

Abstract
The role of B cells and posttranslational modifications in pathogenesis of organ-specific immune diseases is increasingly envisioned but remains poorly understood, particularly in human disorders. In celiac disease, transglutaminase 2–modified (TG2-modified; deamidated) gluten peptides drive disease-specific T cell and B cell responses, and antibodies to deamidated gluten peptides are excellent diagnostic markers. Here, we substantiate by high-throughput sequencing of IGHV genes that antibodies to a disease-specific, deamidated, and immunodominant B cell epitope of gluten (PLQPEQPFP) have biased and stereotyped usage of IGHV3-23 and IGHV3-15 gene segments with modest somatic mutations. X-ray crystal structures of 2 prototype IGHV3-15/IGKV4-1 and IGHV3-23/IGLV4-69 antibodies reveal peptide interaction mainly via germline-encoded residues. In-depth mutational analysis showed restricted selection and substitution patterns at positions involved in antigen binding. While the IGHV3-15/IGKV4-1 antibody interacts with Glu5 and Gln6, the IGHV3-23/IGLV4-69 antibody interacts with Gln3, Pro4, Pro7, and Phe8 — residues involved in substrate recognition by TG2. Hence, both antibodies, despite different interaction with the epitope, recognize signatures of TG2 processing that facilitates B cell presentation of deamidated gluten peptides to T cells, thereby providing a molecular framework for the generation of these clinically important antibodies. The study provides essential insight into the pathogenic mechanism of celiac disease.
Keywords: Autoimmunity
Keywords: Autoimmune diseases, Immunoglobulins
Antibodies to a celiac disease–specific, deamidated epitope of gluten have biased and stereotyped usage of IGHV gene segments due to antigen interaction with germline-encoded residues.
Introduction
Celiac disease (CD) is an immune-mediated disorder with autoimmune features caused by immune reactions to dietary gluten proteins from wheat (gliadin and glutenin), barley (hordein), and rye (secalin) (1). CD has strong genetic basis, and the MHC locus with certain HLA-DQA1 and HLA-DQB1 alleles encoding HLA-DQ2 and HLA-DQ8 molecules is the chief genetic risk factor (2). Hallmarks of the disease are gluten-reactive CD4 T cells that recognize deamidated gluten peptides in the context of the HLA-DQ2 or HLA-DQ8 molecules (3), as well as antibodies to the autoantigen transglutaminase 2 (TG2) (4) and to deamidated gluten peptides (5). Both the anti-TG2 antibodies and the anti-deamidated gluten peptide antibodies are diagnostic markers for CD with extraordinary disease specificity and sensitivity (6, 7). TG2 is not only involved in CD by being the target of autoantibodies, but it is also responsible for the deamidation of gluten peptides rendering them immunogenic for CD4 T cells (8, 9). The TG2 enzyme targets Gln residues in polypeptides in a sequence-specific manner (i.e., QXP[F]) and converts them to Glu (deamidation) or crosslinks them to primary amines (transamidation) (10–12). The T cell response to deamidated gluten peptides and the B cell response to TG2 and deamidated gluten are likely mechanistically linked. Both B cells reactive with deamidated gluten and B cells reactive with TG2, the latter via B cell receptor–mediated (BCR-mediated) uptake of TG2-gluten peptide complexes, may serve as antigen presenting cells for gluten-reactive CD4 T cells and thereby engage an amplifying loop for the pathogenic T cell response (3, 13).
A role for posttranslational modifications in the pathogenesis of organ-specific immune disorders is increasingly envisioned (14), not least because of the highly disease-specific antibodies to citrullinated protein antigens in rheumatoid arthritis (15, 16). Still, many issues remain unsolved. In rheumatoid arthritis, tellingly, the question of which enzyme is involved in the citrullination (i.e., bacterial or human peptidylarginine deiminases) and which T cell epitopes are involved in the antibody formation are still elusive (17). The work from CD, including this report on the detailed molecular basis for antibody recognition of posttranslationally modified antigen, illustrates how critical insight can be obtained.
We recently isolated plasma cells (PCs) of celiac gut lesions reactive with deamidated gluten peptides, and we generated from such cells a panel of 38 human monoclonal antibodies (hmAbs) (18). Many of the hmAbs were derived from PCs that were selected with a peptide containing the sequence PLQPEQPFP (hereafter termed DGP) that harbors the sequence QPEQPF; a previously identified highly disease-specific and immunodominant B cell epitope in CD (5). In general, the 38 gluten-reactive hmAbs expressed IGHV genes with relatively few somatic mutations — a characteristic shared with TG2 antibodies of CD patients (19, 20). Furthermore, among the 38 hmAbs, there was a restricted usage of VH and VL pairs, with PLQPEQPFP-reactive hmAbs typically using the IGHV3-23/IGLV4-69 and IGHV3-15/IGKV4-1 genes (18). Additionally, pull-down and characterization of peptides from complex proteolytic digests of gluten treated with TG2 suggested that the core epitope–recognized -QPEQPFP- is only found within or in proximity of many different CD4 T cell epitopes in gluten proteins (21). This suggests that the B cell response toward dietary gluten in CD is connected to the antigluten T cell response and that the response to DGP plays a central role in the immune reaction to deamidated gluten.
In this study, we have further dissected the CD-specific antibody response to DGP. We first did high-throughput sequencing (HTS) of IGHV genes of PCs from 10 CD patients with active disease. We observed that PCs reactive with the DGP had fewer mutations in the IGHV genes than PCs not reacting with this peptide. These gluten-specific PCs were clonally expanded with a restricted IGHV repertoire and over-usage of the IGHV3-15 and IGHV3-23 gene segments. We then solved the structural basis for recognition of the peptide by 2 prototype antibodies encoded by IGHV3-23/IGLV4-69 (hmAb UCD1002-1E01) and IGHV3-15/IGKV4-1 (hmAb UCD1002-1E03), hereafter termed 1E01 and 1E03, respectively. The structural insight of antibody recognition was used to reinterrogate the HTS sequence data, which together provide insight into the scheme by which CD-specific antibodies are selected in response to an immunodominant and deamidated gluten epitope. Interestingly both antibodies, despite different epitope interaction, recognize signatures of TG2 processing that would allow both B cells to take up and present deamidated gluten peptides to T cells. The study helps to understand, at the molecular level, how the antibody response to post-translationally modified gluten peptides develop in CD.
Results
HTS reveals selection of a stereotypic antibody response to DGP
In order to comprehensively analyze the antibody response to DGP, we undertook HTS of IGHV genes of DGP-reactive and non–DGP-reactive PCs (likely reactive with gut commensals and pathogens) from 10 untreated CD patients. Single cell suspensions of small intestine biopsies were stained and analyzed by flow cytometry. The frequency of DGP-reactive IgA PCs ranged between 0.2% and 2%. DGP-reactive and non–DGP-reactive IgA PCs were sorted, and amplicon libraries were prepared, sequenced on Illumina MiSeq platform, and analyzed. The IGHV repertoire of DGP-reactive PCs was relatively narrow, with a preferential usage the IGHV3-23 and IGHV3-15 gene segments (Figure 1A). There was some variation between individual patients; most patients predominantly used IGHV3-23 (as depicted in Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.93961DS1); however, in one subject (CD1195), IGHV3-15 strongly dominated the response (Supplemental Figure 1B). Furthermore, DGP-reactive PCs had significantly lower numbers of somatic mutations (Figure 1B), as well as lower clonal diversity in comparison with non–DGP-reactive PCs, as measured by Hill’s diversity index (Figure 1C; ref. 22). The lower diversity (qD) values observed for DGP-reactive PCs indicated that this population consisted of clones that were larger than non–DGP-reactive PCs. The finding of consistent and biased usage of IGHV3-23 and IGHV3-15 across CD patients confirms and extends the results obtained by analysis of 38 single celiac gut PCs (18) and clearly demonstrates that there exists a stereotyped antibody response to DGP in CD.
Figure 1. IGHV repertoire, frequency of somatic mutation, and clonal diversity of DGP-reactive PCs.

(A) Bars represent the mean frequencies ± SD of the most abundantly expressed IGHV gene segments by DGP-reactive PCs and non–DGP-reactive PCs in 10 CD patients with active disease. (B) Mutation frequency in the IGHV genes of non–DGP-reactive and DGP-reactive PCs from 10 CD patients. White dots indicate the median frequency of mutation with 1.5 interquartile range. The differences between DGP-reactive PCs and non–DGP-reactive PCs were assessed by 2-tailed Student’s t test (**P < 0.01 and ****P < 0.0001). (C) Diversity and clonal size distribution of DGP-reactive and non–DGP-reactive PCs as measured by Hill’s diversity index. qD values depict the level of diversity for a given value of q; lower qD values represent lower diversity. Shaded area represents 95% percentiles.
