization data in both mouse (http://www.alleninstitute.org/what-we-do/brainscience/research/scientific-publications/) and rat subiculum (Talley et al. 1999), revealed the dominant expression of mRNA for CaV3.1 followed by CaV3.2 isoform, we focused our study on these two T‐channel subtypes. Hence, we first used voltage‐clamp recordings to compare the amplitudes of T‐currents in the mouse subiculum in wild‐type (WT), CaV3.1 KO and CaV3.2 KO mice. Averages of representative traces from these experiments are depicted in Fig. 7 A for WT (black trace), CaV3.1 KO (red trace) and CaV3.2 KO mice (green trace). Average data points presented in Fig. 7 B demonstrate that current amplitudes after −110 mV prepulse were decreased in CaV3.1 KO for about 50% (n = 19 neurons, 4 mice) in comparison to WT neurons (n = 27 neurons, 9 mice), as assessed with one‐way ANOVA followed by Tukey's post hoc test (F(2,60) = 5.40, P = 0.007; post hoc: P = 0.005). Furthermore, T‐current amplitudes in the subiculum of CaV3.1 KO mice were also smaller, although not significantly, than in mice lacking CaV3.2 subtype (n = 17 neurons, 5 mice), while current amplitudes in CaV3.2 KO mice were not significantly different from WT mice. The time constant of macroscopic T‐current inactivation for subicular neurons was relatively fast, with an inactivation τ of 32.6 ± 3.0 ms at V t −40 mV in WT (n = 9), and 34.9 ± 6.6 ms (n = 6) in CaV3.2 KO mice, whereas inactivation τ in CaV3.1 KO was somewhat slower at 46.0 ± 4.7 ms (n = 9, P = 0.029 vs. WT, two‐tailed t test). Taken together, our results strongly suggest the dominant functional expression of CaV3.1 T‐type calcium channels in the WT mouse subiculum.
Figure 7. Decreased T‐current densities and bursting in the subiculum of CaV3.1 KO mice.
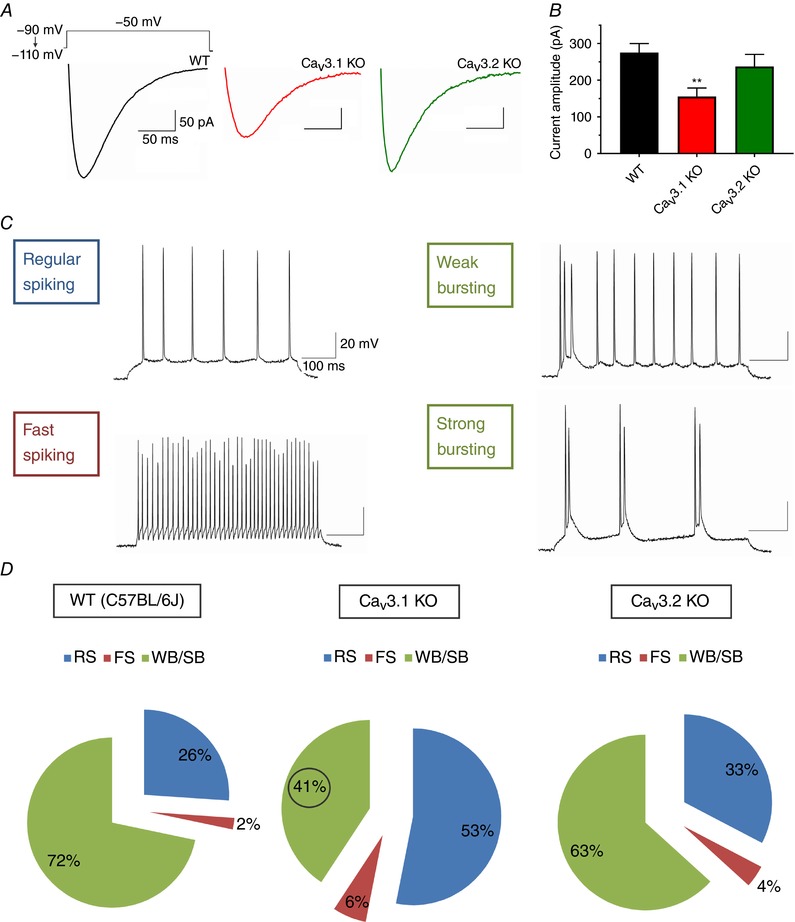
A, representative averaged original traces of subicular T‐currents from WT mice (black line), CaV3.1 KO mice (red line) and CaV3.2 KO mice (green line). B, bar graphs represent averaged current amplitudes after −110 mV prepulse during the steady‐state inactivation protocols in three cohorts as depicted in panel A of this figure. Note that average current amplitudes are significantly decreased only in CaV3.1 KO mice when compared to WT mice. C, original traces from representative of the four spike firing patterns identified in the WT mouse subiculum as a response to a 500‐ms depolarizing current injection. D, pie charts representing the percentages of different firing patterns of subicular neurons in C57BL/6J, CaV3.1 KO and CaV3.2 KO mice: RS, regular‐spiking; FS, fast‐spiking; WB, weak bursting; SB, strong bursting; total n = 222 neurons. Note the smaller portion of burst firing neurons and larger portion of regular spiking neurons in CaV3.1 KO mice when compared to C57BL/6J WT mice.
In the present study, we identified four different firing patterns in the mouse subiculum by injecting a 500‐ms depolarizing current: regular‐spiking, fast‐spiking, weak bursting and strong bursting (Fig. 7 C). The criteria used for classification were taken from Staff et al. (2000), with slight modification. In short, a burst was defined as the occurrence of at least two action potentials firing at high frequency (>100 Hz), followed by ADP. Weak bursting neurons fired one initial burst followed by a regular train of single spikes (similar to regular‐spiking neurons), whereas strong bursting fired at least two bursts during a 500‐ms depolarizing pulse. Consistent with respective T‐current amplitudes, the fraction of bursting neurons out of the total population was 72% in the WT group (66 out of 92 neurons), 41% in CaV3.1 KO (33 out of 81), and 63% in CaV3.2 KO mice (31 out of 49) (Fig. 7 D). The apparently lower percentage of small fast‐spiking neurons in wild‐type mice could be a matter of chance, given our data, and it should not influence the overall conclusion that bursting neurons were less likely to be found in the subiculum proper of CaV3.1 KO mice than of WT (P < 0.001, Fisher's exact test), or CaV3.2 KO mice (P = 0.018). In contrast, the small difference in fraction of bursting neurons in WT (72%) and CaV3.2 KO (63%) mice was not significant (P = 0.342). In rats, the distribution of regular‐spiking and burst firing neurons seemingly follows a simple pattern: the further away from CA1, the more likely the neuron will exhibit bursting (Staff et al. 2000; Kim & Spruston, 2012). To our knowledge, no such studies have been performed on mice; however, to avoid this possibility we recorded randomly throughout the area between CA1 and presubiculum in all three groups of mice.
