

Abstract
Acute myeloid leukemia (AML) is an aggressive hematological malignancy with a poor outcome; overall survival is approximately 35% at two years and some subgroups have a less than 5% two-year survival. Recently, significant improvements have been made in our understanding of AML biology and genetics. These fundamental discoveries are now being translated into new therapies for this disease. This review will discuss recent advances in AML biology and the emerging treatments that are arising from biological studies. Specifically, we will consider new therapies that target molecular mutations in AML and dysregulated pathways such as apoptosis and mitochondrial metabolism. We will also discuss recent advances in immune and cellular therapy for AML.
Introduction
Acute myeloid leukemia (AML) is a hematological malignancy characterized by the accumulation of immature myeloid precursors with resultant peripheral blood cytopenias. The median age at diagnosis of AML is 67 years and outcomes vary according to clinical and laboratory parameters (1). However, for most patients, outcomes remain poor with high rates of relapse. For example, according to data from the Surveillance, Epidemiology, and End Results (SEER) program of the National Cancer Institute, the median overall survival of patients over 66 years with AML is less than 19 months despite intensive therapy (2). For the last 43 years, the standard of care has been 3+7 combination chemotherapy, with three days of an anthracycline and seven days of cytarabine (3). Recently, there have been dramatic advances in our understanding of AML biology and genetics. This new knowledge is now being translated into better predictive markers and novel targeted therapies. The new therapies being developed for AML include drugs targeting specific mutated proteins and dysregulated signaling pathways downstream of the genetic mutations. Epigenetic dysregulation is a key driver of AML biology and new epigenetic therapies are one of many exciting developments for this disease (4). Novel immune- and cell-based therapies are also under development. Here, we will discuss advances in AML biology and the emerging therapies arising from biological studies.
Molecular subgroups of AML have therapeutic implications
Earlier classifications of AML relied primarily on morphology and cytogenetics. While these markers helped predict outcome of therapy, some subgroups, such as patients with AML and normal cytogenetics, remained heterogeneous and their management difficult. Genomic investigations of AML have demonstrated that several genes are recurrently mutated (5–8), leading to new genomic classifications, predictive markers, and new therapeutic targets (5, 7, 9).
Compared with solid tumors, AML has fewer mutations, with an average of 13 mutations per case. For example, The Cancer Genome Atlas (TCGA) study discovered that 23 genes are recurrently mutated, but a further 237 mutations were identified (6). While some mutations such as FMS-related tyrosine kinase 3 (FLT3), nucleophosmin (NPM1), and isocitrate dehydrogenase 1 and 2 (IDH1/2) had already been reported in AML, other mutations such as U2AF1 and others involved in spliceosome pathways were discovered. The pattern of AML mutations was subsequently validated in additional cohorts with larger numbers of adult patients (5, 10). The presence and prognostic significance of genetic mutations have also been examined in pediatric AML and show trends similar to those of adult AML. For example, NPM1 and CEBPA mutations are less frequent in pediatric AML, but similar to adults, these mutations are associated with improved clinical outcomes and lower relapse rates (11, 12).
These sequencing efforts led to the description of nine functional categories of mutations in AML (Table 1). Additional studies have highlighted differences in the frequency of these mutations in de novo and secondary AML (13) (Table 1). For example, NPM1 mutations are more common in de novo AML, while mutations in chromatin modifiers and spliceosome variants are more frequent in secondary AML.
Table 1. Mutations and their incidence in primary and secondary AML grouped according to category subtypes (6, 13).
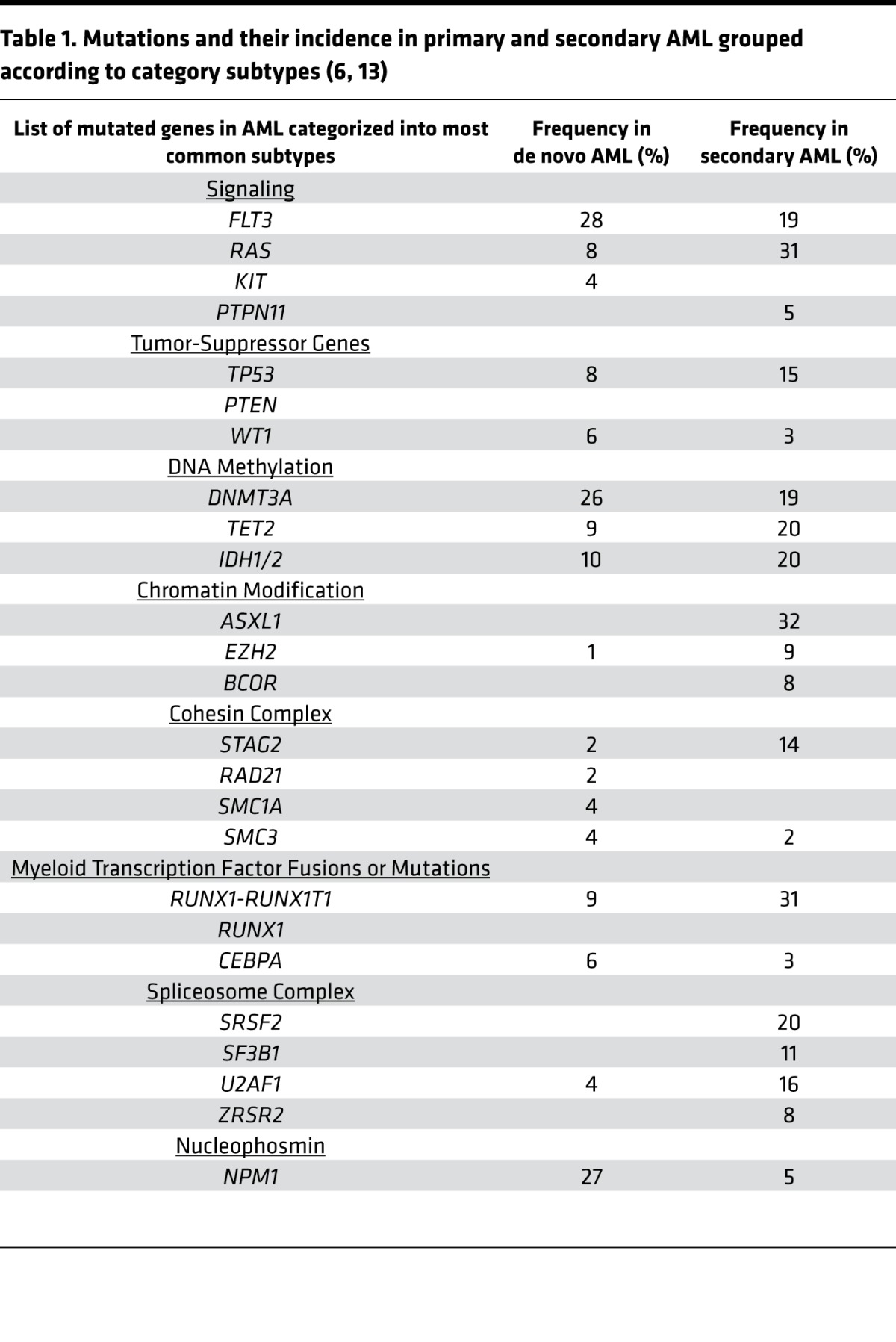
Molecular mutations in genes such as FLT3 have prognostic importance. However, understanding how different combinations of molecular mutations cooperate and predict outcome of an individual patient is challenging. Recently, clinical and genomic variables from multiple studies have been linked to provide an online algorithm that can predict survival and therapy needs based on an individual patient’s clinical and molecular status (http://cancer.sanger.ac.uk/aml-multistage) (14). In the future, such resources may help tailor upfront and postremission therapy, including the need for allotransplant in first remission.
