

Abstract
Background
Hepatitis C virus (HCV) infection and metabolic diseases including nonalcoholic steatohepatitis (NASH) exhibit a complex interplay. Although free fatty acid-mediated apoptosis is a prominent feature of NASH, the impact of HCV infection on hepatocyte lipotoxicity has remained largely unexplored. The study aimed at identifying whether infection by HCV affected the apoptotic pathway in hepatocytes during fatty acid assault.
Material/Methods
OR6 cells, which are derived from human hepatocellular carcinoma Huh-7 cells and harbor a full-length HCV RNA genome replication system, were treated with palmitate. Apoptosis was examined by 4′,6-diamidino-2-phenylindole staining. Activation and expression of JNK, Bim, cIAP-1, and Mcl-1 were examined by immunoblotting. mRNA expression of CHOP, a major player in endoplasmic reticulum stress-mediated apoptosis, was assessed by real-time PCR.
Results
Palmitate-induced hepatocyte apoptosis was significantly enhanced in OR6 cells compared to cured cells, in which the HCV genome had been eradicated by treatment with interferon-α. Although basal expression of CHOP mRNA was enhanced in OR6 cells compared to cured cells, it was similarly upregulated in both cell lines following palmitate treatment. Notably, palmitate-induced JNK phosphorylation was accentuated in OR6 cells compared to cured cells. Inhibition of JNK with SP600125 attenuated palmitate-induced apoptosis. Palmitate-mediated upregulation of BH3-only protein Bim, which acts downstream of JNK, was also enhanced in OR6 cells compared to cured cells. In contrast, Mcl-1 and cIAP-1 were equally reduced in OR6 cells and cured cells following palmitate treatment.
Conclusions
These findings suggest that during lipoapoptosis, HCV infection may enhance hepatocyte toxicity by increasing JNK phosphorylation.
MeSH Keywords: Cell Death; Fatty Acids, Nonesterified; Fatty Liver; Hepatitis C, Chronic
Background
Nonalcoholic fatty liver disease (NAFLD) is currently the most common cause of chronic liver disease worldwide [1]. Free fatty acids, the majority of which are released from subcutaneous fat and enter hepatocytes, represent the source of hepatic triglycerides in NAFLD [2]. Excessive accumulation of free fatty acids results in hepatocyte apoptosis, termed lipoapoptosis, and contributes to the progression of nonalcoholic steatohepatitis (NASH), which is an advanced form of NAFLD. Hepatocyte lipoapoptosis occurs through induction of complex signaling cascades, including endoplasmic stress, activation of c-Jun N-terminal kinase (JNK), degradation of anti-apoptotic Bcl-2 family proteins, and upregulation of pro-apoptotic Bcl-2 homology (BH3)-only proteins [3]. During hepatocyte lipoapoptosis, JNK activation is likely initiated following phosphorylation by glycogen synthase kinase (GSK)-3 [4]. The aforementioned pathways promote mitochondrial cell death, which is regulated by interactions between pro- and anti-apoptotic Bcl-2 family proteins [5–7]. Upon free fatty acid stimulation, pro-apoptotic Bim is activated by JNK to induce mitochondrial dysfunction. At the same time, degradation of anti-apoptotic myeloid cell leukemia sequence 1 (Mcl-1) further enhances cell death signaling [6,8]. Free fatty acids also induce endoplasmic reticulum (ER) membrane saturation and unfolded protein response (UPR) in hepatocytes [9]. The main apoptotic player in this process is the transcription factor CCAAT/enhancer binding protein homologous protein (CHOP), which is transcriptionally activated following free fatty acid exposure and subsequent induction of the ER stress sensor protein kinase R (PKR)-like endoplasmic reticulum kinase (PERK) and/or activating transcription factor 6 (ATF6) [10–12]. Upregulation of CHOP, in turn, promotes activation of downstream apoptotic modulators such as death receptor 5 (DR5), eventually resulting in the aforementioned mitochondrial dysfunction [13]. Additionally, proteasomal degradation of cellular inhibitor of apoptosis protein (cIAP)-1, an inhibitor of DR5, enhances lipoapoptosis [8,14].
