

Abstract
Numerous drug delivery systems have been applied to the problem of providing prolonged duration local anesthesia (PDLA). Here we review the rationale for PDLA, the desirable features for and important attributes of such systems, and specific examples that have been developed.
Keywords: local anesthesia, drug delivery, biomaterials, injectable particles, injectable liquids, triggerable
Introduction
Pain management can be categorized as systemic or local. In systemic treatments, such as with oral or intravenous opioids (narcotics), the drug courses throughout the body, and has most of its effects in the central nervous system. Systemic treatments therefore tend to have many side effects: constipation, urinary retention, itchiness, and perhaps more seriously, sedation, addiction/tolerance, and death from overdose. Moreover, prescribed opioids have a tendency to be diverted from their intended use (i.e. sold for “recreational purposes”.) In local anesthesia, drugs (local anesthetics) that affect axonal impulse conduction are deposited at the desired site of action, interrupting the transmission of pain signals to the central nervous system. This can be applied by injection throughout a tissue to be affected (infiltration anesthesia), or on a specific peripheral nerve leading to anesthesia of all downstream structures (peripheral nerve blocks), or on the around the spinal cord (spinal and epidural blocks). Local anesthetics can be used to treat acute pain (e.g. perioperative pain), or chronic pain (e.g. cancer-related pain). The use of prolonged duration local anesthesia (PDLA) formulations to prevent the development of neuropathic pain (pain attributed to nerve injury) has been explored, with conflicting results.[1-4] Local anesthesia generally results in both sensory and motor (immobility) block of the affected regions. Systemic toxicity can occur with excessive administration or with accidental intravascular injection,[5,6] and can result in loss of consciousness, seizures, cardiac rhythm disturbances, and cardiovascular collapse.
The effects of local anesthetics are generally relatively short in duration, lasting for several hours after a single injection, which is inadequate for the treatment of prolonged acute pain and chronic pain.[7] In such instances, achieving prolonged sensory nerve blockade, lasting days to weeks, is desirable. In current practice, prolonged nerve blockade can be achieved by repeated injections or by placement of indwelling catheters for continuous infusions of drugs.[8,9] Both approaches are resource intensive, necessitating skilled personnel for administration, adjustment of drug levels, and monitoring of drug effects – both intended (pain relief) and unintended (cardiorespiratory effect). This level of effort and monitoring may require hospitalization and requires patients to be tethered to external devices, which presents a risk of infection.
Design considerations (Box 1)
Box. 1. Desirable Features in Prolonged Duration Local Anesthesia.
– Simple to administer to patients
– It should be safe and reliable
– Initiated by a single injection
– Adequate shelf life
– Biodegradable vehicle, with a degradation rate similar to the rate of depletion of the therapeutic compound.
– Does not require general anesthesia or surgery to initiate
– Prolonged duration of nerve block (as needed for the particular context)
– Relatively zero-order release
– Favorable ratio of sensory and motor blockade
– Acceptable local inflammatory response
– No local myotoxicity and neurotoxicity
– Favorable therapeutic index (minimal systemic toxicity)
– Fully reversible (i.e. wears off)
– Easy to make from available materials
– Cost effective
Prolonged duration local anesthesia requires maintenance of high (therapeutic) drug concentrations at the nerve over an extended period without excessive systemic levels, i.e. a good therapeutic index. Controlled release systems lend themselves naturally to achieving such conditions; we will review some representative examples of such systems. Few have successfully and convincingly increased the duration of local anesthesia much beyond what can be achieved by simple local anesthetics in solution.
The generic properties of an ideal PDLA formulation are summarized in Box 1. It should be initiated by a single injection, without needing general anesthesia or surgery. It should be simple to administer, requiring a minimum of special tools or mixing of ingredients. Prolonged duration local anesthesia should last for an extended period compared to commercially available local anesthetics; the target duration would depend on the application – perhaps a day or two for perioperative pain, days to even weeks for chronic pain. Local tissue reaction should be benign, with minimal local inflammatory response, acute or chronic, and no local neurotoxicity. There should be no systemic toxicity. The delivery vehicle should be biodegradable, ideally at a rate similar to the rate of depletion of the delivered compound, so that residual material does not persist for prolonged periods after resolution of drug effect – which would complicate repeated injection. Where the pain is of finite duration, and where the patient is not terminal, anesthesia should eventually wear off completely without lasting sequelae.
The payload
In selecting materials for PDLA formulations it is important to be mindful of the chemical properties of the amino-amide and amino-ester local anesthetics in current clinical practice (which we will refer to as “conventional” local anesthetics). Conventional local anesthetics share a common chemical structure containing a lipophilic aromatic ring, linked by an ester or amide to a tertiary amine (Fig 1).[7] Protonation under acidic pH of the tertiary amine in amino-esters (with a pKa of 9 – 10) and secondary and tertiary amines in amino-amides (with a pKa of 7 – 8) renders the drugs hydrophilic. In clinical practice they are usually dispensed in acidic aqueous solutions. Acidification can be used to solubilize local anesthetics and enable their loading into the aqueous cores of liposomes or hydrogels. The hydrophobic free base of local anesthetics is soluble in organic solvents and can be incorporated into hydrophobic particles and other delivery vehicles.
Figure. 1.
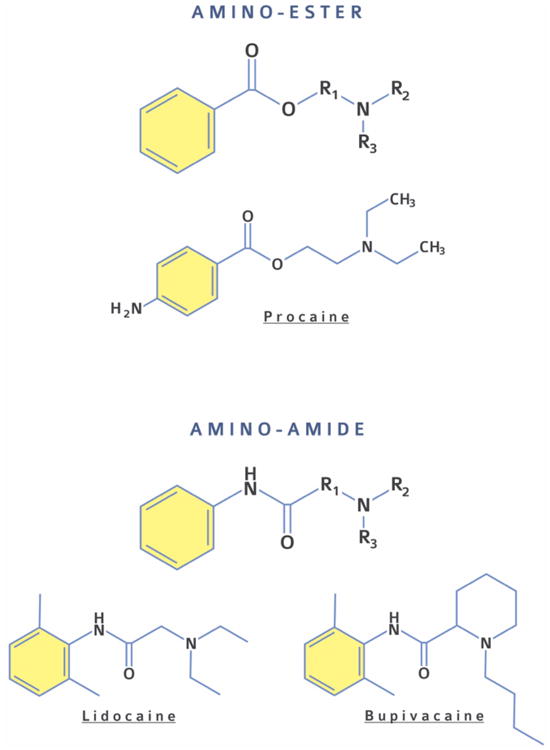
Chemical structures of amino-ester (i.e., procaine) and amino-amide (i.e., lidocaine, bupivacaine) local anesthetics.
The efficacy of local anesthetics can be greatly enhanced by co-delivery of a second or third drug. The concurrent release of such a second drug can greatly increase the duration of block from a sustained release system (Fig. 2). Examples include site 1 sodium channel blockers (e.g., tetrodotoxin and the saxitoxins),[10] and glucocorticoid receptor agonists (e.g. dexamethasone).[11-13] Site 1 sodium channel blockers are themselves ultrapotent local anesthetics, which have the useful property of causing essentially no local tissue injury. [14-16] They are beginning to penetrate into human clinical practice, as seen in the use of neosaxitoxin with and without bupivacaine or epinephrine as a local anesthetic.[17,18] Tetrodotoxin has been used systemically to treat heroin addiction and cancer pain.[19-23] The chemistry of other drugs may not be amenable to simple co-encapsulation with conventional local anesthetics. For example, the site 1 sodium channel blockers are hydrophilic and may have substantial (and toxic) burst release when co-encapsulated within polymeric microspheres in which conventional anesthetics are easily loaded. Such chemical differences may necessitate more sophisticated co-encapsulation schemes, designing co-solvent systems, or delivering separate populations of vehicles, one for each drug type.
Figure 2.