Many patients with AML have preleukemic mutations that developed in their stem cells long before AML is clinically evident (15, 16). These mutations typically occur in genes associated with DNA methylation and chromatin modification, such as DNMT3A and TET2 (17). Age-related clonal hematopoiesis (ARCH), also known as clonal hematopoiesis of indeterminate potential (CHIP), also complicates the interpretation of the significance of these lesions (18–22). Preleukemic mutations associated with AML, such as DNMT3A, are found in up to 10% of the aging population without evidence of cytopenias. While CHIP is associated with an 11- to 13-fold increased risk of developing a hematological malignancy, the overall annual incidence remains low at approximately 0.5% to 1% per year (23). As such, it is currently unclear which patients should be screened for CHIP and which, if any, patients would benefit from therapy to prevent transformation to AML. Understanding the subgroups at highest risk of transformation would help address these questions. Potential high-risk subgroups include patients with CHIP who are receiving high-dose chemotherapy for other malignancies. For example, in a study of 401 patients with non-Hodgkin lymphoma receiving autotransplant, patients with CHIP had a 10-year cumulative risk of 14.1% of developing therapy-related myeloid neoplasms and this risk was 3-fold higher than patients without CHIP (24). Perhaps in the future, these patients will receive closer monitoring for the development of a secondary myeloid malignancy. These patients might also benefit from therapy such as demethylating agents to eradicate the clonal population, although the efficacy of demethylating agents in eradicating these clones is unknown.
While molecular mutations can be used to classify subgroups of AML, other groups have focused on identifying AML subgroups based on gene or protein expression. For example, a stemness score, termed LSC17, based on the gene expression profile of functionally defined leukemic stem cells (LSCs), identifies patients with high and low stemness. Patients with high stemness scores have lower rates of remission after standard 3+7 chemotherapy, higher rates of relapse, and lower overall survival (25). In the future, patients with high LSC17 scores could receive more intensive induction chemotherapy or upfront therapy with novel agents.
Emerging therapies for AML that target molecular mutations
Inhibitors of the FLT3 receptor tyrosine kinase.
Among the molecular therapeutic targets identified for AML, FLT3 and its inhibitors are most advanced. FLT3 encodes a receptor tyrosine kinase containing a characteristic interrupted kinase domain (26). Ligand binding triggers receptor dimerization, tyrosine kinase domain (TKD) activation, autophosphorylation, and activation of signaling pathways, including Ras, PI3-kinase, and STAT5 (Figure 1). FLT3 is primarily expressed in hematopoietic precursors and normally promotes hematopoietic development (27). Activating mutations of FLT3 caused by internal tandem duplications (ITDs) were first reported in 1996 (28). FLT3-ITD mutations represent in-frame tandem duplications found within exon 14 (29). The mutant protein forms hetero- and homodimers in the absence of ligand, resulting in constitutive tyrosine kinase signaling. FLT3-ITD is found in approximately 20% of cases of adult AML (30) and is overrepresented in normal karyotype AML and less common in core binding factor AML or poor cytogenetic risk leukemia. The presence of FLT3-ITD is a marker of adverse prognosis. Some studies (31) suggest that the allele ratio of the FLT3-ITD is important in risk classification. Of note, activating point mutations (e.g., Asp835) in the TKD of FLT3 have also been described (32, 33); however, the prognostic significance of these point mutations is less clear.
Figure 1. FLT3 mutations promote cell proliferation and chemoresistance.

Activating mutations of FLT3 caused by internal tandem duplications (ITDs) or point mutations in the tyrosine kinase domain (TKD) result in constitutive tyrosine kinase signaling. FLT3 signaling recruits pathways, including Ras, PI3-kinase, and STAT5, that promote cellular proliferation and chemoresistance. Illustrated by Rachel Davidowitz.
FLT3 activating mutations have been shown to induce growth factor independence, cell proliferation, and leukemic transformation. Constitutive FLT3 signaling blocks G-CSF–induced differentiation in cell lines and suppresses myeloid transcription factor expression (26). Mouse models demonstrate the potential transforming role of FLT3. Soon after identifying FLT3-ITD mutations in AML, there was interest in developing inhibitors of this kinase. Initially, clinical trials were launched using multi-kinase inhibitors that also targeted FLT3. Subsequently, drugs that more specifically target FLT3 were developed.
Sorafenib.
Sorafenib demonstrates broad spectrum tyrosine kinase inhibition. In addition to FLT3, sorafenib inhibits c-Kit, VEGFR, and PDGFR-α and -β (34). Preclinical studies demonstrated reduced proliferation and impaired FLT3 signaling after sorafenib treatment in cell lines and mouse models (35).
A phase 1 trial of single-agent sorafenib in patients with relapsed/refractory AML or elderly patients with untreated myelodysplastic syndrome (MDS)/secondary AML (36) identified hematological improvements in three of nine patients with the FLT3-ITD mutation with one complete remission (CR). Given these results, a phase 3 study was initiated and randomized 201 patients over 60 years old with newly diagnosed AML to induction/consolidation chemotherapy with or without sorafenib. This trial did not demonstrate benefit in rates of remission, relapse, or survival (37). Adverse events were more common in the sorafenib arm, resulting in higher treatment-related mortality, lower CR rates, and less delivery of consolidation chemotherapy. A similar combination study was conducted in 267 younger patients (18–60 years old) with newly diagnosed AML (38). This trial demonstrated improved event-free survival (21 versus 9 months, P = 0.013) but a high rate of grade III toxicities in sorafenib-treated patients. Given the advent of more potent inhibitors and high incidence of toxicity, sorafenib has a limited role in treatment of AML.
Midostaurin (PKC-412).
Like sorafenib, midostaurin is an inhibitor of multiple kinases including FLT3. Midostaurin was evaluated in a randomized phase 3 study in 717 patients aged 60 or younger with newly diagnosed, FLT3-mutated, AML. In a trial that took over 8 years to complete, patients received midostaurin or placebo in induction, consolidation, and 12 months of maintenance (39). Rates of complete remission and allogeneic bone marrow transplantation were equivalent between arms. Improved overall survival (hazard ratio [HR] 0.77, one-sided P = 0.007) and event-free survival (HR 0.8, P = 0.004) were seen in the midostaurin group. Advantages persisted after censoring at the time of transplant. Improvements in survival may reflect the ability of inhibitor therapy to overcome FLT3-driven survival signaling and allow elimination of clones responsible for relapse. Recently, midostaurin was approved for the treatment of adult patients with newly diagnosed AML who have FLT3 mutations in combination with induction and consolidation chemotherapy. As such, midostaurin in combination with induction chemotherapy should be considered the new standard of care for FLT3-mutated AML patients who are eligible for induction chemotherapy.
An early study demonstrated that midostaurin was capable of inducing responses in FLT3-wild-type patients (40), suggesting other signaling pathways may be impacted. Midostaurin metabolites have since been shown to be active and less specific to FLT3. While lower potency, these metabolites are found at higher levels in plasma and appear to contribute significantly to midostaurin’s activity (41). Studies in breast cancer (42) and systemic mastocytosis (43) have identified SYK as a target of midostaurin. The relative contribution of SYK inhibition remains unclear, but given that SYK is an independent therapeutic target (44), beneficial off-target effects are likely. Therefore, testing the efficacy of midostaurin in combination with induction chemotherapy in newly diagnosed AML with wild-type FLT3 would also be of interest.
Quizartinib (AC220).
Quizartinib is a highly potent inhibitor of FLT3, capable of inhibiting signaling in the low nanomolar range. Inhibition of related tyrosine kinases (e.g., c-Kit, PDGFR-α, PDGFR-β, RET, and CSF1R) occurs, although at potencies at least 10-fold lower than for FLT3 (45). Xenograft tumor models demonstrated complete responses after quizartinib therapy; engraftment models demonstrated dose-dependent prolongation in survival (45), although tumor growth resumed after treatment discontinuation.
Quizartinib demonstrated an overall response rate of 30% in a single-agent phase 1 study enrolling patients with AML irrespective of FLT3 status (46). Nine of 17 patients (53%) with FLT3-ITD responded, including four composite complete responses. Notably, and in contrast to earlier FLT3 inhibitors, QT prolongation emerged as a dose-limiting toxicity (23%–25% with continuous 200 mg dosing) (46). A subsequent phase 2 study evaluated quizartinib monotherapy in 137 patients over 18 years of age with relapsed/refractory AML or relapse after allogeneic stem cell transplantation (47). A composite CR rate of 44% was seen in FLT3-ITD–positive patients, with a median duration of response of 11.3 weeks. FLT3-ITD–negative patients also responded (composite CR rate 34%), although they had a shorter median duration of response at only five weeks. Similar composite CR rates (47%) were seen in another study evaluating 76 FLT3-ITD–positive patients (48) receiving lower doses of quizartinib monotherapy. Adverse events, particularly QT prolongation, were less frequently seen in this latter study.
Quizartinib’s tolerability and potency, along with the benefit seen with midostaurin therapy, make upfront therapy appealing. Clinical trials in this context are ongoing. Secondly, quizartinib’s ability to induce transient remissions in relapsed/refractory AML (with or without systemic chemotherapy) suggests it could potentially induce remission prior to allogeneic transplantation.
