

Abstract
Chronic inflammatory diseases, such as periodontal disease, associate with adverse wound healing in response to myocardial infarction (MI). The goal of this study was to elucidate the molecular basis for impaired cardiac wound healing in the setting of periodontal-induced chronic inflammation. Causal network analysis of 168 inflammatory and extracellular matrix genes revealed that chronic inflammation induced by a subseptic dose of Porphyromonas gingivalis lipopolysaccharide (LPS) exacerbated infarct expression of the proinflammatory cytokine Ccl12. Ccl12 prevented initiation of the reparative response by prolonging inflammation and inhibiting fibroblast conversion to myofibroblasts, resulting in diminished scar formation. Macrophage secretion of Ccl12 directly impaired fibronectin and collagen deposition and indirectly stimulated collagen degradation through upregulation of matrix metalloproteinase-2. In post-MI patients, circulating LPS levels strongly associated with the Ccl12 homologue monocyte chemotactic protein 1 (MCP-1). Patients with LPS levels ≥ 1 endotoxin units (EU)/ml (subseptic endotoxemia) at the time of hospitalization had increased end diastolic and systolic dimensions compared with post-MI patients with < 1 EU/ml, indicating that low yet pathological concentrations of circulating LPS adversely impact post-MI left ventricle (LV) remodeling by increasing MCP-1. Our study provides the first evidence to our knowledge that chronic inflammation inhibits reparative fibroblast activation and generates an unfavorable cardiac–healing environment through Ccl12-dependent mechanisms.
Keywords: Cardiology, Inflammation
Keywords: Cardiovascular disease, Chemokines, Extracellular matrix
Chronic inflammation inhibited reparative fibroblast activation to generate an unfavorable cardiac wound healing environment through Ccl12-dependent mechanisms.
Introduction
Periodontal disease (PD) is associated with adverse wound healing in response to injury across organ systems, including poor cardiac responses to myocardial infarction (MI) (1–6). MI incidence correlates with the number of pockets > 4 mm deep, bleeding sites after probing, and severity of tooth loss, even after adjustment for known cardiovascular risk factors (e.g., smoking, diabetes, or hypertension) (4). Antibodies against the periodontal pathogen Porphyromonas gingivalis are also elevated in MI patients (4), and heart-failure patients exhibit increased bone turnover markers in circulation and a more advanced PD phenotype compared with healthy control patients (6). In addition, severe periodontitis is more prevalent among post-MI heart-failure patients than dilated cardiomyopathy–induced heart-failure patients (5). PD is caused by chronic inflammation of tissues surrounding the teeth, in response to bacterial biofilm accumulation (7). Bacterial products, especially endotoxins, are key drivers of inflammation and PD development (8). It is hypothesized that the link between PD and cardiovascular disease (CVD) is due to chronic inflammatory mechanisms initiated by the bacteria products present within periodontal lesions (9). While the oral health and CVD epidemiological correlation is quite strong, the mechanistic link between oral health and MI response is not fully understood.
Porphyromonas gingivalis is one of the most frequent PD pathogens detected in the gums and circulation of patients with PD (10). Previously, we showed subseptic concentrations (0.8 μg/g body weight/day) of Porphyromonas gingivalis lipopolysaccharide (LPS) accelerated macrophage infiltration and increased cardiac rupture after MI in mice, suggesting PD-induced chronic inflammation may alter scar formation by altering the macrophage population (11).
Fibroblasts are the primary source of extracellular matrix (ECM) in the myocardium. A balanced turnover of ECM through matrix metalloproteinase–mediated (MMP-mediated) degradation and reparative fibroblast ECM synthesis is critical for adequate post-MI scar formation (12). Macrophages regulate both sides of the degradation and synthesis equation. Secretion of proinflammatory molecules such as TNF-α and IL-1β stimulates ECM degradation and tissue clearance by promoting MMP production, while secretion of antiinflammatory molecules such as TGF-β1 promote ECM synthesis and scar deposition (13). Despite our knowledge that macrophages regulate ECM turnover, little is known about the mechanisms behind macrophage activation as a means to regulate scar formation. The goal of the present study was to use a multidimensional systems biology approach to elucidate the molecular basis for impaired cardiac wound healing in the setting of periodontal-induced chronic inflammation.
Results
Chronic inflammation prolonged proinflammatory macrophage infiltration after MI.
To dissect macrophage regulation of post-MI scar formation, we assessed day 7 (d7) post-MI infarcts from LPS preexposed mice (LPS+MI), compared with both MI positive controls and no-MI negative controls (d0). At d7 after MI, macrophages are the predominant inflammatory cell type present in the left ventricle (LV) infarct, with ~1.5 × 104 cells/mg infarct (14, 15). LPS+MI decreased total leukocyte count (CD11b+ cells) and macrophage numbers (Mac3+ and F4/80+ cells) in the infarct at d7 after MI, detected by immunofluorescent staining (Figure 1, A–D), cell counts (Figure 1E), and flow cytometry (Figure 1F). Previously, we have shown LPS exposure increased macrophage infiltration at d1 after MI when compared with controls (11). The data indicate that LPS treatment accelerated the macrophage influx timeline, resulting in a more rapid peak and efflux.
Figure 1. Chronic inflammation decreased reparative M2 macrophage polarization at d7 after myocardial infarction (MI).

(A–D) By immunofluorescence, fewer leukocytes (CD11b+) and macrophages (CD11b+Mac3+) were present in the infarcts of lipopolysaccharide (LPS) preexposed MI mice (LPS+MI). Scale bar: 100 μm. n = 4/group (2 male [M], 2 female [F]); MI = 4.8 ± 0.1 months; LPS+MI = 5.2 ± 0.1 months. (E and F) Cell counts of CD11b+ and F4/80+ cells confirmed fewer leukocytes and macrophages in LPS exposed infarcts. (G and H) The decrease in macrophage numbers in the LPS+MI group was due to decreased reparative M2 macrophages (Q2; F4/80+CD206+). n = 6/group (3M, 3F); MI = 4.2 ± 0.1 months; LPS+MI = 4.8 ± 0.1 months. Data is shown as box and whisker plots with mean ± minimum/maximum; Nonparametric Wilcoxon rank sum test; *P < 0.05 vs. WT MI.