The prototype IGHV3-23/IGLV4-69 and IGHV3-15/IGKV4-1 hmAbs recognize signatures of TG2 processing in the DGP
We aimed to scrutinize the basis for the predominant usage of IGHV3-23 and IGHV3-15 gene segments in DGP-reactive PCs. Among the 38 hmAbs previously generated from gluten-specific gut PCs, there was a striking heavy-chain variable region/light-chain variable region (VH/VL) pairing preference with IGHV3-23 preferentially pairing with IGLV4-69 and IGHV3-15 with IGKV4-1 (18); hmAbs of these configurations in the panel were all reactive with the DGP. Of note, the IGHV3-23/IGLV4-69 hmAbs also recognize the native form of the epitope, whereas IGHV3-15/IGKV4-1 hmAbs are only reactive with the deamidated peptide (18, 21). The differential reactivity to native/deamidated peptides suggested that the 2 types of antibodies might differ in their fine specificity.
We investigated this by assessing binding of 2 prototypical hmAbs, 1E01 (IGHV3-23/IGLV4-69) and 1E03 (IGHV3-15/IGKV4-1), to Ala-substituted variants of the 9-mer peptide PLQPEQPFP. Disassociation constants (Kd) for the 2 hmAbs and the peptide variants were determined by microscale thermophoresis (MST). Impaired binding was observed for Gln3, Pro4, Pro7, and Phe8 for hmAb 1E01 and for Pro4, Glu5, and Gln6 for hmAb 1E03 (Figure 2A). This analysis suggested that the 2 hmAbs make different interactions with the DGP. To obtain insight at atomic level for the antibody-antigen binding, we undertook X-ray crystallography studies. We produced Fab fragments of the 2 antibodies and generated crystal structures of Fab fragments with and without bound peptide antigens — altogether 5 structures: 1E01Fab, 1E01Fab-E, 1E01Fab-Q, 1E03Fab, and 1E03Fab-E.
Figure 2. Recognition of the peptide PLQPEQPFP by IGHV3-23/IGLV4-69 and IGHV3-15/IGKV4-1 hmAbs.

(A) Binding (1/Kd values determined by MST) of hmAbs 1E01 (IGHV3-23/IGLV4-69) and 1E03 (IGHV3-15/IGKV4-1) to Ala-substituted variants of the DGP (upper panel), and B-factor distribution for the cocrystal structures 1E01Fab-E and 1E03Fab-E (lower panel). Residues predicted to be involved in substrate recognition by TG2 are shown in blue, with residues being particularly important in bold. The Glu5 residue formed by TG2-mediated deamidation is shown in red. (B) Docking of DGP to structures of 1E01Fab and 1E03Fab. (C) Two pockets in the surface of 1E01Fab-E accommodate side chains of peptide residues Gln3 (pocket A) and Pro7/Phe8 (pocket B). (D) The interaction surface of 1E03Fab-E displays no major pockets for harboring side chains of DGP residues. In the zoom-in, bonds between Glu5 of the peptide and with R55-H (salt bridge) and T113-L (hydrogen bond) are shown as black dotted lines. Interacting residues are drawn as sticks, with nitrogen atoms in blue and oxygen atoms in red.
The crystal structures of 1E01Fab-E and 1E01Fab-Q were highly similar (Figure 2B and Supplemental Figure 2, A and B). The root-mean-square deviation (rmsd) value for superimposing the 2 structures was 0.374, and the spatial orientations of the peptide residues were very similar (Supplemental Figure 2B). The rmsd values for superimposing 1E01Fab with 1E01Fab-Q and 1E01Fab-E were 0.969 and 0.920, respectively (Supplemental Figure 2, C and D). There were good electron densities for the CDR3 loops of both 1E01Fab-Q and 1E01Fab-E (Supplemental Figure 2, E and F). Further, in both structures, the peptide stretched (N- to C-terminus) in an extended conformation from the CDR-L1, via its CDR-L3 and CDR-H3, to CDR-H2 (Figure 2B and Supplemental Figure 2A). The side chains of the Glu5/Gln5 in both 1E01Fab-E and 1E01Fab-Q faced the solvent and did not interact with the antibody (Supplemental Figure 2, B and G). Residues located in all 3 CDR-H loops, in FR-H2 and FR-H3 as well as in CDR-L1 and CDR-L3, interacted with either main chain or side chain of 8 peptide residues via hydrogen bonds, or hydrophobic or aromatic interactions (Supplemental Figure 2G and Table 1). The interface area for 1E01Fab-E and 1E01Fab-Q were 669 Å2 and 654 Å2, respectively, with about equal contribution by the heavy and light chains in both complexes (Table 2). There were 2 large pockets in the interaction surface of 1E01Fab that accommodated side chains of the peptide, with Gln3 filling pocket A and Pro7 and Phe8 filling pocket B (Figure 2C).
Table 1. A summary of the interactions between 1E01Fab and 1E03Fab and the DGP (PLQPEQPFP).
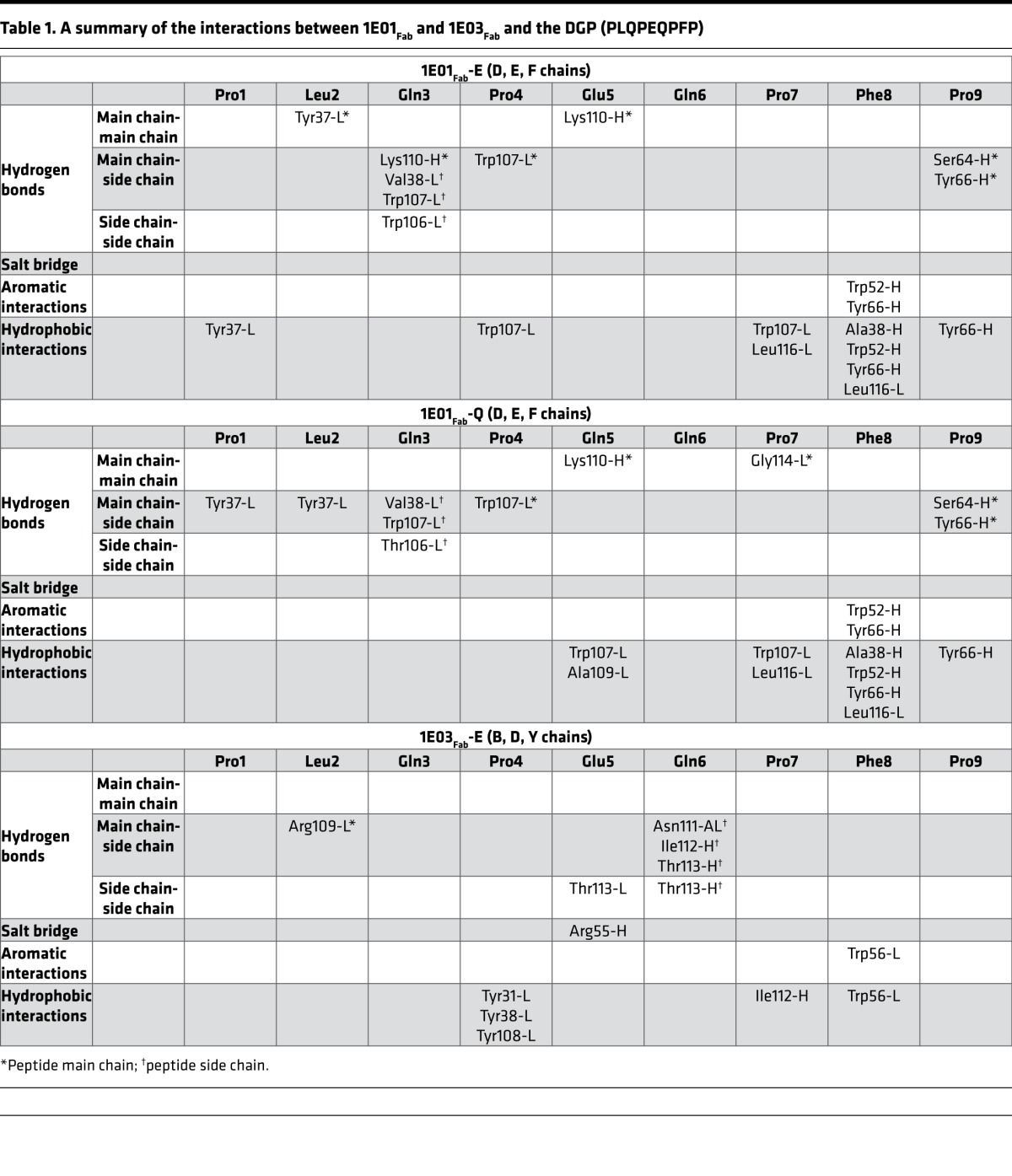
Table 2. Interface characteristics of interactions between the DGP (PLQPEQPFP) and Fab fragments.