Gilteritinib (ASP2215).
Gilteritinib is a highly specific and potent inhibitor of mutant FLT3 (49). Gilteritinib also inhibits AXL, a receptor tyrosine kinase shown to confer a growth advantage to AML cells (50) and contribute to FLT3-inhibitor resistance. Gilteritinib demonstrates lower affinity for c-Kit than other FLT3-directed tyrosine kinase inhibitors (49), potentially resulting in less hematological toxicity. A phase 1/2 study of gilteritinib monotherapy, evaluating a total of 166 patients with relapsed/refractory AML, demonstrated an overall response rate of 57% in 82 patients with FLT3 mutations (51). Subsequent followup from this study, reporting on pharmacokinetic/pharmacodynamic findings in 215 patients, reported significant QT prolongation in only 5% of patients (52). Maintenance and upfront studies (in patients ineligible for high dose chemotherapy) are ongoing.
While capable of inhibiting AXL, this effect is seen at concentrations approximately 20-fold higher than that required for FLT3-ITD inhibition (49). The clinical relevance of this effect at therapeutic doses remains unclear. Gilteritinib’s relative lack of QT prolongation and c-Kit inhibition may contribute to better tolerability and thus clinical utility.
FLT3-inhibitor resistance.
Acquired mutations in the FLT3 TKD confer resistance to FLT3 inhibitors and limit the efficacy of the drug. (53, 54). Strategies to prevent or overcome this mechanism of resistance include using combinations of FLT3 inhibitors, as the mutations that develop are unique for the FLT3 inhibitor tested. Moreover, cells that become resistant to one FLT3 inhibitor can remain sensitive to others (55). Alternatively, agents such as crenolanib, which retain efficacy against specific FLT3 mutations, can overcome some forms of acquired resistance (56, 57). In addition to the acquisition of new mutations, resistance to FLT3 inhibitors may develop through upregulation of downstream signaling pathways (58) and alterations in marrow microenvironment mediated by AXL (59).
Inhibitors of mutant IDH1/IDH2
IDH1 and IDH2 encode enzymes that catalyze the conversion of isocitrate to α-ketoglutarate (αKG) in the cytoplasm and mitochondria, respectively. αKG, along with Fe(II), is an essential cofactor for TET2 and contributes to the active demethylation of DNA. Missense mutations in IDH1 and IDH2 alter the affinity of the enzymes for their substrates, leading to conversion of αKG to R-2-hydroxyglutarate (R-2-HG) (60). Production of R-2-HG inhibits TET2 and other αKG-dependent enzymes (60), either through αKG depletion or interference with Fe(II) binding (61) (Figure 2). The net effect is DNA hypermethylation (60) and altered gene expression. The finding that IDH1/2 mutations are mutually exclusive with TET2 mutations (60) suggests that inhibiting the mutant enzyme and restoring normal IDH function might reverse the aberrant epigenetic changes and restore normal hematopoietic function.
Figure 2. IDH1/2 mutations lead to increased production of the oncometabolite that alters DNA methylation.

(A) Isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) catalyze the conversion of isocitrate to α-ketoglutarate (αKG). αKG, along with Fe(II), is an essential cofactor for TET2 and contributes to the active demethylation of DNA. (B) Mutations in IDH1 and IDH2 alter the affinity of the enzymes for their substrates, leading to conversion of αKG to R-2-hydroxyglutarate (R-2-HG). In addition to αKG depletion, R-2-HG inhibits TET2 and other αKG-dependent enzymes, resulting in DNA hypermethylation. Illustrated by Rachel Davidowitz.
Enasidenib.
Enasidenib (AG-221), a potent, oral, reversible and selective inhibitor of mutant IDH2, has been shown in mouse models of IDH2-mutant leukemia to reduce R-2-HG production and induce differentiation of leukemic cells (62). Treatment reversed aberrant methylation of multiple genes involved in hematopoietic proliferation and differentiation (62). A recent phase 1/2 trial evaluated AG-221 monotherapy in patients with mutant IDH2 and advanced myeloid malignancy. The overall response rate was 40% and 19% achieved CR. For the patients who achieved CR, the median overall survival was almost 20 months (63). Based on these results, the FDA approved enasidenib for patients with relapsed or refractory AML and an IDH2 mutation. Studies of enasidenib in combination with chemotherapy are underway.
AG-120.
AG-120 is similar to AG-221 and is a selective inhibitor of mutant IDH1. In preclinical models, AG-120 reduced R-2-HG levels, inhibited proliferation, and restored differentiation in AML cell lines and primary samples (64). Results from a single-agent dose escalation phase of the first-in-human study are available. As of May 2016, 78 patients with IDH1-mutated hematological malignancy had been treated. The overall response rate was 38.5% (30 of 78) with 17.9% (14) achieving CR (65). The median duration on treatment was 3.2 months. Nine (11.5%) patients remained on therapy. Interestingly, serial DNA sequencing demonstrated that the IDH1 mutant allele fell to undetectable (<1%) levels in three of 11 responders.
IDH305.
IDH305 is a targeted inhibitor of the IDH1 R132 mutant protein. A phase 1 basket study enrolled patients with this mutation in three disease groups — glioma, AML/MDS, and non-CNS solid tumors (66). A total of 81 patients, 24 having hematological malignancy (21 AML, 3 MDS), were enrolled and received IDH305 monotherapy. Objective responses were seen in seven (33%) patients with AML. Responses were reported as durable though clinical updates and pharmacokinetic/dynamic studies are awaited.
Differentiation syndrome with IDH1 and IDH2 inhibitors.
Inhibition of mutant IDH1 or IDH2 leads to differentiation of AML cells and IDH1/2 inhibitors can produce a differentiation syndrome similar to all-trans-retinoic acid (ATRA) treatment in acute promyelocytic leukemia (63, 67). The differentiation syndrome after IDH inhibitors can be severe (leukocytosis requiring hydryoxyurea therapy, hypotension and, in one case, pericardial/pleural effusion requiring admission to intensive care) (67). Like the differentiation syndrome caused by ATRA, differentiation syndrome after treatment with IDH inhibitors is responsive to corticosteroids (68).
Emerging therapies for AML that target dysregulated pathways
Targeting BCL-2 and apoptosis.
While our understanding of the molecular mutations associated with AML has increased dramatically in the last several years, most of these mutations, save for IDH and FLT3, are not directly druggable. Therefore, alternative therapeutic strategies for AML are needed. One approach is to target dysregulated pathways downstream of genetic mutations. Below, we highlight progress in developing inhibitors of BCL-2, DOT1L, and mitochondrial pathways.
The BCL-2 family of proteins play a pivotal role in regulating mitochondria-mediated apoptosis. Different groups of BCL-2 family member proteins, sharing one or more of the four BCL-2 homology (BH) domains (BH1–4), have been identified based on homology to the BCL-2 protein and function. These are the proapoptotic multi-domain effector proteins BAX and BAK, the proapoptotic BH3-only proteins Bid, BIM, and PUMA (the activators), BAD BMF, and NOXA (the sensitizers), and antiapoptotic members BCL-2, BCL-XL, BCL-2–like 2 (BCL-w), MCL-1, and BFL-1/A1. The BCL-2–like proteins maintain cell survival and mitochondrial outer membrane integrity by preventing the activation of BAX and BAK. Under stress conditions, the BH3-only proteins are activated to prompt apoptosis by releasing BAX/BAK from inhibition by the BCL-2–like proteins, or in the case of the activators, by activating BAX/BAK through direct binding. Subsequently, by controlling the permeability of the mitochondrial outer membrane and mitochondrial membrane potential, BAX/BAK regulate the release of proapoptotic factors such as cytochrome c, SMAC, and AIF (69–71).
Of the family members, BCL-2 is the most advanced as a therapeutic target. Preclinical studies of BCL-2 inhibitors have shown activity as single agents and in combination with other antineoplastic agents (72, 73). In addition, BCL-2 is frequently overexpressed in AML compared with normal hematopoietic cells and levels are higher in progenitor/stem cells (74, 75). Finally, using a functional assay termed BH3 profiling, mitochondria from a subset of AML cells are more primed for response to BCL-2 inhibition (76). Although molecular and genetic mutations do not predict for response to BCL-2 inhibitors in vitro, increased BH3 priming predicts for response in vitro and might be a strategy to identify patients who would respond to these therapies.