In addition to fewer macrophages within the infarct, there was also a decrease in CD206+ cells (Figure 1G). This decline was predominantly due to a drop in the reparative M2 macrophage population (F4/80+CD206+; Figure 1H). By d7 after MI, proinflammatory factors subside to facilitate collagen deposition and reparative fibroblast activation (16). In MI controls, 70% ± 3% of macrophages at d7 after MI were reparative M2 macrophages (Figure 1H, second quadrant), consistent with previous findings (14, 17). In contrast, only 54% ± 4% of LPS+MI macrophages were reparative M2 macrophages (P < 0.05). M1 macrophage numbers were not changed, resulting in an increased M1/M2 ratio (Figure 1H).
Proinflammatory M1 marker gene expression for Ccl3, IL-1β, and TNF-α, but not IL-6, were higher in macrophages isolated from LPS+MI mice compared with d7 post-MI controls (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.94207DS1). While the M2 marker CD206 was significantly lower at the protein level, no differences were observed in gene levels of any of the M2 markers evaluated (Arg1, CD206, Fizz, or Ym1). Although d7 post-MI macrophage numbers were decreased with LPS exposure, the net phenotype remained proinflammatory, leading to excessive and prolonged myocardial injury.
A multiomics approach revealed monocyte chemotactic protein-5 (MCP-5; Ccl12) as a potent proinflammatory M1 macrophage marker.
By hierarchal clustering of 168 inflammatory and ECM genes (Supplemental Table 1), the top 25 ranked differences in the LV infarct were linked to macrophage biology and proinflammation. Of these differences, 23 genes were upregulated and 2 downregulated in the LPS+MI infarct (Supplemental Figure 2A), compared with MI controls. Integrated pathway analysis revealed the main targeted pathways in the LPS+MI LV involved leukocyte extravasation (Supplemental Figure 2B). Changes in leukocyte kinetics would tip the balance toward a poor wound healing environment. The proinflammatory cytokine, Ccl12, was identified as the top feature exacerbated at d7 in the LPS+MI LV, by both volcano plot and causal network analyses (Supplemental Figure 2, C and D). In our previous study, we identified MCP-1 and -3 as being significantly elevated in LPS+MI mice compared with MI controls at d1 after MI (11). By d7, both MCP-1 and -3 were no longer significantly elevated in our LPS+MI mice compared with MI controls (Supplemental Table 1). Our results suggest that MCP-1 and -3 are fundamental during the early but not later post-MI phase and explain why we had an acceleration of the macrophage infiltration timeline with chronic LPS exposure. In the current study, Ccl12 is elevated at d1 in the LPS+MI group, and this elevation persisted through d7.
Ccl12 is an M1 marker highly expressed by macrophages and is associated with monocyte-derived macrophage and fibroblast recruitment (18). Ccl12 mRNA increased 150-fold at d5 and d7 in the infarct region, compared with d0, and the increase in Ccl12 mirrored the influx of monocyte-derived macrophages into the infarct area (Figure 2A). Increased Ccl12 was accelerated and doubled in the LPS+MI mice. Previously, we showed macrophage infiltration began earlier with LPS exposure (11). Consistent with the past report, Ccl12 spiked in the LPS+MI group at d1 after MI. The source of Ccl12 was the macrophage, as Ccl12 mRNA expression was 2-fold elevated, and protein secretion was 3-fold elevated, in macrophages isolated from the d7 infarct region (Figure 2, B and C). This was unexpected because total macrophage numbers were decreased in the LPS+MI group at d7 after MI, compared with the MI group. Ccl2, a homologue of Ccl12, was not significantly different in macrophages isolated from the infarct of d7 post-MI LV controls compared with the LPS+MI group. Since macrophages are known to regulate ECM deposition and scar formation (12, 17, 19), we hypothesized that chronic inflammation may prolong M1 macrophage polarization to regulate fibroblast function through increased secretion of Ccl12. We provide here the first evidence to our knowledge that chronic inflammation exacerbated macrophage secretion of Ccl12.
Figure 2. Chronic inflammation enhanced macrophage secretion of Ccl12, a proinflammatory M1 macrophage marker.

At d7 after myocardial infarction (MI), Ccl12 expression was significantly higher in lipopolysaccharide (LPS) preexposed mice (LPS+MI) in both (A) infarct tissue (n = 6/group [3M, 3F]; MI = 5.2 ± 0.1 months; LPS+MI = 6.0 ± 0.1 months) and (B) isolated macrophages (n = 6/group [3M, 3F]; MI = 4.0 ± 0.1 months; LPS+MI = 4.4 ± 0.1 months). (C) By ELISA of the post-MI macrophage secretome, Ccl12 secretion increased 3-fold in LPS+MI macrophages compared with MI macrophages. n = 8/group (4M, 4F); MI = 3.9 ± 0.1 months; LPS+MI = 4.5 ± 0.1 months. Data is shown as box and whisker plots with mean ± minimum/maximum. Time course was analyzed by one-way ANOVA, followed by the Student Newman-Keuls. For two group comparisons, the nonparametric Wilcoxon rank sum test was used. *P < 0.05 vs. d0; †P < 0.05 vs. WT MI.
Chronic inflammation reduced infarct strength by altering ECM composition to destabilize the infarct region.
Ccl12 directly and negatively linked with fibronectin and indirectly and positively linked with collagen degradation through MMP-2, elucidated by causal network analysis (Supplemental Figure 2D). Fibronectin and collagen are key ECM proteins that dictate the infarct scar formation (12, 20). Post-MI, full-length fibronectin, collagen I, and collagen III each increased at least 10-fold at d7 post-MI compared with no-MI controls (Figure 3, A–C). LPS+MI decreased the MI-stimulated increase in full-length fibronectin and collagen III protein more than 2-fold without a concomitant fall in mRNA levels (Supplemental Table 1), demonstrating impaired ECM synthesis or deposition. Accelerated collagen I turnover, as evidenced by enhanced collagen fragmentation, was significantly higher in LPS+MI LV compared with MI controls (Figure 3B).
Figure 3. Chronic inflammation decreased extracellular matrix (ECM) deposition and increased collagen turnover at d7 after myocardial infarction (MI).