Based on these findings, as well as the voltage‐clamp experiments showing a large decrease in current amplitude only in mice lacking the CaV3.1 T‐channel, we decided to focus our subsequent current‐clamp experiments on the comparison between CaV3.1 KO and WT mice.
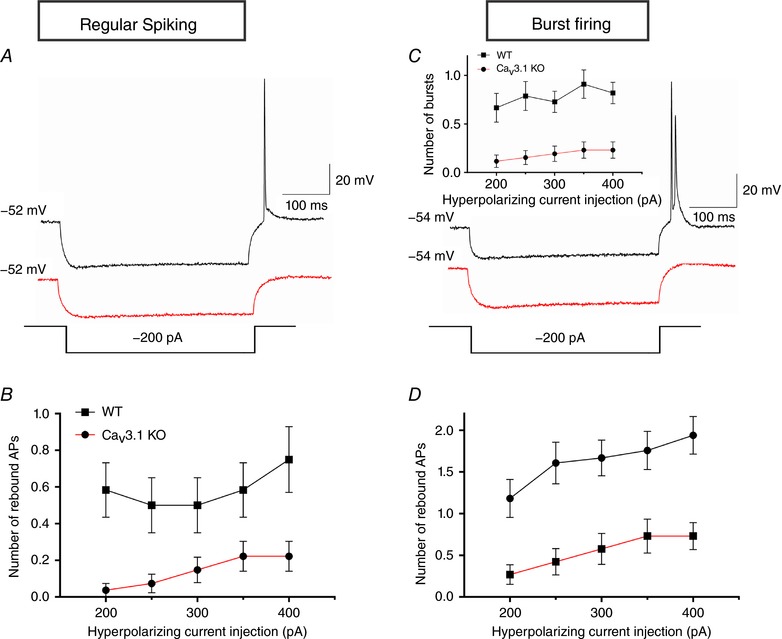
Decreased excitability of regular‐spiking and burst firing neurons in CaV3.1 KO mice
In different brain areas, regular‐spiking neurons always respond to a brief depolarization pulse with a train of repetitive single action potentials. To investigate the possible contribution of CaV3.1 channels to the excitability of regular‐spiking neurons, gradually increasing current injections were applied through the recording pipette. Over the range of current injections between 40 and 190 pA, a significant decrease in AP frequency ranging from about 20% to 70% was noticed in CaV3.1 KO (n = 26 neurons, 14 mice), compared to WT mice (n = 12 neurons, 9 mice) (Fig. 8 A and B). For example, the AP frequency after the strongest current injection was 29.8 ± 1.7 Hz in CaV3.1 KO mice and 37.8 ± 3.3 Hz in WT mice. This difference was even more apparent following weaker current injections (i.e. 70% decrease at 50 pA current injections). Importantly, passive membrane properties were similar in CaV3.1 KO and WT mice (Table 1).
Figure 8. Excitability characteristics of subicular neurons of WT and CaV3.1 KO mice.
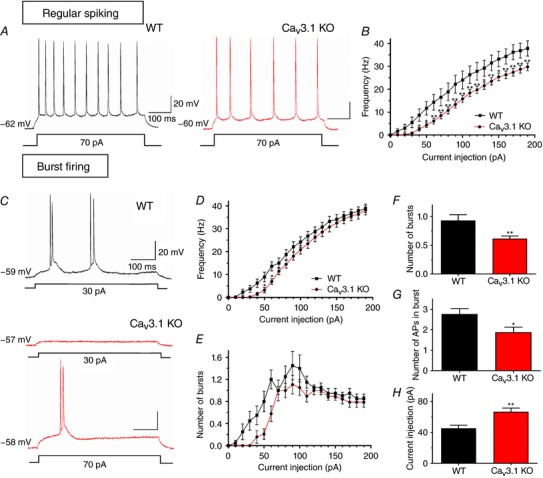
A, original traces from representative regular‐spiking subicular neurons in WT (black) and CaV3.1 KO (red) mice show active membrane responses to a depolarizing current injection of 70 pA. B, a family of escalating current injections resulted in lower frequency of regular‐spiking CaV3.1 KO neurons, compared to WT. C, original traces from representative burst firing neurons in the subiculum of WT (black) and CaV3.1 KO (red) mice show active membrane responses to 30 pA and 70 pA. As opposed to neurons lacking CaV3.1 T‐channels, bursting in WT neurons could be elicited already at 30 pA. D, the firing frequency of CaV3.1 KO bursting neurons shows effects only at smaller current injections. E, a lower number of bursts in CaV3.1 KO than in WT neurons was revealed after a series of escalating current injections. F and G, the average number of bursts (F) and the average number of APs per burst (G) were significantly decreased in CaV3.1 KO, compared to WT neurons. H, a larger current injection was required to elicit burst firing in CaV3.1 KO than in WT neurons (* P < 0.05 and ** P < 0.01, Tukey's post hoc test or two‐tailed t test).
Table 1.
