

ABSTRACT
Matrix metalloproteinases (MMPs) are a family of zinc endopeptidases that cleave nearly all components of the extracellular matrix as well as many other soluble and cell‐associated proteins. MMPs have been implicated in normal physiological processes, including development, and in the acquisition and progression of the malignant phenotype. Disappointing results from a series of clinical trials testing small molecule, broad spectrum MMP inhibitors as cancer therapeutics led to a re‐evaluation of how MMPs function in the tumor microenvironment, and ongoing research continues to reveal that these proteins play complex roles in cancer development and progression. It is now clear that effective targeting of MMPs for therapeutic benefit will require selective inhibition of specific MMPs. Here, we provide an overview of the MMP family and its biological regulators, the tissue inhibitors of metalloproteinases (TIMPs). We then summarize recent research from model systems that elucidate how specific MMPs drive the malignant phenotype of breast cancer cells, including acquisition of cancer stem cell features and induction of the epithelial–mesenchymal transition, and we also outline clinical studies that implicate specific MMPs in breast cancer outcomes. We conclude by discussing ongoing strategies for development of inhibitors with therapeutic potential that are capable of selectively targeting the MMPs most responsible for tumor promotion, with special consideration of the potential of biologics including antibodies and engineered proteins based on the TIMP scaffold. J. Cell. Biochem. 118: 3531–3548, 2017. © 2017 The Authors. Journal of Cellular Biochemistry Published by Wiley Periodicals, Inc.
Keywords: MATRIX METALLOPROTEINASES, TISSUE INHIBITORS OF METALLOPROTEINASES, BREAST CANCER, TUMOR PROGRESSION, EPITHELIAL MESENCHYMAL TRANSITION, MMP INHIBITORS, CANCER BIOMARKERS, TUMOR MICROENVIRONMENT
STRUCTURE AND FUNCTION OF THE MATRIX METALLOPROTEINASE FAMILY
MMPs are a large family of zinc‐dependent endopeptidases found in all kingdoms except protozoa (MEROPS database: http://merops.sanger.ac.uk/) [Rawlings et al., 2012]. Humans express 23 MMPs. These enzymes possess a modular domain structure (Fig. 1A), the minimal form of which consists of a signal peptide for extracellular localization, a prodomain that inhibits the zymogen form of the enzyme until its removal by a separate, activating protease, and a conserved catalytic domain. This most simplified domain organization is found in MMP‐7 and ‐26; additional modules found in other MMPs facilitate localization, association with multiprotein complexes, or selectivity for specific protein substrates. Most MMPs are soluble proteins, although MMP‐14, ‐15, ‐16, and ‐24 are directly tethered to the cell membrane through C‐terminal transmembrane domains, MMP‐17 and ‐25 are localized to the cell membrane via C‐terminal glycophosphatidylinositol (GPI) anchors, and MMP‐23 via an N‐terminal type II transmembrane domain. Recent NMR studies demonstrate the ability of soluble MMP‐7 and MMP‐12 to bind directly to membrane bilayers, which may prove to be a new general mechanism by which these and other soluble MMPs are directed toward pericellular proteolytic activities [Koppisetti et al., 2014; Prior et al., 2015]. Beyond the minimal domain architecture, many MMPs also contain hemopexin‐like (PEX) domains, which assist in localizing MMPs to the cell membrane via interactions with other cell‐surface molecules [Piccard et al., 2007; Murphy and Nagase, 2011; Bauvois, 2012]. The PEX adaptor modules also mediate interactions with other soluble proteins, controlling distinct patterns of localization and substrate specificity [Piccard et al., 2007; Sela‐Passwell et al., 2010]. The three fibronectin type II repeats in MMP‐2 and MMP‐9 further assist in recognition of specific extracellular matrix substrates, including elastin and denatured collagen [Murphy et al., 1994; Steffensen et al., 1995; Shipley et al., 1996; Mikhailova et al., 2012]. Finally, MMP‐23 has several unusual modules, including the unique cysteine array domain that has homology to potassium channel blocking toxins and may modulate ion channel activity [Rangaraju et al., 2010], and an immunoglobulin‐like domain that has been implicated in protein‐protein interactions that affect localization or substrate recognition, a function similar to the PEX domains found in other MMPs [Galea et al., 2014].
Figure 1.
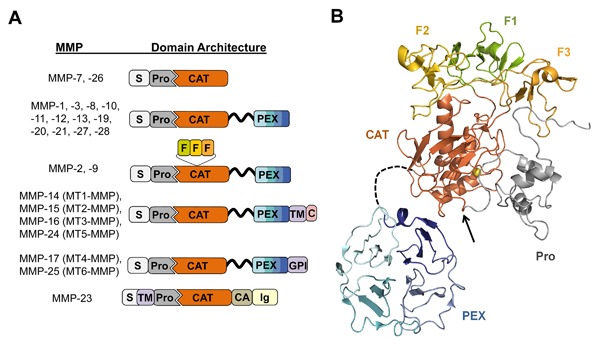
MMP domain structure and protein fold. (A) The domain organization of each human MMP is illustrated schematically; S, signal peptide; Pro, propeptide; CAT, catalytic domain; F, fibronectin type II repeats; PEX, hemopexin domain; TM, transmembrane domain; GPI, glycophosphatidylinositol membrane anchor; C, cytoplasmic domain; CA, cysteine array; Ig, immunoglobulin‐like domain. The flexible, variable length linker between CAT and PEX is shown as a black ribbon. (B) The representative 3D protein fold of proMMP‐2 is illustrated; individual domains are colored as in panel A. The flexible linker between CAT and PEX domains, shown as a black dashed line, varies in length among MMPs. The prodomain (gray) inhibits activity by coordinating the catalytic zinc (yellow sphere) and blocking access to substrates. Activation requires proteolysis within the loop indicated by the black arrow, leading to dissociation of the prodomain. Figure was generated with PyMOL (Schrodinger, LLC) from coordinates of PDB ID: 1GXD [Morgunova et al., 2002].
DETERMINANTS OF CATALYTIC ACTIVITY AND SUBSTRATE SPECIFICITY
The MMP catalytic domain is highly conserved among members of the family, and contains key features of the larger metzincin metallopeptidase clan, including the conserved HExxHxxGxxH motif which functions to coordinate the catalytic zinc ion. The MMP catalytic mechanism involves activation of a water molecule by the zinc ion and a conserved Glu residue for nucleophilic attack on the target peptide bond [Tallant et al., 2010; Cerda‐Costa and Gomis‐Ruth, 2014]. Prior to enzyme activation, the prodomain of the MMP blocks access to the active site cleft (Fig. 1B), usually (with the exception of MMP‐26) through interaction of the “cysteine switch” motif containing a conserved PRCGxPD with the catalytic zinc. Activation of the MMP involves disruption of the cysteine switch interaction and cleavage of the prodomain by an activating protease [Rosenblum et al., 2007a; Tallant et al., 2010]. MMP‐26, which uniquely possesses a mutated and nonfunctional cysteine switch motif, is also activated proteolytically, although the mechanism by which its zymogen retains latency is not clearly understood [Marchenko et al., 2002]. Many of the MMPs, and all of the transmembrane MMPs, can be activated by furin in the secretory pathway of the cell, while others can be activated once localized to the extracellular space by serine proteases or other MMPs [Ra and Parks, 2007; Tallant et al., 2010].
The MMP active site is located within a broad, shallow cleft that is capable of accommodating a peptide substrate in extended conformation, with the target peptide bond positioned for cleavage adjacent to the catalytic zinc. Binding subsites in the catalytic cleft provide a limited degree of substrate sequence specificity [Tallant et al., 2010]; relative to many other proteases, MMPs have less defined intrinsic specificity for the primary sequence of the cleavage site, with critical determinants of specificity located at exosites further removed, both on the surface of the catalytic domain and on adjacent accessory domains [Palmier et al., 2010; Arnold et al., 2011; Robichaud et al., 2011; Mikhailova et al., 2012; Stura et al., 2013]. Beyond enhancing molecular recognition of specific substrates, accessory domains may also help to promote catalysis, as in the case of MMP‐1 collagenolysis, where cooperative reorganization between the catalytic and PEX domains after substrate binding leads to a deformation of the collagen triple helical structure necessary for proteolysis [Bertini et al., 2011; Manka et al., 2012]. Similar interdomain flexibility is present in MMP‐9 and MMP‐12, and may be a general property of MMPs with PEX domains [Rosenblum et al., 2007b; Bertini et al., 2008, 2009]. Such independent domain mobility may facilitate unique MMP functions, as in the case of MMP‐9, where the particularly long and flexible linker domain may allow the catalytic domain access to complex pericellular substrate networks while the PEX domain maintains cell surface localization [Rosenblum et al., 2007b]. A better understanding of the roles of domains, recognition sites, and their cooperative interactions can help to identify new strategies for therapeutic targeting of MMPs; using this understanding, it may be feasible to selectively block a subset of MMP activities responsible for specific protumorigenic functions.
REGULATION BY TISSUE INHIBITORS OF METALLOPROTEINASES
Tissue inhibitors of metalloproteinases (TIMPs) are endogenous protein protease inhibitors that regulate MMPs along with several other families of zinc‐dependent metallopeptidases, forming very tight 1:1 stoichiometric inhibitory complexes. Upon secretion, TIMPs can interact with a variety of membrane‐anchored or secreted MMPs (Fig. 2) [Jackson et al., 2017]. Structurally, TIMPs are comprised of two domains that pack side‐by‐side, each stabilized by three internal disulfide bonds [Gomis‐Ruth et al., 1997; Batra and Radisky, 2014]. The N‐terminal domain is sometimes referred to as the “inhibitory domain” due to the fact that as an isolated recombinant domain it remains capable of MMP inhibition [Murphy et al., 1991]; however, in MMP complexes with intact TIMPs the interaction is more extensive, spanning both domains [Batra et al., 2013; Batra and Radisky, 2014], resulting in stronger inhibition by nearly an order of magnitude in several cases that have been examined [Huang et al., 1996]. The four human TIMPs possess broad, overlapping specificity and each is capable of inhibiting most MMPs, although with a spectrum of affinities spanning many orders of magnitude, from 0.6 fM for the association of TIMP‐2 with MMP‐2 [Hutton et al., 1998], to the high nanomolar range for the relatively poor inhibition of MMP‐14 and MMP‐15 by TIMP‐1 (Table I). The strongest of these interactions, including those of TIMP‐2/MMP‐2 and TIMP‐1/MMP‐9, involve not only the MMP catalytic domain but additional interactions with the PEX domain [O'Connell et al., 1994; Butler et al., 1999a].
Figure 2.
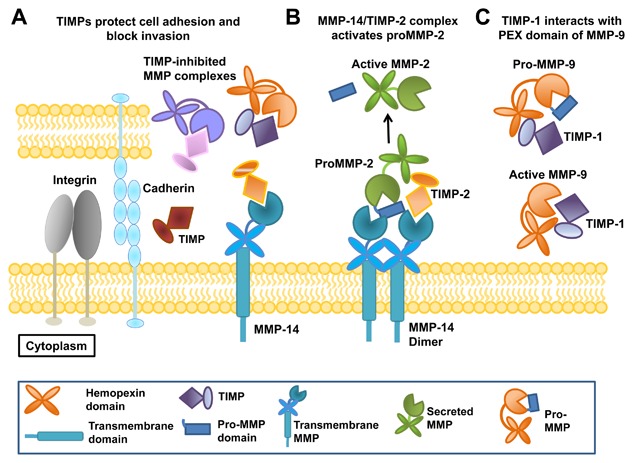
TIMPs regulate MMP function and activation. (A) Soluble and extracellular matrix‐associated TIMPs form inhibitory interactions with the catalytic domains of extracellular and transmembrane MMPs, protecting cell adhesion and blocking invasion. (B) The dimeric form of transmembrane MMP‐14 forms a receptor complex with TIMP‐2 that is responsible for MMP‐2 activation; proMMP‐2 is bound via its C‐terminal PEX domain, allowing proteolysis of the prodomain by the uninhibited MMP‐14 subunit. (C) TIMP‐1 forms C‐terminal domain interactions with the PEX domain of proMMP‐9 or MMP‐9, protecting proMMP‐9 from activation and quenching activity after activation.
Table I.
Reported Apparent Ki Values for TIMP‐Peptidase Interactions
| Enzyme | TIMP‐1 | TIMP‐2 | TIMP‐3 | TIMP‐4 |
|---|---|---|---|---|
| MMP‐1 | 100 pM1 | 650 pM3 | ||
| 250 pM2 | 2.6 nM3, a | |||
| 2.7 nM1, a | ||||
| MMP‐2 | 140 pM2 | 0.6 fM5 | <2 pM6 | 4 pM3 |
| 10 nM4 | 7 nM4 | |||
| MMP‐3 | 240 pM2 | 550 pM7, a | 1.5 nM3 | |
| 250 pM2, a | 1.2 nM3, a | |||
| 110 pM7, a | ||||
| MMP‐7 | 370 pM8 | |||
| MMP‐9 | <50 pM9 | <50 pM9 | ||
| 9 nM4 | 43 nM4 | |||
| MMP‐10 | 1.1 nM7, a | 5.8 nM7, a | ||
| MMP‐14 | 178 nM10, b | 70 pM11, a | 160 pM11, a | 340 pM11, a |
| MMP‐15 | 100 nM12, a | |||
| MMP‐16 | 170 pM11, a | 8.2 pM11, a | 340 pM11, a | |
| MMP‐17 | ∼10 nM13, a | ∼10 nM13, a | ∼10 nM13, a | ∼10 nM13, a |
| MMP‐19 | 57.6 nM14, a | 5 pM14, a | 5 pM14, a | 5 pM14, a |
| MMP‐25 | 200 pM15, a | 2 nM15, a |
Sources: 1Taylor et al. [1996]; 2Huang et al. [1996]; 3Troeberg et al. [2002]; 4Olson et al. [1997]; 5Hutton et al. [1998]; 6Lee et al. [2001]; 7Batra et al. [2012a]; 8O'Shea et al. [1992]; 9O'Connell et al. [1994]; 10Lee et al. [2003]; 11Zhao et al. [2004]; 12Morrison et al. [2001]; 13Amour et al. [2002]; 14Stracke et al. [2000]; 15Sun et al. [2007].
Reflects binding of truncated MMP catalytic domain.
Reflects binding of truncated N‐terminal TIMP domain.
The primary interaction of a TIMP with an MMP catalytic domain utilizes a conserved core epitope centered around the N‐terminal strand of the TIMP, which coordinates to the active site zinc of the MMP, blocking the substrate‐binding cleft (Fig. 3A and B). This interaction also involves adjacent EF and C‐connector loops of the TIMP N‐terminal domain, which are tethered to the N‐terminal strand by disulfide bonds (Fig. 3B). Additional loops flanking the core epitope, including the AB loop of the N‐terminal domain and the multiple turn and GH loops of the C‐terminal domain, can form adventitious contacts with exosites on the surface of the MMP catalytic domain (Fig. 3A) [Batra et al., 2013; Batra and Radisky, 2014]. Whereas the core epitope interaction with different MMPs is highly structurally conserved, the residues involved in peripheral interactions are much less conserved among both TIMPs and MMPs, resulting in considerable variation in the extent and nature of exosite interactions in different TIMP/MMP pairs. These regions of sequence and structural variability contribute to the wide variation in TIMP/MMP binding constants [Williamson et al., 2001; Batra et al., 2013], which span many orders of magnitude (Table I). Additional determinants of differential affinity include subtle packing alterations at the intermolecular interface with the core epitope [Lee et al., 2003; Batra et al., 2012a].