(A) Fibronectin protein levels were significantly lower in LPS+MI mice compared with MI mice. (B) Full-length collagen I was not affected, while collagen I fragmentation was significantly higher in LPS+MI infarcts, indicating accelerated ECM turnover. (C) In the LPS+MI infarcts, collagen III was significantly lower compared with the MI control. n = 6/group (3M, 3F); MI = 5.1 ± 0.1 months; LPS+MI = 5.9 ± 0.1 months. (D) MMP-2 gene levels significantly increased in LPS+MI infarcts and correlated with decreased post-MI LV wall thickness. n = 7–8/group (3–4M, 4F); MI = 5.2 ± 0.1 months; LPS+MI = 6.0 ± 0.1 months. This indicated that chronic inflammation induced insufficient ECM deposition after MI, leading to poor scar formation and excessive wall thinning. Data is shown as box and whisker plots with mean ± minimum/maximum; one-way ANOVA with Student Newman-Keuls post-test; *P < 0.05 vs. d0; †P < 0.05 vs. WT MI.
MMP-2 was the key regulator for the increased collagen turnover in the LPS+MI LV, as MMP-2 was the only one of 12 MMPs measured that was significantly higher and negatively correlated with decreased wall thickness in LPS+MI LV (Supplemental Table 1 and Figure 3D). In addition to ECM synthesis and deposition, ECM cross-linking is a necessary component to generate scars of sufficient strength. Lysyl oxidase (LOX), the major collagen cross-linking enzyme, increases during the MI maturation phase (21, 22). LPS+MI reduced LOX at d7 after MI compared with MI controls (Figure 4A). After MI, fibroblasts differentiate into myofibroblasts and actively produce ECM. Myofibroblasts are more contractile and have increased potential for ECM synthesis than fibroblasts, enabling repair. The myofibroblasts marker, α–smooth muscle actin (αSMA), was significantly decreased in the infarct area of LPS+MI mice (Figure 4B), indicating diminished differentiation capacity.
Figure 4. Chronic inflammation decreased fibroblast differentiation and increased proliferation at d7 after myocardial infarction (MI).

(A) Representative immunoblot. n = 6/group (3M, 3F); MI = 5.1 ± 0.1 months; LPS+MI = 5.9 ± 0.1 months. The collagen cross-linking enzyme, lysyl oxidase (LOX), was lower in infarcts of LPS+MI mice at d7 after MI compared with MI controls. (B) By immunofluorescence, the percent area stained positive for α–smooth muscle actin (αSMA) was reduced in the LPS+MI infarcts at d7 after MI. Scale bar: 60 μm. n = 6/group (3M, 3F); MI = 4.8 ± 0.1 months; LPS+MI = 5.2 ± 0.1 months. By d7 after MI, fibroblasts typically differentiate into the reparative myofibroblast phenotype to increase ECM production. Our data indicate chronic inflammation impaired wound healing by inhibiting myofibroblast activation. Data is shown as box and whisker plots with mean ± minimum/maximum; LOX was analyzed by one-way ANOVA, followed by the Student Newman-Keuls, and αSMA was analyzed by nonparametric Wilcoxon rank sum test. *P < 0.05 vs. d0; †P < 0.05 vs. WT MI.
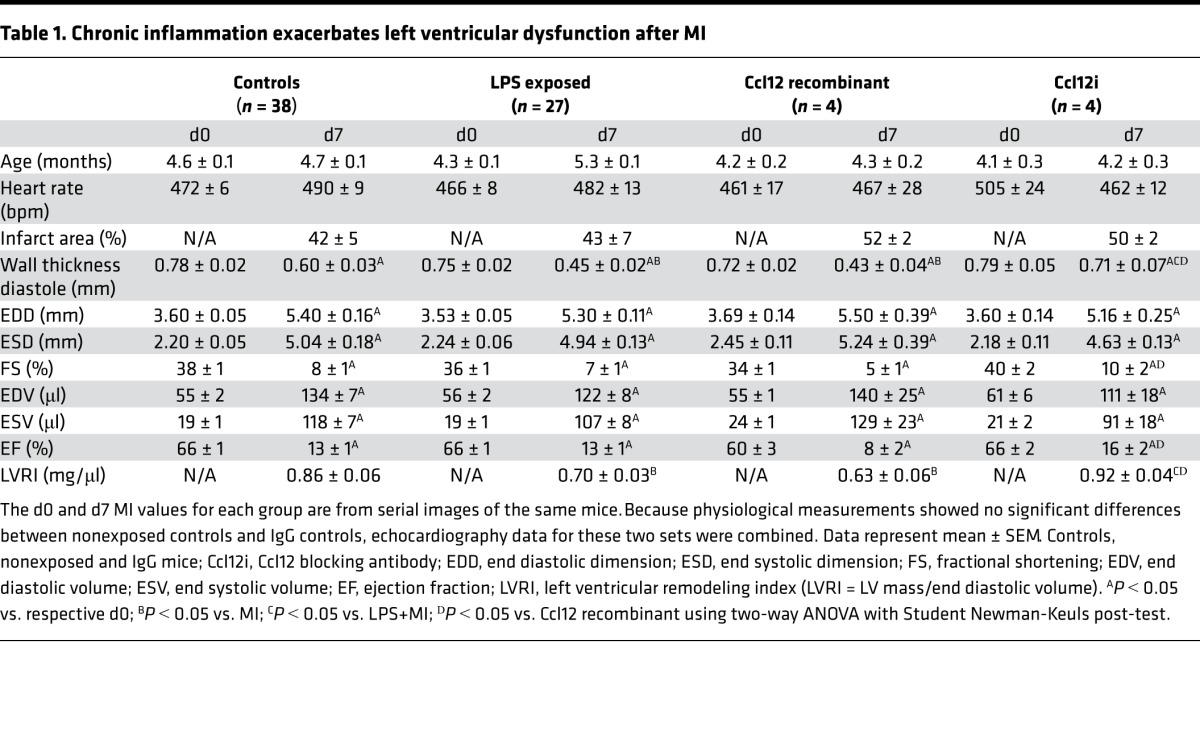
Impaired ECM synthesis and insufficient cross-linking can increase wall stress, initiating excessive infarct expansion and increasing susceptibility to rupture (23). Chronic inflammation accelerated the timing and increased the incidence of cardiac rupture in the current cohort of mice (Figure 5, A and B), consistent with previous findings (11). Post-MI cardiac dysfunction was exacerbated by chronic inflammation, as indicated by increased infarct wall thinning and decreased LV remodeling index (Table 1). Early after MI, the infarct is less extensible in both the circumferential and longitudinal directions (24). LPS+MI exacerbated myocardial fiber rearrangement, resulting in decreased overall radial strain, systolic radial strain, and systolic longitudinal strain (Figure 5, C–F). Impaired strain reflects increased myocardial wall stress, with concomitant decreased cardiac physiology and increased rupture (25). These results implicate Ccl12 as a possible mediator of adverse cardiac wound healing during chronic inflammation by interfering with multiple scar formation components.