Passive membrane properties of subicular neurons from the mice used in our current‐clamp recordings
| WT | CaV3.1 KO | CaV3.2 KO | |||
|---|---|---|---|---|---|
| RMP (mV) | Input res. (MΩ) | RMP (mV) | Input res. (MΩ) | RMP (mV) | Input res. (MΩ) |
| Regular‐spiking neurons (n = 12) | Regular‐spiking neurons (n = 27) | Regular‐spiking neurons (n = 10) | |||
| −57.2 ± 1.1 | 125.2 ± 7.0 | −57.9 ± 0.6 | 128.5 ± 6.6 | −60.1 ± 1.1 | 104.3 ± 5.9 |
| Burst firing neurons (n = 38) | Burst firing neurons (n = 26) | Burst firing neurons (n = 20) | |||
| −57.1 ± 0.6 | 103.3 ± 3.3 | −57.8 ± 0.9 | 111.9 ± 4.8 | −57.6 ± 0.9 | 92.2 ± 5.4** |
Passive membrane properties of subicular neurons from C57BL/6J (WT), CaV3.1 KO and CaV3.2 KO mice. RMP, resting membrane potential; Input res., input resistance (calculated for −200 pA current injection). ** P < 0.01 for CaV3.2 KO vs. CaV3.1 KO group (one‐way ANOVA followed by Tukey's post hoc test). The number of neurons in each cohort is indicated in the parentheses.
Next, we examined the importance of CaV3.1 channels in burst‐firing neurons using the same depolarizing protocol that was applied to regular‐spiking neurons (Fig. 8 C). This experiment revealed a significantly lower AP frequency in CaV3.1 KO (n = 19 neurons, 10 mice) than in WT mice (n = 20 neurons, 13 mice), but only at 50 and 60 pA current injections (Fig. 8 D), as assessed with two‐way ANOVA with repeated measures followed by Tukey's test (interaction: F(19,703) = 1.72, P = 0.029; post hoc: P = 0.025, and P = 0.010, respectively). Most notably, however, the number of bursts was profoundly diminished, or completely abolished, in CaV3.1 KO mice at different current injections (Fig. 8 E; genotype: F(1,37) = 6.22, P = 0.017, current injection: F(19,703) = 26.19, P < 0.001 and their interaction: F(19,703) = 1.58, P = 0.056). The fact that CaV3.1 KO neurons had a lower number of bursts was also evident when we averaged the data across all current injections (Fig. 8 F; P = 0.007, two‐tailed t test, n = 21 neurons per group). Furthermore, these neurons exhibited lower number of APs within the burst than WT neurons (Fig. 8 G; P = 0.013, two‐tailed t test). Finally, a significantly larger current injection was needed to elicit a burst in CaV3.1 KO neurons (66.7 ± 4.8 pA), compared to the control group (45.2 ± 4.2 pA; Fig. 8 H; P = 0.002, two‐tailed t test). The significantly lower excitability of burst firing neurons lacking CaV3.1 channels was also evident when we normalized the current injection over the burst threshold (P = 0.001 vs. WT, two‐tailed t test; data not shown). All parameters measured indicate that the activation of CaV3.1 isoform‐containing T‐type calcium channels is required for a normal function of burst firing neurons in the mouse subiculum.
Rebound firing in the mouse subiculum is heavily dependent on CaV3.1 T‐channels
Representative traces from regular‐spiking neurons from WT (black line) and CaV3.1 KO mice (red line) are presented on Fig. 9 A. We found that the average number of rebound APs was significantly lower in mice lacking the CaV3.1 T‐channels (Fig. 9 B; genotype: F(1,37) = 12.92, P < 0.001; current injection: F(4,148) = 3.96, P = 0.004; their interaction: F(4,148) = 1.22, P = 0.303). This effect was so prominent that, for example, after the 200 pA hyperpolarizing current injection less than 4% of CaV3.1 KO regular‐spiking neurons (1 out of 27) responded with a rebound AP, compared to the WT group (7 out of 12 neurons). Again, it is important to note that the resting membrane potential and input resistance were not significantly affected by genotype (Table 1).
Figure 9. Properties of rebound firing of subicular neurons in WT and CaV3.1 KO mice.
A, original traces from representative regular‐spiking neurons of WT (black) and CaV3.1 KO (red) mice show active membrane responses to a hyperpolarizing current injection of 200 pA. B, escalating current injections resulted in fewer rebound action potentials (APs) in CaV3.1 KO than in WT neurons. C, original traces from representative burst firing neurons of WT (black) and CaV3.1 KO (red) mice, as a response to a hyperpolarizing current injection of 200 pA. The inset graph shows a profound decrease in rebound burst firing across a set of hyperpolarizing current injections. D, the number of rebound APs per burst was also significantly diminished in CaV3.1 KO, compared to WT neurons.
Next, the rebound firing of burst‐firing subicular neurons was investigated using the same protocol (Fig. 9 C). Similar to regular‐spiking neurons, the number of bursts was profoundly affected in CaV3.1 KO (n = 26 neurons, 12 mice), compared to WT mice (n = 33 neurons, 20 mice) (inset of Fig. 9 C; genotype: F(1,57) = 15.19, P < 0.001; current injection: F(4,228) = 3.59, P = 0.007; their interaction: F(4,228) = 0.68, P = 0.605). As expected, the number of rebound APs in the burst firing neurons was also significantly diminished in the KO group (Fig. 9 D; genotype: F(1,57) = 15.58, P < 0.001; current injection: F(4,228) = 12.46, P < 0.001; their interaction: F(4,228) = 0.81, P = 0.520). We did not detect any substantial difference in the resting membrane potential between CaV3.1 KO (−57.8 ± 0.9 mV, n = 26 neurons) and WT mice (−57.1 ± 0.6 mV, n = 38 neurons). In addition, the input resistance of burst‐firing neurons was not significantly affected by genotype (WT 103.3 ± 3.3 MΩ vs. CaV3.1 KO 111.9 ± 4.8 MΩ; P > 0.05). Therefore, it is evident that rebound firing of both regular‐spiking and burst‐firing neurons in the mouse subiculum strongly depends on the CaV3.1 isoform of T‐type calcium channels.