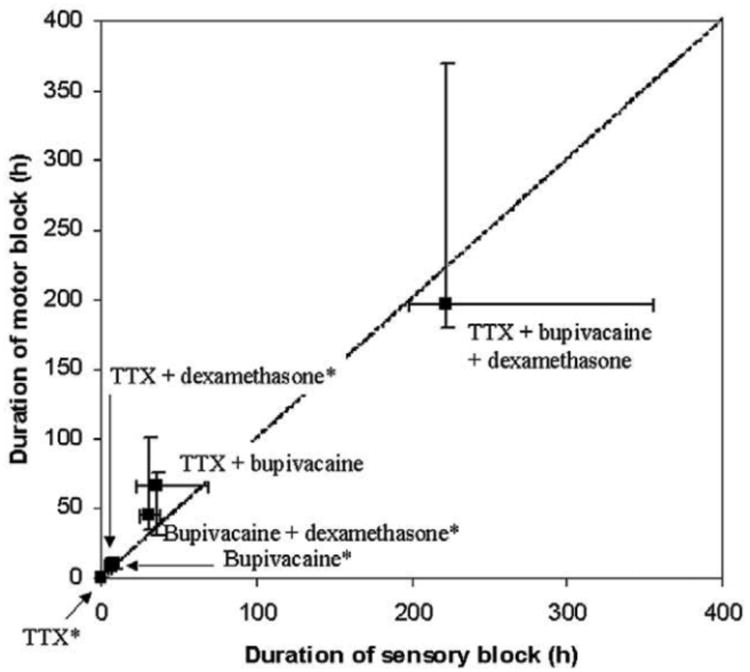
Rats were injected at the sciatic nerve with polymeric (poly(lactic-co-glycolic acid) [PLGA]) microspheres containing tetrodotoxin (TTX) and/or bupivacaine and/or dexamethasone. Two-compound formulations had a longer duration of effect than did single-compound ones, and microspheres containing all three compounds had an even longer duration of effect (>9 days from a single injection). Reprinted with permission from Lippincott Williams. [10] Copyright 2003.
Rationales for drug delivery systems
As alluded to above, local anesthetic formulations are generally deposited directly at the site of intended use, usually by injection, where they act as a depot system. From this, it follows that the formulations should be injectable, either as particles (or larger objects that can fit through a needle) or as a liquid. Unfortunately, the properties of drug delivery systems that provide the longest and slowest release of drug are not ideal for injectables. In general, the larger the particle size, the higher the drug encapsulation, and the slower drug release. These larger particles are likely to remain where deposited, with longer times to complete degradation – all of which will tend to prolong potential nerve block (these properties tend to argue against the use of nanomaterials as the depot formulation). However, the larger the particle, the greater the probability of clogging a needle. Some delivery systems are too large to be injected and must therefore be implanted,[24] although delivery of a shaped material through a special needle is also possible.
Similarly, the more viscous a liquid formulation is, the more slowly drugs will be released. Viscosity makes injectability more difficult; this has engendered a number of formulations which are initially liquids but gel after injection.[25,26]
Biocompatibility
With many drug delivery systems, the focus is on the biocompatibility of the excipients. In the case of PDLA formulations, it is often tissue reaction to the drug itself that is of greatest concern. Inflammation, myotoxicity, and neurotoxicity are well established local side effects of conventional local anesthetics. [27-30] This can be particularly problematic when local anesthetics are delivered by sustained release systems.[31] Site 1 sodium channel blockers do not cause significant neurotoxicity or myotoxicity.[15] In general, the vehicle itself tends to produce inflammation rather than actual tissue injury. The specific type and intensity of inflammation depend on the materials (and their rates and perhaps products of degradation), particle size, time after injection, etc.[31,32] It is unclear whether tissue reaction to the materials per se affects toxicity from the encapsulated drugs. It is crucial to be aware that tissue reaction to PDLA systems can be so severe as to derail otherwise promising formulations.[31]
Systemic toxicity is an important consideration in PDLA. These systems usually contain very large quantities of drugs that would be highly toxic if delivered too rapidly. The “burst” release seen in many drug delivery systems could therefore cause severe toxicity. This is a particularly important consideration with the site 1 sodium channel blockers, which are small hydrophilic molecules. Their therapeutic index is narrow and they can cause hypotension, and profound muscular weakness and phrenic nerve blockade that can lead to respiratory failure.[14] (Undesirable as those adverse events are, they are easy for anesthesiologists to manage, compared to the seizures and arrhythmias seen with conventional local anesthetics.)
Formulations
There is now a substantial literature on PDLA, in which many different devices are described. [33-37] Here we present a subset of formulations representative of the following general categories: injectable particles, injectable liquids, hybrid systems, macroscopic devices, and triggerable formulations. Many ingenious and exotic formulations have not been described here; those presented are selected for illustrative purposes and not as an endorsement.
In theory, it should be possible to predict the duration of nerve block from given formulations based on an understanding of their physicochemical properties (size, hydrophobicity) and the expected effect on characteristics (such as drug release kinetics). In practice, however, such thought experiments are rarely successful, in part because what matters in terms of duration of effect may not be so much the duration of release, but the time during which release exceeds some ill-defined tissue drug level. Thus, it is possible for formulations which achieve complete release in a less than a day to have a duration of nerve block similar to that from a formulation that releases drugs over days to weeks.[38] Moreover, meaningful comparison of the experimental (or clinical) performance of various formulations can be challenging. There is considerable variability in protocols to characterize drug release and many other physical parameters. There are enormous differences in the nature and interpretation of in vivo experimentation because of blockade of different nerves in different animal models, in which different amounts of formulations are injected with different techniques and their effectiveness and toxicity assessed by different metrics. Comparing formulations used for infiltration anesthesia vs peripheral nerve block vs neuraxial block is difficult, as are the results in different pain models (cold, heat, inflammation, etc.). Meaningful comparison of the intensity and duration of sensory and motor block can be difficult, as both are greatly affected by protocol, and are operator-dependent. A full discussion of the study of pain is beyond the scope of this review, but caveat emptor.
Injectable Particles
Polymeric Micro and Nanoparticles
Polymeric particles (Fig. 3) are usually prepared from synthetic hydrophobic polymers such as poly(lactic acid) (PLA), poly(glycolic acid) (PGA), and poly(lactic-co-glycolic acid) (PLGA) owing to their control of drug release, biocompatibility and biodegradability. [39,40] The physicochemical properties (such as molecular weight, polydispersity, hydrophobicity, functional moieties) can be tuned to obtain particles of particular composition, size, and surface-to-volume ratio, which determine the release kinetics and rate of degradation. Polymeric particles can be prepared as a solid polymer matrix, or as core-shell capsules (e.g. hydrophobic core with a polymer shell).[34]
Figure 3.

Schematic representation of (a) a solid polymer matrix sphere, in which the drug is dispersed throughout the polymer matrix and (b) a capsule, in which the drug is in solution and surrounded by a shell-like wall. Adapted with permission from Springer. [35] Copyright 2014.
Conventional local anesthetics can be easily incorporated into such polymeric materials (e.g. by single emulsion), especially since their free bases and the polymer matrix can be dissolved in many common organic solvents. Some amino-esters and amino-amides are believed to form hydrogen bonding with PLA, PGA, and PLGA in aqueous environments, which may also contribute to the high loading and sustained release.[41] Hydrophobic interactions are also likely to be very important given that both drug and polymer are often dissolved in the same organic solvents during encapsulation.[42] Drug loadings over 70% (w/w) have been achieved.[43] Co-encapsulation of a second hydrophilic drug can be more complicated (e.g. requiring a double emulsion method).[44] It can be challenging to achieve high loadings of hydrophilic drugs in polymeric particles, or to avoid burst release with concomitant toxicity.[10]
Polymeric micro and nanoparticles have been used as carrier systems for various local anesthetic agents. These systems can deliver relatively large quantities of drug compared to aqueous solutions and can release them over extended periods. Increasing particle size can allow higher drug loading, slower degradation and larger particles tend to remain where injected.[32] PLGA microspheres 65-75 μm in mean diameter have been loaded with bupivacaine and shown to reduce pain for 3 to 4 days after a skin incision,[45] prolonging the effect of 0.5% bupivacaine solution by more than 20-fold.
Drug combinations have been used to increase the duration of effect with this type of formulation.[10,12,13] PLGA microspheres containing a combination of bupivacaine and dexamethasone improved sciatic nerve block in rats from 7 hours (bupivacaine alone) to 47 hours (bupivacaine-dexamethasone combination),[12] and intercostal nerve block in sheep from 4 days (bupivacaine alone) to 13 days (bupivacaine-dexamethasone combination). [13] A third anesthetic, tetrodotoxin, further prolonged the duration of effect of the bupivacaine-dexamethasone combination.[10] Unfortunately, biocompatibility issues made such formulation commercially non-viable.[31]
Nanoparticles have also been used for the encapsulation of local anesthetic agents. Poly ε-caprolactone nanospheres have been used to encapsulate 0.5% lidocaine demonstrating a two-fold increase in the sensory block duration compared to that achieved with 0.5% lidocaine solution.[46] Alginate/bis (2-ethylhexyl) sulfosuccinate (AOT) and alginate/chitosan nanospheres were used for the entrapment of 0.5% bupivacaine hydrochloride.[47] The duration of motor and sensory blockade in the sciatic nerves of mice was improved to 5-fold of that obtained with 0.5% bupivacaine solution.