Figure 3.
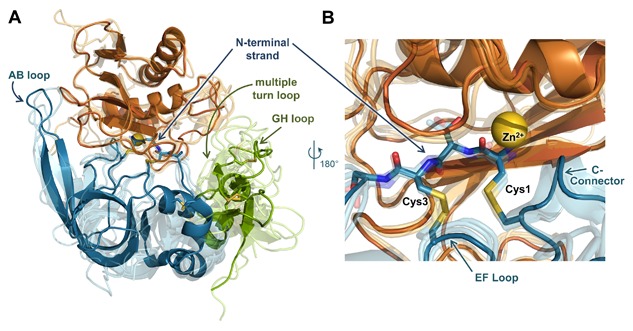
MMP/TIMP interfaces feature a conserved core interaction and diverse peripheral interactions. (A) The structure of the representative complex of MMP‐10CAT (orange) with TIMP‐2 (N‐terminal domain blue, C‐terminal domain green) (PDB ID: 4ILW) [Batra et al., 2013] is shown; the catalytic zinc ion is rendered as a yellow sphere. Four additional MMPCAT/TIMP structures (semi‐transparent) are superposed to highlight regions of structural conservation versus diversity: MMP‐10CAT/TIMP‐1 (PDB ID: 3V96) [Batra et al., 2012a], MMP‐3CAT/TIMP‐1 (PDB ID: 1UEA)[Gomis‐Ruth et al., 1997], MMP‐14CAT/TIMP‐2 (PDB ID: 1BQQ)[Fernandez‐Catalan et al., 1998], and MMP‐13CAT/TIMP‐2 (PDB ID: 2E2D)[Maskos et al., 2007]. Diverse interactions with different MMPs are formed by peripheral TIMP epitopes of the AB, GH, and multiple turn loops. (B) Closer view of the superposed complexes reveals conserved positioning and interactions of the TIMP core epitope, including N‐terminal residues 1–4, the EF loop, and C‐connector loop. The amine terminus of residue Cys‐1 coordinates directly to the catalytic zinc ion. Figure was generated using PyMOL (Schrodinger, LLC).
Beyond the primary inhibitory interaction of TIMPs with MMP catalytic domains, additional, noninhibitory interactions can occur between select TIMP/MMP pairs. The C‐terminal domains of TIMP‐2, TIMP‐3, or TIMP‐4 can bind to the PEX domain of MMP‐2 or pro‐MMP‐2, while the C‐terminal domains of TIMP‐1 or TIMP‐3 can associate with MMP‐9 or pro‐MMP‐9 (Fig. 2) [Brew and Nagase, 2010]. The interaction of pro‐MMP‐2 with TIMP‐2 has been structurally characterized [Morgunova et al., 2002] and shown to be important in MMP‐2 activation by MT1‐MMP (Fig. 2) [Strongin et al., 1995]. This role is unique to TIMP‐2, as TIMP‐3 or TIMP‐4 do not participate in MMP‐2 activation but may serve as negative regulators of the process [Bigg et al., 2001; Hernandez‐Barrantes et al., 2001; English et al., 2006]. The pro‐MMP‐9/TIMP‐1 complex has been less well characterized structurally, but biochemical data support the significance of the pro‐MMP‐9 PEX domain and the TIMP‐1 C‐terminal domain in this interaction [Murphy et al., 1991; O'Connell et al., 1994]. Several studies have implicated pro‐MMP‐9/TIMP‐1 complex formation as a mechanism that protects the secreted proenzyme from activation by other MMPs both in vitro and in vivo (Fig. 2) [Goldberg et al., 1992; Ogata et al., 1995; Ardi et al., 2007, 2009]. Finally, in addition to their role in MMP inhibition, TIMPs are also multifunctional proteins with emerging pleiotropic activities mediated through MMP‐independent protein–protein interactions with other binding partners. These alternative cell signaling activities and their implications in cancer have been reviewed elsewhere [Chirco et al., 2006; Stetler‐Stevenson, 2008; Brew and Nagase, 2010], and will not be described here in detail.
TUMORIGENIC PROCESSES ACTIVATED BY MMPS IN BREAST CANCER
MMPs are so named because they are capable of cleaving nearly every component of the extracellular matrix (ECM); through matrix degradation they directly facilitate cancer invasion and metastasis by clearing pathways for invading cancer cells and for ingressing vasculature to provide oxygen and nutrients for the growing tumor [Kessenbrock et al., 2010]. MMPs also play key roles in the generation of disruptive fibrotic stroma, both through degradation of the native ECM and through MMP‐induced activation of stromal fibroblasts, and further can stimulate tumor‐promoting metabolic switches through interaction with adipocytes [Kessenbrock et al., 2010]. MMPs additionally target receptors on the surface of the tumor cells themselves, activating pathways that promote proliferation or suppress apoptosis [Gialeli et al., 2011]. MMPs can also indirectly drive tumor initiation and progression, as disruption of the ECM can activate cellular processes that cause DNA damage and stimulate genomic instability [Radisky and Bissell, 2006]. Finally, MMPs can directly induce the epithelial–mesenchymal transition (EMT) in target epithelial cells; EMT is a developmental process that is associated with resistance to apoptosis and increased invasiveness, and has been associated with development of a cancer stem cell phenotype [Radisky and Radisky, 2010; Nistico et al., 2012]. Here, we will describe some of the more recently discovered cancer‐associated processes in which MMPs have been implicated.
REGULATION OF PROLIFERATION AND APOPTOSIS
Expansion of tumor mass is associated with increased proliferation and decreased apoptosis. One of the most well‐studied regulators of cellular proliferation and apoptosis in the tumor microenvironment is TGFβ, which has cytostatic and apoptotic function in most nonmalignant/normal cells, but which can also stimulate proliferation and suppress apoptosis through canonical and noncanonical pathways in many transformed or malignant cells [Massague, 2012]. TGFβ is generated and secreted as a component of an inactive complex; MMPs can cleave the TGFβ precursor or other complex components to generate the active cytokine [Yu and Stamenkovic, 2000; Dallas et al., 2002; Mu et al., 2002; Annes et al., 2003; Tatti et al., 2008], and can also modify the ECM to facilitate nonproteolytic activation of TGFβ [Cichon and Radisky, 2014a]. MMPs and members of the related ADAM (a disintegrin and metalloproteinase) family can trigger the epidermal growth factor (EGF) receptor by shedding of cell membrane‐associated ligands, such as HB‐EGF, TGFβ, and amphiregulin [Kessenbrock et al., 2010; Gialeli et al., 2011]. MMPs can also proteolytically inactivate the cell death receptor Fas, leading to inhibition of the intrinsic apoptosis pathway which is involved in chemotherapeutic cell killing [Mitsiades et al., 2001; Crawford et al., 2002]; specifically inhibiting the MMP–Fas interaction is thus an important avenue for development of novel therapeutics [Villa‐Morales and Fernandez‐Piqueras, 2012].
PROMOTION OF INVASION AND METASTASIS
In addition to the well‐characterized functions of MMPs to facilitate cancer cell invasion through degradation of ECM, several MMPs have been implicated as direct activators of invasion through breakdown of cell–cell interactions and activation of cell motility and migration processes. As an example of this phenomenon, MMPs cleave the Wnt/planar cell polarity protein‐tyrosine kinase‐7 (PTK7), stimulating cancer cell invasion and metastasis [Golubkov et al., 2010, 2014; Golubkov and Strongin, 2012]. Many MMPs can also directly activate aspects of the EMT program in epithelial cancer cells [Lamouille et al., 2014]. MMP‐induced EMT has been observed in kidney [Cheng and Lovett, 2003; Cheng et al., 2006; Zheng et al., 2009; Tan et al., 2010], ovary [Cowden Dahl et al., 2008], lens [West‐Mays and Pino, 2007], lung [Illman et al., 2006; Yamashita et al., 2011; Stallings‐Mann et al., 2012], pancreas [Mehner et al., 2014a], and prostate [Cao et al., 2008] cells, although the process of MMP‐induced EMT has been best characterized in mammary epithelial cells [Radisky and Radisky, 2010; Foroni et al., 2012]. MMP‐3, the most upregulated MMP family member during postlactational involution of the breast [Radisky and Hartmann, 2009], was found to induce spontaneous mammary tumors when expressed in transgenic mice by a milk protein promoter [Sympson et al., 1995; Sternlicht et al., 1999, 2000]. Dissection of this process revealed that MMP‐3 directly activates EMT [Lochter et al., 1997a,1997b] by activating alternative splice pathways which result in the expression of Rac1b [Radisky et al., 2005; Pelisch et al., 2012; Cichon et al., 2015]. Rac1b is an activated splice variant with unique tumor‐promoting activities [Orlichenko et al., 2010], which stimulates EMT by increasing levels of cellular reactive oxygen species [Radisky et al., 2005; Nelson et al., 2008; Cichon and Radisky, 2014b], through a process that depends upon cell–ECM interactions [Lee et al., 2012; Chen et al., 2013a]. Thus, MMPs can stimulate invasion through both extrinsic (ECM‐targeting) and intrinsic (cell phenotype alteration) processes.
MMP‐induced EMT can also facilitate increased tumorigenicity through induction and regulation of stem cell characteristics. MMPs play a variety of roles in physiological functions of stem cells, including tissue morphogenesis and regeneration; in the mammary gland, MMP‐3 regulates Wnt signaling and consequent stem cell expansion via a specific capacity to bind and inactivate the noncanonical Wnt ligand Wnt5b [Kessenbrock et al., 2013]. More recently, MMPs have been identified in cancer stem/progenitor cells (CSCs) as well [Kessenbrock et al., 2015]. Similar to normal stem cells, CSCs have low proliferation rates and resistance to apoptosis, characteristics that confer increased resistance to cytotoxic therapies and tumor recurrence; consequently, identification of CSCs within tumors and development of therapies that specifically target CSCs have become an important area of research [Singh and Settleman, 2010]. An emerging body of research has emphasized that activation of the EMT program, even in an incomplete or dysregulated fashion, can stimulate CSC characteristics [Singh and Settleman, 2010]. Targeting MMP‐induced EMT, therefore, is a promising new approach for reducing CSC‐associated malignancy.
CLINICAL STUDIES IDENTIFY SPECIFIC MMPS AS MOST LIKELY THERAPEUTIC TARGETS IN BREAST CANCER
While MMPs clearly play prominent roles in shaping the tumor microenvironment and driving cancer progression and metastasis as summarized above, early efforts to target these proteases therapeutically in clinical trials of more than 50 MMP inhibitors led to widely disappointing results [Coussens et al., 2002; Vandenbroucke and Libert, 2014]. A trial of broad spectrum MMP inhibitor marimastat in advanced stage breast cancer patients found no benefit to progression‐free survival [Sparano et al., 2004], while phase II trials in early stage breast cancer patients found further exploration of the approach to be unfeasible in this population due to issues of toxicity [Miller et al., 2002, 2004]. In the wake of these trials, consensus has emerged that lack of inhibitor specificity was one of the critical problems with the approach. This is because not all MMPs confer exclusively tumor promoting effects, and some have even been shown to provide a primarily protective function [Lopez‐Otin and Matrisian, 2007; Martin and Matrisian, 2007; Decock et al., 2011]. MMP‐8 provides one of the more clear‐cut examples of this phenomenon. In mouse models of breast cancer, high expression of MMP‐8 suppresses metastasis, while MMP‐8 silencing or knockout promotes tumor progression and metastasis [Montel et al., 2004; Decock et al., 2015]. Importantly, the net antitumor effect of MMP‐8 is corroborated by the association of MMP‐8 tumor expression with longer relapse‐free survival in breast cancer patients [Gutiérrez‐Fernández et al., 2008], and by the association of a high expression promoter variant of the MMP‐8 gene with more favorable prognosis [Decock et al., 2007].
For other MMPs, categorization as friend or foe is not as straightforward. Mounting studies reveal that depending on the model system, the animal genetic background, or other experimental factors, an MMP may exert a tumor‐promoting, tumor‐inhibitory, or null effect. One example of this phenomenon is found in MMP‐9. Early studies with MMP‐9 knockout mice in a pancreatic carcinogenesis model identified MMP‐9 as a critical component of an angiogenic switch, driving angiogenesis and tumor progression though release of vascular endothelial growth factor (VEGF) [Bergers et al., 2000]. Proangiogenic, tumor promoting functions of MMP‐9 have subsequently been confirmed in many other models [Deryugina and Quigley, 2010], yet, some studies have also implicated MMP‐9 in the proteolytic generation of angiogenesis inhibitors angiostatin (a fragment of plasminogen) [Pozzi et al., 2000], tumstatin (a fragment of collagen IV) [Hamano et al., 2003], and endostatin (a fragment of collagen XVIII) [Bendrik et al., 2008]. A recent study examined the effect of MMP‐9 knockout on tumorigenesis in several genetically engineered mouse models; the absence of MMP‐9 delayed tumorigenesis in the C3(1)‐Tag model of basal‐like triple‐negative breast cancer, but had no comparable effect on tumorigenesis in the (MMTV)‐Neu model of luminal breast cancer [Park et al., 2015].
The impact of MMP‐9 on metastasis can also differ depending on the model. In an orthotopic mouse model of human breast cancer, silencing of MMP‐9 expression in tumor cells blocked metastasis, demonstrating a critical role for tumor cell‐produced MMP‐9 in metastasis [Mehner et al., 2014b]. On the other hand, a study examining genetic ablation of MMP‐9 in the MMTV‐PyVT mouse mammary tumor model demonstrated that host‐derived (immune cell) MMP‐9 could exert a pro‐metastatic influence under some circumstances, depending on the genetic background of the mouse strain used [Martin et al., 2008].
In light of such inconsistent or paradoxical findings from diverse experimental models, each with its own strengths and limitations, how can we best evaluate the likelihood that a given MMP represents a significant and promising therapeutic target in human breast cancer? We argue that evidence from clinical studies examining associations of specific MMPs with poor prognosis should be used as a framework to guide interpretation of experimental results, and to prioritize MMP candidates for development of therapeutic strategies. Among the large MMP family, relatively few have shown consistent association with poor outcomes across multiple cohorts, narrowing the field. Here, we highlight clinical findings that implicate several MMPs as potential therapeutic targets.