TTA‐P2 and global deletion of CaV3.1 T‐channels induce profound suppression of LTP in the subiculum but not in CA1 region
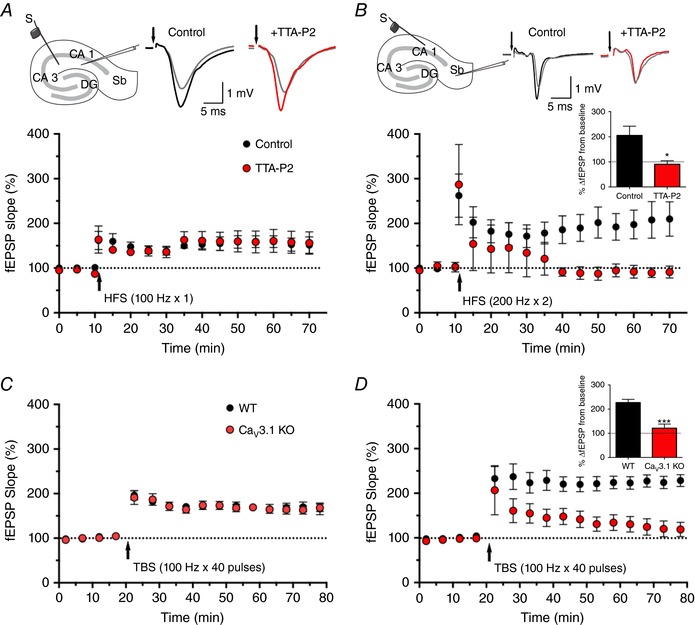
In vitro studies using extracellular field potential recordings have demonstrated that high‐frequency stimulation of CA1 efferents can induce LTP in the subiculum (Boeijinga & Boddeke, 1996), which has been corroborated by in vivo studies (Commins et al. 1998). In contrast to other hippocampal regions (e.g. CA1), the contribution of voltage‐gated calcium channels to long‐term plasticity at CA1–subiculum synapses has not yet been studied. Hence, we examined LTP in both CA3–CA1 and the CA1–subiculum pathway in the absence and presence of 10 μm TTA‐P2. Baseline field excitatory postsynaptic potentials (fEPSPs) elicited from single stimuli did not differ significantly between control and TTA‐P2‐treated slices in either region. Robust LTP induction was observed 60 min after stimulation in controls for both the CA1 region and subiculum electrode locations, as assessed by the input–output curves (with fEPSPs showing increases to 141 ± 12% (n = 5) and 189 ± 28% (n = 5) of baseline, respectively 60 min after HFS). In the CA1 recorded regions (Fig. 10 A), TTA‐P2 treatment failed to suppress LTP (155 ± 8% (n = 5) of baseline). On the other hand, LTP in the subiculum was eliminated by TTA‐P2, as evidenced by the significant decrease in the fEPSP slope, averaged between 50 and 60 min after high‐frequency stimulation (P = 0.020, two‐tailed t test; n = 5; inset of Fig. 10 B).
Figure 10. Selective inhibition with TTA‐P2 and deletion of CaV3.1 channels supress LTP in the subiculum but not in the CA1 region.
A, schematic diagram of hippocampal formation: stimulation probe for CA1 recordings was placed in the Schaffer collateral pathway. Original fEPSP traces in control (grey/black) and 10 μm TTA‐P2‐treated slices (grey/red). Constant current pulses (0.1 ms, 100 Hz) HFS successfully induced LTP in CA1 regardless of TTA‐P2 application measured over 70 min. B, schematic diagram of hippocampal formation: stimulation probe for LTP induction in the subiculum was located towards the end of CA1 in the stratum oriens/alveus region, whereas the recording pipette was in the molecular layer of the subiculum. Original fEPSP traces depict the absence of LTP at the CA1–subiculum synapse after the addition of 10 μm TTA‐P2 (grey/red), as opposed to the control slices (grey/black). The normalized fEPSP slopes show that HFS at 200 Hz x 2 successfully induced subicular LTP in the absence, but not in the presence of TTA‐P2. The bar graph in the inset shows a significant decrease in the average fEPSP (50–60 min after HFS) in slices pre‐incubated with 10 microM TTA‐P2, compared to the control (* P < 0.05, two‐tailed t test). C, time course of normalized fEPSP slope recordings of CA3–CA1 pathway in the hippocampus from WT (black circles) and CaV3.1KO (red circles) mice, showing successful LTP induction in both WT and Cav3.1 KO mice. Arrow indicates timing of TBS (40 pulses). D, time course of normalized fEPSP slope recordings of CA1–subiculum pathway in the hippocampus from WT (black circles) and CaV3.1KO (red circles) mice, showing successful LTP induction in WT, but not Cav3.1 KO mice. The bar graph in the inset shows quantification of change in the normalized fEPSP slope averaged between 50 and 60 min after TBS. *** P < 0.001 compared with WT, two tailed t test.
Finally, we used mouse genetics to investigate if synaptic plasticity in the subiculum is mediated, at least in part, through CaV3.1 channels. At the CA3–CA1 synapse, LTP development after theta burst stimulation was practically identical in WT (Fig. 10 C; 166 ± 13% of baseline 60 min after TBS, n = 6) and CaV3.1 KO mice (168 ± 8%, n = 6). Conversely, the same stimulation protocol revealed striking difference between fEPSP slopes of WT and CaV3.1 KO mice at the CA1–subiculum synapse (inset of Fig. 10 D; P < 0.001, two‐tailed t test, n = 6 in both groups), suggesting that CaV3.1 channels indeed play a crucial role in synaptic plasticity at this particular synapse.
Discussion
Subicular neurons have prominent T‐currents
Our voltage‐clamp experiments revealed similar biophysical properties of T‐currents in subicular neurons of WT mice and rats and correspond well with studies investigating T‐currents in the adjacent CA1 area of the hippocampal formation (Takahashi et al. 1991), or acutely isolated and tissue‐cultured rat hippocampal neurons (O'Dell & Alger, 1991). Using dynamic‐clamp experiments, Tscherter et al. (2011) showed that even subtle differences in biophysical properties of T‐channels could shape the precise firing patterns of thalamic neurons. For example, the alteration of the control slope factor drastically changed the excitability of these neurons. Remarkably, the maximal excitability was observed for the slope factor of k = 6, which corresponds reasonably well with the values obtained in our experiments, thus drawing a direct comparison between the two CaV3.1 channel‐rich populations of neurons (thalamocortical and subicular).