ABT-737 and its clinical analogue ABT-263 (navitoclax) bind BCL-2, BCL-XL, and BCL-w with high affinity (77). In clinical trials of patients with relapsed or refractory lymphoid malignancies, single-agent navitoclax yielded high response rates (21%–100% depending on specific type of malignancy) and resulted in progression-free survival ranging from 15–25 months, but efficacy was limited by thrombocytopenia resulting from inhibition of BCL-XL (78, 79). Further evaluation of ABT-263 was halted and a more specific inhibitor of BCL-2, venetoclax, was developed. Compared with ABT-263, venetoclax binds BCL-2 with 5-fold higher affinity and binds BCL-XL with 4,800-fold lower affinity, thus reducing the risk of thrombocytopenia. In a phase 2 open-label, multicenter trial of venetoclax monotherapy in patients with high-risk relapsed/refractory AML, the overall response rate was 19%, while an additional 19% demonstrated antileukemic activity not meeting International Working Group (IWG) criteria for response (80). Three of five patients in this study who achieved CR/complete remission with incomplete hematological recovery (CRi) had IDH mutations, consistent with preclinical reports demonstrating that mutations in IDH1/2 increase sensitivity to BCL-2 inhibitors (81). While the efficacy of single-agent venetoclax is limited, encouraging results have been observed in a phase 1b clinical trial that combined venetoclax with hypomethylating agents (azacytidine or decitabine). When tested in patients 65 years or older with newly diagnosed AML, the combination produced a CR/CRi rate of 61% (82). Based on these data, phase 3 studies of the combination are underway.
Targeting MLL
Mixed-lineage leukemia 1 (MLL), also known as KMT2A, regulates the transcription of developmental genes including the HOX genes and thereby controls cellular proliferation and differentiation. Rearrangements of MLL at position 11q23 occur in 5%–10% of acute leukemias of lymphoid, myeloid, or mixed lineage and are associated with a poor prognosis. The vast majority of these rearrangements result in oncogenic fusion proteins that interact with the histone methyltransferase DOT1L (disruptor of telomeric silencing-like). As a result of this abnormal interaction, DOT1L is recruited to MLL target genes where it promotes the hypermethylation of H3K79, leading to aberrant expression of HOXA9 and MEIS1 and leukemic transformation (83, 84). As a strategy to target rearranged MLL, DOT1L inhibitors have been developed and advanced into clinical trial. Preliminary results from an ongoing first-in-human phase 1 clinical trial of single-agent EPZ-5676 for adult patients with relapsed/refractory acute leukemia with rearranged MLL1r (clinicaltrials.gov: NCT01684150) have shown early signs of efficacy (85). Among 37 patients enrolled in the dose escalation trial, there were four responders. As a strategy to target rearranged MLL, DOT1L inhibitors have been developed and advanced into clinical trial. Thus, continued investigation of EPZ-5676 in patients with MLL gene rearrangements is warranted.
Other strategies to target acute leukemia with MLL rearrangements include the use of enhancer of zeste homolog (EZH) 1 and 2 inhibitors or inhibitors of lysine-specific demethylase KDM1A (LSD1), as EZH 1/2 and LSD1 are required for the development of MLL-induced leukemia in mice (86, 87).
Mitochondria-targeted therapies
Recent studies have demonstrated that AML cells and stem cells have a unique reliance on oxidative phosphorylation due, in part, to decreased reserve capacity in their respiratory chains (88, 89). Thus, strategies that target respiratory chain activity or other mitochondrial pathways such as mitochondrial translation, mitochondrial DNA replication, or mitochondrial proteases may have efficacy in AML (88, 90, 91). For example, preclinical studies demonstrated that the antimicrobial tigecycline inhibited mitochondrial protein translation at the mitochondrial ribosome and selectively targeted AML cells in vitro and in vivo (88). In a phase 1 clinical trial of escalating doses of once daily tigecyline in patients with relapsed and refractory AML, no clinical responses were observed; however, the half-life of the drug was shorter than that reported in published literature (92). As a result, inhibition of mitochondrial protein translation was not sustained long enough to reduce levels of mitochondrial proteins. Alternate dosing schedules and drug formulations will be required to fully test the efficacy of this therapeutic strategy.
Emerging immunological therapies for AML
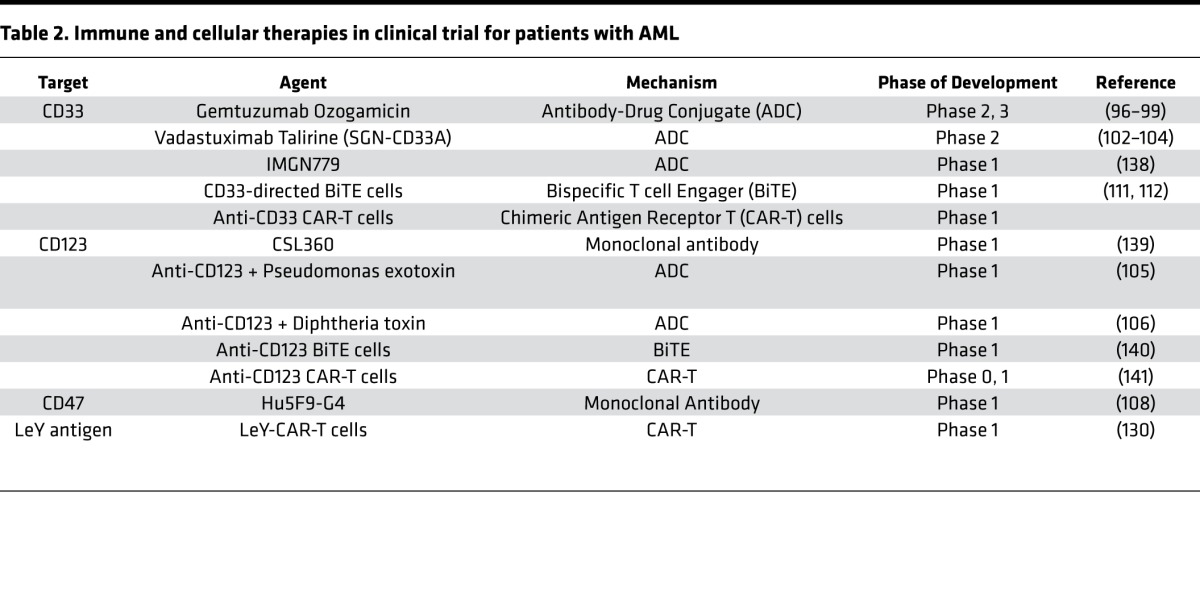
In addition to small-molecule therapeutics, immune- and cell-based therapies for AML are being evaluated in clinical trials with promising results (Table 2).
Table 2. Immune and cellular therapies in clinical trial for patients with AML.
Monoclonal antibodies and antibody-drug conjugates
Anti-CD33.
Sialic acid–binding Ig-like lectin 3 (SIGLEC-3), also known as CD33, is a transmembrane receptor preferentially expressed in myeloid cells. CD33 is frequently overexpressed on leukemic blasts, including the stem cell fraction, suggesting it may be an effective therapeutic target (93). Early studies evaluated unconjugated or naked anti-CD33 monoclonal antibodies (mAbs) in AML and reported preclinical efficacy in vivo due to antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent phagocytosis, and modulatory effects on cytokines in the tumor microenvironment (94). Although effective in preclinical models, unconjugated anti-CD33 was only modestly active in phase 1 and 2 clinical trials. However, greater efficacy was noted when anti-CD33 was conjugated to antitumor antibiotics as in the case of gemtuzumab ozogamicin (GO), where a recombinant, humanized anti-CD33 mAb was conjugated to calicheamicin, an antibiotic with cytotoxic properties (95). In phase 3 studies of GO as monotherapy in patients over age 60 with relapsed AML, an overall response rate of 30% was reported. Based on these data, GO received accelerated approval through the FDA for this indication (96). However, a subsequent multicenter, phase 3, randomized clinical trial comparing induction chemotherapy with and without GO (6 mg/m2) on day 4 of chemotherapy failed to demonstrate differences in survival. In fact, patients receiving GO had a higher rate of mortality during induction due to an increased incidence of veno-occlusive disease (VOD; 5.5% death rate in the combination arm versus 1.4% in the chemotherapy-alone arm) (97). As a result, the drug was voluntarily withdrawn from the market. However, the withdrawal of GO may have been premature. Subsequent randomized trials evaluating lower doses of GO in combination with chemotherapy demonstrated improved overall survival without increased toxicities such as VOD (98, 99). As such, these studies support a role for toxin-conjugated anti-C33 in the treatment AML.