MMP‐9
Consistent with the demonstrated functional roles of MMP‐9 in tumor angiogenesis, invasion, and metastasis [Wang et al., 2010; Bauvois, 2012; Deryugina et al., 2014; Mehner et al., 2014b], in clinical studies of breast cancer patients MMP‐9 has been consistently associated with cancer progression and poorer outcomes. Analyses of transcriptional microarray data have shown MMP‐9 to be one of only a few MMPs consistently prognostic for poor survival in several large cohorts of breast cancer patients [van de Vijver et al., 2002; McGowan and Duffy, 2008; Dufour et al., 2011; Curtis et al., 2012; Roy and Walsh, 2014]. MMP‐9 is also the only MMP gene to be included among the 70 genes in the Rosetta poor prognosis breast cancer signature [van 't Veer et al., 2002], on which the Mammaprint clinical prognostic assay is based (Agendia, Inc., Irvine, CA). At the protein expression level, tissue microarray analyses of primary breast tumors have found MMP‐9 total immunostaining to be associated with shorter relapse‐free survival, and individually scored MMP‐9 staining of tumor cells, stromal fibroblasts, and mononuclear inflammatory cells were also each significantly associated with distant metastasis and poorer relapse‐free survival [Gonzalez et al., 2007; Vizoso et al., 2007]. In other immunohistochemical studies of large breast cancer cohorts, MMP‐9 expression specifically in stromal cells was found to be most strongly prognostic for both recurrence and poor survival [Pellikainen et al., 2004; Mylona et al., 2007]. The significance of MMP‐9 expression in tumor cells, especially in early stages of cancer, is less clear; in studies examining primarily node negative breast cancers, tumor cell MMP‐9 was prognostic for shorter relapse‐free survival in one cohort [Li et al., 2004] but associated with better survival in others [Scorilas et al., 2001; Pellikainen et al., 2004]. Serum levels of MMP‐9 have also been found to be elevated in breast cancer patients and significantly associated with poor outcomes in numerous studies [Wu et al., 2008; Radisky and Radisky, 2015]. The absence of such prognostic associations for circulating MMP‐9/TIMP‐1 complexes [Thorsen et al., 2013] suggests that the presence of free, catalytically active MMP‐9 may be most relevant for predicting cancer progression, consistent with experimental studies demonstrating that TIMP‐free MMP‐9 is required for tumor angiogenesis and tumor cell intravasation [Bekes et al., 2011]. The consensus of clinical studies supports consideration of MMP‐9 as a potential therapeutic target in breast cancer, particularly for specific subsets of patients with aggressive disease. MMP‐9 is most highly expressed in the aggressive basal‐like triple negative and HER2‐positive breast cancer molecular subtypes [Mehner et al., 2014b; Yousef et al., 2014]. Furthermore, several studies have found MMP‐9 protein expression to be significantly prognostic for shorter time to progression and poorer overall survival specifically in basal‐like or triple negative breast cancer patients [Gonzalez et al., 2009; Zhao et al., 2013]. Given the limited therapeutic options for these patients, MMP‐9 may offer an attractive target for antimetastatic therapies.
MMP‐11
MMP‐11, also known as stromelysin‐2, is another MMP that repeatedly appears in the clinical literature in association with poor breast cancer outcomes. At the transcriptional level, it was associated with poor survival in a microarray dataset representing primary tumors from 1500 breast cancer patients [Curtis et al., 2012; Roy and Walsh, 2014]. MMP‐11 is part of the 21 gene signature in the Oncotype DX clinical assay (Genomic Health, Inc., Redwood City, CA), originally developed to predict recurrence of breast cancer after tamoxifen treatment [Paik et al., 2004]. It is also included among the 50 genes of the PAM50 profile used to predict breast cancer intrinsic molecular subtypes and risk of recurrence [Parker et al., 2009]. At the protein level, MMP‐11 is found to be expressed by many cell types in the tumor microenvironment, and generally predicts poor prognosis irrespective of localization. In women with invasive ductal breast cancers, total immunostaining scores for MMP‐11, like MMP‐9, were shown to be significantly prognostic for shorter relapse‐free survival, as were specific expression of MMP‐11 by fibroblasts or mononuclear inflammatory cells [Gonzalez et al., 2007; Vizoso et al., 2007]. In a different cohort of 192 patients, MMP‐11 staining both in tumor cells and in stromal fibroblast‐like cells was found to be significantly associated with poor overall survival [Min et al., 2013]. Yet another study found overall tumor MMP‐11 staining to be significantly associated with advanced tumor stage and lymph node metastasis [Cheng et al., 2010]. Further investigations have identified a subset of poor prognosis breast tumors characterized by infiltration of MMP‐11‐expressing mononuclear inflammatory cells, revealing a distinct profile of associated inflammatory cytokines [Gonzalez et al., 2009; Eiro et al., 2012]. Expression of MMP‐11 by endothelial cells in tumor blood vessels has also been found to be associated with shorter relapse‐free survival and overall survival [Cid et al., 2016]. In one study, MMP‐11 was significantly more strongly expressed by stromal fibroblast‐like cells in tumors of the aggressive basal‐like and HER2‐positive subtypes [Min et al., 2013]. For the basal‐like subtype specifically, overall MMP‐11 expression, fibroblast, or mononuclear inflammatory cell MMP‐11 expression were each individually associated with poorer relapse‐free survival [Gonzalez et al., 2009]. Taken together with experimental studies implicating MMP‐11 in survival and early tumor invasion of breast cancer cells [Lefebvre et al., 1992; Masson et al., 1998; Boulay et al., 2001; Wu et al., 2001; Takeuchi et al., 2011], the available data suggest that MMP‐11 may offer a viable molecular target especially for basal‐like or triple negative breast cancers, where its targeting may be of greatest utility in earlier stages of progression.
TRANSMEMBRANE MMPS: MMP‐14 AND MMP‐15
Consistent with the role of transmembrane MMPs as a key mediators of cancer cell invasion and migration [Koshikawa et al., 2000; Hotary et al., 2003, 2006; Shiomi and Okada, 2003; Sabeh et al., 2004; Cao et al., 2005; Ota et al., 2009; Zarrabi et al., 2011], clinical studies have found both MMP‐14 and MMP‐15 expression to signal poor prognosis, especially at the transcriptional level. In a transcriptional microarray dataset comprised of primary breast tumors from 295 patients [van de Vijver et al., 2002], high expression of MMP‐14 and MMP‐15 were each associated with poorer overall survival [McGowan and Duffy, 2008]. MMP‐15 expression was associated with higher grade tumors, while MMP‐14 was the only MMP to remain a prognosticator of poor survival in multivariate analysis, independent of tumor size, grade, lymph node status, and ER status [McGowan and Duffy, 2008]. MMP‐15 was similarly prognostic for poor overall survival in analysis of another transcriptional microarray dataset of 1500 breast cancers [Curtis et al., 2012; Roy and Walsh, 2014]. In a study employing mRNA in situ hydridization to examine MMP‐14 expression in 539 breast cancers, MMP‐14 was found almost exclusively in stromal cells, and was associated with poor overall survival in univariate analyses as well as multivariate analyses after adjustment for tumor size and lymph node involvement [Tetu et al., 2006]. Studies examining protein expression of MMP‐14 by immunohistochemistry have observed expression in cancer cells as well as stromal cells to be common [Mylona et al., 2007; Vizoso et al., 2007]; however only MMP‐14 expression by mononuclear inflammatory cells was significantly associated with a higher rate of distant metastasis in one study [Vizoso et al., 2007]. Several studies have suggested that MMP‐14, similarly to MMP‐9 and MMP‐11, may be particularly relevant as a target in basal‐like or triple negative breast cancers. In a study combining patient tumor analyses with experimental investigations, tumor staining for MMP‐14 was significantly associated with blood vessel invasion of tumor cells specifically in the subset of triple negative breast cancers, consistent with the demonstration in a mouse orthotopic model that MMP‐14 downregulation in tumor cells reduces blood vessel invasion and spontaneous metastasis [Perentes et al., 2011]. Another recent integrative study evaluated MMP‐14 transcript expression by qRT/PCR in a cohort of 458 breast cancers patients, finding expression to be associated with high grade, with the triple negative subtype, and with shorter metastasis‐free survival [Lodillinsky et al., 2016]. Immunostaining of tissue microarrays for MMP‐14 likewise confirmed associations with grade, triple negative subtype, and with progression from ductal carcinoma in situ to invasive cancer. Consistent with these clinical data, the authors found that knockdown of MMP‐14 expression in tumor cells impaired invasion in multiple intraductal xenograft models of basal‐like breast cancer [Lodillinsky et al., 2016]. These studies help to solidify MMP‐14 as a target of potential interest for treating this aggressive breast cancer subtype. Based on transcriptional associations with breast cancer outcome and functional studies of invasion in cell culture models, MMP‐15 may also be of interest as a therapeutic target. However, investigations of MMP‐15 protein expression and its prognostic significance in breast cancer, along with functional investigations of MMP‐15 in mouse models of breast cancer, are lacking and remain an important area for future investigation.
DEVELOPING MMP INHIBITORS FOR THERAPEUTIC APPLICATIONS
As discussed above, several MMPs are strongly implicated as promising targets for breast cancer therapy, yet MMP inhibitors evaluated in clinical trials to date have failed, causing major musculoskeletal toxicity and failing to improve clinical outcomes [Coussens et al., 2002; Vandenbroucke and Libert, 2014]. The musculoskeletal toxicity was thought to derive from inhibition of MMP‐1, ADAM family metalloproteases, and/or metalloenzymes outside of the MMP and ADAM families, and in some cases could be avoided by inhibitors with restricted selectivity toward a subset of MMPs [Maquoi et al., 2004; Fingleton, 2008; Vandenbroucke and Libert, 2014]. The poor efficacy of broad spectrum inhibitors was likely caused in part by their inhibition of cancer‐protective MMPs such as MMP‐8 along with cancer‐promoting enzymes. Thus, it is anticipated that development of highly selective inhibitors for the best MMP drug targets could solve both problems, minimizing drug toxicity while sparing enzymes that protect the host from tumor progression.
DEVELOPING MORE SELECTIVE SMALL MOLECULE MMP INHIBITORS
Small molecule MMP inhibitors have typically targeted the catalytic domain active site, binding in the variable S1′ pocket of MMPs and chelating the catalytic zinc ion. Because MMPs have similar active site structures, many such inhibitors have limited ability to discriminate among various MMPs, and development of improved selectivity has proved challenging [Overall and Kleifeld, 2006; Devel et al., 2010; Jacobsen et al., 2010; Cathcart et al., 2015]. Nevertheless, progress has been made in the development of selective inhibitors for a few MMPs. For instance, optimizing S1′ pocket interactions as well as recruiting phosphinate or P2′ glutamate as alternatives to more traditional zinc‐chelating groups have resulted in very selective inhibitors of MMP‐12 [Devel et al., 2006, 2010, 2012]. Other studies have focused on MMP‐13, where optimizing interactions with the unusually deep and branched S1′ pocket has resulted in potent and selective inhibitors based on a fused pyrimidine zinc binding group [Nara et al., 2017], or lacking interactions with the zinc altogether [Engel et al., 2005; Nara et al., 2014; Hugenberg et al., 2017]. A recent biochemical and biophysical study demonstrated that the zinc‐binding group can make surprising contributions to inhibitor selectivity, arguing for drug development strategies that would more widely diversify this functionality in combinatorial approaches [Rouanet‐Mehouas et al., 2017].
A unique approach for limiting MMP inhibitor toxicity was reported recently, in a study that aimed to achieve selectivity not only for the target enzyme but also through targeted drug delivery. Although MMP‐2 has not generally been considered a primary drug target in cancer due in part to its ubiquitous presence and involvement in many physiological processes, the importance of this enzyme in progression of bone metastatic breast cancer renders it a target of interest in this specific setting [Tauro et al., 2017]. Bisphosphonic‐based inhibitors selective for MMP‐2 over the similar MMP‐9 were developed, which due to the affinity of bisphosphonate for hydroxyapatite were efficiently localized to the bone microenvironment, where they were shown to inhibit MMP‐2 and to prevent breast cancer growth and associated bone destruction in a mouse model [Tauro et al., 2017].
Other approaches for developing selectivity aim to target MMP exosites beyond the active site, on catalytic domain or accessory domain surfaces, with the rationale that these sites are less conserved among MMPs. A set of branched amphiphilic polymers was used to probe MMP‐12 and ‐14 catalytic domains for identification of hidden allosteric regulatory sites; these polymers compromised catalytic activity by interacting with flexible surfaces of MMP‐12 and ‐14, altering protein dynamics [Udi et al., 2013]. Comparable but unique allosteric regulatory sites were computationally predicted to exist in other MMP catyalytic domains, making these sites potential targets for developing novel therapeutic agents [Udi et al., 2013]. Exosites of accessory domains can also be effective targets for developing MMP inhibitors with greater selectivity. Triple helical peptides (THPs) that mimic collagen substrates of MMPs have been developed as efficient inhibitors that simultaneously target multiple domains of MMPs [Ndinguri et al., 2012]. THP substrate and phosphinate transition state analogues showed high affinity and selectivity toward MMP‐2 and ‐9 [Lauer‐Fields et al., 2007], and could be designed for selective inhibition of proteolytic activity toward specific substrates, by targeting fibronectin domain exosite interactions important for the recognition of those substrates [Lauer‐Fields et al., 2008]. More recently, heterotrimeric THPs were synthesized using click chemistry as highly selective binders to MMP‐13 and ‐14, with complete selectivity between MMP‐1 and ‐14 [Bhowmick et al., 2015]. Other peptide‐based and small molecule inhibitors have succeeded in blocking MMP‐9‐dependent cellular migration and metastasis by targeting exosites on the PEX domain that are important for homodimerization and binding to cell surface recptor CD44 [Dufour et al., 2010, 2011]. Similarly, a small molecule targeting the MMP‐14 PEX domain inhibited MMP‐14 dimerization as well as tumor growth and collagen degradation in an orthotopic mammary tumor model [Remacle et al., 2012].
DEVELOPING ANTIBODY THERAPEUTICS THAT BLOCK MMP FUNCTIONALITY
Many of the preceding examples have achieved selectivity by extending interactions with MMP targets beyond the immediate active site, to encompass broader interaction surfaces mimicking the protein–protein interfaces of natural substrates and effectors. While successful at enhancing selectivity, these larger, often peptide‐based inhibitors suffer from stability issues that may limit drug development. An alternative therapeutic approach takes advantage of the naturally broader interaction surface, high affinity and selectivity offered by proteins themselves. Protein‐based MMP inhibitors such as antibodies can offer superior potential for higher selectivity and reduced toxicity, and importantly, antibodies represent a class of drugs for which a path to clinical translation is well established.
Mouse monoclonal antibodies raised against human MMPs have demonstrated the advantageous selectivity of these agents toward individual MMPs. For example, REGA‐3G12, a function blocking antibody raised against human MMP‐9, recognizes a surface epitope of the MMP‐9 catalytic domain with a Kd of 2.1 nM and with high selectivity [Paemen et al., 1995; Martens et al., 2007]. Several studies have explored different approaches to develop function‐blocking antibodies directed against specific functions of MMP‐14, a target of interest in many types of cancers including breast cancer [Pahwa et al., 2014]. By targeting a loop on the MMP‐14 catalytic domain far from the active site but important for interaction with TIMP‐2, a mouse monoclonal antibody inhibited proMMP‐2 activation and lymphangiogenesis, while other catalytic functions were unaffected [Ingvarsen et al., 2013; Shiryaev et al., 2013]. Another mouse monoclonal antibody, raised against a cyclic peptide recapitulating a different surface loop of the MMP‐14 catalytic domain, inhibited collagenolytic activity via an allosteric mechanism, with little impact on MMP‐14 dimerization or proMMP‐2 activation [Gálvez et al., 2001; Udi et al., 2015]. At present it is not yet clear whether superior therapeutic results are likely to be achieved by drugs selectively targeting only a subset of MMP‐14 activities, but these reagents can be used to explore this question. In another unique approach, MMP neutralizing antibodies were generated in mice immunized with a synthetic MMP active site mimic. Surprisingly, the antibodies produced using this method targeted MMP‐2 and ‐9 preferentially, and blocked catalytic activity with a mechanism similar to inhibition by natural TIMPs [Sela‐Passwell et al., 2012].