Polymeric delivery systems can cause generic acute and chronic inflammation after perineural injection which eventually resolves completely.[29] However, the incorporation of local anesthetics into polymeric systems can cause local toxicity and worsens inflammation.[31] Previous studies have shown that in vivo administration of 0.5% bupivacaine solution caused less myotoxicity than polymeric microspheres releasing similar concentrations of bupivacaine.[27] Myotoxicity appeared to be related to both the release kinetics of bupivacaine (e.g. burst and duration of release) and perhaps the presence of the particles themselves. Moreover, a drawback of many of these polymeric systems is that residual excipient materials persist in tissues long after the drug payload has been delivered.[29,48].
Liposomes
Liposomes are lipid-based particles with an aqueous core and a lipid bilayer shell (Fig 4a).[49,50] Lipid bilayers commonly consist of naturally occurring biocompatible amphiphilic molecules such as phospholipids and cholesterol.[49] Encapsulation of local anesthetics into liposomes results in slow drug release, prolonged duration of anesthetic effect, and reduced side effects.[51,52] As with many other particle types, the duration of in vitro release does not necessarily correlate well with the duration of nerve block.[53]
Figure 4.
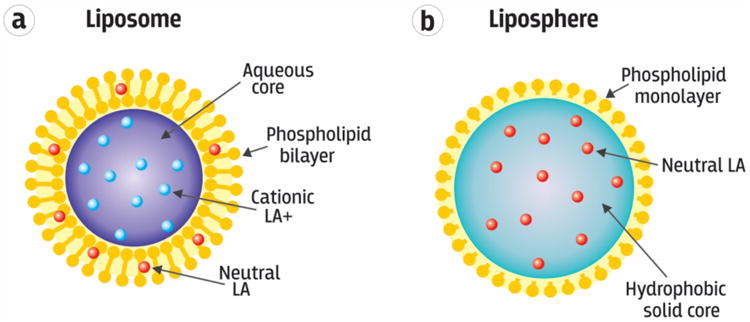
Schematic of (a) a liposome loaded with an amphiphilic local anesthetic drug partitioned into either the internal aqueous space as a charged cation (LA+) or the hydrophobic lipid bilayer as a neutral base (LA). (b) A liposphere comprised of a solid hydrophobic core matrix impregnated with local anesthetic neutral base. Adapted with permission from Springer. [35] Copyright 2014.
Physicochemical properties of the lipids (such as the length of the acyl chain, the degree of saturation of the hydrocarbon chains, the type of polar head group) determine the encapsulation efficiency and release kinetics. Both hydrophilic and hydrophobic molecules can be encapsulated. Hydrophilic drugs are entrapped in the aqueous phase, whereas hydrophobic compounds partition into the lipid bilayers.[54,55] Amphiphilic molecules can be distributed into both compartments. Electrostatic interactions between the lipids and cationic local anesthetics have been used to improve encapsulation. The rate of drug release depends on the permeability of the lipid bilayer. Phase transition temperature, above which a lipid bilayer turns from a solid into a fluid with much greater mobility of lipids and thus greater chance of permeation, is an important determinant of the release rate.[56] The phase transition temperature varies with the molecular structure of the lipid, and the characteristics of the surrounding media (such as pH and ionic strength). Liposomes made from lipids with high phase transition temperature and loaded with saxitoxin demonstrated greater stability during storage and produced longer durations of nerve blockade, compared to lipids with low phase transition temperature (Fig. 5).[28] A number of vesicular configurations with different sizes (varying from nano- to micrometer) have been designed for various drug delivery (Fig. 6)
Figure 5.

Duration of sensory and motor block following sciatic nerve injection with 75 mg of liposomes loaded with bupivacaine, saxitoxin, and/or dexamethasone. “Fluid” liposomes are composed of low-temperature phase transition lipids, whereas “solid” liposomes are prepared with high temperature phase transition lipids. Solid liposomes prolonged the duration of nerve blockade more than did fluid liposomes. Additionally, at lower concentrations dexamethasone greatly extended the duration of nerve blockade, whereas at higher concentration dexamethasone altered the stability of the liposome, resulting in “leakage” and rapid release. Reprinted with permission from the National Academy of Sciences. [28] Copyright 2009.
Figure 6.

A schematic representation of the common vesicle size and lamellarity classification system of liposomes. Small unilamellar vesicles are less than 100 nm in diameter and large unilamellar vesicles are between 100 and 1000 nm. Multilamellar vesicles have more than one membrane layers, and multivesicular vesicles encapsulate smaller vesicles. Adapted with permission from The Royal Society of Chemistry. [50] Copyright 2013.
Drugs can be encapsulated during liposome formation (termed passive loading), or after the liposome has formed (termed active or remote loading).[57] Loading efficiency refers to the mass of drug as a percentage of the total mass of the delivery system. Ideally, loading efficiency should be >90% to avoid the need to remove free drug (typically done by dialysis or molecular exclusion chromatography).[57] Passive loading efficiency for certain hydrophobic drugs can approach 100%, due to their great solubility in the lipid bilayers. Active or remote loading utilizes transmembrane pH or chemical gradients to drive solutes into the lipid or aqueous compartments of the liposome.[58] Amphiphilic drugs, such as local anesthetics, may be encapsulated within the aqueous and/or the lipid bilayer.
Aggregation or fusion of liposomes and hydrolysis or oxidation of the phospholipids and/or encapsulated drugs may occur with some liposome formulations.[59] The use of high-transition temperature lipids can mitigate drug efflux during storage.[60]
Tissue reaction to liposomes is generally benign. 0.5% liposomal bupivacaine resulted in similar myotoxicity to that of 0.5% bupivacaine HCl. Liposomes with higher concentrations of bupivacaine are less myotoxic than the corresponding bupivacaine solutions.[53]
Encapsulation of bupivacaine into liposomes protect against cardiovascular and central nervous system toxicity after intravascular injection. A study in rabbits showed that the doses of bupivacaine inducing seizures and ventricular tachycardia were significantly higher for liposomal bupivacaine (22.43 ± 2.63 mg kg−1) than for plain bupivacaine (15.7 ± 2.5 mg kg−1).[61] A recent report from four preclinical studies evaluating safety and pharmacokinetics of liposomal bupivacaine in dogs confirmed that liposomal bupivacaine has a more favorable safety profile than bupivacaine HCl.[62] The enhanced safety was attributed to the liposome-bound nature of bupivacaine, which reduces the proportion of free (unbound) drug available for systemic distribution. In one liposomal formulation used for the encapsulation of local anesthetics, there was no evidence of neurotoxicity in vitro, in neurobehavioral studies, by high-resolution microscopy, and in analyses of gene expression profiles in dorsal root ganglia.[28] The risk of antigenicity of liposomes is low as their composition can be similar to that of biological membranes.[63,64] However, components and metabolites of some liposomes have been shown to have local neurotoxicity.[65] Note that lipid emulsions are in current clinical use as antidotes for overdose of local anesthetics and other relatively hydrophobic drugs.[66,67] Currently, the only liposomal sustained release formulation in clinical use is DepoFoam bupivacaine (EXPAREL®).[68-74] It continues to be assessed in humans.
Drug combinations have been used to enhance the duration of effect of liposomal PDLA. For example, liposomes containing saxitoxin alone provided a duration of block of ∼ 2 days; co-encapsulation of dexamethasone increased the duration of effect to 7 days.[28]
Lipospheres
Lipospheres are stable microparticles (0.01 to 500 μm-diameter) consisting of a solid, hydrophobic triglyceride or fatty acid core containing the drug, surrounded by a monolayer of phospholipids (Fig 4b).[75] Lipospheres were developed to address the production costs and instability of liposomes.