Hepatitis C virus (HCV) is a major worldwide causative pathogen of chronic hepatitis, cirrhosis, and hepatocellular carcinoma [15]. Upregulation of death receptors and hepatocyte apoptosis is also considered a pivotal feature of liver injury in chronic hepatitis C (CHC) [16,17]. Because of the high prevalence of NAFLD and CHC, it is expected that these diseases may coincide in a certain proportion of patients [18]. In addition, CHC and metabolic diseases including NASH may exhibit a complex interplay [19]. The incidence of hepatic steatosis in CHC patients is over 50%, with the major cause likely to be de novo lipogenesis. This may result in an excess of free fatty acids in hepatocytes [20–22]. The main products of de novo lipogenesis are saturated free fatty acids, which are more toxic than unsaturated free fatty acids [22]. Based on these observations, co-existence of NAFLD with HCV infection might enhance a person’s susceptibility to hepatocyte cell death, which could slow down disease progression. However, the impact of HCV infection on free fatty acid-mediated apoptosis has been largely unexplored [17,23]. In this study, we aimed to elucidate the influence of HCV on hepatocyte apoptosis and its possible mechanisms.
Material and Methods
Cells
Human hepatocellular carcinoma Huh-7 cells were used in this study. OR6 cells, which are derived from Huh-7 cells and harbor a full-length HCV genotype 1 replicon containing the Renilla luciferase gene ORN/C-5B/KE, were a generous gift from Dr. Masanori Ikeda, Persistent and Oncogenic Viruses, Center for Chronic Viral Diseases, Graduate School of Medical and Dental Sciences, Kagoshima University, and were also used in selected experiments [24]. HCV RNA was eradicated from select OR6 cells following treatment with 500 IU/mL interferon (IFN)-α for two weeks, and resulted in “cured cells”. Huh-7 and cured cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (100 g/L) fetal bovine serum and streptomycin. OR6 cells were maintained in the presence of 300 μg/mL of G418. To confirm the effect of IFN-α on cured cells, we used the Renilla luciferase RL assay (Promega, Madison, WI, USA) was performed.
Fatty acid treatment
Palmitate was initially dissolved in isopropanol at a concentration of 100 mM and was added to DMEM containing 1% bovine serum albumin to assure a physiological ratio between bound and unbound palmitate in the medium. The final concentration of palmitate ranged from 200 to 800 μM.
Quantification of apoptosis
Nuclear staining with 4′,6-diamidino-2-phenylindole (DAPI) and fluorescence microscopy were used to assess apoptotic cells, which were expressed as a percentage of total cells (N >100). In addition, apoptosis was confirmed by immunoblotting for poly (ADP-ribose) polymerase (PARP) cleavage.
Real-time polymerase chain reaction (RT-PCR)
Total RNA was isolated from cells using the GenElute™ mammalian total RNA miniprep kit (Sigma-Aldrich, Munich, Germany) and was quantified using a Nanodrop-1000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). RNA was reverse-transcribed into complementary DNA with random primers (Invitrogen, Grand Island, NY, USA). The complementary DNA template was quantified by RT-PCR using SYBR green (Molecular Probes, Eugene, OR, USA) and a Light Cycler 480 (Roche Applied Science, Penzberg, Germany). Human CHOP primers were as follows: 5′-GCGCATGAAGGAGAAAGAAC-3′ (forward), 5′-TCACCATTCGGTCAATCAGA-3′ (reverse). Primers for the 18S ribosomal RNA sequence (Ambion, Austin, TX, USA) were used as an internal control.
Immunoblot analysis
For immunoblot analysis, OR6 cells and cured cells were seeded on 10-cm plates and treated with 200–800 μM palmitate for 24 hours. Samples containing 50 μg protein were resolved by 15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and incubated with primary and then appropriate secondary antibodies. Bound antibodies were visualized with Clarity Western ECL Substrate (Bio RAD, Hercules, CA, USA) using a FluorChem® FC2 chemiluminescent imaging system (Alpha Innotech, San Leandro, CA, USA).