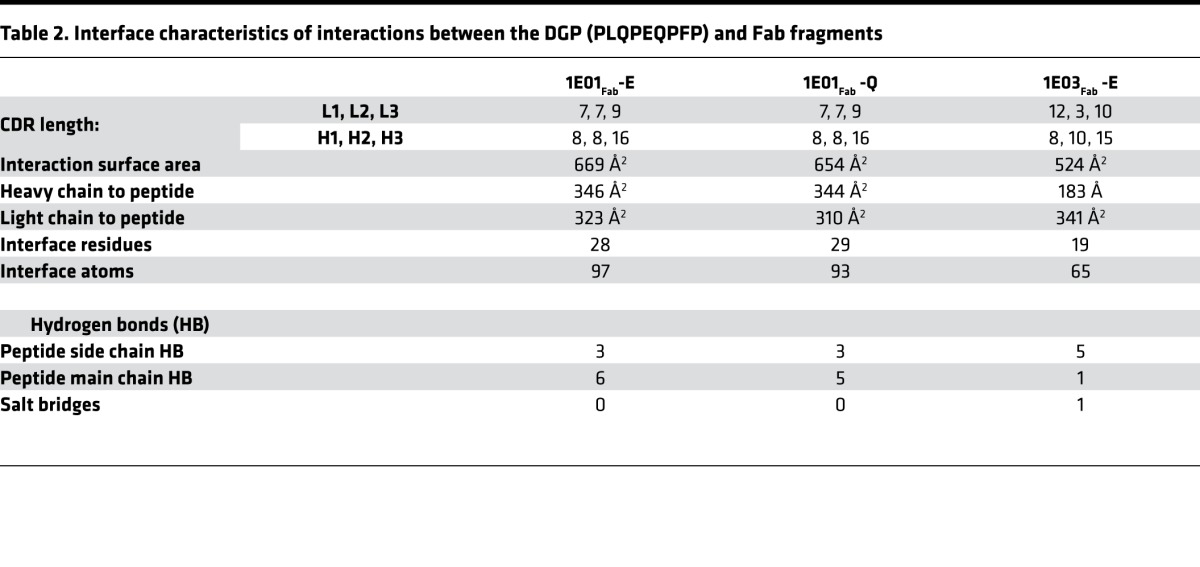
In the structures of 1E03Fab and 1E03Fab-E, electron densities for all CDR loops were intact, with the exception of molecule 1 in the asymmetric unit of 1E03Fab, where 5 residues were missing in the CDR-H3 loop (Supplemental Figure 3, A–D). For 1E03Fab-E, the Fab made an interaction with an extended conformation of the peptide via its FR-H2 and CDR-H3, as well as all 3 CDR-L loops. The orientation of the peptide was distinctly different from that seen in the 1E01Fab-E and 1E01Fab-Q structures (Figure 2B). The interface area was 524 Å2, with the biggest contribution from the light chain (Table 2). The rmsd value for superimposing 1E03Fab with 1E03Fab-E was 0.552 (Supplemental Figure 3E), demonstrating that binding of the peptide antigen, as was also observed for 1E01, did not induce changes in structure of the Fab. No real pockets could be defined in the interface area (Figure 2D). Peptide residues Pro4, Glu5, and Gln6 interacted with the antibody (Supplemental Figure 3F and Table 1). In particular, the salt bridge formed between Arg55-H and Glu5 is likely to give an enthalpy change of paramount importance for binding (Figure 2D and Table 1). Importantly, for both 1E01Fab-E/Q and 1E03Fab-E, all residues of the Fab fragments that made contact with the peptides were germline encoded, apart from V38-L of 1E01Fab and R109-L of 1E03Fab.
Results of the Ala-scan affinity resonated well with the insight obtained from the crystal structures for both 1E01 and 1E03. This is also reflected for the B factors of each residue of the peptides when bound to the two Fab fragments (Figure 2A). Higher B factor values were seen for residues at both ends of the 9-mer peptides. For 1E01Fab-E, an increase in B factor values were seen for the Glu5 and Gln6 compared with their neighboring residues, reflecting less interaction between the side chain of the part of the peptide with this hmAb.
Taken together, the binding analysis and X-ray crystal structure data demonstrated that the 1E01 (IGHV3-23/IGLV4-69) and 1E03 (IGHV3-15/IGKV4-1) hmAbs bind to the same epitope, yet with very different docking modes. While 1E01 (IGHV3-23/IGLV4-69) interacts with Gln3, Pro4, Pro7, and Phe8, 1E03 (IGHV3-15/IGKV4-1) interacts with Glu5 and Gln6. Interestingly, Glu5 is generated by TG2-mediated deamidation and Gln3, Pro4, Pro7, and Phe8 are all involved in recognition by TG2 (11, 12). Thus, both antibodies recognize signatures of TG2 processing.
In-depth mutational analysis of positions involved in binding of the DGP
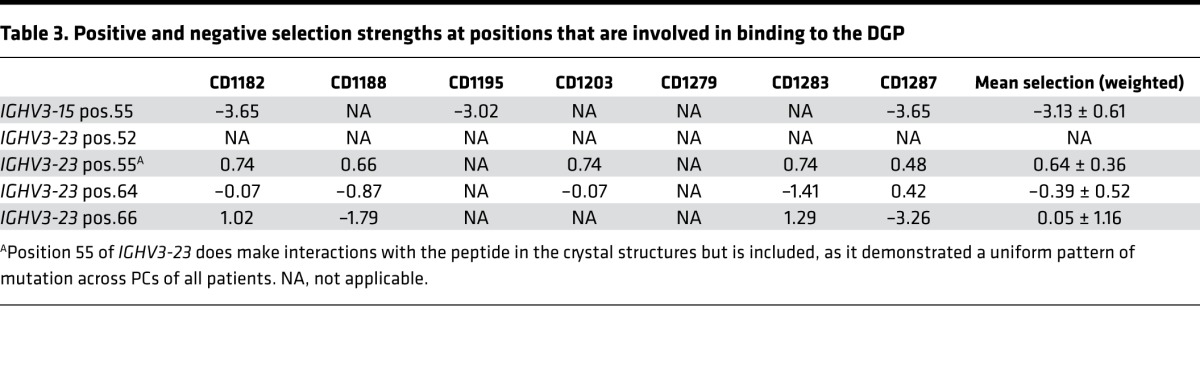
To further understand the basis for antibody-recognition of posttranslational modified gluten antigen in CD and to extend these observations beyond the 2 prototypical hmAbs, we undertook a detailed analysis of antibody mutations. We interrogated HTS data for positions involved in interactions with the DGP in the X-ray crystal structures (Table 1; position 55 for IGHV3-15 and positions 52, 64, and 66 for IGHV3-23) looking at mutations and affinity-depended selection strength. Data from patients from whom we had at least 30 unique sequences of each gene segment were analyzed (i.e., patients CD1182, CD1195, and CD1287 for IGHV3-15 and CD1182, CD1188, CD1203, CD1279, CD1283, and CD1287 for IGHV3-23). Furthermore, we assessed the functional importance of theses positions for binding to DGP by testing single amino acid mutants of the prototype hmAbs 1E03 and 1E01.
Analysis of IGHV3-15.
At position 55 in DGP-reactive PCs of all 3 patients had lower number of mutations than median, with evidence for negative selection in all patients (Figure 3A, Supplemental Figure 4A, and Table 3). Non–DGP-reactive PCs most often mutated to His or Leu at this position. The hmAb 1E03 carries a germline-encoded Arg position, and changing this residue to His abolished binding (Figure 3B). This strong effect is expected, as R55-H in the 1E03Fab-E structure forms a salt bridge with the side chain of the Glu5 peptide residue (Figure 2D and Table 1). The Glu5 side chain also forms a hydrogen bond with Thr113-L (Figure 2D and Table 1). We therefore generated a variant of the 1E03 hmAb carrying Ala at this light chain position and found that this also severely impaired binding (Figure 3B). Our results, thus, demonstrate that the germline-encoded R55-H and T113-L residues are both essential for interactions with the key Glu5 residue of the DGP peptide (Figure 2A).
Figure 3. Somatic mutations in IGHV3-15 and the effect of positions involved in binding on the affinity of 1E03 hmAb.

(A) Frequency of amino acid substitution along the IGHV3-15 gene segment of DGP-reactive and non–DGP-reactive PCs from a representative CD patient with more than 30 sequences available for analysis. (B) The effect of single amino acid substitution on binding affinity of hmAbs 1E03 to DGP as assessed by MST (average of 3 measurements ± SD). Variants at R55-H and T113-L were tested. GL, germline; VH, heavy chain; VΚ, Κ light chain.
Table 3. Positive and negative selection strengths at positions that are involved in binding to the DGP.
Analysis of IGHV3-23.