Figure 5. Chronic inflammation increased myocardial wall stress, resulting in increased cardiac rupture after myocardial infarction (MI).

(A) Survival analysis showed no significant difference in day-7 survival in LPS+MI mice compared with MI controls. (B) All mice that died after MI in the LPS+MI group died of cardiac rupture (37/37). In MI controls, 50% died of cardiac rupture (11/23), indicating chronic inflammation increased the risk of rupture after MI. (C–F) Strain analysis of the mid to apical region (green line; C) of MI and LPS+MI mice showed a significant decrease in (D) overall radial strain, (E) systolic radial strain, and (F) systolic longitudinal strain at d7 after MI. Chronic inflammation exacerbated this response, indicating an increase in myocardial wall stress. n = 8/group (4M, 4F); MI = 4.2 ± 0.1 months; LPS+MI = 4.7 ± 0.1 months. Data is shown as box and whisker plots with mean ± minimum/maximum. The survival rate was analyzed by Kaplan–Meier survival analysis and compared by the log-rank test. Rupture rates were analyzed by Fisher’s exact test. Multiple group comparisons were analyzed by one-way ANOVA, followed by the Student Newman-Keuls. *P < 0.05 vs. MI; †P < 0.05 vs. d0.
Table 1. Chronic inflammation exacerbates left ventricular dysfunction after MI.
Macrophage secretion of Ccl12 regulated ECM gene expression, reparative fibroblast activation, and proliferation.
To determine the mechanisms whereby macrophage secretion of Ccl12 was responsible for decreased ECM deposition, macrophages were isolated from the MI and LPS+MI infarct regions and cultured for 18 hours to collect the secretome. Cells underwent a washing step before culture to remove any residual LPS left from in vivo exposure in order to ensure effects observed were due to MI-stimulated macrophage secretion. Fibroblasts isolated from control unoperated LVs and exposed to the secretomes of macrophages isolated from d7 LPS+MI infarcts were more proliferative than fibroblasts stimulated with the MI macrophage secretome (Figure 6, A and B). No differences were observed in migration between LPS+MI and MI macrophage secretome–stimulated fibroblasts.
Figure 6. Macrophage secretion of Ccl12 regulates wound healing by decreasing myofibroblast activation and increasing proliferation.

(A) Stimulating naive fibroblasts with macrophage conditioned media isolated from lipopolysaccharide-preexposed MI mice (LPS+MI) increased cell proliferation but had no effect on cell migration. n = 4/group (4M); 4.5 ± 0.1 months for all groups. (B) Fibroblasts stimulated with macrophage conditioned media from LPS+MI mice decreased gene levels of α–smooth muscle actin (αSMA), collagen I, and TGFβ1 and increased BrdU uptake to indicate increased fibroblasts proliferation. n = 6/group (4M); 4.0 ± 0.1 months for all groups. (C) Secretion of extracellular matrix (ECM) decreased in fibroblasts stimulated with macrophage media from LPS+MI mice as shown by decreased procollagen and fibronectin protein. Ccl12 blocking antibody (Ccl12i) attenuated the effect of the LPS+MI macrophage secretome. n = 6/group (6M); 4.0 ± 0.1 months for all groups. All in vitro stimulation experiments were paired. Data is shown as box and whisker plots with mean ± minimum/maximum; one-way ANOVA with Student Newman-Keuls post-test; *P < 0.05 vs. MI; †P < 0.05 vs. LPS+MI.
Ccl12 inhibition (Ccl12i) decreased proliferation and increased migration in fibroblasts stimulated with the secretome of macrophages isolated from the LPS+MI infarct, indicating the secretome effect was Ccl12 mediated. When Ccl12 signaling was blocked, proliferation decreased with a concomitant rise in migration. LPS alone did not explain the in vivo fibroblast phenotype, as cardiac fibroblasts stimulated in vitro with the same nonseptic concentrations of LPS demonstrated no direct effect on proliferation or F-actin formation (Supplemental Figure 3). These results are consistent with studies in human gingival and small intestinal lamina propria fibroblasts (26–28). LPS, therefore, was not directly responsible for the in vivo changes in fibroblast function; this effect was via LPS-induced macrophage secretion of Ccl12.
Fibroblasts isolated from control unoperated LVs and stimulated with LPS+MI macrophage secretome exhibited reduced mRNA expression of collagen I, TGFβ1, and αSMA (Figure 6B). Ccl12i restored αSMA gene expression, unmasking the importance of Ccl12 in regulating fibroblast differentiation. The effect was not autocrine because fibroblasts expressed 20-fold lower Ccl12 at all post-MI times examined, compared with macrophage contributions, and therefore the fibroblast was not a relevant Ccl12 source (Supplemental Figure 4A). Gene levels of connective tissue growth factor (Ctgf), collagen III, and fibronectin were not significantly different between LPS+MI and MI controls (Supplemental Figure 4B). Proinflammatory macrophage secretion of Ccl12, therefore, directly regulated post-MI fibroblast activation and ECM deposition.
The secretome of macrophages isolated from d7 LPS+MI infarcts inhibited fibroblast secretion of pro–collagen I and fibronectin protein, demonstrated by immunoblot analysis of fibroblast-conditioned media (Figure 6C and Supplemental Figure 4C). Ccl12i partially restored collagen I but not fibronectin protein in LPS+MI macrophage secretome–exposed fibroblasts. Our results indicate that fibroblasts within a proinflammatory environment further impair wound healing by failing to undergo myofibroblast activation. Of the secretome constituents, our data identified Ccl12 as the contributor to adverse remodeling by increasing fibroblast proliferation and decreasing fibroblast migration and activation to impair collagen production.