Improved stability of the drug in the formulation, controlled particle size, high loading of hydrophobic drugs, controlled drug release and minimal carrier toxicity are some of the desirable features of liposphere systems.[76]
Bupivacaine lipospheres produced 1-3 days of reversible and dose-dependent nerve blockade when applied directly to the sciatic nerve of rats.[77]
Some of these formulations had stability problems within days of manufacture. The use of synthetic phospholipids and the addition of carboxymethyl cellulose yielded bupivacaine liposphere formulations that were physically stable for more than 1 year at room temperature. [78]
Injectable Liquids
Cyclodextrins
Cyclodextrins (CD) are hydrophilic cyclic oligosaccharides with the repeat unit of α-D-glucopyranose connected via carbons at α- 1 and α- 4 positions.[79] Cyclodextrins have a cone-like shape with hydroxyl groups on the outside of the cone and hydrophobic skeletal carbons and etheric oxygens on the surface of the internal cavity, where hydrophobic guest molecules can be hosted.[79] The hydroxyl groups on the surface of the CD cone are easily functionalizable, for reduced toxicity and enhanced solubility in organic solvents.[80]
Therapeutic agents of different molecular weights can be encapsulated by CD with specific cone diameters, which are determined by the number of gluocopyranose units (typically 6 to 8). Cyclodextrins can enhance the aqueous solubility and thus bioavailability of hydrophobic drugs.[79] However, unmodified CD can produce metabolites that are nephrotoxic when administered parenterally,[81] and may remove components of human erythrocytes that may lead to cell membrane disruption and hemolysis.[82]
An aqueous solution of maltosyl-β-CD complexed with levobupivacaine[83] or lidocaine[84] doubled the duration of intrathecal and sciatic nerve blockade. Similar results were achieved by complexation of ropivacaine with hydroxypropyl-β-CD.[85] Hydroxypropyl-β-CD formulations of bupivacaine and ropivacaine produced less local anesthetic myotoxicity and neurotoxicity than did free drug injections.[86]
Injectable Liquid Polymers
Simple mixtures of local anesthetics and polymers can also achieve PDLA. These formulations have the advantage of relatively easy administration (via injection instead of surgical implantation, low probability of clogging).
Biocompatible and biodegradable polyesters, polyanhydrides, poly(ortho esters), and polyphosphazenes have been studied for this purpose.[87] Biodegradability of these compounds results from enzymatic reactions or hydrolysis and can be enhanced by incorporation of labile groups (such as ester or anhydride) into the polymer backbone.[87]
Experiments using 10% (w/v) bupivacaine loaded in the injectable biodegradable poly(sebacic-co-ricinoleic acid) prolonged sciatic nerve blockade from 8 to 30 h.[88] Incorporation of anhydride bonds (which undergo hydrolysis automatically) in the polymer backbone rendered the formulation biodegradable. In vitro, 70% of the incorporated drug was released over the course of 1 week. Bupivacaine has been incorporated into hydrophobic, hydrolysis-resistant polyester-poly(lactic acid-co-castor oil) at 10% (w/v), resulting in a slower degradation rate, prolonged drug release, and 1-2 days of nerve block.[89] However, the formulation suffered significant burst release that led to systemic toxicity. Increasing the bupivacaine concentration to 15% (w/v) prolonged the duration of sensory blockade to 96 h and exhibited less burst release than with 10% bupivacaine.[90] This effect was attributed to increased formulation density and hydrophobicity, resulting in reduced water penetration into the drug-polymer matrix.
Naturally occurring polymers have also been used, such as hyaluronic acid.[26] Unmodified hyaluronic acid has had variable effects on the block duration from co-dissolved local anesthetics (including no effect at all).[26] Its effectiveness in prolonging the effect of local anesthetics depended on the solution viscosity and polymer molecular weight which, it has been argued, affects charge density.[91] Hyaluronic acid is anionic and therefore could bind conventional local anesthetics which are mostly cationic at physiological pH.[26] Hyaluronic acid has also been functionalized so as to cross-link in situ; such formulations can also prolong the duration of effect.[26] Hyaluronic acid exhibits excellent biocompatibility.
Polymer solutions viscosity tends to increase the duration of drug release. However, more viscous solutions are often difficult to inject. Shear-thinning polymers and polymer blends, which exhibit decreased viscosity under shear strain, have been investigated to address this issue. For example, hyaluronic acid and hydroxypropylmethyl cellulose (HPMC) blends demonstrated significantly higher injectability compared with that achievable with either of the polymers alone at the same concentration.[92] The shear thinning effect was attributed to the suppression of the high yield stress of hyaluronic acid solutions by HPMC and the flow instabilities induced by hyaluronic acid during injection. Higher polymer concentrations can thus be used and still remain injectable. As a result of being able to inject higher polymer concentrations, release of drug in vivo was slower. For example a solution of hyaluronic acid and hydroxypropylmethyl cellulose loaded with bupivacaine had a duration of rat sciatic nerve block three times that achieved with a bupivacaine solution.[93]
Hydrogels
Hydrogels are three-dimensional networks of hydrophilic polymers with high water content.[94] They are generally highly biocompatible. In the swollen state, hydrogels are highly porous. Drug loading can be tuned by the molecular weight, cross-linking density, and hydrophilicity of the hydrogel..[94] Molecular weight and crosslinking density also affects the degradation rate of the hydrogel in vivo.[94,95] Despite the tunability, hydrogel formulations yield relatively rapid drug release because of the high water content and large pore size.[94] Also, some hydrogels are not injectable once formed, thus necessitating surgical implantation, which limits their clinical use. Hence there is a growing interest in hydrogels that undergo transitions from liquid to gel upon injection (e.g. by in situ cross-linking or reverse thermal gelation) or ones that display shear-thinning properties.[25,26,96]
Hyaluronic acid (see previous section) has been modified to aldehyde and hydrazide forms that would crosslink upon mixing at the time of injection. With these modifications it formed a gel in situ, trapping co-injected local anesthetics in the vicinity of the nerve for extended release. Two percent (w/v) cross-linked hyaluronic acid doubled the duration of nerve blocks when used in conjunction with 0.1%, 0.25%, and 0.5% (w/v) bupivacaine, without a statistically significant increase in myotoxicity.[26] Tissue reaction to this formulation was comparable to that observed with free bupivacaine at the same concentration.[26]
Poloxamer 407, a synthetic polymer that exhibits reverse-phase thermal gelation (i.e. liquid phase at cold temperatures, gel at body temperature), has been used to extend the duration of lidocaine release.[97] [25] It has also been used to further extend the duration of effect of local anesthetic-loaded polymeric microparticles.[98]
Current hydrogel systems have not achieved nerve block durations on par with those observed with microparticle-based systems. However, the duration of blockade is adequate for many postoperative and/or dental procedures without entailing the sequelae of microparticle injection (e.g. particle debris persisting at the injection site for weeks).
Hybrid Formulations
Different types of controlled-release systems have been combined so that they can combine their best features (and perhaps obviate their undesirable ones). The combination of particles and hydrogels is the most common.
Polymeric Microparticle-Hydrogel
Systems have been developed incorporating microspheres within in situ-forming hydrogels. The hydrogel helps to maintain microparticles at the injection site and prevents their phagocytosis.[99] Moreover, the hydrogel can further slow drug diffusion from the microparticles, decreasing the initial burst release and extending the duration of effect. PLGA microspheres containing 31% (w/w) lidocaine suspended in the thermosensitive Poloxamer 407, resulted in longer sciatic nerve block in rats than did lidocaine in microspheres.[98]
Liposome-Hydrogel
Prolonged drug release can be achieved by combining liposomes and hydrogels, where drug-loaded liposomes are embedded in hydrogel polymer matrices. The hydrogel provides additional diffusion resistance while liposomes act as the drug reservoirs.[100] The hydrogels may also play a role in keeping the particles in place, or controlling the interaction between the particles and surrounding tissues (e.g. it the particles might have an undesirable effect on tissue reaction).[101]
Bupivacaine-loaded multivesicular liposomes have been entrapped in a calcium-alginate cross-linked hydrogel to form injectable beads (3-5 mm). The formulation was stable for 2 years under 4°C. At 37°C, the liposomal hydrogel formulation achieved longer duration of nerve block than the liposomal formulation.[102]
Macroscopic Drug Delivery Systems
Macroscopic polymeric devices can be loaded with large quantities of drug and have a low surface area to volume ratio, so may be able to achieve very prolonged release (weeks to months).[103] The principal disadvantage of such systems is that they may not be injectable. Macroscopic devices are otherwise conceptually similar to other formulations. They are placed at the intended site of action, e.g. placed adjacent to a nerve during surgery. Examples of such devices include drug-eluting pellets[104] and sutures.[105] In theory, almost any suitable polymer could be used; PLGA has been a common choice.[104,106-109] Analgesic bupivacaine-loaded PLGA sutures have provided incisional analgesia in rats for 7 days following a skin wound.[105]
On-Demand Remotely Triggerable Local Anesthesia
One potential problem with the systems described above is that once initiated they will release drug autonomously, irrespective of the patient's condition or wishes. If a patient was administered a formulation that would provide nerve block for 7 days, there would not be much that could be done if she/he would like the block to be turned off on day 3. A system that would allow the patient to determine when local anesthesia is initiated, how long it lasts, and even the intensity of the analgesia would greatly enhance pain management
The toolkit of stimulus-responsive drug delivery could be employed to achieve that end.[110] The basic idea is that an external stimulus (various wavelengths of light, magnetic fields, electric fields, ultrasound, etc.) would induce a change in a drug delivery system that would trigger drug release (Fig. 7). Triggering by near-infrared light has been the primary modality examined to date in PDLA.