At position 52, DGP-reactive PCs of all 6 patients analyzed had a lower-than-median number of mutations, but no information on selection strength could be discerned, due to lack of mutations (Figure 4A, Supplemental Figure 4B, and Table 3). Non–DGP-reactive PCs also had few mutations at this position (Figure 4A and Supplemental Figure 4B), which is why we did not test antibody mutants for this position. At position 64, 5 of 6 patients had mutation frequencies higher than median but with no clear sign of positive or negative selection (Figure 4A, Supplemental Figure 4B, and Table 3). We observed a preference for Ser to Gly or Thr substitutions in DGP-reactive PCs at this position, while non–DGP-reactive PCs had a variety of substitutions, with Arg and Asn being the most frequent residues (Figure 4A and Supplemental Figure 4B). The hmAb 1E01 carries a germline-encoded Ser at this position. No changes in binding were observed by mutation of Ser to Asn or Arg (Figure 4B). S64-H is making a hydrogen bond to the main chain oxygen of peptide residue Pro9 (Table 1). Our findings suggest that this hydrogen bond is of little importance for binding. At position 66, DGP-reactive PCs acquired very little mutations, and only one patient (CD1283) had a mutation number that was higher than median (Figure 4A and Supplemental Figure 4B). There were signs of negative selection in 2 patients and positive selection in 2 patients, as well as a lack of mutations prevented assessment of selection strength in 2 patients. Non–DGP-reactive PCs, by comparison, most often mutated to Phe or Asp at this position (Figure 4A and Supplemental Figure 4B). The hmAb 1E01 carries a germline-encoded Tyr at this position, and mutations to Phe or Asp profoundly impaired binding (Figure 4B). In the 1E01Fab-E structure, Y66-H is involved in binding with a hydrogen bond to the main chain oxygen of peptide residue Pro9, as well as in aromatic and hydrophobic interactions with Phe8. The binding results suggest that this germline-encoded residue is important for the interaction with the DGP.
Figure 4. Somatic mutations in IGHV3-23 and the effect of positions involved in binding on the affinities of hmAb 1E01.

(A) Frequency of amino acid substitution along the IGHV3-23 gene segment of DGP-reactive and non–DGP-reactive PCs from a representative CD patient with more than 30 sequences available for analysis. (B) Binding of DGP by single amino acid substituted variants S64-H (B, left panel) and Y66-H (B, right panel) of hmAb 1E01 as assessed by MST (average of 3 measurements ± SD).
In summary, from our HTS analysis, we found that some positions involved in binding of the DGP peptide had a higher-than-median number of mutations. Other positions, like position 55 of IGHV3-15, had virtually no mutation and had signs of negative selection. The finding for this position is particularly notable as R55-H makes a salt bridge with the peptide and, thus, has a dominant role in binding of DGP. Overall, our results reveal that germline-encoded residues at interacting positions are favorable for binding to DGP. This resonates well with the general low degree of mutations present among DGP-reactive PCs.
Selection at position 55 of IGHV3-23 for small amino acids that allow for binding of DGP
Strikingly, the degree of mutations at position 55 in IGHV3-23 was more pronounced than what we observed for positions 52, 64, and 66, and we observed that DGP-reactive PCs uniformly across all 5 patients had mutation from Ala (germline) to Gly, with evidence for positive selection in all subjects (Figure 4A and Supplemental Figure 4B). Although this residue was not identified to make interactions with the peptide in the crystal structures, the reason for this mutation pattern could potentially relate to binding of the DGP. We hence undertook a further examination, including analysis of intracodon mutation patterns for this position. In comparison with non–DGP-reactive PCs, DGP-reactive PCs had statistically significant substitution patterns, with fewer nucleotide substitutions, and when acquiring a substitution, this typically was a single nucleotide substitution at position 2 of the codon (i.e., nucleotide 164) (Supplemental Figure 5, A and B). By contrast, non–DGP-reactive PCs had multiple nucleotide substitutions or single substitutions at positions 1 and 2. Moreover, the type of substitution at position 2 was highly selective for DGP-reactive PCs, with 84% of the sequences having a substitution at this position only, compared with 34% of non–DGP-reactive PCs. Typically, the cells had a C164G substitution, encoding an A55G mutation (Supplemental Figure 5A). These findings suggest that G55-H encoded by IGHV3-23 is preferred in interaction with DGP.
We tested single amino acid mutants of the 1E01 hmAb to assess impact of position 55 of IGHV3-23 for binding to DGP. The hmAb 1E01 carries Gly at position 55 that is altered from Ala in germline. In contrast to DGP-reactive PCs, non–DGP-reactive PCs often had Ser or Thr, whereas Ala and Gly were observed in both types of PCs. Substitution from Gly to Ala, Ser, or Thr in hmAb 1E01 led to impaired binding — particularly, the Gly to Thr mutation had a big effect (Figure 5A). In the 1E01Fab-E structure, G55-H lines a binding pocket that accommodates the side chain of peptide residue Phe8 (Figure 5B). Conceivably, A55-H and G55-H could be favorable residues at this position, due to the small or absent side chains. S55-H and T55-H, however, have larger side chains that could cause steric hindrance and, hence, impaired binding (Figure 5, B and C). Phe8 is part of the recognition sequence of TG2 (11, 12), and this observation again demonstrates that the DGP-reactive cells carry BCRs that recognize signatures of the peptides that reflect processing by TG2.
Figure 5. Steric hindrance at position 55 of IGHV3-23 interferes with binding of DGP.

(A) Binding of DGP by hmAb 1E01 mutated at heavy chain position 55 as assessed by MST (average of 3 measurements ± SD.). (B and C) Model for a possible steric hindrance following Gly-to-Thr substitution in the 1E01Fab-E structure. (B) The side chain of Phe8 is accommodated in pocket B and is located in proximity of G55-H. (C) The side chain of Thr is predicted to fill into the pocket that should be accommodated by Phe8, thereby causing steric hindrance. GL, germline; VH, heavy chain.
BCR-mediated uptake of DGP facilitates presentation of gluten T cell epitopes
Antigen uptake by specific BCRs can greatly facilitate presentation of T cell epitopes that are physically linked to the B cell epitopes (23). To test whether B cells specific to DGP enhance presentation of gluten T cell epitopes, we designed an antigen presentation assay to T cells using, as antigen presenting cells, A20 B cell lymphoma cells retrovirally transduced with HLA-DQ2.5 encoding genes, as well as genes for 1E03 or a non–DGP-reactive mAb (2F02) in the format of membrane-bound human IgD. IL-2 secretion from transfectant T cells specific for the DQ2.5-glia-ω2 epitope was used as a readout. As antigen, we used a synthetic ω-gliadin 34-mer fragment (ω34mer peptide) that is naturally created by gut enzyme digestion and that contains the DQ2.5-glia-ω2 T cell epitope, as well as 3 copies of the B cell core epitope (QPEQPF) (21). We observed that transfectant B cells expressing 1E03 as BCR rapidly bound the ω34mer peptide on their surface (Figure 6A) and stimulated the T cells more efficiently than B cells with irrelevant BCR (Figure 6B). Hence, cognate T cell and B cell interaction can indeed operate when gluten T cell and B cell epitopes are physically linked. As ω34mer peptide is an antigen that likely binds directly to surface MHC class II molecules, the difference between the specific and nonspecific B cells in our assay is less than what is usually observed for antigens that are completely antigen processing dependent for T cell recognition (23).
Figure 6. DGP-specific B cells efficiently induce IL-2 secretion from gluten-specific T cells.

(A) Binding of deamidated ω34mer gluten peptide by transfectant B cells expressing either (i) HLA-DQ2.5 alone (blue line), (ii) HLA-DQ2.5 and DGP-reactive Ig (BCR) 1E03 (green line), or (iii) DGP-negative Ig (BCR) 693-2F02 (red line). Cells were stained with biotinylated ω34mer that was multimerized with APC-conjugated streptavidin. (B) Antigen presentation by transfectant B cells pulsed with the indicated concentrations of ω34mer peptide and cocultured with transfectant T cells specific for the DQ2.5-glia-ω2 epitope. IL-2 production was assayed (mean ± SEM). One representative of 3 independent experiments is shown.
Discussion
In this study, we have dissected the antibody response against the peptide PLQPEQPFP, which harbors an immunodominant epitope of gluten recognized by highly disease-specific antibodies of CD patients (5, 18, 24, 25). By Ala scan and X-ray crystallographic studies, we defined the core epitope of the peptide to comprise residues QPEQPF. Two stereotyped classes of CD antibodies recognizing this epitope utilize the gene segments IGHV3-23/IGLV4-69 and IGHV3-15/IGKV4-1 for recognition leading to fundamentally distinct docking modes of the Fab fragments. Strikingly, both antibodies recognize signatures reflecting TG2 processing of the antigen, thereby supporting a critical role of the TG2 enzyme in shaping the anti-gluten antibody response in CD.