Ccl12 mimicked effects of chronic inflammation on fibroblast ECM gene expression, reparative fibroblast activation, and proliferation.
To determine how the effects of chronic inflammation on post-MI wound healing were directly due to elevated Ccl12 levels, no MI cardiac fibroblasts were stimulated with Ccl12 recombinant protein. To assess the Ccl12 signaling pathway, Ccr2 — the only known Ccl12 receptor — was inhibited to determine whether Ccl12 actions were dependent on Ccr2 activation (29). Ccr2 inhibition (Ccr2i) totally obliterated fibroblast migration, which was predictable, as Ccr2 is vital for cell migration (18, 30). Fibroblasts stimulated with Ccl12 were proliferative (BrdU-positive) independent of Ccr2, confirming that Ccl12 can directly stimulate fibroblast proliferation through Ccr2-independent signaling (Figure 7A).
Figure 7. Ccr2 activation by Ccl12 induced fibroblast dysfunction and impaired wound healing.

(A) In vitro stimulation of cardiac fibroblasts with Ccl12 attenuated cell migration (by electric cell–substrate impedance sensing, ECIS) and increased cell proliferation (by BrdU uptake) independent of Ccr2. n = 5/group (5M); 4.5 ± 0.1 months for all groups. All in vitro stimulation experiments were paired. (B) Collagen I, fibronectin, TGFβ1, and α–smooth muscle actin (αSMA) significantly decreased in Ccl12-stimulated fibroblasts. Ccr2 inhibition partially restored Ccl12 effects on all ECM genes. n = 5/group (5M); 4.5 ± 0.1 months for all groups. All in vitro stimulation experiments were paired. (C) In vivo Ccl12 exposure in the post-MI LV increased cardiac rupture similar to rates observed for the LPS+MI mice. This was attenuated by administering a Ccl12 blocking antibody (Ccl12i) in vivo. (D) Administration of Ccl12i increased the 25 kDa band by 2-fold, indicating successful Ccl12 inhibition. (E) In addition, Ccl12 upregulation increased collagen turnover and (F) active MMP-2 similar to what was observed in LPS+MI mice. Ccl12i attenuated this effect. No change was observed in IgG controls. n = 4/group (2M, 2F for MI, IgG, and Ccl12i; 1M, 3F for recombinant Ccl12 [rCcl12]); MI = 4.7 ± 0.1 months; IgG = 3.6 ± 0.1 months; rCcl12 = 4.3 ± 0.2 months; Ccl12i = 3.6 ± 0.1 months. Data is shown as box and whisker plots with mean ± minimum/maximum; one-way ANOVA with Student Newman-Keuls post-test; *P < 0.05 vs. 0.1% FBS; †P < 0.05 vs. Ccl12 stimulated cells; ‡P < 0.05 vs. d7 MI.
Providing additional proof that Ccl12 is responsible for LPS+MI effects on fibroblast dysfunction, Ccl12 decreased fibroblast expression of collagen I, fibronectin, TGFβ1, and αSMA (Figure 7B). Stimulating fibroblasts with Ccl12 did not directly affect collagen III or Ctgf (Supplemental Figure 4D). This is consistent with what has been observed with the Ccl12 human isoform, hMCP-1, as stimulation of human fibroblasts from multiple sources (fetal or adult lung or adipose tissue) with hMCP-1 in vitro inhibits collagen expression and decreases αSMA expression (31). While the effects of Ccl12 on proliferation and migration were independent of Ccr2, Ccr2i restored effects on collagen I, fibronectin, TGFβ1, and αSMA protein expression. This suggests that Ccl12 actions are both dependent and independent of Ccr2 engagement.
To dissect Ccl12 effects in vivo, mice were infused with either recombinant Ccl12 or a Ccl12 blocking antibody (Ccl12i) at the time of MI surgery by s.c. osmotic minipump placement. Similar to what was observed in LPS+MI mice, increasing Ccl12 levels increased LV wall thinning and incidence of cardiac rupture (Table 1 and Figure 7C). Bound Ccl12 (25-kDa band) increased 2-fold with Ccl12i infusion, indicating the blocking antibody was successfully binding to Ccl12 and, thus, inhibiting its biological activity (Figure 7D). Collagen I fragmentation and active MMP-2 levels were also enhanced with Ccl12 infusion compared with MI controls (Figure 7, E and F). Administration of Ccl12i attenuated this affect and provided further evidence that Ccl12 is a key regulator of adverse post-MI wound healing. The IgG control antibody group had no significant differences in cardiac physiology (Table 1), bound Ccl12 levels, collagen fragmentation, or active MMP-2 levels (Figure 7, D–F) compared with the MI nonexposed controls. Our data present a crucial role for macrophage secretion of Ccl12 in the setting of chronic inflammation to stimulate fibroblast dysfunction.
In humans, elevated subseptic levels of circulating LPS in post-MI patients linked to circulating hMCP-1 concentrations and increased LV dilation.
Humans express Ccl2 (MCP-1) but not Ccl12 (MCP-5), while mice have both homologues of hMCP-1: mCcl2 (MCP-1) and mCcl12 (MCP-5). Protein alignment showed mCcl2 was 75% homologous (Bit Score = 114, E-value = 1 × 10–38) to hMCP-1, while mCcl12 was 84% homologous (Bit score = 138, E-value = 6 × 10–49) (Figure 8A). The differences in protein sequence resulted in structural changes in mCcl2 that are absent in mCcl12 and hMCP-1 (Figure 8B). While sequence homology is high for both isoforms, hMCP-1 and mCcl12 contain a β-strand at residue 20 that is absent in mCcl2, resulting in changes in binding efficiency and, thus, protein function (32–34). Mouse Ccl12, therefore, is closer in homology and structure to hMCP-1. Because of this, mouse Ccl12 is also termed MCP-1–related chemokine.
Figure 8. Elevated circulating endotoxins resulted in increased monocyte chemotactic protein 1 (MCP-1) levels, leading to exacerbation of cardiac remodeling.