Vadastuximab talirine (SGN-CD33A), a later generation of toxin-conjugated anti-CD33 therapy, is an anti-CD33 mAb conjugated to a pyrrolobenzodiazepine (PBD) dimer (100). Upon internalization into CD33-expressing cells, the PBD dimer is released from the antibody and induces DNA cross-linking and subsequent cell death (101, 102). A phase 1 trial of single-agent SGN-CD33A in 27 older (median age 74 years) treatment-naive AML patients reported responses in 54%, with 14 patients achieving CR/CRi and five achieving a morphologically leukemia-free state (103). Minimal residual disease (MRD) negativity by flow cytometry was noted in six of 13 patients for whom data were available. VOD was not reported with this drug. Current trials are evaluating SGN-CD33A in combination with standard agents in the upfront setting. For example, a trial of SGN-CD33A in combination with decitabine or azacitidine in 24 patients with AML unfit or unwilling for conventional chemotherapy had a response rate (CR+CRi) of 73% among 49 evaluable patients (104). Of note, the combinations were well tolerated with a 30-day mortality rate of 2%.
Anti-CD123.
CD123 (interleukin-3 receptor α [IL-3Rα]) expression is increased in leukemic cells and stem cells compared with normal hematopoietic stem cells and progenitors. CSL360 is a second-generation anti-CD123 mAb with a modified Fc domain to enhance NK cell recruitment and improve ADCC. In early-phase clinical trials, CSL362 produced CR in 10 of 20 evaluable patients with relapsed and refractory AML. Notably, three of six patients became MRD negative after treatment with the antibody. Related therapies under development include CD123 mAb conjugated to Pseudomonas exotoxin (105) as well as IL-3Rα ligand conjugated to diphtheria toxin (106).
Anti-CD47.
CD47 is overexpressed in AML blasts and LSCs, enabling these cells to escape phagocytosis through SIRPα binding (107). Preclinical studies of a CD47-blocking mAb (Hu5F9-G4) have been shown to induce macrophage phagocytosis of AML cells in vitro and in vivo (108). Human phase 1 trials of Hu5F9-G4 and another agent CC-90002 are ongoing (NCT02678338 and NCT02641002). Likewise, fragments of the SIRPα Fc are also in clinical trials for AML and other hematological diseases.
Bispecific T cell–engaging antibodies (BiTEs)
Bispecific T cell–engaging antibodies (BiTEs) are a novel class of biologics constructed by linking two single-chain variable fragments (scFv’s) with a peptide chain, giving them the ability to simultaneously bind the tumor cell and T cell and thereby recruit T cells to the tumor, leading to direct cytotoxicity (109). The CD19/CD3 BiTE construct blinatumomab has demonstrated efficacy in acute lymphocytic leukemia (ALL) and has recently been approved for patients with CD19+ relapsed ALL (110). A CD33/CD3 BiTE antibody has been developed for AML and showed efficacy in preclinical ex vivo studies (111, 112). A phase 1 trial in relapsed/refractory AML is currently underway (NCT02520427). Dual-affinity retargeting (DART) molecules have also been developed and may be potentially more active than BiTEs due to greater efficiency of B and T cell cross-linking (113). Preclinical in vitro and primate models have demonstrated efficacy (114, 115) and phase 1 trials in relapsed/refractory AML and intermediate-2/high-risk MDS are underway (NCT02152956).
Adoptive cellular therapy
Adoptive immunotherapy is based on ex vivo engineering and expansion of immunologically active T cells that are then infused into the recipient with the intention of inducing direct cytotoxicity against tumor cells. These approaches emerged from our understanding of graft versus leukemia (GVL) observed after allotransplant and donor lymphocyte infusions (DLIs) as well as changes in NK cells after allotransplant (116).
NK cell–based therapy has been utilized in the setting of an allotransplant to enhance the GVL effect without compromising graft integrity (117, 118). Advances in cellular processing and increased access to good manufacturing practices–compliant (GMP-compliant) laboratories has enabled ex vivo activation and expansion (using IL-2 or IL-15) of donor-derived NK cells for use in patients with active disease (119). These strategies have been limited by donor availability and the risk of graft-versus-host disease (GVHD) (120). The advent of off-the-shelf products such as the NK-92 and KHYG-1 cell lines is a promising development, with demonstrable safety in a variety of cancers including hematological malignancies (121, 122).
Preclinical studies of donor-derived CD3+CD4–CD8– double-negative T cells (DNTs) have shown dose-dependent cytotoxicity against AML cells in vitro as well as in murine xenograft models (123). Cytotoxicity occurs primarily through a perforin-dependent pathway and DNTs derived from AML patients in CR can be effectively expanded ex vivo (124). A first-in-human study of the safety and tolerability of healthy donor-derived DNTs as adjuvant therapy in AML patients is expected to begin recruitment shortly (NCT03027102).
T cells with engineered chimeric antigen receptors (CAR-T cells) have shown remarkable activity in B cell malignancies (125, 126). CAR-T cells possess chimeric immunoreceptors targeted to a tumor cell surface antigen (such as CD19 in B cell diseases) in addition to costimulatory domains like CD28 in second-generation CAR-T cells (127). Despite the success of CD19-CAR-T cells, studies of CD33- or CD123-targeted CAR-T cells in AML have been less prominent, primarily owing to profound myeloablation (128, 129). Thus, alternative CAR-T cell targets are being sought for AML. Examples of newer CAR-T products for AML include cells directed to the LeY antigen. In a phase 1 trial, LeY-CAR-T cells reduced peripheral blood and marrow blasts without profound myelosuppression in a small phase 1 trial in relapsed AML (130). Future roles of CAR-T cells in AML may involve their use as conditioning prior to allotransplant or by employing gene-edited donor stem cells (131).
Cytokine release syndrome
Cytokine release syndrome (CRS) is a unique toxicity associated with immunotherapies such as CAR-T cells, BiTES, and DARTs. CRS is characterized by high fevers, increased inflammatory biomarkers, and multi-organ dysfunction (including cerebral edema). CRS can be severe and require ICU support. Current management of CRS includes intensive supportive care, corticosteroids, and tocilizumab, an mAb that inhibits the IL-6 receptor (132).
Future directions in immunotherapy: Tumor vaccines and checkpoint inhibitors
Peptide vaccines against PR1, WT1, and CD168 have demonstrated immunological responses in early-study settings and may have a role in postremission therapy (133). Dendritic cell vaccines may prove to be more effective by overcoming some of the limitations of peptide vaccines, although clinical benefit has been minimal in trial settings. Potential use as a postremission therapy to prevent or delay relapse is under investigation (134). Finally, PD-1 and PD-L1 immune check point inhibitors that have efficacy in solid tumors such as melanoma may also have efficacy alone or in combination with hypomethylating agents in AML and MDS patients (135).
Conclusions and future directions
Genetic studies have significantly expanded our knowledge of the molecular mutations that underlie AML and have led to better understanding of AML biology and the development of novel therapeutics. However, many patients are not eligible for these targeted therapies, as most AML mutations are not currently druggable. As such, additional therapeutic approaches are urgently needed.
While new drugs are improving outcomes for some patients with AML, most patients either do not respond or ultimately relapse. As such, it will be important to identify biomarkers that can best predict patients’ responses. In addition, the lower rates of remission and high rates of relapse with these newer agents suggest that the leukemic subclones driving relapse are not targeted by monotherapy. Genetic studies of matched leukemic samples collected at diagnosis and relapse demonstrated that the genetic clones responsible for relapse are often present at very low frequency in the diagnostic sample (17). In other cases, the AML clones responsible for relapse are quiescent LSCs (136, 137). As we improve our understanding of the origins of relapse, we will increase our ability to identify and monitor the genetic clones and stem cells that cause relapse. In the future, we predict that therapy will continue postremission until these clones are eliminated and that new therapies that are more effective at eradicating the clones responsible for relapse will be developed.
In developing new therapeutic strategies for AML, extrinsic factors that are important for the pathogenesis and progression of AML, such as the immune environment, must also be considered. A variety of immune and cellular strategies have been developed and have entered clinical studies for AML. Given the success of immunotherapy in other hematological malignancies and solid tumors, we anticipate that immune therapies will also play an important role in the treatment of AML.