Novel directed evolution and high‐throughput screening approaches have been applied to engineer human antibodies with high affinity and selectivity toward MMP‐14 for potential therapeutic applications. A phage display approach was used to develop a fully human monoclonal antibody targeting the catalytic domain of MMP‐14; this antibody inhibited tumor growth, metastasis, and angiogenesis in an orthotopic xenograft model of breast cancer [Devy et al., 2009]. In a different study, single chain antibodies selectively targeting the MMP‐14 catalytic domain exclusive of the active site were selected from a naïve library and affinity matured using mutagenesis and phage display. Although not inhibitory of general catalytic activity, the antibodies potently blocked cell surface activities of MMP‐14 including collagenolysis and proMMP‐2 activation, and inhibited tumor growth and metastasis in an orthotopic xenograft model of breast cancer [Botkjaer et al., 2016]. Recently, a systematic approach using next‐generation sequencing was applied for in‐depth analysis of a Fab library enriched for MMP‐14 binding using phage display [Lopez et al., 2017]. Interestingly, highly potent and selective Fabs against MMP‐14 were found using next‐generation sequencing which could not be identified by ELISA. Another recent study generated synthetic Fabs with high selectivity toward MMP‐14 by incorporating a previously identified cyclic MMP‐14 inhibitory peptide (peptide G) [Suojanen et al., 2009] into the complementary determining region CDR‐H3 of a human antibody Fab library [Nam et al., 2017]. A selective binder Fab was isolated using phage panning that possessed more than 1000‐fold enhancement in MMP‐14 inhibition relative to the inhibitory peptide alone [Nam et al., 2017]. Another novel approach toward human Fab library construction incorporated an extended CDR‐H3 length to mimic the convex shape normally found in camelid single chain antibodies, with the rationale that a convex paratope is more amenable to recognition of recessed protease active sites [Nam et al., 2016]. An MMP‐14 function‐blocking antibody selected from this library was shown to inhibit MMP‐14 mediated collagenolysis, cellular invasion, and metastasis in a mouse model of melanoma [Remacle et al., 2017]. Engineering antibodies by directed evolution, using cell display platforms and high‐throughput screening methods to improve affinity, selectivity, and physicochemical characteristics, has developed as a fertile area of research [Boder et al., 2012; Doerner et al., 2014], and these approaches are anticipated to lead to yet greater advances toward selective MMP inhibitors with therapeutic potential.
IMPROVING TIMP SELECTIVITY FOR POWERFUL THERAPEUTIC MMP INHIBITION
An alternative to antibodies as scaffolds for engineering therapeutic protein inhibitors of MMPs is presented by the natural human TIMPs [Nagase and Brew, 2003; Radisky and Radisky, 2015]. The TIMPs have essential anticancer functions, as evidenced by the observation that simultaneous knockout of the four TIMPs conferred powerful cancer‐promoting properties upon fibroblasts [Shimoda et al., 2014]. Preclinical studies suggest that TIMP‐based therapeutics are likely to be well tolerated; for example, systemic gene transfer of each of the TIMPs into mice in a large number of studies has caused no TIMP‐related toxicity [Brand, 2002], and recombinant TIMP‐1 has been used to treat mice at doses up to 50 mg/kg without reported toxicity [Schultz et al., 1988; Alvarez et al., 1990]. Studies of TIMP gene transfer or treatment with recombinant TIMP proteins have in most models shown antitumor responses for TIMP‐1, ‐2, and ‐3, whereas systemic gene transfer of TIMP‐4 increased tumor incidence and growth in an orthotopic breast cancer model [Jiang et al., 2001; Brand, 2002]. However, despite promising early results in multiple animal models, it may be unlikely that the native TIMPs will advance as therapeutics, given that they show very limited discrimination among different MMP targets, but instead have evolved to accommodate “multispecific” recognition across the spectrum of MMPs [Sharabi et al., 2014]. Additionally, the natural TIMPs have been found to possess alternative, MMP‐independent activities [Chirco et al., 2006; Stetler‐Stevenson, 2008; Brew and Nagase, 2010; Jackson et al., 2017] that may be undesirable in MMP‐targeted therapeutics. On the other hand, at about 21 kDa in size and with a compact globular protein fold, the TIMPs, like single chain antibodies, could offer versatile scaffolds for protein engineering of altered binding functionality, so as to achieve exquisite selectivity toward individual MMPs for therapeutic applications.
Intriguingly, structural examination reveals similarities between the protein surfaces used for molecular recognition by antibodies and TIMPs (Fig. 4). The antigen‐binding variable fragment (Fv) of an antibody is comprised of two domains, one from the heavy chain and one from the light chain; in single chain Fvs these two domains are linked in a single protein. Each Fv domain contains three hypervariable complementarity determining regions (CDRs), flexible loops through which the antibody binds to its antigen (Fig. 4A). Similarly, TIMPs are also comprised of two domains which pack side‐by‐side, with each domain contributing multiple flexible loops to the MMP‐binding surface (Fig. 4B). The six contact regions of a TIMP present a broad binding surface, creating a TIMP/MMP interface area nearly double that of typical contact regions between antibodies and protein antigens. This large intermolecular surface area, involving ∼20 TIMP amino acid residues, offers significant opportunity for mutational fine‐tuning to modulate selectivity [Sharabi et al., 2014]. Importantly, high‐throughput screening approaches used for in vitro affinity maturation of therapeutic monoclonal antibodies can also be harnessed for the directed evolution of highly selective TIMPs, as described below.
Figure 4.
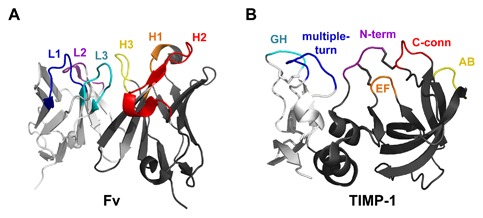
Similarities between antibody Fv and TIMP binding interfaces. (A) An Fv binds to an antigen through three loops of the heavy chain (dark gray): complementarity‐determining regions (CDRs) H1 (orange), H2 (red), and H3 (yellow), and three loops of the light chain (light gray): CDRs L1 (blue), L2 (purple), and L3 (cyan). Coordinates are from PDB ID: 1YQV [Cohen et al., 2005]. (B) TIMPs, like antibodies, recognize the MMP target using a broad interface comprised of multiple loops spread across two domains, including segments of the N‐terminal domain (dark gray): N‐terminus (purple), AB loop (yellow), C‐connector loop (red), and EF loop (orange), and segments of the C‐terminal domain (light gray): GH loop (cyan) and multiple‐turn loop (blue). Coordinates are from PDB ID: 1UEA [Gomis‐Ruth et al., 1997]. Figure was generated using PyMOL (Schrodinger, LLC).
Various studies employing targeted site‐directed mutagenesis, focusing primarily on the isolated N‐terminal TIMP domains, have demonstrated that multiple TIMP residues located in the N‐terminal strand, AB‐loop, C‐connector loop, and EF‐loop are capable of modulating selectivity [Butler et al., 1999b; Meng et al., 1999; Williamson et al., 2001; Lee et al., 2003, 2004; Wei et al., 2003; Hamze et al., 2007]. For example, a combination of three mutations in the TIMP‐1 N‐terminal domain, initially a poor inhibitor of MMP‐14, improved affinity toward MMP‐14 by ∼100‐fold [Lee et al., 2003], resulting in a molecule capable of blocking collagenase activity and shedding of CD44 in cell culture models of breast cancer and fibrosarcoma [Lee et al., 2010]. While the effect of C‐terminal domain mutations of full‐length TIMPs have not yet been explored, it is likely that these too may help to fine‐tune selectivity. However, given the large number of residues involved in the TIMP‐MMP binding interaction, a challenge comes in identifying the best combination of mutations to optimize a TIMP for selectivity toward an individual MMP. The use of structural knowledge to predict the best mutations is made problematic by the high degree of backbone flexibility evidenced in some regions of the TIMP interface [Wu et al., 2000; Grossman et al., 2010; Batra et al., 2013; Batra and Radisky, 2014], and by the complex and unpredictable nature of entropic contributions to MMP‐TIMP binding energies [Arumugam et al., 2003; Wu et al., 2011; Zou et al., 2016]. Fortunately, we are not limited to approaches strictly dependent on the ability to predict advantageous mutations, as directed evolution approaches have now shown substantial promise for development of selective TIMPs.
An early proof‐of‐principle for combinatorial screening as a route to TIMP selectivity was demonstrated by phage display of a TIMP‐2 library, incorporating diversity into the N‐terminal strand, AB‐loop, and C‐connector loop, to evolve variants selective for MMP‐1 in preference to MMP‐3 [Bahudhanapati et al., 2011]. More recent efforts have employed the yeast surface display platform to evolve variants of the N‐terminal domain of TIMP‐2 with high affinity and selectivity toward MMP‐14 [Arkadash et al., 2017]. This recent study combined the strengths of structure‐based design with directed evolution, by using computational predictions of the most promising residues for optimization to guide design of the yeast surface display library. The best variant identified showed 900‐fold improvement in MMP‐14 affinity with an inhibitory constant of 0.9 pM, greatly enhanced selectivity, and improvements in binding to MMP‐14 on the cell surface and inhibition of breast cancer cell invasion [Arkadash et al., 2017]. The encouraging results from these studies point to the utility of integrating structural and computational insights with directed evolution approaches [Rosenfeld et al., 2016], and we anticipate that this could be a general path forward for developing TIMP‐based drugs selectively targeting the different MMPs most strongly implicated as therapeutic targets in breast cancer. Keeping in mind the MMP‐independent activities of the natural TIMPs [Chirco et al., 2006; Stetler‐Stevenson, 2008; Brew and Nagase, 2010], an additional important aspect of developing TIMP‐based drugs will be to better define the TIMP epitopes responsible for some of these activities, and to remove unwanted off‐target activities through protein engineering. Directed evolution strategies incorporating negative selection may prove to be useful for these efforts as well. A final challenge, given the short serum half‐life of unmodified recombinant TIMPs in vivo [Batra et al., 2012b], will be to develop formulations and delivery methods capable of achieving sustained therapeutic concentrations. Promising approaches reported to date include PEGylation [Batra et al., 2012b; Chen et al., 2013b], fusion to serum albumin [Kang et al., 2007; Lee et al., 2011], nanoparticle delivery [Chaturvedi et al., 2012, 2014], and encapsulation in peptide hydrogels [Chowdhury et al., 2017] to enhance recombinant TIMP availability, stability and efficacy in vivo.
PERSPECTIVES
The investigation of MMPs as cancer mediators and therapeutics has now spanned multiple decades, with periods of collective advances and intervening setbacks. The considerable enthusiasm following studies that implicated MMPs in so many different cancer‐associated processes prompted a series of inhibitor‐based clinical trials that, in hindsight, did not account for the incredible complexity of this protein family and its roles in diverse physiological processes. Now, with the development of a much more sophisticated understanding of how individual MMPs act in different cancers and at distinct stages of cancer progression, in combination with newly developed methods for discovery and refinement of highly selective inhibitors, we are poised for a renaissance of therapeutically valuable MMP inhibition in breast cancer. These approaches will prove especially relevant for triple negative/basal subtype cancers, in which specific MMPs have been shown to play critical roles, and for which targeted therapeutics have been slow to develop.
ACKNOWLEDGMENTS
This work was supported by NCI grants (R01CA154387 and R21CA205471 to ESR) and (R01CA187692 to DCR), the Bankhead‐Coley Foundation (5BC02 to DCR), and the Mayo Clinic SPORE in Breast Cancer grant P50CA116201 (PI James Ingle).
Conflicts of interest: The authors have no conflicts of interest to disclose.