It is noteworthy that practically all pyramidal neurons investigated had detectable T‐currents with prominent amplitudes. On the other hand, we detected T‐currents in less than half of GABAergic interneurons, in accordance with the likelihood of rebound spiking. Therefore, we speculate that this portion of fast‐spiking GABA interneurons is able to produce a robust T‐channel‐dependent rebound burst, since TTA‐P2 drastically suppressed this firing pattern. Further support for this hypothesis is provided by an immunohistochemistry study showing that CaV3.1 channels are moderately expressed in GABAergic interneurons, throughout hippocampal layers (Vinet & Sik, 2006).
T‐channels mediate burst firing in both mouse and rat subiculum
A transient depolarization of the membrane potential following a single spike (i.e. ADP) seems to be necessary for mediating burst firing of subicular neurons (Menendez de la Prida et al. 2003). Although the ionic mechanisms responsible for ADP have not been fully revealed yet, an important role of low‐threshold calcium currents in creating ADP has been already established in different brain regions (Zhang et al. 1993; Geijo‐Barrientos, 2000). In the CA1 region, it appears that the bursting firing pattern of developing neurons results from a complex mechanism consisting of a back‐propagating spike that activates T/R‐ and L‐type calcium channels in the distal apical dendrites and persistent voltage‐gated Na+ channels in the somatic region (Chen et al. 2005). A similar mechanism was suggested in the rat subiculum (Jung et al. 2001; Wellmer et al. 2002; Cooper et al. 2005). Our present results showed that the ADP was largely suppressed by blocking T‐channels with TTA‐P2, thus promoting a switch from bursting to regular‐spiking. Furthermore, the findings from pharmacological experiments have been largely supported by our study with KO mice, where neurons lacking CaV3.1 channels exhibited significantly lower bursting compared to WT mice.
Interestingly, the fact that bursting in subicular neurons may occur at more depolarized membrane potentials was used in prior studies as a basis to rule out T‐channels as significant contributors to subicular excitability (Jung et al. 2001; Cooper et al. 2005). Hence, a fundamental role of T‐channels in depolarization‐induced burst firing in the subiculum reported in our current study is somewhat surprising since the predominant population of T‐channels is inactivated at the more depolarized potentials. Nonetheless, Swensen & Bean (2003) found that T‐current is responsible for most of the total calcium current between the spikes in Purkinje neurons, where bursting after short depolarization strongly resembles that elicited in subicular pyramidal neurons. Our finding is also in agreement with a study by Deleuze et al. (2012), which showed that the activation of T‐channels plays a crucial role in tonic firing of thalamocortical neurons, and thus regulates the transfer of sensory inputs during wakefulness.
What about the contribution of R‐type calcium channels? All studies investigating calcium currents underlying burst firing in the subiculum have used nickel (Ni2+) in an effort to discern between low‐ and high‐voltage activated calcium currents. However, Ni2+ blocks both CaV3.2 T‐type and CaV2.3 (R‐type) channels at similar, low micromolar concentrations (Zamponi et al. 1996; Lee et al. 1999). Our experiments showed that depolarization‐induced burst firing was not affected by a concentration of Ni2+ thought to be selective for CaV2.3 and CaV3.2 currents. Furthermore, our experiments revealed that SNX‐482 failed to block bursting in rats, thus strongly suggesting that R‐type calcium channels are not an essential component of a burst in the subiculum. Alternatively, it is possible that the current underlying the ADP could be supported, at least partly, by an SNX‐resistant isoform of CaV2.3 channels (Metz et al. 2005).
CaV3.1 channels have a prominent role in the excitability of subicular neurons
Our voltage‐clamp experiments in mice revealed a large decrease in current amplitude in CaV3.1 when compared to WT and CaV3.2 KO mice. This finding has been corroborated by current‐clamp data where neurons lacking CaV3.1 channels had a lower fraction of burst‐firing neurons. In addition, the relatively fast time of macroscopic T‐current inactivation and relatively short duration of bursts strongly argues against prominent contribution of CaV3.3 channels. However, mRNA expression of the CaV3.3 isoform observed in our study suggests a possibility of distal dendritic localization of this T‐channel subtype in the subiculum, as proposed by McKay et al. (2006). Further molecular, electrophysiological and immuno‐histochemical studies, as well as the use of genetically altered mice, are needed to investigate this notion in subicular neurons.
CaV3.1 channels are necessary for LTP expression in the subiculum
It is known that postsynaptic bursting may be necessary and sufficient for LTP induction within the CA1 area of the hippocampus (reviewed in Paulsen & Sejnowski, 2000). The recruitment of dendritic Ca2+ currents by backpropagating action potentials is a crucial component of this phenomenon. If this is also the case in the subiculum, LTP induction and/or expression might be suppressed by eliminating burst firing. Indeed, our results demonstrate a complete block of LTP expression in the subiculum by TTA‐P2 at a concentration that also blocks burst firing. Nonetheless, we cannot exclude possible effects of TTA‐P2 on presynaptic components in the mechanisms underlying LTP at these synapses, although a lack of effect of TTA‐P2 on basal transmission in either CA1 or subiculum argues against a major presynaptic effect.
Our LTP study in mice replicates findings in rats and supports the idea that CaV3.1 isoform is necessary for the induction of LTP at the CA1–subiculum pathway. Furthermore, our findings suggest that blocking T‐channels or deleting the CaV3.1 isoform alone has no effect on initial LTP induction at CA3–CA1 synapses. Our finding is consistent with previous research showing an important link between T‐type calcium channels and LTP in a number of neuronal circuits, including the visual cortex (Yoshimura et al. 2008), Purkinje cells (Ly et al. 2013) and CA1 (Ito et al. 1995; Chen et al. 2012). It is noteworthy that LTP was completely absent in parallel fibre Purkinje cells lacking CaV3.1 channels, whereas blocking T‐type calcium channels with TTA‐P2 equally prevented LTP induction (Ly et al. 2013). In another study using CaV3.2 KO mice, only the expression of late‐phase LTP (180 min after high frequency stimulation) was lost in hippocampal slices (Chen et al. 2012). This study showed that early phase LTP was not significantly altered in the CaV3.2 KO mice, which may explain the lack of effect of TTA‐P2 on LTP induction at the CA3–CA1 synapse, observed in our experiment. In the related experiment at this synapse, the application of 25 μm Ni2+ inhibited the induction of LTP likely by blocking calcium influx through both CaV3.2 T‐type and CaV2.3 R‐type calcium channels (Isomura et al. 2002), indicating that CaV3.2 isoform of T‐channels may play a role in stabilizing LTP after initial induction.