Until recently, induction therapy for AML had been unchanged for decades. We are now seeing the fundamental discoveries related to AML biology being translated into new therapies and drugs, such as midostaurin, being combined with upfront standard chemotherapy to improve patient survival. As our ability to produce long-term remissions and cures in AML improves, greater attention will focus on the late effects of chemotherapy in AML survivors, similar to the situation in childhood acute lymphoblastic leukemia. To address the late effects, we predict that future therapeutic strategies will focus on developing less toxic induction regimens, and for some patients might ultimately lead to treatment with combinations of novel agents without standard cytotoxic chemotherapy. Likewise, we predict improvements in supportive care will reduce the short-term and long-term toxicity of chemotherapy. Thus, advanced in AML biology is leading to a surge in novel therapeutics for this disease.
Acknowledgments
This work was supported by the Leukemia and Lymphoma Society, the Canadian Institutes of Health Research, the Princess Margaret Cancer Centre Foundation, and the Ministry of Long Term Health and Planning in the Province of Ontario. ADS holds the Barbara Baker Chair in Leukemia and Related Diseases.
Version 1. 09/21/2017
Electronic publication
Footnotes
Conflict of interest: S. Chan has received research support from Celgene Corporation and AbbVie Corporation. A.D. Schimmer has received an honorarium from Novartis Pharmaceuticals and research support from Medivir and Takeda Pharmaceuticals.
Published: September 21, 2017
Reference information: JCI Insight. 2017;2(18):e95679. https://doi.org/10.1172/jci.insight.95679.
Contributor Information
Simon Kavanagh, Email: simon.kavanagh@uhn.ca.
Tracy Murphy, Email: tracy.murphy@uhn.ca.
Arjun Law, Email: arjun.law@uhn.ca.
Dana Yehudai, Email: Dana.Yehudai@uhn.ca.
Jenny M. Ho, Email: Jenny.Ho@uhn.ca.
References
- 1.Arber DA, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 2.Medeiros BC, Satram-Hoang S, Hurst D, Hoang KQ, Momin F, Reyes C. Big data analysis of treatment patterns and outcomes among elderly acute myeloid leukemia patients in the United States. Ann Hematol. 2015;94(7):1127–1138. doi: 10.1007/s00277-015-2351-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 4.Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127(1):42–52. doi: 10.1182/blood-2015-07-604512. [DOI] [PubMed] [Google Scholar]
- 5.Papaemmanuil E, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walter MJ, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Döhner H, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eisfeld AK, et al. The mutational oncoprint of recurrent cytogenetic abnormalities in adult patients with de novo acute myeloid leukemia. Leukemia. doi: 10.1038/leu.2017. [published online ahead of print April 18, 2017]. https://doi.org/10.1038/leu.2017.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rau R, Brown P. Nucleophosmin (NPM1) mutations in adult and childhood acute myeloid leukaemia: towards definition of a new leukaemia entity. Hematol Oncol. 2009;27(4):171–181. doi: 10.1002/hon.904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ho PA, et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2009;113(26):6558–6566. doi: 10.1182/blood-2008-10-184747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindsley RC, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376. doi: 10.1182/blood-2014-11-610543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerstung M, et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat Genet. 2017;49(3):332–340. doi: 10.1038/ng.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shlush LI, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Corces-Zimmerman MR, Hong WJ, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111(7):2548–2553. doi: 10.1073/pnas.1324297111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krönke J, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122(1):100–108. doi: 10.1182/blood-2013-01-479188. [DOI] [PubMed] [Google Scholar]
- 18.McKerrell T, et al. Leukemia-associated somatic mutations drive distinct patterns of age-related clonal hemopoiesis. Cell Rep. 2015;10(8):1239–1245. doi: 10.1016/j.celrep.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jaiswal S, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Busque L, et al. Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet. 2012;44(11):1179–1181. doi: 10.1038/ng.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie M, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. doi: 10.1038/ncomms12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Steensma DP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. doi: 10.1182/blood-2015-03-631747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gibson CJ, et al. Clonal hematopoiesis associated with adverse outcomes after autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35(14):1598–1605. doi: 10.1200/JCO.2016.71.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng SW, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 26.Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003;17(9):1738–1752. doi: 10.1038/sj.leu.2403099. [DOI] [PubMed] [Google Scholar]
- 27.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100(5):1532–1542. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 28.Nakao M, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10(12):1911–1918. [PubMed] [Google Scholar]
- 29.Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Genomic structure of human FLT3: implications for mutational analysis. Br J Haematol. 2001;113(4):1076–1077. doi: 10.1046/j.1365-2141.2001.02821.x. [DOI] [PubMed] [Google Scholar]
- 30.Marcucci G, Haferlach T, Döhner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29(5):475–486. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]
- 31.Schlenk RF, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124(23):3441–3449. doi: 10.1182/blood-2014-05-578070. [DOI] [PubMed] [Google Scholar]
- 32.Abu-Duhier FM, Goodeve AC, Wilson GA, Care RS, Peake IR, Reilly JT. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br J Haematol. 2001;113(4):983–988. doi: 10.1046/j.1365-2141.2001.02850.x. [DOI] [PubMed] [Google Scholar]
- 33.Yamamoto Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97(8):2434–2439. doi: 10.1182/blood.V97.8.2434. [DOI] [PubMed] [Google Scholar]
- 34.Kumar R, et al. Myelosuppression and kinase selectivity of multikinase angiogenesis inhibitors. Br J Cancer. 2009;101(10):1717–1723. doi: 10.1038/sj.bjc.6605366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Auclair D, et al. Antitumor activity of sorafenib in FLT3-driven leukemic cells. Leukemia. 2007;21(3):439–445. doi: 10.1038/sj.leu.2404508. [DOI] [PubMed] [Google Scholar]
- 36.Crump M, et al. A randomized phase I clinical and biologic study of two schedules of sorafenib in patients with myelodysplastic syndrome or acute myeloid leukemia: a NCIC (National Cancer Institute of Canada) Clinical Trials Group Study. Leuk Lymphoma. 2010;51(2):252–260. doi: 10.3109/10428190903585286. [DOI] [PubMed] [Google Scholar]
- 37.Serve H, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31(25):3110–3118. doi: 10.1200/JCO.2012.46.4990. [DOI] [PubMed] [Google Scholar]
- 38.Röllig C, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16(16):1691–1699. doi: 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 39.Stone RM, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin O, et al. Pharmacokinetics (PK) and pharmacodynamics (PD) of midostaurin (PKC412) in patients with acute myeloid leukemia (AML) J Clin Oncol. 2008;26(15_suppl):7064. doi: 10.1200/jco.2008.26.15_suppl.7064. [DOI] [Google Scholar]
- 41.Levis M, et al. Pharmacokinetic and pharmacodynamic studies of lestaurtinib (CEP-701) and PKC-412: cytotoxicity is often dependent on non-FLT3-mediated effects. Blood. 2005;106(11):2463 [Google Scholar]
- 42.Muellner MK, et al. Targeting a cell state common to triple-negative breast cancers. Mol Syst Biol. 2015;11(1):789. doi: 10.15252/msb.20145664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peter B, et al. Target interaction profiling of midostaurin and its metabolites in neoplastic mast cells predicts distinct effects on activation and growth. Leukemia. 2016;30(2):464–472. doi: 10.1038/leu.2015.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu J, et al. Anti-tumor activity of TAK-659, a dual inhibitor of SYK and FLT-3 kinases, in AML models. J Clin Oncol. 2016;34(15_suppl):e14091 [Google Scholar]