Antibodies and reagents
Palmitic acid (P5585) was purchased from Sigma-Aldrich. The JNK inhibitor SP600125 (420119) was purchased from Calbiochem (San Diego, CA, USA). Anti-Bim (C34C5) rabbit monoclonal antibody (mAb) (#2933), anti-PARP (#9542), anti-JNK (#9252), anti-phospho-JNK (Thr183/Tyr185) (#9251), and anti-phospho-glycogen synthase (Ser641) (#3891) rabbit polyclonal antibodies were purchased from Cell Signaling, Inc. (Danvers, MA, USA). Anti-Mcl-1 (S-19) (sc-819) and anti-actin (C-11) (sc-1615) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX, USA). Anti-cIAP-1 (AF8181) antibody was purchased from R&D Systems (Minneapolis, MN, USA).
Statistical analysis
Data are expressed as mean ± standard error (SE) of at least three independent experiments. Statistical review of the study was performed by a biomedical statistician. SAS 8.2 software (SAS Institute, Cary, NC, USA) was employed for statistical analysis. For apoptosis measurements, we first confirmed the homogeneity of the data within experimental conditions using the Chi-squared test. The dose-dependent increase in apoptosis among palmitate-treated cells was then assessed by the Cochran-Armitage test. Finally, statistical differences between OR6 cells and cured cells under each experimental condition were determined by Fisher’s exact test. For RT-PCR, differences between groups were compared using one-way analysis of variance (ANOVA) followed by a post hoc test (Bonferroni’s method), or a two-way ANOVA followed by Bonferroni’s method. A p value <0.05 was considered statistically significant.
Results
Palmitate induces elevated hepatocyte apoptosis in OR6 cells
First, we tested differences in sensitivity between OR6 cells and cured cells to free fatty acid treatment. Prior to palmitate treatment, OR6 cells and cured cells presented a similar rate of apoptosis (Figure 1A). However, palmitate led to dose-dependent induction of apoptosis in OR6 cells but not in HCV-cured cells (p<0.05), as revealed by characteristic morphological nuclear changes (Figure 1A) and elevated levels of cleaved PARP in OR6 cells (Figure 1B). To confirm that suppression of apoptosis exhibited by cured cells was not solely due to exposure to IFN-α, Huh-7 cells were treated with IFN-α for two weeks. The apoptosis assay revealed that IFN-α treatment did not attenuate lipoapoptosis in Huh-7 cells (Figure 1C). Thus, the presence of HCV, but not exposure to IFN-α, appears to enhance palmitate-induced apoptosis in Huh-7 cells.
Figure 1.

Induction of hepatocyte lipoapoptosis in HCV replicon cells. (A) Upper panel: OR6 cells and Huh-7 cells were treated with palmitate for 24 hours. Nuclear staining with DAPI and fluorescence microscopy was used to assess apoptotic cells. Palmitate-induced hepatocyte apoptosis was enhanced in OR6 cells, * p<0.05. Data are expressed as mean ±SE. Lower panel: Images of DAPI staining overlaid with bright field view. Each image is representative of at least three independently taken photographs. Arrows indicate apoptotic cells. Original magnification was 400×. (B) Cleaved and uncleaved PARP were examined by immunoblotting after 24 hours of palmitate treatment in OR6 cells and cured cells. (C) Huh-7 cells were treated with IFN-α for two weeks. Cells were then treated by palmitate for 24 hours and lipoapoptosis was examined by staining cells with DAPI. Data are expressed as mean ±SE. For apoptosis measurements, we first confirmed the homogeneity of the data within experimental conditions using the Chi-squared test. The dose-dependent increase in apoptosis among palmitate-treated cells was then assessed by the Cochran-Armitage test. Finally, statistical differences between OR6 cells and cured cells, as well as differences between IFN-α treated cells and untreated cells under each experimental condition were determined by Fisher’s exact test.
Accentuated JNK phosphorylation in PA-treated OR6 cells
Free fatty acids induce JNK-mediated hepatocyte apoptosis [25]. Thus, we investigated whether JNK activation was enhanced during free fatty acid assault in OR6 cells. At basal level, phosphorylation of JNK did not differ between OR6 cells and cured cells. However, JNK phosphorylation increased in OR6 cells following palmitate treatment (Figure 2A), indicating that elevated JNK activation in HCV-infected cells may contribute to free fatty acid-induced apoptosis. Previous studies have suggested that GSK inhibition attenuates palmitate-induced cell death in HCV-uninfected hepatocytes via the JNK pathway [4]. Interestingly, palmitate did not enhance GSK phosphorylation in OR6 cells, implying that alternative pathways were involved in JNK activation in HCV-infected OR6 cells during free fatty acid toxicity (Figure 2B). To further confirm that JNK contributed to lipoapoptosis in HCV-infected cells, OR6 cells were treated with palmitate and SP600125, a JNK inhibitor. In these cells, lipoapoptosis was significantly suppressed by the JNK inhibitor (Figure 2C). We then investigated the pro-apoptotic BH3-only protein Bim, which is activated downstream of JNK [5,26]. palmitate-mediated up-regulation of Bim was enhanced in OR6 compared to cured cells (Figure 3A). This suggests that in HCV-positive hepatocytes, palmitate enhances apoptosis by increasing JNK/Bim activation independently of GSK-3 phosphorylation.