Our study provides the first evidence that T‐channels are important for synaptic plasticity in the subiculum. Similar to other brain areas, LTP induction at the CA1–subiculum pathway is NMDA receptor‐dependent (Boeijinga & Boddeke, 1996). Thus, the removal of the voltage‐dependent Mg2+ block of NMDA receptors by T‐channel‐induced depolarization may be a critical step in this process. Finally, our findings implicate the bursting mechanism as a crucial element of synaptic plasticity in the subiculum, which has already been suggested for CA1 region (Thomas et al. 1998; Pike et al. 1999).
In conclusion, deciphering the ionic mechanisms underlying bursting in the subiculum could help clarify how this region plays a role in different processes dependent on hippocampal function. Besides memory processing, the subiculum, with its predominance of burst firing neurons, appears to play an important role in the initiation, propagation, and/or termination of epileptic seizures of the temporal lobe origin (reviewed in Stafstrom, 2005). T‐channels may present a central mechanism underlying the excitability of subicular neurons, and thus prove to be an important drug target for the treatment of temporal lobe epilepsy. On the other hand, promoting subicular excitability through T‐channel potentiation could be beneficial for treatment of cognitive deficits such as those seen in schizophrenia or Alzheimer's disease.
Additional information
Competing interests
The authors declare no conflicts of interests.
Author contributions
S.M.J., P.E. and M.R.D. performed all patch‐clamp experiments. S.Lj.J., V.T. and S.M.J. performed qRT‐PCR experiments. LTP study in rats was designed by Y.I. and C.F.Z., and performed by Y.I.; the LTP study in mice was designed by P.S.H. and performed by R.M.D. and J.E.O. S.M.J. and S.M.T. planned the entire study, analysed the data, and wrote the paper with input from Y.I., V.J.‐T., P.S.H. and C.F.Z. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the National Institutes of Health (R01GM102525 to S.M.T., R01GM118197 to V.J.‐T. and MH077791 to C.F.Z.) and funds from the Department of Anesthesiology, University of Virginia and Department of Anesthesiology University of Colorado Anschutz Medical Camus (S.M.T.), as well as the Bantly Foundation (C.F.Z.).
Acknowledgements
VGAT‐Venus transgenic rats were generated by Drs Y. Yanagawa, M. Hirabayashi and Y. Kawaguchi in National Institute for Physiological Sciences, Okazaki, Japan, using pCS2‐Venus provided by Dr A. Miyawaki. We thank Dr Charles Adrian Handforth for donating breeding pairs of CaV3.1 knockout mice to our laboratory.
Linked articles This article is highlighted by a Perspective by Turner. To read this Perspective, visit https://doi.org/10.1113/JP274981.
References
- Amaral DG & Witter MP (1995). Hippocampal formation In The Rat Nervous System, 2nd edn, ed. Paxinos G, pp. 247–291. Academic Press, New York. [Google Scholar]
- Boeijinga PH & Boddeke HWGM (1996). Activation of 5‐HT1B receptors suppresses low but not high frequency synaptic transmission in the rat subicular cortex in vitro. Brain Res 721, 59–65. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrère C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A & Nargeot J (2005). Silencing of the Cav3.2 T‐type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 24, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commins S, Gigg J, Anderson M & O'Mara SM (1998). Interaction between paired‐pulse facilitation and long‐term potentiation in the projection from hippocampal area CA1 to the subiculum. Neuroreport 9, 4109–4113. [DOI] [PubMed] [Google Scholar]
- Chen C‐C, Lamping KG, Nuno DW, Barresi R, Prouty SJ, Lavoie JL, Cribbs LL, England SK, Sigmund CD, Weiss RM, Williamson RA, Hill JA & Campbell KP (2003). Abnormal coronary function in mice deficient in α1H T‐type Ca2+ channels. Science 302, 1416–1418. [DOI] [PubMed] [Google Scholar]
- Chen C‐C, Shen J‐W, Chung N‐C, Min M‐Y, Cheng S‐J & Liu IY (2012). Retrieval of context‐associated memory is dependent on the Cav3.2 T‐s. PLoS One 7, e29384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yue C & Yaari Y (2005). A transitional period of Ca2+‐dependent spike afterdepolarization and bursting in developing rat CA1 pyramidal cells. J Physiol 567, 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe W, Messinger RB, Leach E, Eckle VS, Obradovic A, Salajegheh R, Jevtovic‐Todorovic V & Todorovic SM (2011). TTA‐P2 is a potent and selective blocker of T‐type calcium channels in rat sensory neurons and a novel antinociceptive agent. Mol Pharmacol 80, 900–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Eliot LS, Ito K, Miyakawa H & Johnston D (1995). Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike‐induced Ca2+ influx. J Neurophysiol 73, 2553–2557. [DOI] [PubMed] [Google Scholar]
- Cooper DC, Chung S & Spruston N (2005). Output‐mode transitions are controlled by prolonged inactivation of sodium channels in pyramidal neurons of subiculum. PLoS Biology 3, e175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleuze C, David F, Béhuret S, Sadoc G, Shin HS, Uebele VN, Renger JJ, Lambert RC, Leresche N & Bal T (2012). T‐type calcium channels consolidate tonic action potential output of thalamic neurons to neocortex. J Neurosci 32, 12228–12236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz RM, Deng G, Orfila JE, Hui X, Traystman RJ & Herson PS (2016). Therapeutic hypothermia protects against ischemia‐induced impairment of synaptic plasticity following juvenile cardiac arrest in sex‐dependent manner. Neuroscience 325, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G, Orfila JE, Dietz RM, Moreno‐Garcia M, Rodgers KM, Coultrap SJ, Quillinan N, Traystman RJ, Bayer KU & Herson PS (2017). Autonomous CaMKII Activity as a drug target for histological and functional neuroprotection after resuscitation from cardiac arrest. Cell Rep 18, 1109–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGruccio MR, Joksimovic S, Joksovic PM, Lunardi N, Salajegheh R, Jevtovic‐Todorovic V, Beenhakker MP, Goodkin HP & Todorovic SM (2015). Hyperexcitability