Figure 7.
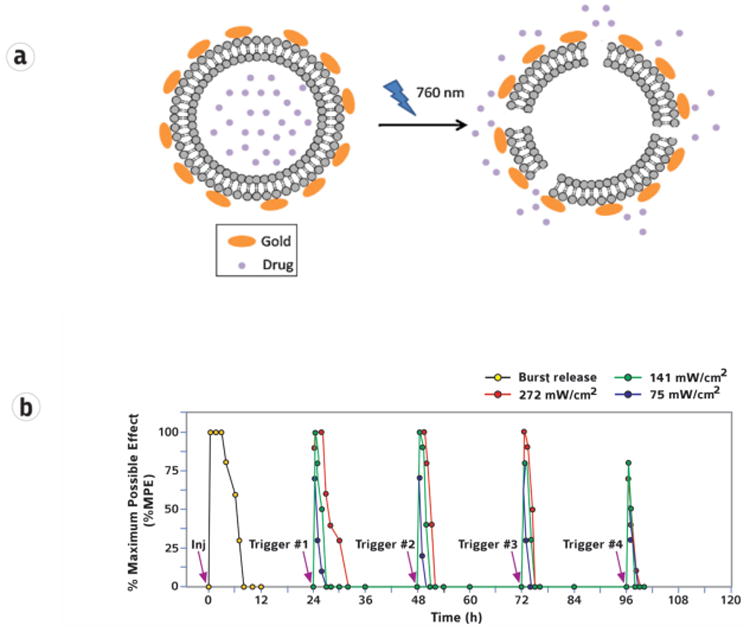
Light-triggered release from thermoresponsive liposomes coated with gold nanoparticles. Irradiation with 760 nm light induced local heating, which released encapsulated drug from the liposome. (b) Triggerable local anesthesia in the rat footpad following injection (Inj) of thermoresponsive liposomes. Subsequent irradiation (purple arrows) with an 808 nm continuous wave NIR laser at 75, 141, and 272 mW/cm2 for 10 min led to adjustable degrees of local anesthesia. Local anesthesia is presented as % maximum possible effect. Reprinted with permission from the National Academy of Sciences [111] and the American Chemical Society. [112] Copyright 2015 and 2016 respectively.
In one approach a photosensitizer capable of releasing singlet oxygen in response to near infrared (NIR) light was incorporated into liposomes. The singlet oxygen caused peroxidation of unsaturated lipids in the liposome, which made the liposome membrane more permeable so that drug was released. Such liposomes containing tetrodotoxin caused an untriggered nerve block immediately after injection. Once the initial block wore off, irradiation with 730 nm light caused release of TTX and produced effective nerve blockade. The triggered nerve block was repeatable, and its duration and intensity were tunable by adjusting the irradiance and duration of irradiation.[111]
In an alternative approach, gold nanoparticles (nanorods) were conjugated to liposomes containing tetrodotoxin and dexmedetomidine. Exposure to 808 nm light caused heating of the gold nanorods by surface plasmon resonance, which increased liposomal permeability. In vivo, triggered drug release led to repeatable infiltration anesthesia (Fig 8).[112]
Phototriggered systems are limited by the depth of tissue penetration that can be achieved, and the irradiance and/or duration of exposure that is required to achieve a given effect at a particular tissue depth. High irradiances can cause thermal injury [113,114]. Moreover, light outside of the NIR range has relatively poor tissue penetration, and in the case of UV light may be carcinogenic. Phototriggered systems have been reviewed.[111,112]
Oscillating magnetic fields have also been used for triggered release of local anesthetics.[115] A triggerable membrane was created which acted as a drug reservoir. It was composed of an impermeable ethylcellulose matrix containing an interconnecting network of thermosensitive poly(N-isopropylacrylamide) (poly NIPAAM) nanogels and, separately, superparamagnetic ferromagnetic nanoparticles. Under an oscillating magnetic field, the ferromagnetic nanoparticles heated up, causing the nanogels to collapse, opening pores in the membrane that allowed flux of bupivacaine [115]. Effectiveness in vivo was not demonstrated for PDLA, but was shown for control of hyperglycemia in diabetic rats using a very similar system where gold nanoshells were used instead of the ferromagnetic nanoparticles and NIR light was used as the stimulus instead of magnetic fields.[113]
Summary and Prospects
Prolonged duration local anesthesia has been achieved by a wide range of drug delivery systems, albeit with widely differing effects in terms of duration and intensity of anesthesia. In some cases, systemic toxicity is problematic, but local toxicity is a bigger issue with most, particularly with delivery of amino-amide and amino-ester local anesthetics. As has been the case for decades, comparison of results from different preclinical formulations in preclinical work will remain difficult. The hope that such studies will become standardized – or that the same will happen in human trials – is likely a forlorn one.
As PDLA formulations start to enter the market, emphasis will be not only on intensity and duration of nerve block but also on local tissue reaction. It is likely, at least for conventional local anesthetics, that efficacy and tissue reaction will be related. That being said, there is relatively little literature on the biocompatibility of local anesthetic products in clinical development.
Despite the substantial number of formulations that have been developed, few have progressed to clinical trials or commercialization. Recent examples have included a liposomal bupivacaine formulation (Exparel®) from Pacira Pharmaceuticals; [68-74] bupivacaine formulation (Posimir®) described as an esterified sugar derivative on the Durect website;[116] a poly(ortho ester) formulation of bupivacaine and the anti-inflammatory drug meloxicam (HTX-011) from Heron Therapeutics;[117,118] and a collagen-based implantable bupivacaine release system (XaraColl ®) from Innocoll.[119-121] A review focused on the engineering of sustained release systems is not the best place to review the comparative merits of commercial products, their tribulations with regulatory bodies, or to review clinical trials. Moreover, the abovementioned difficulties in comparing formulations in animals can be true of human trials as well. Irrespective of the clinical prospects of products under development, the fact that relatively many are currently under development is a clear indication of a perceived need in the clinical community.
This need is likely to drive innovation: as sustained release local anesthetics enter standard clinical practice, there will likely be both clinical and economic pressure for better, more differentiated products. This may take the form of formulations that are optimized for specific medical indications or anatomic locations, or that provide specific intensities of anesthesia, or specific durations (e.g. ultra-prolonged nerve block for chronic pain). As local anesthetic systems evolve, they may allow some controversial subjects to be revisited, such as the role of PDLA in preventing the development of neuropathic pain.[1-4] They may also be used to provide extended yet reversible treatment of other excitable tissues (heart, brain, etc.).[122]
Conversely, independent advances in the general field of drug delivery are likely to become applied to PDLA formulations. Therefore, as drug delivery systems become more sophisticated, it is likely that those developed for PDLA will become so also. Systems that provide on-demand local anesthesia in response to patient needs are already being developed. It can be envisioned that electrically and/or chemically responsive systems capable of delivering compounds in response to abnormal electrical or chemical signals will be able to control pain autonomously, perhaps acting through closed-loop systems. The introduction of new types of drug delivery systems may also introduce new and relatively exotic materials (e.g. inorganic nanomaterials), which may bring with them attendant biomedical and regulatory issues.
Acknowledgments
This work was supported by NIH GM073626.