Figure 2.

HCV infection induces JNK phosphorylation. (A) JNK phosphorylation was examined by immunoblotting after treatment with palmitate for eight hours. (B) Expression of GSK was examined by immunoblotting after treatment with palmitate for eight hours. (C) Left panel: OR6 cells were treated with JNK inhibitor SP600125, and lipoapoptosis was examined by staining cells with DAPI after treatment for 24 hours, * p<0.05. Data are expressed as mean ±SE. Right panel: images of DAPI staining overlaid with bright field view. Lower panel: images of DAPI staining overlaid with bright field view. Each image is representative of at least three independently taken photographs. Arrows indicate apoptotic cells. Original magnification was 400×.
Figure 3.

Expression of CHOP and Bim in OR6 cells increases following palmitate treatment. OR6 cells and cured cells were treated with palmitate for eight hours. (A) Bim expression was examined by immunoblotting. Data are expressed as mean ±SE. (B) CHOP expression was assessed by RT-PCR. Differences between groups were compared using ANOVA followed by a post hoc test (Bonferroni’s method), or a two-way ANOVA followed by Bonferroni’s method. A p value <0.05 was considered statistically significant.
ER stress may not contribute to enhanced lipoapoptosis in HCV-infected hepatocytes
Next, we investigated whether ER stress contributed to increased apoptosis in OR6 cells. mRNA levels of CHOP, a critical ER stress-induced death mediator during hepatocyte apoptosis, were enhanced in OR6 cells compared to cured cells at basal level. This finding was consistent with previous observations whereby HCV infection increased ER stress. However, upon palmitate exposure, CHOP was upregulated identically in the two cell lines (Figure 3B). These results imply that although ER stress is constitutively elevated in OR6 cells, the increased lipoapoptosis exhibited by these cells may not be due to enhanced activation of ER stress-mediated CHOP.
Mcl-1 and cIAP-1 are reduced in both OR6 and cured cells following palmitate treatment
Previous studies have demonstrated that hepatocyte apoptosis is partially mediated by proteasomal degradation of anti-apoptotic proteins Mcl-1 and cIAP-1, which are involved in the lipoapoptosis pathway independently of JNK activation or ER stress. Mcl-1 and cIAP-1 levels were reduced in equal measure in OR6 cells and cured cells following palmitate treatment (Figure 4). These results indicate that HCV infection may not affect the degradation of either Mcl-1 or cIAP-1 during lipoapoptosis.
Figure 4.
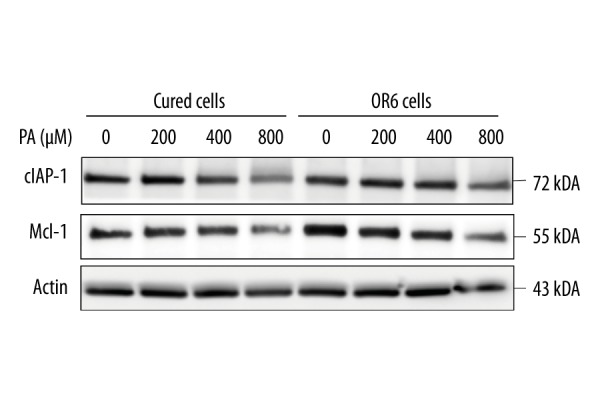
Expression of anti-apoptotic proteins Mcl-1 and cIAP-1 in both OR6 cells and cured cells decreases following palmitate treatment. OR6 cells and Huh-7 cells were treated with palmitate for eight hours. Expression of Mcl-1 and cIAP-1 was examined by immunoblotting. Homogeneity of the data within experimental conditions was confirmed by the Chi-squared test. The dose-dependent increase in apoptosis among palmitate-treated cells was then assessed by the Cochran-Armitage test. Finally, statistical differences between JNK-treated and untreated cells under each experimental condition were determined by Fisher’s exact test.