of rat thalamocortical networks after exposure to general anesthesia during brain development. J Neurosci 35, 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus FM, Tscherter A, Errington AC, Renger JJ, Shin HS, Uebele VN, Crunelli V, Lambert RC & Leresche N (2010). Selective T‐type calcium channel block in thalamic neurons reveals channel redundancy and physiological impact of I Twindow . J Neurosci 30, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle VS & Todorovic SM (2010). Mechanisms of inhibition of CaV3.1 T‐type calcium current by aliphatic alcohols. Neuropharmacology 59, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Zhao X, Smith A, Troyer MD, Uebele VN, Pidkorytov V, Cox K, Murphy M, Snavely D, Lines C & Michelson D (2013). Randomized controlled study of the T‐type calcium channel antagonist MK‐8998 for the treatment of acute psychosis in patients with schizophrenia. Hum Psychopharmacol 28, 124–133. [DOI] [PubMed] [Google Scholar]
- Eller J, Zarnadze S, Bäuerle P, Dugladze T & Gloveli T (2015). Cell type‐specific separation of subicular principal neurons during network activities. PLoS One 10, e0123636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith S, Daniel JA & Vissel B (2010). A study of clustered data and approaches to its analysis. J Neurosci 30, 10601–10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geijo‐Barrientos E (2000). Subthreshold inward membrane currents in guinea‐pig frontal cortex neurons. Neuroscience 95, 965–972. [DOI] [PubMed] [Google Scholar]
- Isomura Y, Fujiwara‐Tsukamoto Y, Imanishi M, Nambu A & Takada M (2002). Distance‐dependent Ni2+‐sensitivity of synaptic plasticity in apical dendrites of hippocampal CA1 pyramidal cells. J Neurophysiol 87, 1169–1174. [DOI] [PubMed] [Google Scholar]
- Ito K, Miura M, Furuse H, Zhixiong C, Kato H, Yasutomi D, Inoue T, Mikoshiba K, Kimura T, Sakakibara S et al (1995). Voltage‐gated Ca2+ channel blockers, omega‐AgaIVA and Ni2+, suppress the induction of theta‐burst induced long‐term potentiation in guinea‐pig hippocampal CA1 neurons. Neurosci Lett 183, 112–115. [DOI] [PubMed] [Google Scholar]
- Izhikevich EM, Desai NS, Walcott EC & Hoppensteadt FC (2003). Bursts as a unit of neural information: selective communication via resonance. Trends Neurosci 26, 161–167. [DOI] [PubMed] [Google Scholar]
- Jagodic MM, Pathirathna S, Nelson MT, Mancuso S, Joksovic PM, Rosenberg ER, Bayliss DA, Jevtovic‐Todorovic V & Todorovic SM (2007). Cell‐specific alterations of T‐type calcium current in painful diabetic neuropathy enhance excitability of sensory neurons. J Neurosci 27, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joksovic PM, Brimelow BC, Murbartián J, Perez‐Reyes E & Todorovic SM (2005). Contrasting anesthetic sensitivities of T‐type Ca2+ channels of reticular thalamic neurons and recombinant Cav3.3 channels. Br J Pharmacol 144, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HY, Staff NP & Spruston N (2001). Action potential bursting in subicular pyramidal neurons is driven by a calcium tail current. J Neurosci 21, 3312–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y & Spruston N (2012). Target‐specific output patterns are predicted by the distribution of regular‐spiking and bursting pyramidal neurons in the subiculum. Hippocampus 22, 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus RL, Li Y, Gregan Y, Gotter AL, Uebele VN, Fox SV, Doran SM, Barrow JC, Yang ZQ, Reger TS, Koblan KS & Renger JJ (2010). In vitro characterization of T‐type calcium channel antagonist TTA‐A2 and in vivo effects on arousal in mice. J Pharmacol Exp Ther 335, 409–417. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL & Perez‐Reyes E (1999). Nickel block of three cloned T‐type calcium channels: low concentrations selectively block alpha1H. Biophys J 77, 3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE (1997). Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci 20, 38–43. [DOI] [PubMed] [Google Scholar]
- Llinás RR & Steriade M (2006). Bursting of thalamic neurons and states of vigilance. J Neurophysiol 95, 3297–3308. [DOI] [PubMed] [Google Scholar]
- Ly R, Bouvier G, Schonewille M, Arabo A, Rondi‐Reig L, Léna C, Casado M, De zeeuw CI & Feltz A (2013). T‐type channel blockade impairs long‐term potentiation at the parallel fiber–Purkinje cell synapse and cerebellar learning. Proc Natl Acad Sci USA 110, 20302–20307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BE, McRory JE, Molineux ML, Hamid J, Snutch TP, Zamponi GW & Turner RW (2006). CaV3 T‐type calcium channel isoforms differentially distribute to somatic and dendritic compartments in rat central neurons. Eur J Neurosci 24, 2581–2594. [DOI] [PubMed] [Google Scholar]
- Magee JC (1998). Dendritic hyperpolarization‐activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci 18, 7613–7624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC & Carruth M (1999). Dendritic voltage‐gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J Neurophysiol 82, 1895–901. [DOI] [PubMed] [Google Scholar]
- Menendez de la Prida L, Suarez F & Pozo MA (2003). Electrophysiological and morphological diversity of neurons from the rat subicular complex in vitro. Hippocampus 13, 728–744. [DOI] [PubMed] [Google Scholar]
- Metz AE, Jarsky T, Martina M & Spruston N (2005). R‐type calcium channels contribute to afterdepolarization and bursting in hippocampal CA1 pyramidal neurons. J Neurosci 25, 5763–5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newcomb R, Szoke B, Palma A, Wang G, Chen X, Hopkins W, Cong R, Miller J, Urge L, Tarczy‐Hornoch K, Loo JA, Dooley DJ, Nadasdi L, Tsien RW, Lemos J & Miljanich G (1998). Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas . Biochemistry 37, 15353–15362. [DOI] [PubMed] [Google Scholar]