(A) Basic local alignment search tool (BLAST) exhibited human MCP-1 is 75% homologous with mouse Ccl2 and 84% homologous with mouse Ccl12. Bit Score was also higher for mCcl12 compared with mCcl2, indicating closer homology. This was especially true at the N-terminal region. (B) The N-terminus of mCcl2 has a β sheet that is absent in hMCP-1 and mCcl12. Structural similarities in the N-terminus result in functional similarities in hMCP-2 and mCcl12. (C) Post-MI patients with elevated circulating endotoxin levels (˃1 EU/ml) had increased left ventricular end systolic and diastolic diameters(LVIDD). (D) Endotoxin levels correlate with increased MCP-1, revealing chronic inflammation induced by periodontal disease induces amplified MCP-1 production after MI. Matrix metalloproteinase 12 (MMP-12) correlated with increased left ventricular internal systolic diameter (LVISD). MMP-12 was normalized to white blood cell (WBC) count to assess macrophage MMP-12 contribution. n = 84/low endotoxin group (53M, 31F; 58 ± 13 years) n = 13/high-endotoxin group (8M, 5F; 57 ± 8 years). Data is shown as box and whisker plots with mean ± minimum/maximum. For two group comparisons, the nonparametric Wilcoxon rank sum test was used. *P < 0.05 vs. 0–1 EU/ml; n = 83 in the 0–1 EU/ml group and n = 13 in the > 1 EU/ml group.
In patients with acute coronary syndrome, high plasma hMCP-1 concentrations (˃238 pg/ml) increased the risk of death and recurrent ischemic events by 2-fold, independent of standard risk predictors (35). To determine if chronic inflammation could be linked to elevated hMCP-1 levels and clinical indices of adverse remodeling, we measured circulating plasma endotoxin levels in post-MI patients (n = 97; age, median 58 years and range 33–86 years; 38% women; 48% African American), using plasma collected during hospitalization and compared with echocardiography and clinical characteristics (Table 2). The average time for sample collection was 22 ± 12 hours after presentation. There was no significant difference in sample collection times between the high and low endotoxin groups (P = 0.75). The majority of MI patients underwent percutaneous coronary intervention (PCI; 78/97; 81%). The rest were treated either with a thrombolytic (2/97; 2%), a combination of anticoagulant and antiplatelet (15/97; 15%), or coronary bypass surgery (2/97; 2%). There were no significant differences in interventions between groups (P = 0.94). Out of the 97 MI patients enrolled, 34 had a history of diabetes (29 in low [37%] and 5 in high [38%] endotoxin groups; P = 0.96).
Table 2. Post-MI patient characteristics.
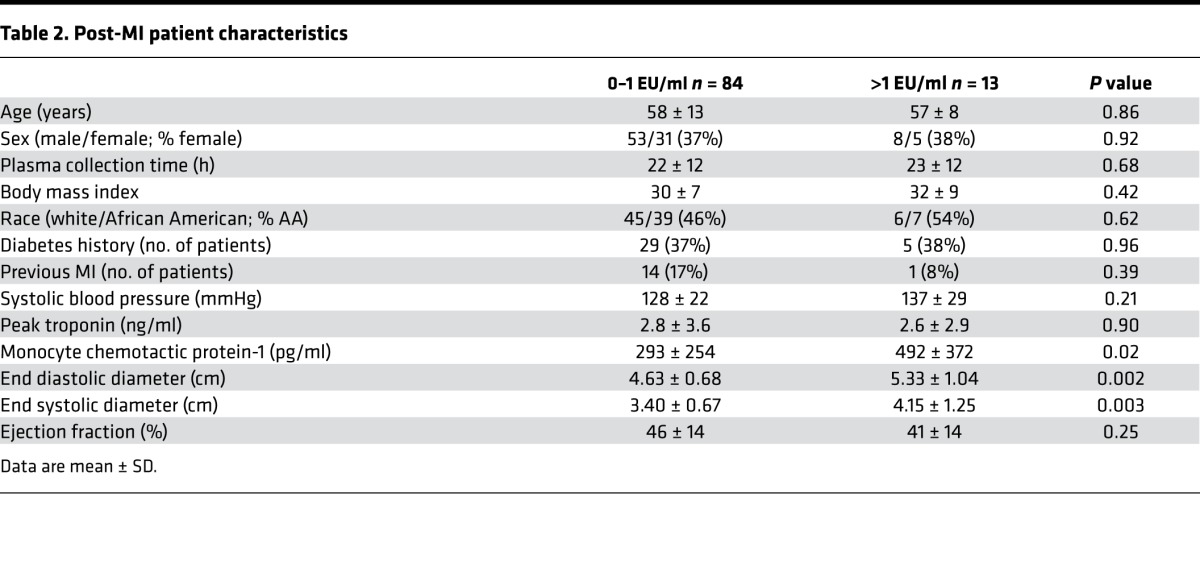
LPS levels in plasma from healthy donors have a range of < 1 EU/ml. In periodontal patients, LPS circulating levels can range from 1–4 EU/ml (10). MI patients with significantly elevated nonseptic endotoxemia (>1 EU/ml) had higher end systolic and diastolic LV internal diameters and higher circulating hMCP-1 (Table 2 and Figure 8C). There were no signs of acute complications (e.g., respiratory tract or urinary tract infection, or infection due to indwelling line) in any of the patients enrolled. Age, sex, BMI, systolic blood pressure, smoking history, diabetes history, and troponin, triglyceride, and cholesterol levels did not explain the effect, as each of these variables was not significantly different between groups. Elevated endotoxin levels significantly correlated with increased circulating hMCP-1 levels (Figure 8D). In addition, MMP-12, a macrophage marker, correlated with increased left ventricular internal systolic diameter (LVISD), demonstrating the impact of macrophage-mediated inflammation on cardiac physiology. For 83 patients (86%), this was their first MI. Of the 14 patients with a previous MI, only 1 was in the high-endotoxin group. Removal of these 14 samples did not affect significance for MCP-1 levels (P = 0.01), LVISD (P = 0.005), LVIDD (P = 0.004), or correlation of endotoxin to MCP-1 (r = 0.23, P = 0.04).