Discussion
This study sought to determine the mechanism of lipoapoptosis in HCV-infected hepatocytes. Results indicate that 1) palmitate-induced hepatocyte apoptosis is enhanced in HCV-infected cells; 2) HCV augments palmitate-induced apoptosis at least in part in a JNK-dependent manner; and 3) increased lipoapoptosis in HCV-infected cells may not involve ER stress or the anti-apoptotic proteins Mcl-1 and cIAP-1.
Altered lipid homeostasis in HCV patients likely influences the prognosis of HCV. It has been shown that steatosis is independently associated with advanced fibrosis in the HCV-infected liver [17]. Dysfunction of insulin-regulated glucose homeostasis, lipolysis of subcutaneous fat, and hepatic steatosis is frequently observed in CHC patients and has also been shown in a cell culture model [16,17,27]. Previous reports have demonstrated that fatty acid assault affects HCV-infected hepatocytes, inhibiting the antiviral activity of IFN-α [20]. However, free fatty acid-related apoptosis in HCV-infected hepatocytes has been relatively unexplored. Our findings show that palmitate-induced apoptosis is enhanced in HCV-infected compared to HCV-cured cells. Our results imply that a normally sub-lethal concentration of palmitate may be toxic to HCV-infected cells. Because these cells are already saturated with free fatty acids, an additional relatively low dose of palmitate could be sufficient to trigger apoptosis.
Although prior studies showed sustained activation of JNK in HCV-infected cells, it was unclear how JNK would play a role in lipoapoptosis in this condition [28]. Here, palmitate treatment triggered phosphorylation of JNK and activation of Bim. Cell death was attenuated by a JNK inhibitor, implying that JNK plays a role in the high sensitivity of OR6 cells to free fatty acids. HCV-uninfected hepatocytes are also known to undergo apoptosis by JNK-dependent Bim expression, which is controlled by GSK-3 [4]. However, GSK was not activated in palmitate-treated OR6 cells, indicating that other, as yet unknown, mechanisms may contribute to JNK phosphorylation. This should be further examined in future experiments. Interestingly, recent observations have shown that infection with HCV-2a alone may induce activation of JNK/Bim and apoptosis in Huh-7.5 cells, although we did not observe increased JNK or Bim activation in OR6 cells without palmitate treatment [28]. Likewise, in our study, constitutive apoptosis was not elevated in OR6 cells compared to cured cells. Differences in basal apoptotic activity between the previous study and ours may have resulted from multiple factors, including the different viral genotypes used. In addition, we compared HCV-infected cells to those treated with IFN; however, we did not assess differences between pre-and post-infection states. Our findings indicate that recovery from HCV infection restores threshold levels of free fatty acid-induced toxicity.
HCV infection also induces ER stress, which likely leads to multiple outcomes [29]. HCV has been shown to form a replication complex that associates with host ER membranes. Interaction of HCV with the ER lumen induces the UPR and activates the ER stress response causing, among others, elevated levels of nuclear factor κB and transforming growth factor β1, both of which are crucial for hepatitis progression [29]. In contrast, HCV-NS2-induced ER stress has been reported to suppress HCV replication through phosphorylation of the translation initiation factor eIF2α and reduction of de novo protein synthesis. Interestingly, the HCV-ER interaction is also associated with hepatic steatosis; the majority of HCV-associated lipid droplets likely bud off from the ER membrane [30]. In line with these studies, OR6 cells did exhibit an elevated ER stress response prior to palmitate treatment. However, CHOP mRNA levels increased identically in OR6 cells and cured cells upon free fatty acid assault. While the role of CHOP in hepatocyte lipoapoptosis is generally accepted, upregulation of CHOP may not be the specific cause of increased apoptosis in OR6 cells [31]. Thus, we speculate that free fatty acid-induced ER stress signals are not specific to HCV-infected cells [29]. A limitation of our study is represented by the use of HCV replicon cells. Further, although in the present study we employed only the HCV genotype 1 replicon, it would be interesting to understand how the different HCV genotypes affect free fatty acid-induced apoptosis. Finally, studies including in vitro HCV infection experiments, as well as in vivo experiments with animal models, aimed at assessing the influence of inflammatory cells, would provide further insights on the dynamics of HCV infection and hepatocyte lipoapoptosis.