- O'Dell TJ & Alger BE (1991). Single calcium channels in rat and guinea‐pig hippocampal neurons. J Physiol 436, 739–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Mara S (2005). The subiculum: what it does, what it might do, and what neuroanatomy has yet to tell us. J Anat 207, 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orfila JE, Shimizu K, Garske AK, Deng G, Maylie J, Traystman RJ, Quillinan N, Adelman JP & Herson PS (2014). Increasing small conductance Ca2+‐activated potassium channel activity reverses ischemia‐induced impairment of long‐term potentiation. Eur J Neurosci 40, 3179–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen O & Sejnowski TJ (2000). Natural patterns of activity and long‐term synaptic plasticity. Curr Opin Neurobiol 10, 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E (2003). Molecular physiology of low‐voltage‐activated T‐type calcium channels. Physiol Rev 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Petilla Interneuron Nomenclature Group , Ascoli GA, Alonso‐Nanclares L, Anderson SA, Barrionuevo G, Benavides‐Piccione R, Burkhalter A, Buzsáki G, Cauli B, Defelipe J, Fairén A et al (2008). Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat Rev Neurosci 9, 557–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko AB, Tsujita M, Kohno T, Sakimura K, Baba H (2007). Mutation of α1GT‐type calcium channels in mice does not change anesthetic requirements for loss of the righting reflex and minimum alveolar concentration but delays the onset of anesthetic induction. Anesthesiology 106, 1177–1185. [DOI] [PubMed] [Google Scholar]
- Pike FG, Meredith RM, Olding AW & Paulsen O (1999). Postsynaptic bursting is essential for ‘Hebbian’ induction of associative longterm potentiation at excitatory synapses in rat hippocampus. J Physiol 518, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipe WD, Barrow JC, Yang ZQ, Lindsley CW, Yang FV, Schlegel KA, Shu Y, Rittle KE, Bock MG, Hartman GD et al (2008). Design, synthesis, and evaluation of a novel 4‐aminomethyl‐4‐fluoropiperidine as a T‐type Ca2+ channel antagonist. J Med Chem 51, 3692–3695. [DOI] [PubMed] [Google Scholar]
- Staff NP, Jung HY, Thiagarajan T, Yao M & Spruston N (2000). Resting and active properties of pyramidal neurons in subiculum and CA1 of rat hippocampus. J Neurophysiol 84, 2398–2408. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE (2005). The role of the subiculum in epilepsy and epileptogenesis. Epilepsy Curr 5, 121–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford IM, Traub RD & Jefferys JG (1998). Limbic gamma rhythms. II. Synaptic and intrinsic mechanisms underlying spike doublets in oscillating subicular neurons. J Neurophysiol 80, 162–171. [DOI] [PubMed] [Google Scholar]
- Stewart M & Wong RK (1993). Intrinsic properties and evoked responses of guinea pig subicular neurons in vitro. J Neurophysiol 70, 232–245. [DOI] [PubMed] [Google Scholar]
- Swensen AM & Bean BP (2003). Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci 23, 9650–9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Ueno S & Akaike N (1991). Kinetic properties of T‐type Ca2+ currents in isolated rat hippocampal CA1 pyramidal neurons. J Neurophysiol 65, 148–155. [DOI] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez‐Reyes E & Bayliss DA (1999). Differential distribution of three members of a gene family encoding low voltage‐activated (T‐type) calcium channels. J Neurosci 19, 1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taube JS (1993). Electrophysiological properties of neurons in the rat subiculum in vitro. Exp Brain Res 96, 304–318. [DOI] [PubMed] [Google Scholar]
- Thomas MJ, Watabe AM, Moody TD, Makhinson M & O'Dell TJ (1998). Postsynaptic complex spike bursting enables the induction of LTP by theta frequency synaptic stimulation. J Neurosci 18, 7118–7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic SM & Lingle CJ (1998). Pharmacological properties of T‐type Ca2+ current in adult rat sensory neurons: effects of anticonvulsant and anesthetic agents. J Neurophysiol 79, 240–252. [DOI] [PubMed] [Google Scholar]
- Tokuda K, O'Dell KA, Izumi Y & Zorumski CF (2010). Midazolam inhibits hippocampal long‐term potentiation and learning through dual central and peripheral benzodiazepine receptor activation and neurosteroidogenesis. J Neurosci 30, 16788–16795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tscherter A, David F, Ivanova T, Deleuze C, Renger JJ, Uebele VN, Shin HS, Bal T, Leresche N & Lambert RC (2011). Minimal alterations in T‐type calcium channel gating markedly modify physiological firing dynamics. J Physiol 589, 1707–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uematsu M, Hirai Y, Karube F, Ebihara S, Kato M, Abe K, Obata K, Yoshida S, Hirabayashi, M , Yanagawa Y & Kawaguchi Y (2008). Quantitative chemical composition of cortical GABAergic neurons revealed in transgenic Venus‐expressing rats. Cereb Cortex 18, 315–330. [DOI] [PubMed] [Google Scholar]
- Vinet J & Sík A (2006). Expression pattern of voltage‐dependent calcium channel subunits in hippocampal inhibitory neurons in mice. Neuroscience 143, 189–212. [DOI] [PubMed] [Google Scholar]
- Wellmer J, Su H, Beck H & Yaari Y (2002). Long‐lasting modification of intrinsic discharge properties in subicular neurons following status epilepticus. Eur J Neurosci 16, 259–266. [DOI] [PubMed] [Google Scholar]
- Yoshimura Y, Inaba M, Yamada K, Kurotani T, Begum T, Reza F, Maruyama T & Komatsu Y (2008). Involvement of T‐type Ca2+ channels in the potentiation of synaptic and visual responses during the critical period in rat visual cortex. Eur J Neurosci 28, 730–743. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E & Snutch TP (1996). Nickel block of a family of neuronal calcium channels: subtype‐ and subunit‐dependent action at multiple sites. J Membr Biol 151, 77–90. [DOI] [PubMed] [Google Scholar]
- Zhang L, Valiante TA & Carlen PL (1993). Contribution of the low‐threshold T‐type calcium current in generating the post‐spike depolarizing afterpotential in dentate granule neurons of immature rats. J Neurophysiol 70, 223–231. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Duan WR, An G, Thomas JW & Nothaft W (2015). A randomized double‐blind, placebo‐, and active‐controlled study of T‐type calcium channel blocker ABT‐639 in patients with diabetic peripheral neuropathic pain. Pain 156, 2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]