Using HTS of IGHV genes, we characterized the repertoire and clonal diversity of IgA DGP-reactive PCs from the gut of CD patients. These PCs had low clonal diversity and a narrow IGHV repertoire with bias in usage of the IGHV3-23 and IGHV3-15 genes, as compared with non–DGP-reactive PCs. This finding confirms and extends finding of 38 hmAbs from single-sorted PCs of CD patients that showed a paring preference of IGHV3-23 with IGLV4-69 and IGHV3-15 with IGKV4-1 (18). Studying 2 prototypic hmAbs 1E01 (IGHV3-23/IGLV4-69) and 1E03 (IGHV3-15/IGKV4-1) with epitope/paratope mutational analysis and X-ray crystallography, we found that the 2 hmAbs, despite interacting with extended peptide conformations with no structure adaptation, employ fundamentally different binding modes. Residues Pro4, Glu5, and Gln6 are essential for recognition of DGP by the 1E03 hmAb, whereas Gln3, Pro4, Pro7, and Phe8 are involved in recognition by the 1E01 hmAb. In both 1E01Fab-E/Q structures, key interactions of the antibody are mediated by peptide side chains (Pro4 and Pro7/Phe8) being buried in pockets in a knob-hole fashion, which is a type of interaction often seen in peptide-protein binding (26). Importantly, no interaction takes place between 1E01Fab and the side chain of Glu5 in the peptide. In the interface of 1E03Fab-E structure, which was slightly smaller than in the 1E01Fab-E/Q structures, no clear pockets could be defined. The Glu5 peptide residue makes critical contacts with the antibody residues, including a salt bridge with residue Arg55-H. Overall, the results provide an explanation for the differential recognition of deamidated and native variants of the PLQPEQPFP peptide by the 1E01 (IGHV3-15/IGKV4-1) and 1E03 (IGHV3-23/IGLV4-69) antibodies.
Only the 1E03 (IGHV3-15/IGKV4-1) hmAb is truly deamidation specific and hence, in a strict sense, qualifies to be termed an anti-deamidated gliadin antibody. Nonetheless, the 1E01 (IGHV3-23/IGLV4-69) antibody recognizes a sequence that intimately associates with deamidation of the gluten peptide. The antibody recognition sequence QPQ/EXPF is highly similar to the preferred substrate recognition sequence of TG2, which spans 5 residues from –1 to +3 (targeted Q at position 0) and harbors the core sequence QXP(F) at residues 0 to +3 (11, 12). As we demonstrate, B cells with BCR of this specificity will bind, internalize, and present peptides that are deamidated by TG2 for presentation to CD4+ gluten–reactive T cells and, hence, be in position to receive T cell help. This will be the case for the 1E03 (IGHV3-15/IGKV4-1) type of antibodies, which recognize the introduced glutamate residue itself. Even though the 1E01 (IGHV3-23/IGLV4-69) type of antibodies can also bind native peptides, this would not result in productive T cell help, as most gluten-reactive T cells of CD patients are strictly deamidation dependent (27). Importantly, both types of antibodies recognize residues in a peptide that intimately reflect productive and pathogenic T cell and B cell interaction in CD. Both types of antibodies, thus, serve as proxies for a key step in the pathogenesis of CD; hence, they become excellent surrogate markers for the disease.
The binding of the immunodominant gliadin epitope by both 1E01 (IGHV3-23/IGLV4-69) and 1E03 (IGHV3-15/IGKV4-1) hmAbs was essentially mediated by germline-encoded heavy and light chain residues. This would explain the selection for usage of certain VH and VL gene segments and also the preferential VH/VL paring among PCs reactive with the DGP. Moreover, our findings suggest that the reactivities of IGHV3-23/IGLV4-69 and IGHV3-15/IGKV4-1 stereotyped antibodies are already established with germline configurations, negating a need for naive B cells to undergo substantial somatic hypermutation to differentiate to effector cells. The HTS data demonstrating low levels of mutations further revealed that most sequences were in germline configuration at the positions that involved bonding of DGP. However, evidence of selection at several of these positions was observed. Most notable was the negative selection at position 55 of IGHV3-15. In the 1E03Fab-E structure, the germline-encoded Arg served as a lynchpin for the binding of the DGP peptide, explaining why preservation of germline configuration is favorable at this position. Position 55 of IGHV3-23, despite not bonding to the DGP in the crystal structure, was found to be frequently mutated in the HTS data, with evidence for positive selection. DGP-reactive PCs typically mutated from germline Ala to Gly while non–DGP-reactive PCs often also mutated to Ser or Thr. Analysis of the 1E01 crystal structures suggests that this position, which lines a binding pocket harboring the peptide Phe8 side chain, has a requirement for small residues. Ser and Thr with larger side chains could cause steric hindrance and, thus, impaired binding as was also experimentally observed. Taken together, it seems that B cells reactive with the peptide PLQPEQPFP undergo some selection in CD patients. However, at positions that are involved in peptide binding, there is preservation of germline-encoded residues of the antibodies.
In conclusion, in this study, we have combined HTS and X-ray crystallography to decipher the antibody response toward the immunodominant deamidated gliadin B cell epitope PLQPEQPFP in CD. We demonstrate that 2 stereotyped forms of antibodies bind to the same epitope fundamentally differently; one targets the recognition sequence of TG2, while the other targets the Glu residue formed by TG2-mediated deamidation. In essence, these 2 types of recognition lead to the same results: uptake of the deamidated peptide by BCR of gluten-reactive B cells, which in turn can receive T cell help from gluten-reactive T cells and thereby undergo clonal expansion. Interestingly, the fine specificity of the 2 stereotyped antibodies recognizing the immunodominant B cell epitope reflects key events in the pathogenesis of CD. On a general level, the isolation of antigen-specific PCs/B cells followed by detailed structural analysis of epitopes illustrates a strategy by how key aspects of disease that involve posttranslational modification and generation B cell and of T cell epitopes can be identified.
Methods
Subjects and cells.
Ten patients with untreated, active CD were enrolled in this study. All were diagnosed according to the guidelines of the British Society of Gastroenterology (28). Single cell suspensions were obtained from small intestinal biopsies of the duodenum obtained by endoscopy as previously described and cryopreserved until use (18, 20).
Isolation of DGP-reactive PCs and library preparation of IGHV HTS.
PCs reactive with the immunodominant gliadin epitope PLQPEQPFP were stained using antigen tetramers as previously described (18). In short, biotinylated-deamidated gliadin peptide (biotin-GSGSGS-PLQPEQPFP, GL Biochem) were incubated with SA-APC (PhycoLink) at a 4:1 molar ratio for 1 hour in PBS supplemented with 3% FCS on ice to create PLQPEQPFP-tetramers. Similarly, TG2 was tetramerized with SA-PE (Invitrogen) and used to exclude TG2-specific PCs from the analysis. Single cell suspensions were incubated with the 2 tetramers for 45 minutes, and the following PC markers were added for the last 20 minutes to stain for specific cellular lineage markers: IgA-FITC (SouthernBiotech, catalog 2050-02), Brilliant Violet 570 CD3 and CD14 (BioLegend, clones UCHT1 and M5E2, respectively), CD19 Pacific Blue (BioLegend, clone HIB19) and CD27-PE-Cy7 (eBioscience, clone LG.7F9). IgA intestinal PCs were defined as live, large, and CD14/CD3-negative, with high expression of CD27. PCs were sorted for being either negative or positive for the PLQPEQPFP-tetramers into 25 μl catch buffer (10 mM DTT and RNAsin 2 U/μl [Promega] in PBS) and kept in –70°C until cDNA synthesis. For cDNA synthesis, 13.5 μl of sorted PC extracts were added to the mix containing indexed IGJH reverse primer (1.4 μM), 0.45% v/v NP-40, and 0.5 μl RNasin 40 U/μl (Promega). DEPC-treated water was added to the final volume of 25 μl, incubated at 65°C for 5 minutes, and put on ice. Next, 10 μl of 5× reverse transcription (RT) buffer, 3.5 μl of 100 mM DTT, 4.3 μl of 10 mM dNTP-Mix (Promega), 0.7 μl of 40 U/μl RNasin (Promega), 0.85 μl superscript III (Invitrogen), and 4.5 μl DEPC-treated water were added. cDNA was synthesized at 42°C for 10 minutes, 25°C for 10 minutes, 50°C for 60 minutes, and 94°C for 5 minutes and stored at –20°C. Second-strand cDNA was synthesized using AmpliTaq Gold polymerase (Applied Biosystems) with forward IGHV1-6 framework region (FR) 2 (29), which were indexed and included 6 random nucleotides (further used as unique molecular identifiers [UMIs]) and a part of Illumina adapters. Second-strand synthesis was carried out at 95°C for 7.5 minutes, 52°C for 2 minutes, and 72°C for 10 minutes. Double-stranded cDNA was further purified using AMPure XP (Beckman Coulter) at a 1:1 ratio according to the manufacturer’s instructions. Next, the second part of the Illumina adapter was connected using R1 and R2 primers. PCR was performed using Qiagen Multiplex PCR at 95°C for 15 minutes, ×25 (95°C for 30 seconds, 60°C for 45 seconds, 72°C for 90 seconds), and 72°C for 10 minutes. Primers were previously described (20). Final IGHV amplicon libraries were purified using AMPure XP (Beckman Coulter) and further run and extracted from agarose gel. Paired-end sequencing of 250 × 2 or 300 × 2 bp was performed using Illumina MiSeq at the Norwegian Sequencing Centre in Oslo, Norway (http://www.sequencing.uio.no). Raw sequencing data was processed using pRESTO (30) as previously described (20, 31). Unique sequences to which at least 2 copies were present were further aligned using ImMunoGeneTics (IMGT) database (32), and their IGHV and IVHJ segments and CDR3 were determined. The number of unique sequences is given in Supplemental Table 1.