Conclusions
In conclusion, our results show that increased apoptosis through JNK activation may at least in part contribute to the rapid progression of fatty liver diseases in chronic HCV hepatitis patients.
Acknowledgements
We thank Dr. Masanori Ikeda, Persistent and Oncogenic Viruses, Center for Chronic Viral Diseases, Graduate School of Medical and Dental Sciences, Kagoshima University for providing OR6 cells. We thank Dr. Gregory Gores, Mayo Clinic, Rochester, MN, for critical input on the study.
Footnotes
Conflicts of interest
The authors have no conflict of interest to declare.
Source of support: Japan Society for the Promotion of Science (JSPS) #24790709
References
- 1.Satapathy SK, Sanyal AJ. Epidemiology and natural history of nonalcoholic fatty liver disease. Semin Liver Dis. 2015;35:221–35. doi: 10.1055/s-0035-1562943. [DOI] [PubMed] [Google Scholar]
- 2.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–89. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akazawa Y, Nakao K. Lipotoxicity pathways intersect in hepatocytes: Endoplasmic reticulum stress, c-Jun N-terminal kinase-1, and death receptors. Hepatol Res. 2016;46(10):977–84. doi: 10.1111/hepr.12658. [DOI] [PubMed] [Google Scholar]
- 4.Ibrahim SH, Akazawa Y, Cazanave SC, et al. Glycogen synthase kinase-3 (GSK-3) inhibition attenuates hepatocyte lipoapoptosis. J Hepatol. 2011;54:765–72. doi: 10.1016/j.jhep.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barreyro FJ, Kobayashi S, Bronk SF, et al. Transcriptional regulation of Bim by FoxO3A mediates hepatocyte lipoapoptosis. J Biol Chem. 2007;282:27141–54. doi: 10.1074/jbc.M704391200. [DOI] [PubMed] [Google Scholar]
- 6.Masuoka HC, Mott J, Bronk SF, et al. Mcl-1 degradation during hepatocyte lipoapoptosis. J Biol Chem. 2009;284:30039–48. doi: 10.1074/jbc.M109.039545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feldstein AE, Werneburg NW, Li Z, et al. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1339–46. doi: 10.1152/ajpgi.00509.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akazawa Y, Guicciardi ME, Cazanave SC, et al. Degradation of cIAPs contributes to hepatocyte lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2013;305:G611–19. doi: 10.1152/ajpgi.00111.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volmer R, van der Ploeg K, Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc Natl Acad Sci USA. 2013;110:4628–33. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cazanave SC, Elmi NA, Akazawa Y, et al. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299:G236–43. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cao J, Dai DL, Yao L, et al. Saturated fatty acid induction of endoplasmic reticulum stress and apoptosis in human liver cells via the PERK/ATF4/CHOP signaling pathway. Mol Cell Biochem. 2012;364:115–29. doi: 10.1007/s11010-011-1211-9. [DOI] [PubMed] [Google Scholar]
- 13.Cazanave SC, Mott JL, Bronk SF, et al. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J Biol Chem. 2011;286:39336–48. doi: 10.1074/jbc.M111.280420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guicciardi ME, Mott JL, Bronk SF, et al. Cellular inhibitor of apoptosis 1 (cIAP-1) degradation by caspase 8 during TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis. Exp Cell Res. 2011;317:107–16. doi: 10.1016/j.yexcr.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calabrese F, Pontisso P, Pettenazzo E, et al. Liver cell apoptosis in chronic hepatitis C correlates with histological but not biochemical activity or serum HCV-RNA levels. Hepatology. 2000;31:1153–59. doi: 10.1053/he.2000.7123. [DOI] [PubMed] [Google Scholar]