Footnotes
Disclosure: Dr. Daniel S. Kohane has patents relating to prolonged duration local anesthetics, particularly involving site 1 sodium channel blockers. He has a financial interest in a licensed site 1 blocker formulation that does not use drug delivery technology.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xie W, Strong JA, Zhang JM. Neuroscience. 2009;160:847–857. doi: 10.1016/j.neuroscience.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xie W, Strong JA, Meij JTA, Zhang JM, Yu L. Pain. 2005;116:243–256. doi: 10.1016/j.pain.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suter MR, Papaloïzos M, Berde CB, Woolf CJ, Gilliard N, Spahn DR, Decosterd I. Anesthesiology. 2003;99:1402–1408. doi: 10.1097/00000542-200312000-00025. [DOI] [PubMed] [Google Scholar]
- 4.Shankarappa SA, Tsui JH, Kim KN, Reznor G, Dohlman JC, Langer R, Kohane DS. Proc Natl Acad Sci USa. 2012;109:17555–17560. doi: 10.1073/pnas.1214634109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Albright GA. Anesthesiology. 1979;51:285–287. doi: 10.1097/00000542-197910000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita A, Matsumoto M, Matsumoto S, Itoh M, Kawai K, Sakabe T. Anesthesia & Analgesia. 2003;97:512–519. doi: 10.1213/01.ANE.0000068885.78816.5B. [DOI] [PubMed] [Google Scholar]
- 7.McLure HA, Rubin AP. Minerva Anestesiol. 2005;71:59–74. [PubMed] [Google Scholar]
- 8.Xu B, Ren L, Tu W, Wu Z, Ai F, Zhou D, Chen B, Zhang X. Eur Spine J. 2015:1–7. doi: 10.1007/s00586-015-3979-x. [DOI] [PubMed] [Google Scholar]
- 9.Liu FF, Liu XM, Liu XY, Tang J, Jin L, Li WY, Zhang LD. Int J Clin Exp Med. 2015;8:5438–5445. [PMC free article] [PubMed] [Google Scholar]
- 10.Kohane DS, Smith SE, Louis DN, Colombo G, Ghoroghchian P, Hunfeld NGM, Berde CB, Langer R. Pain. 2003;104:415–421. doi: 10.1016/s0304-3959(03)00049-6. [DOI] [PubMed] [Google Scholar]
- 11.Colombo G, Padera R, Langer R, Kohane DS. Journal of Biomedical Materials Research Part A. 2005;75:458–464. doi: 10.1002/jbm.a.30443. [DOI] [PubMed] [Google Scholar]
- 12.Castillo J, Curley J, Hotz J, Uezono M, Tigner J, Chasin M, Wilder R, Langer R, Berde C. Anesthesiology. 1996;85:1157–1166. doi: 10.1097/00000542-199611000-00025. [DOI] [PubMed] [Google Scholar]
- 13.Dräger C, Benziger D, Gao F, Berde CB. Anesthesiology. 1998;89:969–979. doi: 10.1097/00000542-199810000-00022. [DOI] [PubMed] [Google Scholar]
- 14.Kohane DS, Yieh J, Lu NT, Langer R, Strichartz GR, Berde CB. Anesthesiology. 1998;89:119–131. doi: 10.1097/00000542-199807000-00019. [DOI] [PubMed] [Google Scholar]
- 15.Padera RF, Tse JY, Bellas E, Kohane DS. Muscle Nerve. 2006;34:747–753. doi: 10.1002/mus.20618. [DOI] [PubMed] [Google Scholar]
- 16.Lahaye LA, Butterworth JF. Anesthesiology. 2015;123:741–742. doi: 10.1097/ALN.0000000000000833. [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez-Navarro AJ, Lagos M, Figueroa C, Garcia C, Recabal P, Silva P, Iglesias V, Lagos N. Neurotox Res. 2009;16:408–415. doi: 10.1007/s12640-009-9092-3. [DOI] [PubMed] [Google Scholar]
- 18.Lobo K, Donado C, Cornelissen L, Kim J, Ortiz R, Peake RWA, Kellogg M, Alexander ME, Zurakowski D, Kurgansky KE, Peyton J, Bilge A, Boretsky K, McCann ME, Berde CB. J Cravero, Anesthesiology. 2015;123:873–885. doi: 10.1097/ALN.0000000000000831. [DOI] [PubMed] [Google Scholar]
- 19.Song H, Li J, Lu CL, Kang L, Xie L, Zhang YY, Zhou XB, Zhong S. Clin Exp Pharmacol Physiol. 2011;38:510–514. doi: 10.1111/j.1440-1681.2011.05539.x. [DOI] [PubMed] [Google Scholar]
- 20.Shi J, Liu TT, Wang X, Epstein DH, Zhao LY, Zhang XL, Lu L. Pharmacol Biochem Behav. 2009;92:603–607. doi: 10.1016/j.pbb.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagen NA, du Souich P, Lapointe B, Ong-Lam M, Dubuc B, Walde D, Love R, Ngoc AH. Journal of Pain and Symptom Management. 2008;35:420–429. doi: 10.1016/j.jpainsymman.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 22.Hagen NA, Fisher KM, Lapointe B, du Souich P, Chary S, Moulin D, Sellers E, Ngoc AH. Journal of Pain and Symptom Management. 2007;34:171–182. doi: 10.1016/j.jpainsymman.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 23.Hagen NA, Lapointe B, Ong-Lam M, Dubuc B, Walde D, Gagnon B, Love R, Goel R, Hawley P, Ngoc AH, du Souich P. Curr Oncol. 2011;18:e109–16. doi: 10.3747/co.v18i3.732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Masters DB, Berde CB, Dutta SK, Griggs CT, Hu D, Kupsky W, Langer R. Anesthesiology. 1993;79:340–346. doi: 10.1097/00000542-199308000-00020. [DOI] [PubMed] [Google Scholar]
- 25.Paavola A, Tarkkila P, Xu M, Wahlström T, Yliruusi J, Rosenberg P. Pharm Res. 1998;15:482–487. doi: 10.1023/a:1011992702604. [DOI] [PubMed] [Google Scholar]
- 26.Jia X, Colombo G, Padera R, Langer R, Kohane DS. Biomaterials. 2004;25:4797–4804. doi: 10.1016/j.biomaterials.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Padera R, Bellas E, Tse JY, Hao D, Kohane DS. Anesthesiology. 2008;108:921–928. doi: 10.1097/ALN.0b013e31816c8a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Epstein-Barash H, Shichor I, Kwon AH, Hall S, Lawlor MW, Langer R, Kohane DS. Proc Natl Acad Sci USa. 2009;106:7125–7130. doi: 10.1073/pnas.0900598106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kohane DS, Lipp M, Kinney RC, Anthony DC, Louis DN, Lotan N, Langer R. J Biomed Mater Res. 2002;59:450–459. doi: 10.1002/jbm.1261. [DOI] [PubMed] [Google Scholar]
- 30.Neal JM, Salinas FV, Choi DS. Regional Anesthesia and Pain Medicine. 2016:1. doi: 10.1097/AAP.0000000000000466. [DOI] [PubMed] [Google Scholar]
- 31.Kohane DS, Langer R. Chem Sci. 2010;1:441–446. [Google Scholar]
- 32.Kohane DS. Biotechnol Bioeng. 2007;96:203–209. doi: 10.1002/bit.21301. [DOI] [PubMed] [Google Scholar]
- 33.Weiniger CF, Golovanevski L, Domb AJ, Ickowicz D. Anaesthesia. 2012;67:906–916. doi: 10.1111/j.1365-2044.2012.07168.x. [DOI] [PubMed] [Google Scholar]
- 34.E. de Paula, C.M. Cereda, L.F. Fraceto, D.R. de Araujo, M. Franz-Montan, G.R. Tofoli, J. Ranali, M.C. Volpato, F.C. Groppo, Www.Expertopin.com/Edd 9 (2012) 1505–1524.