Diversity of B cell clone measurements.
Sequences were clustered into clones originating from common ancestors using Change-O (33) as described previously (20). Diversity scores for clones from DGP-reactive and non–DGP-reactive PCs were calculated using the Hill diversity index, which measures diversity in a population (qD) as a smooth function of a single parameter (q) (22, 34).
qD = (Σsi=1piq)1/(1–q)
S is the number of clones, pi is the frequency of the i’th clone, and the parameter q determines how clonal frequencies are weighted (34).
Using the Hill diversity index, we determined whether the repertoire diversity indices differed between these subsets. To eliminate sampling effects, repertoires were subsampled (1,000 times) according to the number of sequences in the smallest sample (i.e., the DGP-reactive PCs), and Hill’s curve was calculated in 1,000 equally spaced q values between 0 and 15. At a given value of q (x axis in Figure 1D), lower values of qD (y-axis) indicate lower diversity (i.e., more focused repertoire).
Fab fragments and antibody mutants.
The hmAbs 1E01 (IGHV3-23/IGLV4-69) and 1E03 and (IGHV3-15/IGKV4-1) were studied (18). Fab fragments of these 2 hmAbs were generated by introducing a stop codon in the plasmid after residue 232 (228PKSCD232). PCR was performed by using forward primer 5′-TTTCTAGTAGCAACTGCAAC-3′ and reverse primer 5′-gtggacaag aaagttgagcccaaatcttgtgactgaagcttggat-3′, and the products were inserted into the expression vector IgG1 between the AgeI and HindIII restriction sites. The light chains of the 2 antibodies have been previously cloned (18). Plasmids encoding the heavy and light chains were cotransfected into HEK 293-F (ThermoFisher Scientific, R79007) cells by using 293fectin (Invitrogen) or polyethyleneimine (Polysciences Inc.). Transfected HEK 293-F cells were cultured at 37°C with shaking for 6 days. Anti-gliadin autoantibodies were purified from culture supernatants. The 1E01 Fab fragment was affinity purified by using CaptureSelect LC-lambda (Hu) Affinity Matrix (Life Technologies), whereas the 1E03 Fab fragment was affinity purified using a protein L column (GE Healthcare). Purity of the proteins was confirmed by SDS-PAGE.
Site-directed mutagenesis in 1E01 and 1E03 hmAbs was induced using Quickchange Multisite-Directed Mutagenesis Kit (Agilent) following the manufacture’s instructions. Mutations were verified by sequencing of the expression vectors (GATC Biotech). Primers that were used to induce mutations are listed in Supplemental Table 2. Expression vectors containing the original or the mutated genes were cotransfected into HEK293-F cells as previously described (35). Culture supernatants were collected 6 days after transfections, and antibodies were purified on Protein G Sepharose column (GE Healthcare).
Crystallization and data collection.
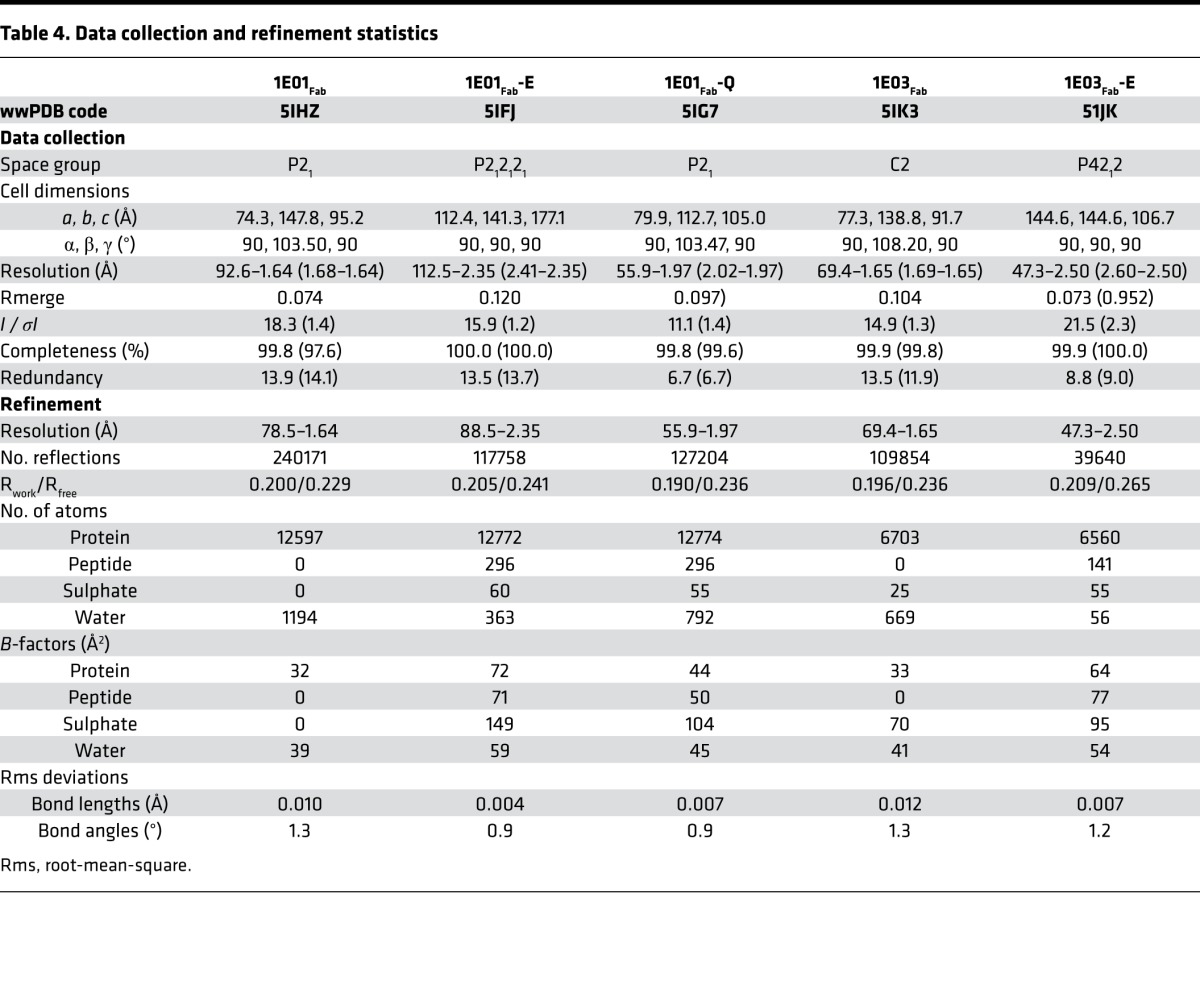
The purified Fab fragments were concentrated to 12 mg/ml in 20 mM Tris, pH 7.2, 150 mM NaCl, 1 mM EDTA. Complexes between the Fab fragments and peptide were formed by incubating them at 1:8 molar ratio overnight at 4°C. Initial large-scale screenings were performed by the sitting drop vapor diffusion method using Cartesian Dispensing system (Genomic Solutions, Harvard University, Boston, Massachusetts, USA). Promising hits were chosen for crystallization optimization and data collection. Altogether, 5 crystals or cocrystals were generated and resolved: 1E01Fab alone (1E01Fab), 1E01Fab in complex with deamidated peptide (1E01Fab-E), 1E01Fab in complex with native peptide (1E01Fab-Q), 1E03Fab alone (1E03Fab), and 1E03Fab-E in complex with deamidated peptide (1E03Fab-E) (Supplemental Table 3). Crystals of 1E03Fab in complex with native peptide could not be generated, as 1E03 does not bind the native peptide. The cryo conditions for freezing the crystals were 25% v/v glycerol in the mother liquor, except for 1E03Fab-E, which required 20% v/v glycerol for cryo solutions. Each single crystal was transferred to the cryo solution and flash-frozen in liquid nitrogen for data collection. Data were processed and scaled by Xia2 (36) for all crystals, except that of 1E03Fab-E, which was processed by XDS (37), POINTLESS (38), and AIMLESS packages (39).
Structure determination and refinement.