REFERENCES
- Alvarez OA, Carmichael DF, DeClerck YA. 1990. Inhibition of collagenolytic activity and metastasis of tumor cells by a recombinant human tissue inhibitor of metalloproteinases. J Natl Cancer Inst 82(7):589–595. [DOI] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. 2003. Making sense of latent TGFbeta activation. J Cell Sci 116(Pt 2):217–224. [DOI] [PubMed] [Google Scholar]
- Amour A, Knight CG, English WR, Webster A, Slocombe PM, Knauper V, Docherty AJ, Becherer JD, Blobel CP, Murphy G. 2002. The enzymatic activity of ADAM8 and ADAM9 is not regulated by TIMPs. FEBS Lett 524(1‐3):154–158. [DOI] [PubMed] [Google Scholar]
- Ardi VC, Kupriyanova TA, Deryugina EI, Quigley JP. 2007. Human neutrophils uniquely release TIMP‐free MMP‐9 to provide a potent catalytic stimulator of angiogenesis. Proc Natl Acad Sci USA 104(51):20262–20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardi VC, Van den Steen PE, Opdenakker G, Schweighofer B, Deryugina EI, Quigley JP. 2009. Neutrophil MMP‐9 proenzyme, unencumbered by TIMP‐1, undergoes efficient activation in vivo and catalytically induces angiogenesis via a basic fibroblast growth factor (FGF‐2)/FGFR‐2 pathway. J Biol Chem 284(38):25854–25866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkadash V, Yosef G, Shirian J, Cohen I, Horev Y, Grossman M, Sagi I, Radisky ES, Shifman JM, Papo N. 2017. Development of high‐affinity and high‐specificity inhibitors of metalloproteinase 14 through computational design and directed evolution. J Biol Chem 292(8):3481–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold LH, Butt LE, Prior SH, Read CM, Fields GB, Pickford AR. 2011. The interface between catalytic and hemopexin domains in matrix metalloproteinase‐1 conceals a collagen binding exosite. J Biol Chem 286(52):45073–45082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam S, Gao G, Patton BL, Semenchenko V, Brew K, Van Doren SR. 2003. Increased backbone mobility in beta‐barrel enhances entropy gain driving binding of N‐TIMP‐1 to MMP‐3. J Mol Biol 327(3):719–734. [DOI] [PubMed] [Google Scholar]
- Bahudhanapati H, Zhang Y, Sidhu SS, Brew K. 2011. Phage display of tissue inhibitor of metalloproteinases‐2 (TIMP‐2): Identification of selective inhibitors of collagenase‐1 (metalloproteinase 1 (MMP‐1)). J Biol Chem 286(36):31761–31770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra J, Robinson J, Soares AS, Fields AP, Radisky DC, Radisky ES. 2012a. Matrix metalloproteinase‐10 (MMP‐10) interaction with tissue inhibitors of metalloproteinases TIMP‐1 and TIMP‐2: Binding studies and crystal structure. J Biol Chem 287(19):15935–15946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra J, Robinson J, Mehner C, Hockla A, Miller E, Radisky DC, Radisky ES. 2012b. PEGylation extends circulation half‐life while preserving in vitro and in vivo activity of tissue inhibitor of metalloproteinases‐1 (TIMP‐1). PLoS ONE 7(11):e50028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra J, Soares AS, Mehner C, Radisky ES. 2013. Matrix metalloproteinase‐10/TIMP‐2 structure and analyses define conserved core interactions and diverse exosite interactions in MMP/TIMP complexes. PLoS ONE 8(9):e75836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra J, Radisky ES. 2014. Tissue inhibitors of metalloproteinases (TIMPs): inhibition of Zn‐dependent metallopeptidases In: Scott RA, editor. Encyclopedia of inorganic and bioinorganic chemistry. Chichester: John Wiley & Sons. [Google Scholar]
- Bauvois B. 2012. New facets of matrix metalloproteinases MMP‐2 and MMP‐9 as cell surface transducers: Outside‐in signaling and relationship to tumor progression. Biochim Biophys Acta 1825(1):29–36. [DOI] [PubMed] [Google Scholar]
- Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, Deryugina EI. 2011. Tumor‐recruited neutrophils and neutrophil TIMP‐free MMP‐9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol 179(3):1455–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendrik C, Robertson J, Gauldie J, Dabrosin C. 2008. Gene transfer of matrix metalloproteinase‐9 induces tumor regression of breast cancer in vivo. Cancer Res 68(9):3405–3412. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Tanzawa K, Thorpe P, Itohara S, Werb Z, Hanahan D. 2000. Matrix metalloproteinase‐9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2(10):737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I, Calderone V, Fragai M, Jaiswal R, Luchinat C, Melikian M, Mylonas E, Svergun DI. 2008. Evidence of reciprocal reorientation of the catalytic and hemopexin‐like domains of full‐length MMP‐12. J Am Chem Soc 130(22):7011–7021. [DOI] [PubMed] [Google Scholar]
- Bertini I, Fragai M, Luchinat C. 2009. Intra‐ and interdomain flexibility in matrix metalloproteinases: Functional aspects and drug design. Curr Pharm Des 15(31):3592–3605. [DOI] [PubMed] [Google Scholar]
- Bertini I, Fragai M, Luchinat C, Melikian M, Toccafondi M, Lauer JL, Fields GB. 2011. Structural basis for matrix metalloproteinase 1‐catalyzed collagenolysis. J Am Chem Soc 134(4):2100–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhowmick M, Stawikowska R, Tokmina‐Roszyk D, Fields GB. 2015. Matrix metalloproteinase inhibition by heterotrimeric triple‐helical peptide transition state analogues. Chembiochem 16(7):1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigg HF, Morrison CJ, Butler GS, Bogoyevitch MA, Wang Z, Soloway PD, Overall CM. 2001. Tissue inhibitor of metalloproteinases‐4 inhibits but does not support the activation of gelatinase A via efficient inhibition of membrane type 1‐matrix metalloproteinase. Cancer Res 61(9):3610–3618. [PubMed] [Google Scholar]
- Boder ET, Raeeszadeh‐Sarmazdeh M, Price JV. 2012. Engineering antibodies by yeast display. Arch Biochem Biophys 526(2):99–106. [DOI] [PubMed] [Google Scholar]
- Botkjaer KA, Kwok HF, Terp MG, Karatt‐Vellatt A, Santamaria S, McCafferty J, Andreasen PA, Itoh Y, Ditzel HJ, Murphy G. 2016. Development of a specific affinity‐matured exosite inhibitor to MT1‐MMP that efficiently inhibits tumor cell invasion in vitro and metastasis in vivo. Oncotarget 7(13):16773–16792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay A, Masson R, Chenard MP, El Fahime M, Cassard L, Bellocq JP, Sautes‐Fridman C, Basset P, Rio MC. 2001. High cancer cell death in syngeneic tumors developed in host mice deficient for the stromelysin‐3 matrix metalloproteinase. Cancer Res 61(5):2189–2193. [PubMed] [Google Scholar]
- Brand K. 2002. Cancer gene therapy with tissue inhibitors of metalloproteinases (TIMPs). Curr Gene Ther 2(2):255–271. [DOI] [PubMed] [Google Scholar]
- Brew K, Nagase H. 2010. The tissue inhibitors of metalloproteinases (TIMPs): An ancient family with structural and functional diversity. Biochim Biophys Acta 1803(1):55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler GS, Apte SS, Willenbrock F, Murphy G. 1999a. Human tissue inhibitor of metalloproteinases 3 interacts with both the N‐ and C‐terminal domains of gelatinases A and B. Regulation by polyanions. J Biol Chem 274(16):10846–10851. [DOI] [PubMed] [Google Scholar]
- Butler GS, Hutton M, Wattam BA, Williamson RA, Knauper V, Willenbrock F, Murphy G. 1999b. The specificity of TIMP‐2 for matrix metalloproteinases can be modified by single amino acid mutations. J Biol Chem 274(29):20391–20396. [DOI] [PubMed] [Google Scholar]
- Cao J, Chiarelli C, Kozarekar P, Adler HL. 2005. Membrane type 1‐matrix metalloproteinase promotes human prostate cancer invasion and metastasis. Thromb Haemost 93(4):770–778. [DOI] [PubMed] [Google Scholar]
- Cao J, Chiarelli C, Richman O, Zarrabi K, Kozarekar P, Zucker S. 2008. Membrane type 1 matrix metalloproteinase induces epithelial‐to‐mesenchymal transition in prostate cancer. J Biol Chem 283(10):6232–6240. [DOI] [PubMed] [Google Scholar]
- Cathcart J, Pulkoski‐Gross A, Cao J. 2015. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis 2(1):26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerda‐Costa N, Gomis‐Ruth FX. 2014. Architecture and function of metallopeptidase catalytic domains. Protein Sci 23(2):123–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaturvedi M, Figiel I, Sreedhar B, Kaczmarek L. 2012. Neuroprotection from tissue inhibitor of metalloproteinase‐1 and its nanoparticles. Neurochem Int 61(7):1065–1071. [DOI] [PubMed] [Google Scholar]
- Chaturvedi M, Molino Y, Sreedhar B, Khrestchatisky M, Kaczmarek L. 2014. Tissue inhibitor of matrix metalloproteinases‐1 loaded poly (lactic‐co‐glycolic acid) nanoparticles for delivery across the blood–brain barrier. Int J Nanomed 9:575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Lovett DH. 2003. Gelatinase A (MMP‐2) is necessary and sufficient for renal tubular cell epithelial‐mesenchymal transformation. Am J Pathol 162(6):1937–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Pollock AS, Mahimkar R, Olson JL, Lovett DH. 2006. Matrix metalloproteinase 2 and basement membrane integrity: A unifying mechanism for progressive renal injury. FASEB J 20(11):1898–1900. [DOI] [PubMed] [Google Scholar]
- Cheng CW, Yu JC, Wang HW, Huang CS, Shieh JC, Fu YP, Chang CW, Wu PE, Shen CY. 2010. The clinical implications of MMP‐11 and CK‐20 expression in human breast cancer. Clin Chim Acta 411(3–4):234–241. [DOI] [PubMed] [Google Scholar]
- Chen QK, Lee K, Radisky DC, Nelson CM. 2013a. Extracellular matrix proteins regulate epithelial‐mesenchymal transition in mammary epithelial cells. Differentiation 86(3):126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Radisky ES, Das P, Batra J, Hata T, Hori T, Baine AM, Gardner L, Yue MY, Bu G, del Zoppo G, Patel TC, Nguyen JH. 2013b. TIMP‐1 attenuates blood‐brain barrier permeability in mice with acute liver failure. J Cereb Blood Flow Metab 33(7):1041–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirco R, Liu XW, Jung KK, Kim HR. 2006. Novel functions of TIMPs in cell signaling. Cancer Metastasis Rev 25(1):99–113. [DOI] [PubMed] [Google Scholar]
- Chowdhury A, Wei B, Yamada Y, Castro N, Salomon D, Schneider JP, Stetler‐Stevenson WG. 2017. Recombinant human Tissue Inhibitor of Metalloprotease‐2; a novel anti‐cancer therapeutic. FASEB J 31(1 Supplement):823.810. [Google Scholar]
- Cichon MA, Radisky DC. 2014a. Extracellular matrix as a contextual determinant of transforming growth factor‐beta signaling in epithelial‐mesenchymal transition and in cancer. Cell Adh Migr 8(6):588–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon MA, Radisky DC. 2014b. ROS‐induced epithelial‐mesenchymal transition in mammary epithelial cells is mediated by NF‐kB‐dependent activation of Snail. Oncotarget 5(9):2827–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichon MA, Nelson CM, Radisky DC. 2015. Regulation of epithelial‐mesenchymal transition in breast cancer cells by cell contact and adhesion. Cancer Inform 14(Suppl 3):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid S, Eiro N, Gonzalez LO, Beridze N, Vazquez J, Vizoso FJ. 2016. Expression and clinical significance of metalloproteases and their inhibitors by endothelial cells from invasive breast carcinomas. Clin Breast Cancer 16(4):e83–e91. [DOI] [PubMed] [Google Scholar]
- Cohen GH, Silverton EW, Padlan EA, Dyda F, Wibbenmeyer JA, Willson RC, Davies DR. 2005. Water molecules in the antibody‐antigen interface of the structure of the Fab HyHEL‐5‐lysozyme complex at 1.7 A resolution: Comparison with results from isothermal titration calorimetry. Acta Crystallogr D 61(5):628–633. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B, Matrisian LM. 2002. Matrix metalloproteinase inhibitors and cancer: Trials and tribulations. Science 295(5564):2387–2392. [DOI] [PubMed] [Google Scholar]
- Cowden Dahl KD, Symowicz J, Ning Y, Gutierrez E, Fishman DA, Adley BP, Stack MS, Hudson LG. 2008. Matrix metalloproteinase 9 is a mediator of epidermal growth factor‐dependent e‐cadherin loss in ovarian carcinoma cells. Cancer Res 68(12):4606–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford HC, Scoggins CR, Washington MK, Matrisian LM, Leach SD. 2002. Matrix metalloproteinase‐7 is expressed by pancreatic cancer precursors and regulates acinar‐to‐ductal metaplasia in exocrine pancreas. J Clin Invest 109(11):1437–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Graf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, Langerod A, Green A, Provenzano E, Wishart G, Pinder S, Watson P, Markowetz F, Murphy L, Ellis I, Purushotham A, Borresen‐Dale AL, Brenton JD, Tavare S, Caldas C, Aparicio S. 2012. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486(7403):346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas SL, Rosser JL, Mundy GR, Bonewald LF. 2002. Proteolysis of latent transforming growth factor‐beta (TGF‐beta)‐binding protein‐1 by osteoclasts. A cellular mechanism for release of TGF‐beta from bone matrix. J Biol Chem 277(24):21352–21360. [DOI] [PubMed] [Google Scholar]
- Decock J, Long JR, Laxton RC, Shu XO, Hodgkinson C, Hendrickx W, Pearce EG, Gao YT, Pereira AC, Paridaens R, Zheng W, Ye S. 2007. Association of matrix metalloproteinase‐8 gene variation with breast cancer prognosis. Cancer Res 67(21):10214–10221. [DOI] [PubMed] [Google Scholar]
- Decock J, Thirkettle S, Wagstaff L, Edwards DR. 2011. Matrix metalloproteinases: Protective roles in cancer. J Cell Mol Med 15(6):1254–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decock J, Hendrickx W, Thirkettle S, Gutiérrez‐Fernández A, Robinson SD, Edwards DR. 2015. Pleiotropic functions of the tumor‐ and metastasis‐suppressing matrix metalloproteinase‐8 in mammary cancer in MMTV‐PyMT transgenic mice. Breast Cancer Res 17(1):38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, Quigley JP. 2010. Pleiotropic roles of matrix metalloproteinases in tumor angiogenesis: Contrasting, overlapping and compensatory functions. Biochim Biophys Acta 1803(1):103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, Zajac E, Juncker‐Jensen A, Kupriyanova TA, Welter L, Quigley JP. 2014. Tissue‐infiltrating neutrophils constitute the major in vivo source of angiogenesis‐inducing MMP‐9 in the tumor microenvironment. Neoplasia 16(10):771–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devel L, Rogakos V, David A, Makaritis A, Beau F, Cuniasse P, Yiotakis A, Dive V. 2006. Development of selective inhibitors and substrate of matrix metalloproteinase‐12. J Biol Chem 281(16):11152–11160. [DOI] [PubMed] [Google Scholar]
- Devel L, Garcia S, Czarny B, Beau F, LaJeunesse E, Vera L, Georgiadis D, Stura E, Dive V. 2010. Insights from selective non‐phosphinic inhibitors of MMP‐12 tailored to fit with an S1′ loop canonical conformation. J Biol Chem 285(46):35900–35909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devel L, Beau F, Amoura M, Vera L, Cassar‐Lajeunesse E, Garcia S, Czarny B, Stura EA, Dive V. 2012. Simple pseudo‐dipeptides with a P2′ glutamate: A novel inhibitor family of matrix metalloproteases and other metzincins. J Biol Chem 287(32):26647–26656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devy L, Huang L, Naa L, Yanamandra N, Pieters H, Frans N, Chang E, Tao Q, Vanhove M, Lejeune A, van Gool R, Sexton DJ, Kuang G, Rank D, Hogan S, Pazmany C, Ma YL, Schoonbroodt S, Nixon AE, Ladner RC, Hoet R, Henderikx P, Tenhoor C, Rabbani SA, Valentino ML, Wood CR, Dransfield DT. 2009. Selective inhibition of matrix metalloproteinase‐14 blocks tumor growth, invasion, and angiogenesis. Cancer Res 69(4):1517–1526. [DOI] [PubMed] [Google Scholar]