- 45.Zarrinkar PP, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114(14):2984–2992. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cortes JE, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–3687. doi: 10.1200/JCO.2013.48.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levis MJ, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia after second-line chemotherapy or hematopoietic stem cell transplantation. Blood. 2012;120(21):673 [Google Scholar]
- 48.Schiller GJ, et al. Final results of a randomized phase 2 study showing the clinical benefit of quizartinib (AC220) in patients with FLT3-ITD positive relapsed or refractory acute myeloid leukemia. J Clin Oncol. 2014;32(1 5_suppl):7100 [Google Scholar]
- 49.Lee LY, et al. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2017;129(2):257–260. doi: 10.1182/blood-2016-10-745133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ben-Batalla I, et al. Axl, a prognostic and therapeutic target in acute myeloid leukemia mediates paracrine crosstalk of leukemia cells with bone marrow stroma. Blood. 2013;122(14):2443–2452. doi: 10.1182/blood-2013-03-491431. [DOI] [PubMed] [Google Scholar]
- 51.Levis MJ, et al. Results of a first-in-human, phase I/II trial of ASP2215, a selective, potent inhibitor of FLT3/Axl in patients with relapsed or refractory (R/R) acute myeloid leukemia (AML) J Clin Oncol. 2015;33(15_suppl):7003 [Google Scholar]
- 52.Smith CC, et al. Pharmacokinetic profile and pharmacodynamic effects of ASP2215, a selective, potent inhibitor of FLT3/AXL, in patients with relapsed or refractory acute myeloid leukemia: results from a first-in-human phase 1/2 study. Blood. 2015;126(23):4836 [Google Scholar]
- 53.Heidel F, et al. Clinical resistance to the kinase inhibitor PKC412 in acute myeloid leukemia by mutation of Asn-676 in the FLT3 tyrosine kinase domain. Blood. 2006;107(1):293–300. doi: 10.1182/blood-2005-06-2469. [DOI] [PubMed] [Google Scholar]
- 54.Smith CC, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485(7397):260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.von Bubnoff N, Engh RA, Aberg E, Sänger J, Peschel C, Duyster J. FMS-like tyrosine kinase 3-internal tandem duplication tyrosine kinase inhibitors display a nonoverlapping profile of resistance mutations in vitro. Cancer Res. 2009;69(7):3032–3041. doi: 10.1158/0008-5472.CAN-08-2923. [DOI] [PubMed] [Google Scholar]
- 56.Zimmerman EI, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122(22):3607–3615. doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Galanis A, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123(1):94–100. doi: 10.1182/blood-2013-10-529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wander SA, Levis MJ, Fathi AT. The evolving role of FLT3 inhibitors in acute myeloid leukemia: quizartinib and beyond. Ther Adv Hematol. 2014;5(3):65–77. doi: 10.1177/2040620714532123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Park IK, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29(12):2382–2389. doi: 10.1038/leu.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Figueroa ME, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18(6):553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu C, et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature. 2012;483(7390):474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shih AH, et al. AG-221, a small molecule mutant IDH2 inhibitor, remodels the epigenetic state of IDH2-mutant cells and induces alterations in self-renewal/differentiation in IDH2-mutant AML model in vivo. Blood. 2014;124(21):437 [Google Scholar]
- 63.Stein EM, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hansen E, et al. AG-120, an oral, selective, first-in-class, potent inhibitor of mutant IDH1, reduces intracellular 2HG and induces cellular differentiation in TF-1 R132H cells and primary human IDH1 mutant AML patient samples treated ex vivo. Blood. 2014;124(21):3734 [Google Scholar]
- 65.DiNardo CD, et al. Determination of IDH1 mutational burden and clearance via next-generation sequencing in patients with IDH1 mutation-positive hematologic malignancies receiving AG-120, a first-in-class inhibitor of mutant IDH1. Blood. 2016;128(22):1070 [Google Scholar]
- 66.DiNardo CD, et al. A phase I study of IDH305 in patients with advanced malignancies including relapsed/refractory AML and MDS that harbor IDH1R132 mutations. Blood. 2016;128(22):1073 [Google Scholar]
- 67.Birendra KC, DiNardo CD. Evidence for clinical differentiation and differentiation syndrome in patients with acute myeloid leukemia and IDH1 mutations treated with the targeted mutant IDH1 inhibitor, AG-120. Clin Lymphoma Myeloma Leuk. 2016;16(8):460–465. doi: 10.1016/j.clml.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fathi A, et al. Differentiation syndrome associated with enasidenib, a selective inhibitor of mutant isocitrate dehydrogenase 2 (mIDH2) J Clin Oncol. 2017;35(15_suppl):7015. doi: 10.1001/jamaoncol.2017.4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McClintock DS, et al. Bcl-2 family members and functional electron transport chain regulate oxygen deprivation-induced cell death. Mol Cell Biol. 2002;22(1):94–104. doi: 10.1128/MCB.22.1.94-104.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ. From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov. 2017;16(4):273–284. doi: 10.1038/nrd.2016.253. [DOI] [PubMed] [Google Scholar]
- 71.Bhola PD, Letai A. Mitochondria-judges and executioners of cell death sentences. Mol Cell. 2016;61(5):695–704. doi: 10.1016/j.molcel.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pan R, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4(3):362–375. doi: 10.1158/2159-8290.CD-13-0609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kohl TM, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21(8):1763–1772. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- 74.Porwit-MacDonald A, Ivory K, Wilkinson S, Wheatley K, Wong L, Janossy G. Bcl-2 protein expression in normal human bone marrow precursors and in acute myelogenous leukemia. Leukemia. 1995;9(7):1191–1198. [PubMed] [Google Scholar]
- 75.Andreeff M, et al. Expression of Bcl-2-related genes in normal and AML progenitors: changes induced by chemotherapy and retinoic acid. Leukemia. 1999;13(11):1881–1892. doi: 10.1038/sj.leu.2401573. [DOI] [PubMed] [Google Scholar]
- 76.Vo TT, et al. Relative mitochondrial priming of myeloblasts and normal HSCs determines chemotherapeutic success in AML. Cell. 2012;151(2):344–355. doi: 10.1016/j.cell.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435(7042):677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 78.Roberts AW, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488–496. doi: 10.1200/JCO.2011.34.7898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wilson WH, et al. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010;11(12):1149–1159. doi: 10.1016/S1470-2045(10)70261-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Konopleva M, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov. 2016;6(10):1106–1117. doi: 10.1158/2159-8290.CD-16-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chan SM, et al. Isocitrate dehydrogenase 1 and 2 mutations induce BCL-2 dependence in acute myeloid leukemia. Nat Med. 2015;21(2):178–184. doi: 10.1038/nm.3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pollyea DA, et al. Results of a phase 1b study of venetoclax plus decitabine or azacitidine in untreated acute myeloid leukemia patients ≥ 65 years ineligible for standard induction therapy. J Clin Oncol. 2016;34(15_suppl):7009 [Google Scholar]
- 83.Okada Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121(2):167–178. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 84.Nguyen AT, Taranova O, He J, Zhang Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood. 2011;117(25):6912–6922. doi: 10.1182/blood-2011-02-334359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Stein EM, et al. A phase 1 study of the DOT1L inhibitor, pinometostat (EPZ-5676), in adults with relapsed or refractory leukemia: safety, clinical activity, exposure and target inhibition. Presented at: ASH 57th Annual Meeting and Exposition; December 6, 2015; Orlando, Florida, USA. Abstract 2547. https://ash.confex.com/ash/2015/webprogramscheduler/Paper78493.html.