All the 5 structures were determined by molecular replacement using CCP4 Phaser (40), and the models used for searching are listed in Supplemental Table 3. Manual buildings of all the 5 structures were carried out by Coot (41), and refinements were implemented by Refmac (42) and PHENIX (43). Validation was done by the validation servers in Protein Data Bank (PDB) and MolProbity (44). The residues were numbered according to the IMGT numbering system (32). Data processing and refinement statistics are summarized in Table 4. Ramachandran statistics indicated that, for all structures, 95.5%–97.8% of the residues had a favored geometry, with 0.4%–0.7% of the residues being outliers. Interactions between the peptide and Fab fragments were analyzed by PIC (45). The determination of interaction surface area, was analyzed by PISA (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) (46). Analysis of pockets on the protein monomers of bound structures was done using the CASTp server (47), and the pockets in the interface area were selected. Each asymmetric unit had several molecules (Supplemental Table 3) and the interaction analysis was made on the following chains: 1E01Fab (PDB 5IHZ), chains A, B; 1E01Fab-E (PDB 5IFJ), chains D, E, F; 1E01Fab-Q (PDB 5IG7), chains D, E, F; 1E03Fab (PDB 5IK3), chains C, D; and 1E03Fab-E (PDB 51JK), chains B, D, Y. Crystallographic figures were generated by PyMOL (http://www.pymol.org).
Table 4. Data collection and refinement statistics.
Antibody affinity measurement using MST.
Binding affinity between hmAbs (1E01, 1E03, or their mutants) and the peptide PLQPEQPFP or Ala-substituted variants of the same peptide were determined using MST. Analyses were performed on a Monolith NT.LabelFree Instrument using Monolith NT.LabelFree capillaries (Nano Temper Technologies). For MST measurements, hmAbs were used at a constant concentration of 0.25 μM. Gliadin peptides were diluted 16 times in a 1:1 serial dilution from 0.5 mM to a final concentration of 15.25 nM and incubated at room temperature with the antibodies for at least 5 minutes before measurement. All measurements were performed in triplicate, and data analysis was performed with the Monolith software (Nano Temper Technologies).
Data alignment and genotyping.
Sequences were aligned to the IMGT database using IMGT/HighV-QUEST (32). Allele frequency was calculated for each gene segment. To be included in an individual’s reference, an allele had to appear in at least 20% of nonmutated sequences assigned to the same gene or at least 20 times, regardless of the frequency. Realignment was accomplished using IgBLAST (48) with the personalized germline reference.
Analysis of mutations, selection, and intracodon nucleotide substitution patterns.
Analysis of mutations in HTS data of IGHV3-23 and IGHV3-15 was done using data from patients from whom we had at least 30 unique sequences of each gene segment with more that 5 clones that were not dominated by a single clone (i.e., patients CD1182, CD1188, CD1203, CD1279, CD1283, and CD1287 for IGHV3-23 and CD1182, CD1195, and CD1287 for IGHV3-15). Mutational analysis was done in the context of the X-ray crystal structures interrogating positions involved in direct bonding with the peptide. Sequences of DGP-reactive and non–DGP-reactive PCs were analyzed by assigning each amino acid position along the gene segments to low/high mutation rates relative to the median mutation rate for each IGHV gene of each sample. Affinity-dependent selection strength among DGP-reactive PCs was quantified for selected residue in the IGHV gene segments using a tailored version of BASELINe (49, 50), adapted to single codons. A local test that compares frequency of observed nonsynonymous mutations with their expected frequency was applied with the S5F targeting model to account for somatic hyper mutation hot/cold spots (31). To avoid biases that may have emerged due to unknown order of mutation acquirement, only sequences with no more than 1 mutation per codon were used. Intracodon mutation pattern was analyzed for position 55 of IGHV3-23. This analysis is based on the fact that, for each codon, 8 distinct combinations of substitutions are possible. For each patient, intracodon substitution patterns were summarized for DGP-reactive and non–DGP-reactive PCs and were compared using a χ2 test for goodness of fit. P values were adjusted using Benjamini-Hochberg correction.
T cell and B cell collaboration assay.
Murine A20 B lymphoma cells were engineered by retroviral transduction to express HLA-DQ2.5 and 1002-1E03 BCR in an IgD format as previously described (19). To assess capacity of these transfectant B cells to bind deamidated gluten peptide, cells were stained with tetramers for 30 minutes on ice and analyzed on a FACSCalibur flow cytometer (BD Biosciences). Gluten peptide tetramers were generated by incubating the biotinylated 34-mer ω gliadin peptide (QPQQPFPEQPQQPEQPFPQPEQPFPWQPEQPFPQ, GL Biochem) with SA-APC (PhycoLink) at a 4:1 molar ratio for 2 hours in PBS supplemented with 2% FCS on ice. Murine BW58α-β cells devoid of endogenous TCR were engineered to express human CD4 and T cell receptor from a DQ2.5-glia-ω2–reactive CD4 T cell clone (TCC737.30) (51) as previously described (52). Tranfectant B cells (1 × 106 cells/ml) expressing HLA-DQ2.5 alone, HLA-DQ2.5 with DGP-negative (693-2F02) BCR, or HLA-DQ2.5 with DGP-reactive BCR (1002-1E03) were incubated in RPMI supplemented with 10% FCS with indicated concentrations of ω34-mer peptide for 10 minutes on ice and washed with RPMI to remove unbound peptide. Tranfectant B cells (50,000) were incubated for 4 hours in 96-well U-bottom plates at 37°C before transfectant T cells (25,000) were added. After 18 hours, culture supernatants were collected and assayed for murine IL-2 production by ELISA as previously described (52). IL-2 concentrations were calculated from a standard curve generated using recombinant murine IL-2 (BioLegend).
Accession codes.
HTS data have been deposited at the National Center for Biotechnology Information Sequence Read Archive (SRA) (accession code SRP098633, bioproject PRJNA368623). Protein structure data have been deposited in the worldwide PDB (wwPDB) under accession codes 5IHZ, 5IFJ, 5IG7, 5IK3 and 51JK.
Statistics.
A two-tailed t test was applied to compare the number of acquired mutations in non–DGP-reactive and DGP-reactive PCs. P < 0.05 was considered significant. χ2 test for goodness of fit followed by Benjamini-Hochberg correction for the adjustment of the P values were used to compared the calculated intracodon mutations in non–DGP-reactive and DGP-reactive PCs. Hill’s diversity index (22) was used to determine the repertoire diversity as reflected by the clonal size distribution. The repertoire was subsampled 1,000 times according to the number of sequences in the smallest subsets (as described above). Statistically significant differences between samples can be visualized when curves describing the diversity of 2 PC subsets are separated and fall outside the 95% that resulted from random sampling.
Study approval.
All subjects had signed informed consents, and the study was approved by the Regional Ethics Committee of South-Eastern Norway (2010/2720).
Author contributions
ØS, OS, and LMS conceived the project. OS processed gut biopsies, sorted PCs, prepared the amplicon library for MiSeq sequencing, performed binding affinity studies, and produced mutants of hmAbs. XC produced and purified proteins for crystallography, generated crystals, solved protein structures by X-ray crystallography, and performed binding affinity studies. MG processed and analyzed sequence data. MFDP performed T cell and B cell collaboration assays. YZ was involved in crystallography work. ØS performed PC sorting. KEAL performed the endoscopies and provided small-intestinal biopsies. LMS and OS wrote the manuscript with contributions from other authors. GY and LMS supervised the study.
Supplementary Material
Acknowledgments
We thank Chaitan Khosla, Bana Jabri, and Jorunn Stamnaes for critical reading of the manuscript; Bjørg Simonsen, Marie K. Johannesen, and Stine Rosenqvist Lund for excellent technical assistance; Karl Harlos for assisting with crystal mounting and data collection; Jingshan Ren for advice on solving crystal structures; and Bjørn Dalhus for help and for providing access to crystallization screening and the MST instrument at the Regional Core Facility for Structural Biology and Bioinformatics at the South-Eastern Norway Regional Health Authority. We are indebted to Diamond Light Source for access to beamline I03 and I24 and the European Synchrotron Radiation Facility (ESRF) for access to beamline ID29. Work in the laboratory of LMS was supported by grants from the South-Eastern Norway Regional Health Authority, the European Commission (grant ERC-2010-Ad-268541), the Research Council of Norway through its Centers of Excellence funding scheme (project number 179573/V40), and Stiftelsen KG Jebsen. Work in the laboratory of GY was supported by the Israel Science Foundation (grant 832/16), and work in the laboratory of YZ was supported by the European Community’s Seventh Framework Program (FP7/2007-2013) under BioStruct-X (grant 283570).
Version 1. 09/05/2017
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(16):e93961. https://doi.org/10.1172/jci.insight.93961.
Contributor Information
Omri Snir, Email: snir.omri@medisin.uio.no.
Xi Chen, Email: sealasting@hotmail.com.