- 35.Mc Alvin JB, Kohane DS. Focal Controlled Drug Delivery. Springer US; Boston, MA: 2013. pp. 653–677. [Google Scholar]
- 36.Weiniger CF, Golovanevski M, Sokolsky-Papkov M, Domb AJ. Expert Opin Drug Deliv. 2010;7:737–752. doi: 10.1517/17425241003767383. [DOI] [PubMed] [Google Scholar]
- 37.Beiranvand S, Eatemadi A, Karimi A. Nanoscale Res Lett. 2016;11:307. doi: 10.1186/s11671-016-1520-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohane DS, Lipp M, Kinney RC, Lotan N, Langer R. Pharm Res. 2000;17:1243–1249. doi: 10.1023/a:1026470831256. [DOI] [PubMed] [Google Scholar]
- 39.Kohane DS, Tse JY, Yeo Y, Padera R, Shubina M, Langer R. Journal of Biomedical Materials Research Part A. 2006;77:351–361. doi: 10.1002/jbm.a.30654. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Chan HF, Leong KW. Advanced Drug Delivery Reviews. 2013;65:104–120. doi: 10.1016/j.addr.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holgado MA, Arias JL, Cózar MJ, Alvarez-Fuentes J, Gañán-Calvo AM, Fernández-Arévalo M. Int J Pharm. 2008;358:27–35. doi: 10.1016/j.ijpharm.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 42.de Melo NFS, Grillo R, Guilherme VA, de Araújo DR, de Paula E, Rosa AH, Fraceto LF. Pharm Res. 2011;28:1984–1994. doi: 10.1007/s11095-011-0425-6. [DOI] [PubMed] [Google Scholar]
- 43.Curley J, Castillo J, Hotz J, Uezono M, Hernandez S, Lim JO, Tigner J, Chasin M, Langer R, Berde C. Anesthesiology. 1996;84:1401–1410. doi: 10.1097/00000542-199606000-00017. [DOI] [PubMed] [Google Scholar]
- 44.Giri TK, Choudhary C, Ajazuddin A, Alexander H, Badwaik DK. Tripathi, Saudi Pharm J. 2013;21:125–141. doi: 10.1016/j.jsps.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ohri R, Wang JCF, Blaskovich PD, Pham LN, Costa DS, Nichols GA, Hildebrand WP, Scarborough NL, Herman CJ, Strichartz GR. Anesthesia & Analgesia. 2013;117:717–730. doi: 10.1213/ANE.0b013e3182a00851. [DOI] [PubMed] [Google Scholar]
- 46.Ramos Campos EV, Silva de Melo NF, Guilherme VA, de Paula E, Rosa AH, de Araújo DR, Fraceto LF. Journal of Pharmaceutical Sciences. 2013;102:215–226. doi: 10.1002/jps.23350. [DOI] [PubMed] [Google Scholar]
- 47.Grillo R, de Melo NFS, de Araújo DR, de Paula E, Rosa AH, Fraceto LF. Journal of Drug Targeting. 2010;18:688–699. doi: 10.3109/10611861003649738. [DOI] [PubMed] [Google Scholar]
- 48.McAlvin JB, Reznor G, Shankarappa SA, Stefanescu CF, Kohane DS. Anesthesia & Analgesia. 2013;116:794–803. doi: 10.1213/ANE.0b013e31828174a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Allen TM, Cullis PR. Advanced Drug Delivery Reviews. 2013;65:36–48. doi: 10.1016/j.addr.2012.09.037. [DOI] [PubMed] [Google Scholar]
- 50.van Swaay D, de Mello A. Lab Chip. 2013;13:752–767. doi: 10.1039/c2lc41121k. [DOI] [PubMed] [Google Scholar]
- 51.Viscusi ER, Sinatra R, Onel E, Ramamoorthy SL. Clin J Pain. 2014;30:102–110. doi: 10.1097/AJP.0b013e318288e1f6. [DOI] [PubMed] [Google Scholar]
- 52.Ilfeld BM, Malhotra N, Furnish TJ, Donohue MC, Madison SJ. Anesthesia & Analgesia. 2013;117:1248–1256. doi: 10.1213/ANE.0b013e31829cc6ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McAlvin JB, Padera RF, Shankarappa SA, Reznor G, Kwon AH, Chiang HH, Yang J, Kohane DS. Biomaterials. 2014;35:4557–4564. [Google Scholar]
- 54.Lian T, Ho RJY. Journal of Pharmaceutical Sciences. 2001;90:667–680. doi: 10.1002/jps.1023. [DOI] [PubMed] [Google Scholar]
- 55.Torchilin V. Fundamentals and Applications of Controlled Release Drug Delivery. Springer US; Boston, MA: 2011. pp. 289–328. [Google Scholar]
- 56.Taylor KMG, Morris RM. Thermochimica Acta. 1995;248:289–301. [Google Scholar]
- 57.Cullis PR, Mayer LD, Bally MB, Madden TD, Hope MJ. Advanced Drug Delivery Reviews. 1989;3:267–282. [Google Scholar]
- 58.J. Gubernator, Www.Expertopin.com/Edd 8 (2011) 565–l580.
- 59.Crommelin DJA, Fransen GJ, Salemink PJM. In: Targeting of Drugs with Synthetic Systems. Gregoriadis G, Senior J, Poste G, editors. Springer US; Boston, MA: 1986. pp. 277–287. [Google Scholar]
- 60.Wang B, Hu L, Siahaan TJ. Drug Delivery. John Wiley & Sons; Hoboken, NJ: 2016. [Google Scholar]
- 61.Boogaerts J, Declercq A, Lafont N, Benameur H, Akodad EM, Dupont JC, Legros FJ. Anesthesia & Analgesia. 1993;76:553–555. doi: 10.1213/00000539-199303000-00018. [DOI] [PubMed] [Google Scholar]
- 62.Joshi G, Patou G, Kharitonov V. Journal of Pain Research. 2015;8:781–789. doi: 10.2147/JPR.S85424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.J.S. Rose, J.M. Neal, D.J. Kopacz, (2005).
- 64.Torchilin VP. Nat Rev Drug Discov. 2005;4:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 65.Grant SA. Best Pract Res Clin Anaesthesiol. 2002;16:345–352. doi: 10.1053/bean.2002.0242. [DOI] [PubMed] [Google Scholar]
- 66.Weinberg GL. Anesthesiology. 2012;117:180–187. doi: 10.1097/ALN.0b013e31825ad8de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cave G, Harvey M. Acad Emerg Med. 2009;16:815–824. doi: 10.1111/j.1553-2712.2009.00499.x. [DOI] [PubMed] [Google Scholar]
- 68.Golf M, Daniels SE, Onel E. Adv Ther. 2011;28:776–788. doi: 10.1007/s12325-011-0052-y. [DOI] [PubMed] [Google Scholar]
- 69.Gorfine SR, Onel E, Patou G, Krivokapic ZV. Diseases of the Colon & Rectum. 2011;54:1552–1559. doi: 10.1097/DCR.0b013e318232d4c1. [DOI] [PubMed] [Google Scholar]
- 70.Bramlett K, Onel E, Viscusi ER, Jones K. Knee. 2012;19:530–536. doi: 10.1016/j.knee.2011.12.004. [DOI] [PubMed] [Google Scholar]
- 71.Haas E, Onel E, Miller H, Ragupathi M, White PF. Am Surg. 2012;78:574–581. doi: 10.1177/000313481207800540. [DOI] [PubMed] [Google Scholar]
- 72.Cohen SM. Journal of Pain Research. 2012;5:567–572. doi: 10.2147/JPR.S38621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feierman DE, Kronenfeld M, Gupta PM, Younger N, Logvinskiy E. Journal of Pain Research. 2014;7:477–482. doi: 10.2147/JPR.S65151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hutchins J, Delaney D, Vogel RI, Ghebre RG, Downs LS, Carson L, Mullany S, Teoh D, Geller MA. Gynecol Oncol. 2015;138:609–613. doi: 10.1016/j.ygyno.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dudala TB, Yalavarthi PR, Vadlamudi HC, Thanniru J, Yaga G, Mudumala NL, Pasupati VK. International Journal of Pharmaceutical Investigation. 2014;4:149–155. doi: 10.4103/2230-973X.143112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patra CN. Journal of Pharmaceutics & Drug Development. 2013;1:1. [Google Scholar]
- 77.Masters DB, Domb AJ. Pharm Res. 1998;15:1038–1045. doi: 10.1023/a:1011978010724. [DOI] [PubMed] [Google Scholar]
- 78.Toongsuwan S, Li LC, Erickson BK, Chang HC. Int J Pharm. 2004;280:57–65. doi: 10.1016/j.ijpharm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 79.Loftsson T, Jarho P, Másson M, Järvinen T. Expert Opin Drug Deliv. 2005;2:335–351. doi: 10.1517/17425247.2.1.335. [DOI] [PubMed] [Google Scholar]
- 80.de Paula E, Cereda CMS, Tofoli GR, Franz-Montan M, Fraceto LF, de Araujo DR. Recent Pat Drug Deliv Formul. 2010;4:23–34. doi: 10.2174/187221110789957228. [DOI] [PubMed] [Google Scholar]
- 81.Frank DW, Gray JE, Weaver RN. Am J Pathol. 1976;83:367–382. [PMC free article] [PubMed] [Google Scholar]
- 82.Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Eur J Biochem. 1989;186:17–22. doi: 10.1111/j.1432-1033.1989.tb15171.x. [DOI] [PubMed] [Google Scholar]