- Doerner A, Rhiel L, Zielonka S, Kolmar H. 2014. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett 588(2):278–287. [DOI] [PubMed] [Google Scholar]
- Dufour A, Zucker S, Sampson NS, Kuscu C, Cao J. 2010. Role of matrix metalloproteinase‐9 dimers in cell migration: Design of inhibitory peptides. J Biol Chem 285(46):35944–35956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour A, Sampson NS, Li J, Kuscu C, Rizzo RC, Deleon JL, Zhi J, Jaber N, Liu E, Zucker S, Cao J. 2011. Small‐molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase‐9. Cancer Res 71(14):4977–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiro N, Gonzalez L, Gonzalez LO, Fernandez‐Garcia B, Lamelas ML, Marin L, Gonzalez‐Reyes S, del Casar JM, Vizoso FJ. 2012. Relationship between the inflammatory molecular profile of breast carcinomas and distant metastasis development. PLoS ONE 7(11):e49047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel CK, Pirard B, Schimanski S, Kirsch R, Habermann J, Klingler O, Schlotte V, Weithmann KU, Wendt KU. 2005. Structural basis for the highly selective inhibition of MMP‐13. Chem Biol 12(2):181–189. [DOI] [PubMed] [Google Scholar]
- English JL, Kassiri Z, Koskivirta I, Atkinson SJ, Di Grappa M, Soloway PD, Nagase H, Vuorio E, Murphy G, Khokha R. 2006. Individual Timp deficiencies differentially impact pro‐MMP‐2 activation. J Biol Chem 281(15):10337–10346. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H, Maskos K. 1998. Crystal structure of the complex formed by the membrane type 1‐matrix metalloproteinase with the tissue inhibitor of metalloproteinases‐2, the soluble progelatinase A receptor. EMBO J 17(17):5238–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingleton B. 2008. MMPs as therapeutic targets‐still a viable option? Semin Cell Dev Biol 19(1):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foroni C, Broggini M, Generali D, Damia G. 2012. Epithelial‐mesenchymal transition and breast cancer: Role, molecular mechanisms and clinical impact. Cancer Treat Rev 38(6):689–697. [DOI] [PubMed] [Google Scholar]
- Galea CA, Nguyen HM, George Chandy K, Smith BJ, Norton RS. 2014. Domain structure and function of matrix metalloprotease 23 (MMP23): Role in potassium channel trafficking. Cell Mol Life Sci 71(7):1191–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gálvez BG, Matías‐Román S, Albar JP, Sánchez‐Madrid F, Arroyo AG. 2001. Membrane type 1‐matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J Biol Chem 276(40):37491–37500. [DOI] [PubMed] [Google Scholar]
- Gialeli C, Theocharis AD, Karamanos NK. 2011. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 278(1):16–27. [DOI] [PubMed] [Google Scholar]
- Goldberg GI, Strongin A, Collier IE, Genrich LT, Marmer BL. 1992. Interaction of 92‐kDa type IV collagenase with the tissue inhibitor of metalloproteinases prevents dimerization, complex formation with interstitial collagenase, and activation of the proenzyme with stromelysin. J Biol Chem 267(7):4583–4591. [PubMed] [Google Scholar]
- Golubkov VS, Chekanov AV, Cieplak P, Aleshin AE, Chernov AV, Zhu W, Radichev IA, Zhang D, Dong PD, Strongin AY. 2010. The Wnt/planar cell polarity protein‐tyrosine kinase‐7 (PTK7) is a highly efficient proteolytic target of membrane type‐1 matrix metalloproteinase: Implications in cancer and embryogenesis. J Biol Chem 285(46):35740–35749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubkov VS, Strongin AY. 2012. Insights into ectodomain shedding and processing of protein‐tyrosine pseudokinase 7 (PTK7). J Biol Chem 287(50):42009–42018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubkov VS, Prigozhina NL, Zhang Y, Stoletov K, Lewis JD, Schwartz PE, Hoffman RM, Strongin AY. 2014. Protein‐tyrosine pseudokinase 7 (PTK7) directs cancer cell motility and metastasis. J Biol Chem 289(35):24238–24249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis‐Ruth FX, Maskos K, Betz M, Bergner A, Huber R, Suzuki K, Yoshida N, Nagase H, Brew K, Bourenkov GP, Bartunik H, Bode W. 1997. Mechanism of inhibition of the human matrix metalloproteinase stromelysin‐1 by TIMP‐1. Nature 389(6646):77–81. [DOI] [PubMed] [Google Scholar]
- Gonzalez LO, Pidal I, Junquera S, Corte MD, Vazquez J, Rodriguez JC, Lamelas ML, Merino AM, Garcia‐Muniz JL, Vizoso FJ. 2007. Overexpression of matrix metalloproteinases and their inhibitors in mononuclear inflammatory cells in breast cancer correlates with metastasis‐relapse. Br J Cancer 97(7):957–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez LO, Corte MD, Junquera S, Gonzalez‐Fernandez R, del Casar JM, Garcia C, Andicoechea A, Vazquez J, Perez‐Fernandez R, Vizoso FJ. 2009. Expression and prognostic significance of metalloproteases and their inhibitors in luminal A and basal‐like phenotypes of breast carcinoma. Hum Pathol 40(9):1224–1233. [DOI] [PubMed] [Google Scholar]
- Grossman M, Tworowski D, Dym O, Lee MH, Levy Y, Murphy G, Sagi I. 2010. The intrinsic protein flexibility of endogenous protease inhibitor TIMP‐1 controls its binding interface and affects its function. Biochemistry 49(29):6184–6192. [DOI] [PubMed] [Google Scholar]
- Gutiérrez‐Fernández A, Fueyo A, Folgueras AR, Garabaya C, Pennington CJ, Pilgrim S, Edwards DR, Holliday DL, Jones JL, Span PN, Sweep FCGJ, Puente XS, López‐Otín C. 2008. Matrix metalloproteinase‐8 functions as a metastasis suppressor through modulation of tumor cell adhesion and invasion. Cancer Res 68(8):2755–2763. [DOI] [PubMed] [Google Scholar]
- Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, Hynes RO, Werb Z, Sudhakar A, Kalluri R. 2003. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP‐9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell 3(6):589–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamze AB, Wei S, Bahudhanapati H, Kota S, Acharya KR, Brew K. 2007. Constraining specificity in the N‐domain of tissue inhibitor of metalloproteinases‐1; gelatinase‐selective inhibitors. Protein Sci 16(9):1905–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Barrantes S, Shimura Y, Soloway PD, Sang QA, Fridman R. 2001. Differential roles of TIMP‐4 and TIMP‐2 in pro‐MMP‐2 activation by MT1‐MMP. Biochem Biophys Res Commun 281(1):126–130. [DOI] [PubMed] [Google Scholar]
- Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. 2003. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the three‐dimensional extracellular matrix. Cell 114(1):33–45. [DOI] [PubMed] [Google Scholar]
- Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ. 2006. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev 20(19):2673–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Suzuki K, Nagase H, Arumugam S, Van Doren SR, Brew K. 1996. Folding and characterization of the amino‐terminal domain of human tissue inhibitor of metalloproteinases‐1 (TIMP‐1) expressed at high yield in E. coli . FEBS Lett 384(2):155–161. [DOI] [PubMed] [Google Scholar]
- Hugenberg V, Wagner S, Kopka K, Schafers M, Schuit RC, Windhorst AD, Hermann S. 2017. Radiolabeled selective matrix metalloproteinase 13 (MMP‐13) inhibitors: (Radio)syntheses and in vitro and first in vivo evaluation. J Med Chem 60(1):307–321. [DOI] [PubMed] [Google Scholar]
- Hutton M, Willenbrock F, Brocklehurst K, Murphy G. 1998. Kinetic analysis of the mechanism of interaction of full‐length TIMP‐2 and gelatinase A: Evidence for the existence of a low‐affinity intermediate. Biochemistry 37(28):10094–10098. [DOI] [PubMed] [Google Scholar]
- Illman SA, Lehti K, Keski‐Oja J, Lohi J. 2006. Epilysin (MMP‐28) induces TGF‐beta mediated epithelial to mesenchymal transition in lung carcinoma cells. J Cell Sci 119(Pt 18):3856–3865. [DOI] [PubMed] [Google Scholar]
- Ingvarsen S, Porse A, Erpicum C, Maertens L, Jurgensen HJ, Madsen DH, Melander MC, Gardsvoll H, Hoyer‐Hansen G, Noel A, Holmbeck K, Engelholm LH, Behrendt N. 2013. Targeting a single function of the multifunctional matrix metalloprotease MT1‐MMP: Impact on lymphangiogenesis. J Biol Chem 288(15):10195–10204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson HW, Defamie V, Waterhouse P, Khokha R. 2017. TIMPs: Versatile extracellular regulators in cancer. Nat Rev Cancer 17(1):38–53. [DOI] [PubMed] [Google Scholar]
- Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. 2010. To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition. Biochim Biophys Acta 1803(1):72–94. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Wang M, Celiker MY, Liu YE, Sang QX, Goldberg ID, Shi YE. 2001. Stimulation of mammary tumorigenesis by systemic tissue inhibitor of matrix metalloproteinase 4 gene delivery. Cancer Res 61(6):2365–2370. [PubMed] [Google Scholar]
- Kang WK, Park EK, Lee HS, Park BY, Chang JY, Kim MY, Kang HA, Kim JY. 2007. A biologically active angiogenesis inhibitor, human serum albumin–TIMP‐2 fusion protein, secreted from Saccharomyces cerevisiae . Protein Expr Purif 53(2):331–338. [DOI] [PubMed] [Google Scholar]
- Kessenbrock K, Plaks V, Werb Z. 2010. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 141(1):52–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Dijkgraaf GJ, Lawson DA, Littlepage LE, Shahi P, Pieper U, Werb Z. 2013. A role for matrix metalloproteinases in regulating mammary stem cell function via the Wnt signaling pathway. Cell Stem Cell 13(3):300–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K, Wang CY, Werb Z. 2015. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol 44–46:184–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppisetti RK, Fulcher YG, Jurkevich A, Prior SH, Xu J, Lenoir M, Overduin M, Van Doren SR. 2014. Ambidextrous binding of cell and membrane bilayers by soluble matrix metalloproteinase‐12. Nat Commun 5:5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V. 2000. Role of cell surface metalloprotease MT1‐MMP in epithelial cell migration over laminin‐5. J Cell Biol 148(3):615–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. 2014. Molecular mechanisms of epithelial‐mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer‐Fields J, Brew K, Whitehead JK, Li S, Hammer RP, Fields GB. 2007. Triple‐helical transition state analogues: A new class of selective matrix metalloproteinase inhibitors. J Am Chem Soc 129(34):10408–10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer‐Fields JL, Whitehead JK, Li S, Hammer RP, Brew K, Fields GB. 2008. Selective modulation of matrix metalloproteinase 9 (MMP‐9) functions via exosite inhibition. J Biol Chem 283(29):20087–20095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MH, Knäuper V, Becherer JD, Murphy G. 2001. Full‐length and N‐TIMP‐3 display equal inhibitory activities toward TNF‐alpha convertase. Biochem Biophys Res Commun 280(3):945–950. [DOI] [PubMed] [Google Scholar]
- Lee MH, Rapti M, Murphy G. 2003. Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase‐1 inactive against membrane type 1‐matrix metalloproteinase. J Biol Chem 278(41):40224–40230. [DOI] [PubMed] [Google Scholar]
- Lee MH, Rapti M, Knauper V, Murphy G. 2004. Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)‐1 in metalloproteinase recognition. J Biol Chem 279(17):17562–17569. [DOI] [PubMed] [Google Scholar]
- Lee MH, Atkinson S, Rapti M, Handsley M, Curry V, Edwards D, Murphy G. 2010. The activity of a designer tissue inhibitor of metalloproteinases (TIMP)‐1 against native membrane type 1 matrix metalloproteinase (MT1‐MMP) in a cell‐based environment. Cancer Lett 290(1):114–122. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kim YH, Kim YJ, Kwon SH, Bang JK, Lee SM, Song YS, Hahm DH, Shim I, Han D, Her S. 2011. Pharmacokinetics and biodistribution of human serum albumin‐TIMP‐2 fusion protein using near‐infrared optical imaging. J Pharm Pharm Sci 14(3):368–377. [DOI] [PubMed] [Google Scholar]
- Lee K, Chen QK, Lui C, Cichon MA, Radisky DC, Nelson CM. 2012. Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, and epithelial‐mesenchymal transition. Mol Biol Cell 23(20):4097–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre O, Wolf C, Limacher JM, Hutin P, Wendling C, LeMeur M, Basset P, Rio MC. 1992. The breast cancer‐associated stromelysin‐3 gene is expressed during mouse mammary gland apoptosis. J Cell Biol 119(4):997–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HC, Cao DC, Liu Y, Hou YF, Wu J, Lu JS, Di GH, Liu G, Li FM, Ou ZL, Jie C, Shen ZZ, Shao ZM. 2004. Prognostic value of matrix metalloproteinases (MMP‐2 and MMP‐9) in patients with lymph node‐negative breast carcinoma. Breast Cancer Res Treat 88(1):75–85. [DOI] [PubMed] [Google Scholar]
- Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. 1997a. Matrix metalloproteinase stromelysin‐1 triggers a cascade of molecular alterations that leads to stable epithelial‐to‐mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol 139(7):1861–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lochter A, Srebrow A, Sympson CJ, Terracio N, Werb Z, Bissell MJ. 1997b. Misregulation of stromelysin‐1 expression in mouse mammary tumor cells accompanies acquisition of stromelysin‐1‐dependent invasive properties. J Biol Chem 272(8):5007–5015. [DOI] [PubMed] [Google Scholar]
- Lodillinsky C, Infante E, Guichard A, Chaligne R, Fuhrmann L, Cyrta J, Irondelle M, Lagoutte E, Vacher S, Bonsang‐Kitzis H, Glukhova M, Reyal F, Bieche I, Vincent‐Salomon A, Chavrier P. 2016. P63/MT1‐MMP axis is required for in situ to invasive transition in basal‐like breast cancer. Oncogene 35(3):344–357. [DOI] [PubMed] [Google Scholar]
- Lopez‐Otin C, Matrisian LM. 2007. Emerging roles of proteases in tumour suppression. Nat Rev Cancer 7(10):800–808. [DOI] [PubMed] [Google Scholar]
- Lopez T, Nam DH, Kaihara E, Mustafa Z, Ge X. 2017. Identification of highly selective MMP‐14 inhibitory Fabs by deep sequencing. Biotechnol Bioeng 114(6):1140–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manka SW, Carafoli F, Visse R, Bihan D, Raynal N, Farndale RW, Murphy G, Enghild JJ, Hohenester E, Nagase H. 2012. Structural insights into triple‐helical collagen cleavage by matrix metalloproteinase 1. Proc Natl Acad Sci USA 109(31):12461–12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquoi E, Sounni NE, Devy L, Olivier F, Frankenne F, Krell HW, Grams F, Foidart JM, Noel A. 2004. Anti‐invasive, antitumoral, and antiangiogenic efficacy of a pyrimidine‐2,4,6‐trione derivative, an orally active and selective matrix metalloproteinases inhibitor. Clin Cancer Res 10(12 Pt 1):4038–4047. [DOI] [PubMed] [Google Scholar]
- Marchenko ND, Marchenko GN, Strongin AY. 2002. Unconventional activation mechanisms of MMP‐26, a human matrix metalloproteinase with a unique PHCGXXD cysteine‐switch motif. J Biol Chem 277(21):18967–18972. [DOI] [PubMed] [Google Scholar]