Discussion
In this study, we combined genomic, proteomic, biochemical, and pathophysiological evaluations in mice and human MI subjects to elucidate the role of PD-induced chronic inflammation on cardiac wound healing after MI. Our study is the first to our knowledge to reveal that chronic inflammation increases macrophage secretion of Ccl12 to inhibit reparative fibroblast activation and ECM deposition (Figure 9). This resulted in decreased cardiac function and increased cardiac rupture, revealing that fibroblast activation is a critical step for stable scar formation.
Figure 9. Chronic inflammation induced adverse wound healing after myocardial infarction (MI) through Ccl12.

Preexisting chronic inflammation (LPS+MI) led to an overabundance of Ccl12 secretion by macrophages. This, in turn, induced fibroblast dysfunction and excessive matrix metalloproteinase 2 (MMP-2) levels. Ccl12-stimulated fibroblasts did not differentiate into myofibroblasts and therefore did not produce sufficient extracellular matrix, resulting in disproportionate collagen turnover, excessive left ventricular dilation, and cardiac rupture. The figure was generated using Servier Medical Art (http://www.servier.com).
Approximately 45% of the population in the US and Europe have been diagnosed with PD (36). Chronic inflammation associates with increased fibrosis across a variety of organs, including the lung and intestine (37, 38). When superimposed on MI, chronic inflammation prolongs wound healing and generates a potentially inadequate scar. Our findings are consistent with studies in early-stage diabetic hearts, where cardiac fibroblasts were highly proliferative and proinflammatory, resulting in compromised wound healing and increased LV dilation (39). Understanding how chronic inflammation alters cardiac wound healing provides insight into how to limit post-MI progression to congestive heart failure.
Activated fibroblasts are the primary source of ECM proteins that comprise the infarct scar, and dysregulation of fibroblast activation can lead to poor healing through excessive or insufficient cardiac scar formation (12, 23). In a proinflammatory environment driven by Ccl12, fibroblast activation is initiated at the proliferation step but does not continue to progress to the differentiated reparative myofibroblast phenotype. Having incompletely activated fibroblasts that are less responsive to apoptotic stimuli compared with myofibroblasts would set the stage for excessive continued fibroblast presence that would alter long-term homeostasis of the cardiac wound. Controlling myofibroblast activation and function in order to stimulate proper wound healing will attenuate adverse or excessive LV remodeling and preserve LV function after MI (12).
The human homologue of Ccl12, hMCP-1, is a regulator of macrophage chemotaxis and has been linked to diseases characterized by monocyte-rich infiltrates (40–42). In patients, hMCP-1 is an immune diagnostic biomarker in the pathogenesis of chronic PD (43). Both mCcl2 and mCcl12 are homologues of hMCP-1, with mCcl2 being 75% homologous (Bit Score = 114) and mCcl12 being 84% homologous (Bit score = 138) to the human form. The overlapping and distinct activities of mCcl2 and mCcl12 allow subcomponents of the hMCP-1 protein to be dissected. After MI, mCcl2 has been shown to regulate macrophage recruitment and activation (44, 45). In addition, cardiac myofibroblasts isolated from WT but not from Ccl2-deleted animals undergoing repetitive myocardial ischemia and reperfusion demonstrated enhanced proliferative capacity (45). Both mCcl2 and mCcl12, therefore, stimulate increased proliferation and decreased migration in cardiac fibroblasts. In addition, in vitro hMCP-1 and mCcl12 — but not mCcl2 — inhibited collagen and αSMA expression in human fetal lung fibroblasts, human pulmonary fibroblasts, and human preadipocytes (31, 45). While the mouse has two homologues of hMCP-1, the mCcl12 homologue matches more closely to human Ccl2 than the mCcl2, both in terms of structure and function. Structurally, both human Ccl2 and mouse Ccl12 lack the β-strand that is present in the mCcl2, indicating that mCcl12 is structurally closer and, thus, better mimics hMCP-1.
The chemokine receptor Ccr2 is the only known receptor for Ccl12 (29). Ccr2i after MI attenuates LV remodeling by reducing inflammation through decreased macrophage recruitment (30, 46). Our study revealed Ccr2 plays a crucial role in LV remodeling by regulating fibroblast activation. Macrophage secretion of Ccl12 activated Ccr2 to inhibit myofibroblast activation and decrease ECM production. Unexpectedly, Ccl12 actions on fibroblast proliferation and migration were independent of Ccr2, consistent with reports that Ccl12 has unidentified receptors (29). Ccl12 interaction with these receptors may be responsible for stimulating a proliferative yet inflammatory fibroblast.
After MI, monocytes are recruited to the infarct in two phases. The first phase is dominated by Ly6chigh monocytes that give rise to early inflammatory macrophages, which remove necrotic tissue by phagocytosis and secrete proteolytic enzymes including MMP-2 (47). In the second phase, Ly6clow monocytes facilitate wound healing and regeneration to promote myofibroblast accumulation, collagen deposition, and angiogenesis (17, 47). Chronic inflammation induced by LPS exposure exacerbated LV adverse remodeling by accelerating macrophage infiltration and inhibiting resolution of M1 macrophages. The combined early removal of macrophages and decreased M2 activation provides a mechanism for impaired LV wound healing. Similar to what we observed, studies performed by both De Jesus et al. (Ripplinger lab) and Panizzi et al. (Nahrendorf lab) have shown that acute LPS exposure at the time of MI increases protease activity, phagocytic activity, macrophage infiltration, and LV dilation at d5 after MI (48, 49). In contrast, macrophages numbers were elevated at d5 after acute LPS exposure, whereas, after chronic exposure, macrophage numbers are attenuated at the later phase after MI. In addition, acute LPS exposure increases cardiac fibrosis without the onset of cardiac injury, suggesting that the mechanisms behind LV dysfunction may have distinct signaling attributes that distinguish acute and chronic inflammation mechanisms (50).
In our clinical study, the post-MI patients were not assessed for PD; rather, plasma endotoxemia levels were measured. We reviewed the clinical data, and there were no indications of active infection. Only one patient was prescribed an antibiotic (as a preventative after coronary bypass surgery); of note, this patient was not part of the high-endotoxin group. Our clinical data demonstrate that chronic inflammation due to a low-grade infection can induce adverse remodeling. This is consistent with Ismahil et al., who showed that increased proinflammatory macrophages led to development of chronic heart failure (51). Additional studies evaluating whether the source of endotoxemia changes response are warranted.