- 83.Karashima K, Taniguchi M, Nakamura T, Takasaki M, Matsuo K, Irikura M, Irie T. Anesthesia & Analgesia. 2007;104:1121–8. doi: 10.1213/01.ane.0000260309.15034.52. – tables of contents. [DOI] [PubMed] [Google Scholar]
- 84.Suzuki R, Arai YCP, Hamayasu K, Fujita K, Hara K, Yamaguchi T, Sasaguri S. J Anesth. 2009;23:295–297. doi: 10.1007/s00540-008-0720-5. [DOI] [PubMed] [Google Scholar]
- 85.de Araujo DR, Tsuneda SS, Cereda CMS, Del F, Carvalho GF, Preté PSC, Fernandes SA, Yokaichiya F, Franco MKKD, Mazzaro I, Fraceto LF, de A, Braga FA, de Paula E. Eur J Pharm Sci. 2008;33:60–71. doi: 10.1016/j.ejps.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 86.Cereda CMS, Tofoli GR, Maturana LG, Pierucci A, Nunes LAS, Franz-Montan M, de Oliveira ALR, Arana S, de Araújo DR, de Paula E. Anesthesia & Analgesia. 2012;115:1234–1241. doi: 10.1213/ANE.0b013e318266f3d9. [DOI] [PubMed] [Google Scholar]
- 87.Ulery BD, Nair LS, Laurencin CT. J Polym Sci B Polym Phys. 2011;49:832–864. doi: 10.1002/polb.22259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shikanov A, Domb AJ, Weiniger CF. Journal of Controlled Release. 2007;117:97–103. doi: 10.1016/j.jconrel.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 89.Sokolsky-Papkov M, Golovanevski L, Domb AJ, Weiniger CF. Pharm Res. 2009;26:32–39. doi: 10.1007/s11095-008-9699-8. [DOI] [PubMed] [Google Scholar]
- 90.Sokolsky-Papkov M, Golovanevski L, Domb AJ, Weiniger CF. Journal of Pharmaceutical Sciences. 2010;99:2732–2738. doi: 10.1002/jps.22025. [DOI] [PubMed] [Google Scholar]
- 91.Doherty MM, Hughes PJ, Korszniak NV, Charman WN. Anesthesia & Analgesia. 1995;80:740–746. doi: 10.1097/00000539-199504000-00016. [DOI] [PubMed] [Google Scholar]
- 92.Hoare T, Zurakowski D, Langer R, Kohane DS. Journal of Biomedical Materials Research Part A. 2010;92:575–585. doi: 10.1002/jbm.a.32392. [DOI] [PubMed] [Google Scholar]
- 93.Hoare T, Bellas E, Zurakowski D, Kohane DS. Journal of Biomedical Materials Research Part A. 2010;92:586–595. doi: 10.1002/jbm.a.32420. [DOI] [PubMed] [Google Scholar]
- 94.Hoare TR, Kohane DS. Polymer. 2008 [Google Scholar]
- 95.Sivakumaran D, Maitland D, Hoare T. Biomacromolecules. 2011;12:4112–4120. doi: 10.1021/bm201170h. [DOI] [PubMed] [Google Scholar]
- 96.Guvendiren M, Lu HD, Burdick JA. Soft Matter. 2012;8:260–272. [Google Scholar]
- 97.Paavola A, Yliruusi J, Kajimoto Y, Kalso E, Wahlström T, Rosenberg P. Pharm Res. 1995;12:1997–2002. doi: 10.1023/a:1016264527738. [DOI] [PubMed] [Google Scholar]
- 98.Chen PC, Kohane DS, Park YJ, Bartlett RH, Langer R, Yang VC. Journal of Biomedical Materials Research Part A. 2004;70:459–466. doi: 10.1002/jbm.a.30101. [DOI] [PubMed] [Google Scholar]
- 99.Gao SQ, Maeda T, Okano K, Palczewski K. Invest Ophthalmol Vis Sci. 2012;53:6314–6323. doi: 10.1167/iovs.12-10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Glavas-Dodov M, Goracinova K, Mladenovska K, Fredro-Kumbaradzi E. Int J Pharm. 2002;242:381–384. doi: 10.1016/s0378-5173(02)00221-1. [DOI] [PubMed] [Google Scholar]
- 101.Yeo Y, Ito T, Bellas E, Highley CB, Marini R, Kohane DS. Ann Surg. 2007;245:819–824. doi: 10.1097/01.sla.0000251519.49405.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cohen R, Kanaan H, Grant GJ, Barenholz Y. J Control Release. 2012;160:346–352. doi: 10.1016/j.jconrel.2011.12.030. [DOI] [PubMed] [Google Scholar]
- 103.Solorio L, Carlson A, Zhou H, Exner AA. Engineering Polymer Systems for Improved Drug Delivery. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2014. pp. 189–225. [Google Scholar]
- 104.Masters DB, Berde CB, Dutta S, Turek T, Langer R. Pharm Res. 1993;10:1527–1532. doi: 10.1023/a:1018995913972. [DOI] [PubMed] [Google Scholar]
- 105.Weldon CB, Tsui JH, Shankarappa SA, Nguyen VT, Ma M, Anderson DG, Kohane DS. J Control Release. 2012;161:903–909. doi: 10.1016/j.jconrel.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang CF, Djalali AG, Gandhi A, Knaack D, De Girolami U, Strichartz G, Gerner P. Anesthesia & Analgesia. 2009;108:1027–1033. doi: 10.1213/ane.0b013e318193596a. [DOI] [PubMed] [Google Scholar]
- 107.Wang CF, Pancaro C, Gerner P, Strichartz G. Anesthesiology. 2011;114:135–149. doi: 10.1097/ALN.0b013e3182001996. [DOI] [PubMed] [Google Scholar]
- 108.Gerner P, Wang CF, Lee BS, Suzuki S, Degirolami U, Gandhi A, Knaack D, Strichartz G. Anesthesia & Analgesia. 2010;111:221–229. doi: 10.1213/ANE.0b013e3181dd2690. [DOI] [PubMed] [Google Scholar]
- 109.Tobe M, Obata H, Suto T, Yokoo H, Nakazato Y, Tabata Y, Saito S. Anesthesiology. 2010;112:1473–1481. doi: 10.1097/ALN.0b013e3181d4f66f. [DOI] [PubMed] [Google Scholar]
- 110.Timko BP, Dvir T, Kohane DS. Adv Mater Weinheim. 2010;22:4925–4943. doi: 10.1002/adma.201002072. [DOI] [PubMed] [Google Scholar]
- 111.Rwei AY, Lee JJ, Zhan C, Liu Q, Ok MT, Shankarappa SA, Langer R, Kohane DS. Proc Natl Acad Sci USa. 2015;112:201518791–15724. doi: 10.1073/pnas.1518791112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhan C, Wang W, McAlvin JB, Guo S, Timko BP, Santamaria C, Kohane DS. Nano Lett. 2015;16:177–181. doi: 10.1021/acs.nanolett.5b03440. [DOI] [PubMed] [Google Scholar]
- 113.Timko BP, Arruebo M, Shankarappa SA, McAlvin JB, Okonkwo OS, Mizrahi B, Stefanescu CF, Gomez L, Zhu J, Zhu A, Santamaria J, Langer R, Kohane DS. Proc Natl Acad Sci USa. 2014;111:1349–1354. doi: 10.1073/pnas.1322651111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Museux N, Perez L, Autrique L, Agay D. Burns. 2012;38:658–667. doi: 10.1016/j.burns.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 115.Hoare T, Santamaria J, Goya GF, Irusta S, Lin D, Lau S, Padera R, Langer R, Kohane DS. Nano Lett. 2009;9:3651–3657. doi: 10.1021/nl9018935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hadj A, Hadj A, Hadj A, Rosenfeldt F, Nicholson D, Moodie J, Turner R, Watts R, Fletcher I, Abrouk N, Lissin D. ANZ J Surg. 2012;82:251–257. doi: 10.1111/j.1445-2197.2011.05754.x. [DOI] [PubMed] [Google Scholar]
- 117.P. Winkle, H.S. Minkowitz, E. Onel, G. Boccia, A. Chu, N.J. Clendeninn, M.R. Keller, T. Ottoboni, S.S. Patel, B. Quart. http://www.herontx.com/sites/default/files/pdfs/posters-presentations/Heron_PainWeek_2016_Hernia_Results_L2c_091516.pdf
- 118.H.S. Minkowitz, P. Winkle, E. Onel, G. Boccia, A. Chu, N.J. Clendeninn, M.R. Keller, T. Ottoboni, S.S. Patel, B. Quart. http://www.herontx.com/sites/default/files/pdfs/posters-presentations/150102000_PainWeek_2016_Hernia_Administration_L2d.pdf
- 119.Cusack SL, Minkowitz HS, Kuss M, Jaros M, Hemsen L. Journal of Pain Research. 2012;5:453–461. doi: 10.2147/JPR.S37310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cusack SL, Jaros M, Kuss M, Minkowitz HS, Winkle P, Hemsen L. Journal of Pain Research. 2012;5:217–225. doi: 10.2147/JPR.S33453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hemsen L, Cusack SL, Minkowitz HS, Kuss ME. Journal of Pain Research. 2013;6:79–85. doi: 10.2147/JPR.S40158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Graber KD, Prince DA. Ann Neurol. 1999;46:234–242. doi: 10.1002/1531-8249(199908)46:2<234::aid-ana13>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]