- Martens E, Leyssen A, Van Aelst I, Fiten P, Piccard H, Hu J, Descamps FJ, Van den Steen PE, Proost P, Van Damme J, Liuzzi GM, Riccio P, Polverini E, Opdenakker G. 2007. A monoclonal antibody inhibits gelatinase B/MMP‐9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim Biophys Acta 1770(2):178–186. [DOI] [PubMed] [Google Scholar]
- Martin MD, Matrisian LM. 2007. The other side of MMPs: Protective roles in tumor progression. Cancer Metastasis Rev 26(3–4):717–724. [DOI] [PubMed] [Google Scholar]
- Martin MD, Carter KJ, Jean‐Philippe SR, Chang M, Mobashery S, Thiolloy S, Lynch CC, Matrisian LM, Fingleton B. 2008. Effect of ablation or inhibition of stromal matrix metalloproteinase‐9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res 68(15):6251–6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskos K, Lang R, Tschesche H, Bode W. 2007. Flexibility and variability of TIMP binding: X‐ray structure of the complex between collagenase‐3/MMP‐13 and TIMP‐2. J Mol Biol 366(4):1222–1231. [DOI] [PubMed] [Google Scholar]
- Massague J. 2012. TGFbeta signalling in context. Nat Rev Mol Cell Biol 13(10):616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masson R, Lefebvre O, Noel A, Fahime ME, Chenard MP, Wendling C, Kebers F, LeMeur M, Dierich A, Foidart JM, Basset P, Rio MC. 1998. In vivo evidence that the stromelysin‐3 metalloproteinase contributes in a paracrine manner to epithelial cell malignancy. J Cell Biol 140(6):1535–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan PM, Duffy MJ. 2008. Matrix metalloproteinase expression and outcome in patients with breast cancer: Analysis of a published database. Ann Oncol 19(9):1566–1572. [DOI] [PubMed] [Google Scholar]
- Mehner C, Miller E, Khauv D, Nassar A, Oberg AL, Bamlet WR, Zhang L, Waldmann J, Radisky ES, Crawford HC, Radisky DC. 2014a. Tumor cell‐derived MMP‐3 orchestrates rac1b and tissue alterations that promote pancreatic adenocarcinoma. Mol Cancer Res 12(10):1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehner C, Hockla A, Miller E, Ran S, Radisky DC, Radisky ES. 2014b. Tumor cell‐produced matrix metalloproteinase 9 (MMP‐9) drives malignant progression and metastasis of basal‐like triple negative breast cancer. Oncotarget 5(9):2736–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Q, Malinovskii V, Huang W, Hu Y, Chung L, Nagase H, Bode W, Maskos K, Brew K. 1999. Residue 2 of TIMP‐1 is a major determinant of affinity and specificity for matrix metalloproteinases but effects of substitutions do not correlate with those of the corresponding P1' residue of substrate. J Biol Chem 274(15):10184–10189. [DOI] [PubMed] [Google Scholar]
- Mikhailova M, Xu X, Robichaud TK, Pal S, Fields GB, Steffensen B. 2012. Identification of collagen binding domain residues that govern catalytic activities of matrix metalloproteinase‐2 (MMP‐2). Matrix Biol 31(7–8):380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KD, Gradishar W, Schuchter L, Sparano JA, Cobleigh M, Robert N, Rasmussen H, Sledge GW. 2002. A randomized phase II pilot trial of adjuvant marimastat in patients with early‐stage breast cancer. Ann Oncol 13(8):1220–1224. [DOI] [PubMed] [Google Scholar]
- Miller KD, Saphner TJ, Waterhouse DM, Chen TT, Rush‐Taylor A, Sparano JA, Wolff AC, Cobleigh MA, Galbraith S, Sledge GW. 2004. A randomized phase II feasibility trial of BMS‐275291 in patients with early stage breast cancer. Clin Cancer Res 10(6):1971–1975. [DOI] [PubMed] [Google Scholar]
- Min KW, Kim DH, Do SI, Pyo JS, Kim K, Chae SW, Sohn JH, Oh YH, Kim HJ, Choi SH, Choi YJ, Park CH. 2013. Diagnostic and prognostic relevance of MMP‐11 expression in the stromal fibroblast‐like cells adjacent to invasive ductal carcinoma of the breast. Ann Surg Oncol 20(Suppl 3):S433–S442. [DOI] [PubMed] [Google Scholar]
- Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I. 2001. Matrix metalloproteinase‐7‐mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res 61(2):577–581. [PubMed] [Google Scholar]
- Montel V, Kleeman J, Agarwal D, Spinella D, Kawai K, Tarin D. 2004. Altered metastatic behavior of human breast cancer cells after experimental manipulation of matrix metalloproteinase 8 gene expression. Cancer Res 64(5):1687–1694. [DOI] [PubMed] [Google Scholar]
- Morgunova E, Tuuttila A, Bergmann U, Tryggvason K. 2002. Structural insight into the complex formation of latent matrix metalloproteinase 2 with tissue inhibitor of metalloproteinase 2. Proc Natl Acad Sci USA 99(11):7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CJ, Butler GS, Bigg HF, Roberts CR, Soloway PD, Overall CM. 2001. Cellular activation of MMP‐2 (gelatinase A) by MT2‐MMP occurs via a TIMP‐2‐independent pathway. J Biol Chem 276(50):47402–47410. [DOI] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. 2002. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1‐MMP‐dependent activation of TGF‐beta1. J Cell Biol 157(3):493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G, Houbrechts A, Cockett MI, Williamson RA, O'Shea M, Docherty AJ. 1991. The N‐terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry 30(33):8097–8102. [DOI] [PubMed] [Google Scholar]
- Murphy G, Nguyen Q, Cockett MI, Atkinson SJ, Allan JA, Knight CG, Willenbrock F, Docherty AJ. 1994. Assessment of the role of the fibronectin‐like domain of gelatinase A by analysis of a deletion mutant. J Biol Chem 269(9):6632–6636. [PubMed] [Google Scholar]
- Murphy G, Nagase H. 2011. Localizing matrix metalloproteinase activities in the pericellular environment. FEBS J 278(1):2–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mylona E, Nomikos A, Magkou C, Kamberou M, Papassideri I, Keramopoulos A, Nakopoulou L. 2007. The clinicopathological and prognostic significance of membrane type 1 matrix metalloproteinase (MT1‐MMP) and MMP‐9 according to their localization in invasive breast carcinoma. Histopathology 50(3):338–347. [DOI] [PubMed] [Google Scholar]
- Nagase H, Brew K. 2003. Designing TIMP (tissue inhibitor of metalloproteinases) variants that are selective metalloproteinase inhibitors. Biochem Soc Symp 70:201–212. [DOI] [PubMed] [Google Scholar]
- Nam DH, Rodriguez C, Remacle AG, Strongin AY, Ge X. 2016. Active‐site MMP‐selective antibody inhibitors discovered from convex paratope synthetic libraries. Proc Natl Acad Sci USA 113(52):14970–14975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam DH, Fang K, Rodriguez C, Lopez T, Ge X. 2017. Generation of inhibitory monoclonal antibodies targeting matrix metalloproteinase‐14 by motif grafting and CDR optimization. Protein Eng Des Sel 30(2):113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nara H, Sato K, Naito T, Mototani H, Oki H, Yamamoto Y, Kuno H, Santou T, Kanzaki N, Terauchi J, Uchikawa O, Kori M. 2014. Discovery of novel, highly potent, and selective quinazoline‐2‐carboxamide‐based matrix metalloproteinase (MMP)‐13 inhibitors without a zinc binding group using a structure‐Based design approach. J Med Chem 57(21):8886–8902. [DOI] [PubMed] [Google Scholar]
- Nara H, Kaieda A, Sato K, Naito T, Mototani H, Oki H, Yamamoto Y, Kuno H, Santou T, Kanzaki N, Terauchi J, Uchikawa O, Kori M. 2017. Discovery of novel, highly potent, and selective matrix metalloproteinase (MMP)‐13 inhibitors with a 1,2,4‐triazol‐3‐yl moiety as a zinc binding group using a structure‐based design approach. J Med Chem 60(2):608–626. [DOI] [PubMed] [Google Scholar]
- Ndinguri MW, Bhowmick M, Tokmina‐Roszyk D, Robichaud TK, Fields GB. 2012. Peptide‐based selective inhibitors of matrix metalloproteinase‐mediated activities. Molecules 17(12):14230–14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Khauv D, Bissell MJ, Radisky DC. 2008. Change in cell shape is required for matrix metalloproteinase‐induced epithelial‐mesenchymal transition of mammary epithelial cells. J Cell Biochem 105(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistico P, Bissell MJ, Radisky DC. 2012. Epithelial‐mesenchymal transition: General principles and pathological relevance with special emphasis on the role of matrix metalloproteinases. Cold Spring Harb Perspect Biol 4(2):pii:a011908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell JP, Willenbrock F, Docherty AJ, Eaton D, Murphy G. 1994. Analysis of the role of the COOH‐terminal domain in the activation, proteolytic activity, and tissue inhibitor of metalloproteinase interactions of gelatinase B. J Biol Chem 269(21):14967–14973. [PubMed] [Google Scholar]
- O'Shea M, Willenbrock F, Williamson RA, Cockett MI, Freedman RB, Reynolds JJ, Docherty AJ, Murphy G. 1992. Site‐directed mutations that alter the inhibitory activity of the tissue inhibitor of metalloproteinases‐1: Importance of the N‐terminal region between cysteine 3 and cysteine 13. Biochemistry 31(42):10146–10152. [DOI] [PubMed] [Google Scholar]
- Ogata Y, Itoh Y, Nagase H. 1995. Steps involved in activation of the pro‐matrix metalloproteinase 9 (progelatinase B)‐tissue inhibitor of metalloproteinases‐1 complex by 4‐aminophenylmercuric acetate and proteinases. J Biol Chem 270(31):18506–18511. [DOI] [PubMed] [Google Scholar]
- Olson MW, Gervasi DC, Mobashery S, Fridman R. 1997. Kinetic analysis of the binding of human matrix metalloproteinase‐2 and ‐9 to tissue inhibitor of metalloproteinase (TIMP)‐1 and TIMP‐2. J Biol Chem 272(47):29975–29983. [DOI] [PubMed] [Google Scholar]
- Orlichenko L, Geyer R, Yanagisawa M, Khauv D, Radisky ES, Anastasiadis PZ, Radisky DC. 2010. The 19‐amino acid insertion in the tumor‐associated splice isoform Rac1b confers specific binding to p120 catenin. J Biol Chem 285(25):19153–19161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota I, Li XY, Hu Y, Weiss SJ. 2009. Induction of a MT1‐MMP and MT2‐MMP‐dependent basement membrane transmigration program in cancer cells by Snail1. Proc Natl Acad Sci USA 106(48):20318–20323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall CM, Kleifeld O. 2006. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer 94(7):941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paemen L, Martens E, Masure S, Opdenakker G. 1995. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two‐site ELISA and inhibition of enzymatic activity. Eur J Biochem FEBS 234(3):759–765. [DOI] [PubMed] [Google Scholar]
- Pahwa S, Stawikowski MJ, Fields GB. 2014. Monitoring and inhibiting MT1‐MMP during cancer initiation and progression. Cancers (Basel) 6(1):416–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. 2004. A multigene assay to predict recurrence of tamoxifen‐treated, node‐negative breast cancer. N Engl J Med 351(27):2817–2826. [DOI] [PubMed] [Google Scholar]
- Palmier MO, Fulcher YG, Bhaskaran R, Duong VQ, Fields GB, Van Doren SR. 2010. NMR and bioinformatics discovery of exosites that tune metalloelastase specificity for solubilized elastin and collagen triple helices. J Biol Chem 285(40):30918–30930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Rasch MG, Qiu J, Lund IK, Egeblad M. 2015. Presence of insulin‐like growth factor binding proteins correlates with tumor‐promoting effects of matrix metalloproteinase 9 in breast cancer. Neoplasia 17(5):421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. 2009. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27(8):1160–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Khauv D, Risso G, Stallings‐Mann M, Blaustein M, Quadrana L, Radisky DC, Srebrow A. 2012. Involvement of hnRNP A1 in the matrix metalloprotease‐3‐dependent regulation of Rac1 pre‐mRNA splicing. J Cell Biochem 113(7):2319–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellikainen JM, Ropponen KM, Kataja VV, Kellokoski JK, Eskelinen MJ, Kosma VM. 2004. Expression of matrix metalloproteinase (MMP)‐2 and MMP‐9 in breast cancer with a special reference to activator protein‐2, HER2, and prognosis. Clin Cancer Res 10(22):7621–7628. [DOI] [PubMed] [Google Scholar]
- Perentes JY, Kirkpatrick ND, Nagano S, Smith EY, Shaver CM, Sgroi D, Garkavtsev I, Munn LL, Jain RK, Boucher Y. 2011. Cancer cell‐associated MT1‐MMP promotes blood vessel invasion and distant metastasis in triple‐negative mammary tumors. Cancer Res 71(13):4527–4538. [DOI] [PubMed] [Google Scholar]
- Piccard H, Van den Steen PE, Opdenakker G. 2007. Hemopexin domains as multifunctional liganding modules in matrix metalloproteinases and other proteins. J Leukoc Biol 81(4):870–892. [DOI] [PubMed] [Google Scholar]
- Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. 2000. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci USA 97(5):2202–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior SH, Fulcher YG, Koppisetti RK, Jurkevich A, Van Doren SR. 2015. Charge‐triggered membrane insertion of matrix metalloproteinase‐7, supporter of innate immunity and tumors. Structure 23(11):2099–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ra HJ, Parks WC. 2007. Control of matrix metalloproteinase catalytic activity. Matrix Biol 26(8):587–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. 2005. Rac1b and reactive oxygen species mediate MMP‐3‐induced EMT and genomic instability. Nature 436(7047):123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Bissell MJ. 2006. Matrix metalloproteinase‐induced genomic instability. Curr Opin Genet Dev 16(1):45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky DC, Hartmann LC. 2009. Mammary involution and breast cancer risk: Transgenic models and clinical studies. J Mammary Gland Biol Neoplasia 14(2):181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky ES, Radisky DC. 2010. Matrix metalloproteinase‐induced epithelial‐mesenchymal transition in breast cancer. J Mammary Gland Biol Neoplasia 15(2):201–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radisky ES, Radisky DC. 2015. Matrix metalloproteinases as breast cancer drivers and therapeutic targets. Front Biosci (Landmark Ed) 20:1144–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Khoo KK, Feng ZP, Crossley G, Nugent D, Khaytin I, Chi V, Pham C, Calabresi P, Pennington MW, Norton RS, Chandy KG. 2010. Potassium channel modulation by a toxin domain in matrix metalloprotease 23. J Biol Chem 285(12):9124–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ, Bateman A. 2012. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40(Database issue):D343–D350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle AG, Golubkov VS, Shiryaev SA, Dahl R, Stebbins JL, Chernov AV, Cheltsov AV, Pellecchia M, Strongin AY. 2012. Novel MT1‐MMP small‐molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res 72(9):2339–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remacle AG, Cieplak P, Nam DH, Shiryaev SA, Ge X, Strongin AY. 2017. Selective function‐blocking monoclonal human antibody highlights the important role of membrane type‐1 matrix metalloproteinase (MT1‐MMP) in metastasis. Oncotarget 8(2):2781–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robichaud TK, Steffensen B, Fields GB. 2011. Exosite interactions impact matrix metalloproteinase collagen specificities. J Biol Chem 286(43):37535–37542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblum G, Meroueh S, Toth M, Fisher JF, Fridman R, Mobashery S, Sagi I. 2007a. Molecular structures and dynamics of the stepwise activation mechanism of a matrix metalloproteinase zymogen: Challenging the cysteine switch dogma. J Am Chem Soc 129(44):13566–13574. [DOI] [PubMed] [Google Scholar]