- 16.Wedemeyer I, Bechmann LP, Odenthal M, et al. Adiponectin inhibits steatotic CD95/Fas up-regulation by hepatocytes: therapeutic implications for hepatitis C. J Hepatol. 2009;50:1405–9. doi: 10.1016/j.jhep.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 17.Walsh MJ, Vanags DM, Clouston AD, et al. Steatosis and liver cell apoptosis in chronic hepatitis C: A mechanism for increased liver injury. Hepatology. 2004;39:1230–38. doi: 10.1002/hep.20179. [DOI] [PubMed] [Google Scholar]
- 18.Patel A, Harrison SA. Hepatitis C virus infection and nonalcoholic steatohepatitis. Gastroenterol Hepatol (NY) 2012;8:305–12. [PMC free article] [PubMed] [Google Scholar]
- 19.Bernsmeier C, Calabrese D, Heim MH, Duong HT. Hepatitis C virus dysregulates glucose homeostasis by a dual mechanism involving induction of PGC1alpha and dephosphorylation of FoxO1. J Viral Hepat. 2014;21:9–18. doi: 10.1111/jvh.12208. [DOI] [PubMed] [Google Scholar]
- 20.Gunduz F, Aboulnasr FM, Chandra PK, et al. Free fatty acids induce ER stress and block antiviral activity of interferon alpha against hepatitis C virus in cell culture. Virol J. 2012;9:143. doi: 10.1186/1743-422X-9-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bressler BL, Guindi M, Tomlinson G, Heathcote J. High body mass index is an independent risk factor for nonresponse to antiviral treatment in chronic hepatitis C. Hepatology. 2003;38:639–44. doi: 10.1053/jhep.2003.50350. [DOI] [PubMed] [Google Scholar]
- 22.Lambert JE, Bain VG, Ryan EA, et al. Elevated lipogenesis and diminished cholesterol synthesis in patients with hepatitis C viral infection compared to healthy humans. Hepatology. 2013;57:1697–704. doi: 10.1002/hep.25990. [DOI] [PubMed] [Google Scholar]
- 23.Tse E, Helbig KJ, Van der Hoek K, et al. Fatty acids induce a pro-inflammatory gene expression profile in Huh-7 cells that attenuates the Anti-HCV action of interferon. J Interferon Cytokine Res. 2015;35:392–400. doi: 10.1089/jir.2014.0165. [DOI] [PubMed] [Google Scholar]
- 24.Ikeda M, Abe K, Dansako H, et al. Efficient replication of a full-length hepatitis C virus genome, strain O, in cell culture, and development of a luciferase reporter system. Biochem Biophys Res Commun. 2005;329:1350–59. doi: 10.1016/j.bbrc.2005.02.138. [DOI] [PubMed] [Google Scholar]
- 25.Malhi GS, Lagopoulos J, Owen AM, et al. Reduced activation to implicit affect induction in euthymic bipolar patients: An fMRI study. J Affect Disord. 2007;97:109–22. doi: 10.1016/j.jad.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 26.Malhi H, Barreyro FJ, Isomoto H, et al. Free fatty acids sensitise hepatocytes to TRAIL mediated cytotoxicity. Gut. 2007;56:1124–31. doi: 10.1136/gut.2006.118059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piodi A, Chouteau P, Lerat H, et al. Morphological changes in intracellular lipid droplets induced by different hepatitis C virus genotype core sequences and relationship with steatosis. Hepatology. 2008;48:16–27. doi: 10.1002/hep.22288. [DOI] [PubMed] [Google Scholar]
- 28.Deng L, Chen M, Tanaka M, et al. HCV upregulates Bim through the ROS/JNK signalling pathway, leading to Bax-mediated apoptosis. J Gen Virol. 2015;96:2670–83. doi: 10.1099/jgv.0.000221. [DOI] [PubMed] [Google Scholar]
- 29.Chusri P, Kumthip K, Hong J, et al. HCV induces transforming growth factor beta1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci Rep. 2016;6:22487. doi: 10.1038/srep22487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dubuisson J, Penin F, Moradpour D. Interaction of hepatitis C virus proteins with host cell membranes and lipids. Trends Cell Biol. 2002;12:517–23. doi: 10.1016/s0962-8924(02)02383-8. [DOI] [PubMed] [Google Scholar]
- 31.Cazanave SC, Elmi NA, Akazawa Y, et al. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am J Physiol Gastrointest Liver Physiol. 2010;299(1):G236–43. doi: 10.1152/ajpgi.00091.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]