- 86.Neff T, et al. Polycomb repressive complex 2 is required for MLL-AF9 leukemia. Proc Natl Acad Sci USA. 2012;109(13):5028–5033. doi: 10.1073/pnas.1202258109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Harris WJ, et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell. 2012;21(4):473–487. doi: 10.1016/j.ccr.2012.03.014. [DOI] [PubMed] [Google Scholar]
- 88.Skrtić M, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20(5):674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sriskanthadevan S, et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood. 2015;125(13):2120–2130. doi: 10.1182/blood-2014-08-594408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cole A, et al. Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2015;27(6):864–876. doi: 10.1016/j.ccell.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liyanage SU, et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood. 2017;129(19):2657–2666. doi: 10.1182/blood-2016-10-741207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Reed GA, et al. A phase 1 study of intravenous infusions of tigecycline in patients with acute myeloid leukemia. Cancer Med. 2016;5(11):3031–3040. doi: 10.1002/cam4.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ehninger A, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. 2014;4:e218. doi: 10.1038/bcj.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sutherland MK, et al. Anti-leukemic activity of lintuzumab (SGN-33) in preclinical models of acute myeloid leukemia. MAbs. 2009;1(5):481–490. doi: 10.4161/mabs.1.5.9288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Der Velden VH, et al. Targeting of the CD33-calicheamicin immunoconjugate Mylotarg (CMA-676) in acute myeloid leukemia: in vivo and in vitro saturation and internalization by leukemic and normal myeloid cells. Blood. 2001;97(10):3197–3204. doi: 10.1182/blood.V97.10.3197. [DOI] [PubMed] [Google Scholar]
- 96.Bross PF, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. 2001;7(6):1490–1496. [PubMed] [Google Scholar]
- 97.Petersdorf SH, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. 2013;121(24):4854–4860. doi: 10.1182/blood-2013-01-466706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Burnett AK, et al. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: results of the MRC AML15 trial. J Clin Oncol. 2011;29(4):369–377. doi: 10.1200/JCO.2010.31.4310. [DOI] [PubMed] [Google Scholar]
- 99.Burnett AK, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J Clin Oncol. 2012;30(32):3924–3931. doi: 10.1200/JCO.2012.42.2964. [DOI] [PubMed] [Google Scholar]
- 100.Sutherland MSK, et al. SGN-CD33A: A novel CD33-directed antibody-drug conjugate, utilizing pyrrolobenzodiazepine dimers, demonstrates preclinical antitumor activity against multi-drug resistant human AML. Blood. 2012;120(21):3589 [Google Scholar]
- 101.Smellie M, Bose DS, Thompson AS, Jenkins TC, Hartley JA, Thurston DE. Sequence-selective recognition of duplex DNA through covalent interstrand cross-linking: kinetic and molecular modeling studies with pyrrolobenzodiazepine dimers. Biochemistry. 2003;42(27):8232–8239. doi: 10.1021/bi034313t. [DOI] [PubMed] [Google Scholar]
- 102.Kung Sutherland MS, et al. SGN-CD33A: a novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood. 2013;122(8):1455–1463. doi: 10.1182/blood-2013-03-491506. [DOI] [PubMed] [Google Scholar]
- 103.Bixby DL, et al. Vadastuximab talirine monotherapy in older patients with treatment naive CD33-positive acute myeloid leukemia (AML) Blood. 2016;128(22):590. doi: 10.1182/blood-2017-06-789800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fathi AT, et al. Vadastuximab talirine plus hypomethylating agents: a well-tolerated regimen with high remission rate in frontline older patients with acute myeloid leukemia (AML) Blood. 2016;128(22):591 [Google Scholar]
- 105.Han L, et al. Antileukemia efficacy and mechanisms of action of SL-101, a novel anti-CD123 antibody conjugate, in acute myeloid leukemia. Clin Cancer Res. 2017;23(13):3385–3395. doi: 10.1158/1078-0432.CCR-16-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Frankel AE, et al. Activity of SL-401, a targeted therapy directed to interleukin-3 receptor, in blastic plasmacytoid dendritic cell neoplasm patients. Blood. 2014;124(3):385–392. doi: 10.1182/blood-2014-04-566737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jaiswal S, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138(2):271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu J, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE. 2015;10(9):e0137345. doi: 10.1371/journal.pone.0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Baeuerle PA, Kufer P, Bargou R. BiTE: Teaching antibodies to engage T-cells for cancer therapy. Curr Opin Mol Ther. 2009;11(1):22–30. [PubMed] [Google Scholar]
- 110.Kantarjian H, et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–847. doi: 10.1056/NEJMoa1609783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Aigner M, et al. T lymphocytes can be effectively recruited for ex vivo and in vivo lysis of AML blasts by a novel CD33/CD3-bispecific BiTE antibody construct. Leukemia. 2013;27(5):1107–1115. doi: 10.1038/leu.2012.341. [DOI] [PubMed] [Google Scholar]
- 112.Laszlo GS, et al. Cellular determinants for preclinical activity of a novel CD33/CD3 bispecific T-cell engager (BiTE) antibody, AMG 330, against human AML. Blood. 2014;123(4):554–561. doi: 10.1182/blood-2013-09-527044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Rader C. DARTs take aim at BiTEs. Blood. 2011;117(17):4403–4404. doi: 10.1182/blood-2011-02-337691. [DOI] [PubMed] [Google Scholar]
- 114.Chichili GR, et al. A CD3xCD123 bispecific DART for redirecting host T cells to myelogenous leukemia: preclinical activity and safety in nonhuman primates. Sci Transl Med. 2015;7(289):289ra82. doi: 10.1126/scitranslmed.aaa5693. [DOI] [PubMed] [Google Scholar]
- 115.Al-Hussaini M, et al. Targeting CD123 in acute myeloid leukemia using a T-cell-directed dual-affinity retargeting platform. Blood. 2016;127(1):122–131. doi: 10.1182/blood-2014-05-575704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Warren EH, Deeg HJ. Dissecting graft-versus-leukemia from graft-versus-host-disease using novel strategies. Tissue Antigens. 2013;81(4):183–193. doi: 10.1111/tan.12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Passweg JR, et al. Purified donor NK-lymphocyte infusion to consolidate engraftment after haploidentical stem cell transplantation. Leukemia. 2004;18(11):1835–1838. doi: 10.1038/sj.leu.2403524. [DOI] [PubMed] [Google Scholar]
- 118.Lee DA, et al. Haploidentical natural killer cells infused before allogeneic stem cell transplantation for myeloid malignancies: a phase I trial. Biol Blood Marrow Transplant. 2016;22(7):1290–1298. doi: 10.1016/j.bbmt.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Knorr DA, Bachanova V, Verneris MR, Miller JS. Clinical utility of natural killer cells in cancer therapy and transplantation. Semin Immunol. 2014;26(2):161–172. doi: 10.1016/j.smim.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shah NN, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125(5):784–792. doi: 10.1182/blood-2014-07-592881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tonn T, Becker S, Esser R, Schwabe D, Seifried E. Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92. J Hematother Stem Cell Res. 2001;10(4):535–544. doi: 10.1089/15258160152509145. [DOI] [PubMed] [Google Scholar]
- 122.Suck G, Branch DR, Keating A. Irradiated KHYG-1 retains cytotoxicity: potential for adoptive immunotherapy with a natural killer cell line. Int J Radiat Biol. 2006;82(5):355–361. doi: 10.1080/09553000600649653. [DOI] [PubMed] [Google Scholar]
- 123.Lee JB, et al. Efficacy and safety of allogeneic double negative T cell as a cellular therapy for AML and its underlying mechanism. Blood. 2015;126(23):1355 [Google Scholar]
- 124.Merims S, et al. Anti-leukemia effect of ex vivo expanded DNT cells from AML patients: a potential novel autologous T-cell adoptive immunotherapy. Leukemia. 2011;25(9):1415–1422. doi: 10.1038/leu.2011.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Maude SL, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kochenderfer JN, et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J Clin Oncol. 2015;33(6):540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao Z, et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Gill S, et al. Preclinical targeting of human acute myeloid leukemia and myeloablation using chimeric antigen receptor-modified T cells. Blood. 2014;123(15):2343–2354. doi: 10.1182/blood-2013-09-529537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kenderian SS, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29(8):1637–1647. doi: 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ritchie DS, et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol Ther. 2013;21(11):2122–2129. doi: 10.1038/mt.2013.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Qasim W, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9(374):eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 132.Fitzgerald JC, et al. Cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukemia. Crit Care Med. 2017;45(2):e124–e131. doi: 10.1097/CCM.0000000000002053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rezvani K, et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111(1):236–242. doi: 10.1182/blood-2007-08-108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Weinstock M, Rosenblatt J, Avigan D. Dendritic cell therapies for hematologic malignancies. Mol Ther Methods Clin Dev. 2017;5:66–75. doi: 10.1016/j.omtm.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. doi: 10.1080/10428194.2017.1344905. Boddu P, Kantarjian H, Garcia-Manero G, Allison J, Sharma P, Daver N. The emerging role of immune checkpoint based approaches in AML and MDS. Leuk Lymphoma. 2017:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ishikawa F, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–1321. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- 137.Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129(12):1577–1585. doi: 10.1182/blood-2016-10-696054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Whiteman KR, et al. The antibody-drug conjugate (ADC) IMGN779 is highly active in vitro and in vivo against acute myeloid leukemia (AML) with FLT3-ITD mutations. Blood. 2014;124(21):2321 [Google Scholar]
- 139.Busfield SJ, et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia. 2014;28(11):2213–2221. doi: 10.1038/leu.2014.128. [DOI] [PubMed] [Google Scholar]
- 140.Bonifant CL, et al. CD123-engager T cells as a novel immunotherapeutic for acute myeloid leukemia. Mol Ther. 2016;24(9):1615–1626. doi: 10.1038/mt.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tettamanti S, et al. Targeting of acute myeloid leukaemia by cytokine-induced killer cells redirected with a novel CD123-specific chimeric antigen receptor. Br J Haematol. 2013;161(3):389–401. doi: 10.1111/bjh.12282. [DOI] [PubMed] [Google Scholar]