- Rosenblum G, Van den Steen PE, Cohen SR, Grossmann JG, Frenkel J, Sertchook R, Slack N, Strange RW, Opdenakker G, Sagi I. 2007b. Insights into the structure and domain flexibility of full‐length pro‐matrix metalloproteinase‐9/gelatinase B. Structure 15(10):1227–1236. [DOI] [PubMed] [Google Scholar]
- Rosenfeld L, Heyne M, Shifman JM, Papo N. 2016. Protein engineering by combined computational and In vitro evolution approaches. Trends Biochem Sci 41(5):421–433. [DOI] [PubMed] [Google Scholar]
- Rouanet‐Mehouas C, Czarny B, Beau F, Cassar‐Lajeunesse E, Stura EA, Dive V, Devel L. 2017. Zinc‐Metalloproteinase inhibitors: Evaluation of the complex role played by the zinc‐binding group on potency and selectivity. J Med Chem 60(1):403–414. [DOI] [PubMed] [Google Scholar]
- Roy DM, Walsh LA. 2014. Candidate prognostic markers in breast cancer: Focus on extracellular proteases and their inhibitors. Breast Cancer 6:81–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeh F, Ota I, Holmbeck K, Birkedal‐Hansen H, Soloway P, Balbin M, Lopez‐Otin C, Shapiro S, Inada M, Krane S, Allen E, Chung D, Weiss SJ. 2004. Tumor cell traffic through the extracellular matrix is controlled by the membrane‐anchored collagenase MT1‐MMP. J Cell Biol 167(4):769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz RM, Silberman S, Persky B, Bajkowski AS, Carmichael DF. 1988. Inhibition by human recombinant tissue inhibitor of metalloproteinases of human amnion invasion and lung colonization by murine B16‐F10 melanoma cells. Cancer Res 48(19):5539–5545. [PubMed] [Google Scholar]
- Scorilas A, Karameris A, Arnogiannaki N, Ardavanis A, Bassilopoulos P, Trangas T, Talieri M. 2001. Overexpression of matrix‐metalloproteinase‐9 in human breast cancer: A potential favourable indicator in node‐negative patients. Br J Cancer 84(11):1488–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela‐Passwell N, Rosenblum G, Shoham T, Sagi I. 2010. Structural and functional bases for allosteric control of MMP activities: Can it pave the path for selective inhibition? Biochim Biophys Acta 1803(1):29–38. [DOI] [PubMed] [Google Scholar]
- Sela‐Passwell N, Kikkeri R, Dym O, Rozenberg H, Margalit R, Arad‐Yellin R, Eisenstein M, Brenner O, Shoham T, Danon T, Shanzer A, Sagi I. 2012. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat Med 18(1):143–147. [DOI] [PubMed] [Google Scholar]
- Sharabi O, Shirian J, Grossman M, Lebendiker M, Sagi I, Shifman J. 2014. Affinity‐ and specificity‐enhancing mutations are frequent in multispecific interactions between TIMP2 and MMPs. PLoS ONE 9(4):e93712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda M, Principe S, Jackson HW, Luga V, Fang H, Molyneux SD, Shao YW, Aiken A, Waterhouse PD, Karamboulas C, Hess FM, Ohtsuka T, Okada Y, Ailles L, Ludwig A, Wrana JL, Kislinger T, Khokha R. 2014. Loss of the Timp gene family is sufficient for the acquisition of the CAF‐like cell state. Nat Cell Biol 16(9):889–901. [DOI] [PubMed] [Google Scholar]
- Shiomi T, Okada Y. 2003. MT1‐MMP and MMP‐7 in invasion and metastasis of human cancers. Cancer Metastasis Rev 22(2–3):145–152. [DOI] [PubMed] [Google Scholar]
- Shipley JM, Doyle GA, Fliszar CJ, Ye QZ, Johnson LL, Shapiro SD, Welgus HG, Senior RM. 1996. The structural basis for the elastolytic activity of the 92‐kDa and 72‐kDa gelatinases. Role of the fibronectin type II‐like repeats. J Biol Chem 271(8):4335–4341. [DOI] [PubMed] [Google Scholar]
- Shiryaev SA, Remacle AG, Golubkov VS, Ingvarsen S, Porse A, Behrendt N, Cieplak P, Strongin AY. 2013. A monoclonal antibody interferes with TIMP‐2 binding and incapacitates the MMP‐2‐activating function of multifunctional, pro‐tumorigenic MMP‐14/MT1‐MMP. Oncogenesis 2:e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Settleman J. 2010. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 29(34):4741–4751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparano JA, Bernardo P, Stephenson P, Gradishar WJ, Ingle JN, Zucker S, Davidson NE. 2004. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first‐line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J Clin Oncol 22(23):4683–4690. [DOI] [PubMed] [Google Scholar]
- Stallings‐Mann ML, Waldmann J, Zhang Y, Miller E, Gauthier ML, Visscher DW, Downey GP, Radisky ES, Fields AP, Radisky DC. 2012. Matrix metalloproteinase induction of Rac1b, a key effector of lung cancer progression. Sci Transl Med 4(142):142ra95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffensen B, Wallon UM, Overall CM. 1995. Extracellular matrix binding properties of recombinant fibronectin type II‐like modules of human 72‐kDa gelatinase/type IV collagenase. High affinity binding to native type I collagen but not native type IV collagen. J Biol Chem 270(19):11555–11566. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Lochter A, Sympson CJ, Huey B, Rougier JP, Gray JW, Pinkel D, Bissell MJ, Werb Z. 1999. The stromal proteinase MMP3/stromelysin‐1 promotes mammary carcinogenesis. Cell 98(2):137–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternlicht MD, Bissell MJ, Werb Z. 2000. The matrix metalloproteinase stromelysin‐1 acts as a natural mammary tumor promoter. Oncogene 19:1102–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler‐Stevenson WG. 2008. Tissue inhibitors of metalloproteinases in cell signaling: Metalloproteinase‐independent biological activities. Sci Signal 1(27):re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracke JO, Hutton M, Stewart M, Pendás AM, Smith B, López‐Otin C, Murphy G, Knäuper V. 2000. Biochemical characterization of the catalytic domain of human matrix metalloproteinase 19. Evidence for a role as a potent basement membrane degrading enzyme. J Biol Chem 275(20):14809–14816. [DOI] [PubMed] [Google Scholar]
- Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. 1995. Mechanism of cell surface activation of 72‐kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 270(10):5331–5338. [DOI] [PubMed] [Google Scholar]
- Stura EA, Visse R, Cuniasse P, Dive V, Nagase H. 2013. Crystal structure of full‐length human collagenase 3 (MMP‐13) with peptides in the active site defines exosites in the catalytic domain. FASEB J 27(11):4395–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Weber CR, Sohail A, Bernardo MM, Toth M, Zhao H, Turner JR, Fridman R. 2007. MMP25 (MT6‐MMP) is highly expressed in human colon cancer, promotes tumor growth, and exhibits unique biochemical properties. J Biol Chem 282(30): 21998–22010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suojanen J, Salo T, Koivunen E, Sorsa T, Pirilä E. 2009. A novel and selective membrane type‐1 matrix metalloproteinase (MT1‐MMP) inhibitor reduces cancer cell motility and tumor growth. Cancer Biol Ther 8(24):2362–2370. [DOI] [PubMed] [Google Scholar]
- Sympson CJ, Bissell MJ, Werb Z. 1995. Mammary gland tumor formation in transgenic mice overexpressing stromelysin‐1. Semin Cancer Biol 6(3):159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Adachi Y, Nagayama T, Furihata M. 2011. Matrix metalloproteinase‐11 overexpressed in lobular carcinoma cells of the breast promotes anoikis resistance. Virchows Arch 459(3):291–297. [DOI] [PubMed] [Google Scholar]
- Tallant C, Marrero A, Gomis‐Ruth FX. 2010. Matrix metalloproteinases: Fold and function of their catalytic domains. Biochim Biophys Acta 1803(1):20–28. [DOI] [PubMed] [Google Scholar]
- Tan TK, Zheng G, Hsu TT, Wang Y, Lee VW, Tian X, Wang Y, Cao Q, Wang Y, Harris DC. 2010. Macrophage matrix metalloproteinase‐9 mediates epithelial‐mesenchymal transition in vitro in murine renal tubular cells. Am J Pathol 176(3):1256–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatti O, Vehvilainen P, Lehti K, Keski‐Oja J. 2008. MT1‐MMP releases latent TGF‐beta1 from endothelial cell extracellular matrix via proteolytic processing of LTBP‐1. Exp Cell Res 314(13):2501–2514. [DOI] [PubMed] [Google Scholar]
- Tauro M, Shay G, Sansil SS, Laghezza A, Tortorella P, Neuger AM, Soliman H, Lynch CC. 2017. Bone seeking matrix metalloproteinase‐2 inhibitors prevent bone metastatic breast cancer growth. Mol Cancer Ther 16(3):494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor KB, Windsor LJ, Caterina NC, Bodden MK, Engler JA. 1996. The mechanism of inhibition of collagenase by TIMP‐1. J Biol Chem 271(39):23938–23945. [DOI] [PubMed] [Google Scholar]
- Tetu B, Brisson J, Wang CS, Lapointe H, Beaudry G, Blanchette C, Trudel D. 2006. The influence of MMP‐14, TIMP‐2 and MMP‐2 expression on breast cancer prognosis. Breast Cancer Res BCR 8(3):R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsen SB, Christensen SL, Wurtz SO, Lundberg M, Nielsen BS, Vinther L, Knowles M, Gee N, Fredriksson S, Moller S, Brunner N, Schrohl AS, Stenvang J. 2013. Plasma levels of the MMP‐9:TIMP‐1 complex as prognostic biomarker in breast cancer: A retrospective study. BMC Cancer 13:598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L, Tanaka M, Wait R, Shi YE, Brew K, Nagase H. 2002. E. coli expression of TIMP‐4 and comparative kinetic studies with TIMP‐1 and TIMP‐2: Insights into the interactions of TIMPs and matrix metalloproteinase 2 (gelatinase A). Biochemistry 41(50):15025–15035. [DOI] [PubMed] [Google Scholar]
- Udi Y, Fragai M, Grossman M, Mitternacht S, Arad‐Yellin R, Calderone V, Melikian M, Toccafondi M, Berezovsky IN, Luchinat C, Sagi I. 2013. Unraveling hidden regulatory sites in structurally homologous metalloproteases. J Mol Biol 425(13):2330–2346. [DOI] [PubMed] [Google Scholar]
- Udi Y, Grossman M, Solomonov I, Dym O, Rozenberg H, Moreno V, Cuniasse P, Dive V, Arroyo AG, Sagi I. 2015. Inhibition mechanism of membrane metalloprotease by an exosite‐swiveling conformational antibody. Structure 23(1):104–115. [DOI] [PubMed] [Google Scholar]
- van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415(6871):530–536. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, Parrish M, Atsma D, Witteveen A, Glas A, Delahaye L, van der Velde T, Bartelink H, Rodenhuis S, Rutgers ET, Friend SH, Bernards R. 2002. A gene‐expression signature as a predictor of survival in breast cancer. N Engl J Med 347(25):1999–2009. [DOI] [PubMed] [Google Scholar]
- Vandenbroucke RE, Libert C. 2014. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov 13(12):904–927. [DOI] [PubMed] [Google Scholar]
- Villa‐Morales M, Fernandez‐Piqueras J. 2012. Targeting the Fas/FasL signaling pathway in cancer therapy. Expert Opin Ther Targets 16(1):85–101. [DOI] [PubMed] [Google Scholar]
- Vizoso FJ, Gonzalez LO, Corte MD, Rodriguez JC, Vazquez J, Lamelas ML, Junquera S, Merino AM, Garcia‐Muniz JL. 2007. Study of matrix metalloproteinases and their inhibitors in breast cancer. Br J Cancer 96(6):903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Nagase H, Watanabe T, Nobusue H, Suzuki T, Asami Y, Shinojima Y, Kawashima H, Takagi K, Mishra R, Igarashi J, Kimura M, Takayama T, Fukuda N, Sugiyama H. 2010. Inhibition of MMP‐9 transcription and suppression of tumor metastasis by pyrrole‐imidazole polyamide. Cancer Sci 101(3):759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei S, Chen Y, Chung L, Nagase H, Brew K. 2003. Protein engineering of the tissue inhibitor of metalloproteinase 1 (TIMP‐1) inhibitory domain. In search of selective matrix metalloproteinase inhibitors. J Biol Chem 278(11):9831–9834. [DOI] [PubMed] [Google Scholar]
- West‐Mays JA, Pino G. 2007. Matrix metalloproteinases as mediators of primary and secondary cataracts. Expert Rev Ophthalmol 2(6):931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Hutton M, Vogt G, Rapti M, Knauper V, Carr MD, Murphy G. 2001. Tyrosine 36 plays a critical role in the interaction of the AB loop of tissue inhibitor of metalloproteinases‐2 with matrix metalloproteinase‐14. J Biol Chem 276(35):32966–32970. [DOI] [PubMed] [Google Scholar]
- Wu B, Arumugam S, Gao G, Lee GI, Semenchenko V, Huang W, Brew K, Van Doren SR. 2000. NMR structure of tissue inhibitor of metalloproteinases‐1 implicates localized induced fit in recognition of matrix metalloproteinases. J Mol Biol 295(2):257–268. [DOI] [PubMed] [Google Scholar]
- Wu E, Mari BP, Wang F, Anderson IC, Sunday ME, Shipp MA. 2001. Stromelysin‐3 suppresses tumor cell apoptosis in a murine model. J Cell Biochem 82(4):549–555. [DOI] [PubMed] [Google Scholar]
- Wu ZS, Wu Q, Yang JH, Wang HQ, Ding XD, Yang F, Xu XC. 2008. Prognostic significance of MMP‐9 and TIMP‐1 serum and tissue expression in breast cancer. Int J Cancer 122(9):2050–2056. [DOI] [PubMed] [Google Scholar]
- Wu Y, Wei S, Van Doren SR, Brew K. 2011. Entropy increases from different sources support the high‐affinity binding of the N‐terminal inhibitory domains of tissue inhibitors of metalloproteinases to the catalytic domains of matrix metalloproteinases‐1 and ‐3. J Biol Chem 286(19):16891–16899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita CM, Dolgonos L, Zemans RL, Young SK, Robertson J, Briones N, Suzuki T, Campbell MN, Gauldie J, Radisky DC, Riches DW, Yu G, Kaminski N, McCulloch CA, Downey GP. 2011. Matrix metalloproteinase 3 is a mediator of pulmonary fibrosis. Am J Pathol 179(4):1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousef EM, Tahir MR, St‐Pierre Y, Gaboury LA. 2014. MMP‐9 expression varies according to molecular subtypes of breast cancer. BMC Cancer 14(1):609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Stamenkovic I. 2000. Cell surface‐localized matrix metalloproteinase‐9 proteolytically activates TGF‐beta and promotes tumor invasion and angiogenesis. Genes Dev 14(2):163–176. [PMC free article] [PubMed] [Google Scholar]
- Zarrabi K, Dufour A, Li J, Kuscu C, Pulkoski‐Gross A, Zhi J, Hu Y, Sampson NS, Zucker S, Cao J. 2011. Inhibition of matrix metalloproteinase 14 (MMP‐14)‐mediated cancer cell migration. J Biol Chem 286(38):33167–33177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Bernardo MM, Osenkowski P, Sohail A, Pei D, Nagase H, Kashiwagi M, Soloway PD, DeClerck YA, Fridman R. 2004. Differential inhibition of membrane type 3 (MT3)‐matrix metalloproteinase (MMP) and MT1‐MMP by tissue inhibitor of metalloproteinase (TIMP)‐2 and TIMP‐3 rgulates pro‐MMP‐2 activation. J Biol Chem 279(10):8592–8601. [DOI] [PubMed] [Google Scholar]
- Zhao S, Ma W, Zhang M, Tang D, Shi Q, Xu S, Zhang X, Liu Y, Song Y, Liu L, Zhang Q. 2013. High expression of CD147 and MMP‐9 is correlated with poor prognosis of triple‐negative breast cancer (TNBC) patients. Med Oncol 30(1):335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Lyons JG, Tan TK, Wang Y, Hsu TT, Min D, Succar L, Rangan GK, Hu M, Henderson BR, Alexander SI, Harris DC. 2009. Disruption of E‐cadherin by matrix metalloproteinase directly mediates epithelial‐mesenchymal transition downstream of transforming growth factor‐beta1 in renal tubular epithelial cells. Am J Pathol 175(2):580–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Wu Y, Brew K. 2016. Thermodynamic basis of selectivity in the interactions of tissue inhibitors of metalloproteinases N‐domains with matrix metalloproteinases‐1, ‐3, and ‐14. J Biol Chem 291(21):11348–11358. [DOI] [PMC free article] [PubMed] [Google Scholar]