Moriah Gidoni, Email: moriahcohen@gmail.com.
Yuguang Zhao, Email: yuguang@strubi.ox.ac.uk.
Øyvind Steinsbø, Email: oyvind.steinsbo@outlook.com.
Gur Yaari, Email: gur.yaari@biu.ac.il.
Ludvig M. Sollid, Email: l.m.sollid@medisin.uio.no.
References
- 1.Kagnoff MF. Celiac disease: pathogenesis of a model immunogenetic disease. J Clin Invest. 2007;117(1):41–49. doi: 10.1172/JCI30253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sollid LM, Thorsby E. HLA susceptibility genes in celiac disease: genetic mapping and role in pathogenesis. Gastroenterology. 1993;105(3):910–922. doi: 10.1016/0016-5085(93)90912-V. [DOI] [PubMed] [Google Scholar]
- 3.Sollid LM. Coeliac disease: dissecting a complex inflammatory disorder. Nat Rev Immunol. 2002;2(9):647–655. doi: 10.1038/nri885. [DOI] [PubMed] [Google Scholar]
- 4.Dieterich W, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3(7):797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 5.Osman AA, et al. B cell epitopes of gliadin. Clin Exp Immunol. 2000;121(2):248–254. doi: 10.1046/j.1365-2249.2000.01312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leffler D, et al. Kinetics of the histological, serological and symptomatic responses to gluten challenge in adults with coeliac disease. Gut. 2013;62(7):996–1004. doi: 10.1136/gutjnl-2012-302196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Husby S, et al. European Society for Pediatric Gastroenterology, Hepatology, and Nutrition guidelines for the diagnosis of coeliac disease. J Pediatr Gastroenterol Nutr. 2012;54(1):136–160. doi: 10.1097/MPG.0b013e31821a23d0. [DOI] [PubMed] [Google Scholar]
- 8.Molberg O, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells in celiac disease. Nat Med. 1998;4(6):713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 9.van de Wal Y, et al. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161(4):1585–1588. [PubMed] [Google Scholar]
- 10.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4(2):140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 11.Fleckenstein B, et al. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation process. J Biol Chem. 2002;277(37):34109–34116. doi: 10.1074/jbc.M204521200. [DOI] [PubMed] [Google Scholar]
- 12.Vader LW, et al. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med. 2002;195(5):643–649. doi: 10.1084/jem.20012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stamnaes J, Sollid LM. Celiac disease: Autoimmunity in response to food antigen. Semin Immunol. 2015;27(5):343–352. doi: 10.1016/j.smim.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Doyle HA, Yang ML, Raycroft MT, Gee RJ, Mamula MJ. Autoantigens: novel forms and presentation to the immune system. Autoimmunity. 2014;47(4):220–233. doi: 10.3109/08916934.2013.850495. [DOI] [PubMed] [Google Scholar]
- 15.Schellekens GA, et al. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101(1):273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Girbal-Neuhauser E, et al. The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol. 1999;162(1):585–594. [PubMed] [Google Scholar]
- 17.Malmström V, Catrina AI, Klareskog L. The immunopathogenesis of seropositive rheumatoid arthritis: from triggering to targeting. Nat Rev Immunol. 2017;17(1):60–75. doi: 10.1038/nri.2016.124. [DOI] [PubMed] [Google Scholar]
- 18.Steinsbø Ø, et al. Restricted VH/VL usage and limited mutations in gluten-specific IgA of coeliac disease lesion plasma cells. Nat Commun. 2014;5:4041. doi: 10.1038/ncomms5041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Niro R, et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med. 2012;18(3):441–445. doi: 10.1038/nm.2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snir O, Mesin L, Gidoni M, Lundin KE, Yaari G, Sollid LM. Analysis of celiac disease autoreactive gut plasma cells and their corresponding memory compartment in peripheral blood using high-throughput sequencing. J Immunol. 2015;194(12):5703–5712. doi: 10.4049/jimmunol.1402611. [DOI] [PubMed] [Google Scholar]
- 21.Dørum S, Steinsbø Ø, Bergseng E, Arntzen MØ, de Souza GA, Sollid LM. Gluten-specific antibodies of celiac disease gut plasma cells recognize long proteolytic fragments that typically harbor T-cell epitopes. Sci Rep. 2016;6:25565. doi: 10.1038/srep25565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hill MO. Diversity and evenness — unifying notation and its consequences. Ecology. 1973;54(2):427–432. doi: 10.2307/1934352. [DOI] [Google Scholar]
- 23.Lanzavecchia A. Antigen-specific interaction between T and B cells. Nature. 1985;314(6011):537–539. doi: 10.1038/314537a0. [DOI] [PubMed] [Google Scholar]
- 24.Aleanzi M, Demonte AM, Esper C, Garcilazo S, Waggener M. Celiac disease: antibody recognition against native and selectively deamidated gliadin peptides. Clin Chem. 2001;47(11):2023–2028. [PubMed] [Google Scholar]
- 25.Pantazes RJ, Reifert J, Bozekowski J, Ibsen KN, Murray JA, Daugherty PS. Identification of disease-specific motifs in the antibody specificity repertoire via next-generation sequencing. Sci Rep. 2016;6:30312. doi: 10.1038/srep30312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.London N, Movshovitz-Attias D, Schueler-Furman O. The structural basis of peptide-protein binding strategies. Structure. 2010;18(2):188–199. doi: 10.1016/j.str.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 27.Sollid LM, Qiao SW, Anderson RP, Gianfrani C, Koning F. Nomenclature and listing of celiac disease relevant gluten T-cell epitopes restricted by HLA-DQ molecules. Immunogenetics. 2012;64(6):455–460. doi: 10.1007/s00251-012-0599-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ludvigsson JF, et al. Diagnosis and management of adult coeliac disease: guidelines from the British Society of Gastroenterology. Gut. 2014;63(8):1210–1228. doi: 10.1136/gutjnl-2013-306578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Dongen JJ, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17(12):2257–2317. doi: 10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 30.Vander Heiden JA, et al. pRESTO: a toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 2014;30(13):1930–1932. doi: 10.1093/bioinformatics/btu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yaari G, et al. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front Immunol. 2013;4:358. doi: 10.3389/fimmu.2013.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lefranc MP, et al. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev Comp Immunol. 2003;27(1):55–77. doi: 10.1016/S0145-305X(02)00039-3. [DOI] [PubMed] [Google Scholar]
- 33.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH. Change-O: a toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics. 2015;31(20):3356–3358. doi: 10.1093/bioinformatics/btv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuomisto H. A diversity of beta diversities: straightening up a concept gone awry. Part 1. Defining beta diversity as a function of alpha and gamma diversity. Ecography. 2010;33(1):2–22. doi: 10.1111/j.1600-0587.2009.05880.x. [DOI] [Google Scholar]
- 35.Smith K, et al. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat Protoc. 2009;4(3):372–384. doi: 10.1038/nprot.2009.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winter G. xia2: an expert system for macromolecular crystallography data reduction. J Appl Crystallogr. 2010;43(pt 7):186–190. [Google Scholar]
- 37.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Evans PR. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr D Biol Crystallogr. 2011;67(pt 4):282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69(pt 7):1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(pt 12 pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 42.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53(pt 3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 43.Adams PD, Mustyakimov M, Afonine PV, Langan P. Generalized X-ray and neutron crystallographic analysis: more accurate and complete structures for biological macromolecules. Acta Crystallogr D Biol Crystallogr. 2009;65(pt 6):567–573. doi: 10.1107/S0907444909011548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen VB, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(pt 1):12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tina KG, Bhadra R, Srinivasan N. PIC: Protein Interactions Calculator. Nucleic Acids Res. 2007;35(Web Server issue):W473–W476. doi: 10.1093/nar/gkm423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 47.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34(Web Server issue):W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41(Web Server issue):W34–W40. doi: 10.1093/nar/gkt382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yaari G, Uduman M, Kleinstein SH. Quantifying selection in high-throughput Immunoglobulin sequencing data sets. Nucleic Acids Res. 2012;40(17):e134. doi: 10.1093/nar/gks457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uduman M, Yaari G, Hershberg U, Stern JA, Shlomchik MJ, Kleinstein SH. Detecting selection in immunoglobulin sequences. Nucleic Acids Res. 2011;39(Web Server issue):W499–W504. doi: 10.1093/nar/gkr413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahal-Koirala S, et al. TCR sequencing of single cells reactive to DQ2.5-glia-α2 and DQ2.5-glia-ω2 reveals clonal expansion and epitope-specific V-gene usage. Mucosal Immunol. 2016;9(3):587–596. doi: 10.1038/mi.2015.147. [DOI] [PubMed] [Google Scholar]
- 52.Qiao SW, et al. Posttranslational modification of gluten shapes TCR usage in celiac disease. J Immunol. 2011;187(6):3064–3071. doi: 10.4049/jimmunol.1101526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.