In conclusion, chronic inflammation prolonged the post-MI proinflammatory macrophage response, which in turn attenuated myofibroblast activation and impaired cardiac wound healing to increase cardiac rupture and dilation. Macrophage secretion of Ccl12 mediated these effects. These findings give us insight into the cellular events that increase the risk of mortality and heart failure in post-MI patients with a chronic inflammatory disease and provide global mechanisms whereby chronic inflammation impairs wound healing.
Methods
Supplemental Methods are available online with this article.
Experimental design.
C57BL/6J WT mice, 3–7 months of age (4.6 ± 0.1 months) and equal male and female numbers, were obtained from Jackson Laboratories for this study. Groups were examined simultaneously in a random experimental design, with the evaluator blinded to groups for all data acquisition and analyses. Porphyromonas gingivalis LPS was infused by osmotic minipumps (models 2004 and 1007D, Durect) to induce chronic inflammation as described previously (11). After 28 days, MI was induced through permanent ligation of the left anterior descending coronary artery (LPS+MI). Mice were continually infused with LPS until tissue collection (11, 52). Recombinant Ccl12, Ccl12 blocking antibody, or goat IgG antibody were infused by osmotic pump at the time of MI surgery to dissect Ccl12 mechanisms on cardiac wound healing in vivo.
Coronary artery ligation.
Coronary artery ligation was performed as described previously (11, 53). Mice were anesthetized with 1%–2% isoflurane in oxygen, intubated, and put on a standard rodent ventilator. An 8-0 suture was used to ligate the left coronary artery, and MI was confirmed by LV blanching and ECG changes showing ST segment elevation. Prior to the surgery, buprenorphine (0.1 mg/kg, UMMC Pharmacy) was administered.
Macrophage and fibroblast phenotyping.
To test the effect of periodontal-induced chronic inflammation, genetic markers were used to assess cell phenotype, as shown in Supplemental Tables 1 and 2.
Protein alignment and structure analyses.
Alignment of human MCP-1 (UniProt, P13500), mouse Ccl2 (UniProt, P10148), and mouse Ccl12 (UniProt, Q62401) was performed using Basic Local Alignment Search Tool (BLAST) and Constraint-based Multiple Alignment Tool (COBALT) as previously described (54–56). Structural 3D models were built using SWISS-MODEL Workspace (57–59).
Human subjects.
Plasma was collected 22 ± 12 hours (range of 4–66 hours) after hospitalization in post-MI patients (n = 97; age, median 58 years and range 33–86 years; 38% women; 48% African American). Patient characteristics are listed in Table 2. Echocardiographic data was collected within 20 ± 14 hours after admission. Plasma LPS levels were detected and semiquantified by a chromogenic assay (Endpoint Chromogenic LAL Assay, Lonza). EU/ml were calculated by a standard curve and best-fit linear trend line. No patient had septic levels of endotoxin.
Statistics.
Multiple group comparisons were analyzed by one-way ANOVA, followed by the Student Newman-Keuls when the Bartlett’s variation test passed, or by the nonparametric Kruskal-Wallis ANOVA test followed by Dunn post-hoc test when normal distribution of data was not met. For two group comparisons, the nonparametric Wilcoxon rank sum test was used. The survival rate was analyzed by Kaplan–Meier survival analysis and compared by the log-rank test. Rupture rates were analyzed by Fisher’s exact test. A value of P < 0.05 was considered statistically significant. The heat map and volcano plot were constructed using statistical software available in Metaboanalyst 3.0 (www.metaboanalyst.ca/) (60, 61). The top features were ranked by two-tailed t tests to retain the most contrasting patterns. Power analysis was performed to ensure all experiments were sufficiently powered to assess changes.
Study approval.
This study complies with the Declaration of Helsinki (62). All animal procedures were approved by the IACUC at the University of Mississippi Medical Center in accordance with the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011) and followed the ARRIVE guidelines (63). Written informed consent was collected from all patients under protocols approved by the ethics committee of the University of Mississippi Medical Center (IRB protocol 2013-0164).
Author contributions
KYDP and MLL conceived the hypothesis. KYDP analyzed all data and performed mouse surgeries, echocardiography, and flow cytometry; CAC, ERF, and KYDP performed cell culture; CAC, DS, MJ, and KYDP performed immunofluorescence; ERF, PLC, and KYDP performed immunoblotting; ERF performed MMP-12 dot blots; KYDP and PLC performed gene analysis; MEH, KYDP, and OKE analyzed patient characteristics; and KYDP, RPI, YM, MRG, WB, MEH, and MLL designed experimental procedures, interpreted data, and drafted manuscript. All authors read, edited, and approved the final manuscript.
Supplementary Material
Acknowledgments
This work was supported by the American Heart Association (15SDG22930009 to YM), by the NIH (T32HL105324 to OKE and HL075360, HL129823, and GM114833 to MLL), by the Biomedical Laboratory Research and Development Service of the Department of Veterans Affairs (5I01BX000505 to MLL and IK2BX003922 to KYDP), and by NIH HL051971, GM104357, GM115428, GM103476, and GM103328. The content is solely the responsibility of the authors and does not necessarily represent the official views of the American Heart Association, the NIH, or the Veterans Administration.
Version 1. 09/21/2017
Electronic publication
Footnotes
License: This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(18):e94207. https://doi.org/10.1172/jci.insight.94207.
Contributor Information
Mira Jung, Email: mjung@umc.edu.
William Buchanan, Email: wibuchanan@umc.edu.
Yonggang Ma, Email: yma@umc.edu.
References
- 1.Authors/Task Force Members, et al. 2016 European Guidelines on cardiovascular disease prevention in clinical practice: The Sixth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of 10 societies and by invited experts): Developed with the special contribution of the European Association for Cardiovascular Prevention & Rehabilitation (EACPR) Eur J Prev Cardiol. 2016;23(11):NP1–NP96. doi: 10.1177/2047487316653709. [DOI] [PubMed] [Google Scholar]
- 2.Blaizot A, Vergnes JN, Nuwwareh S, Amar J, Sixou M. Periodontal diseases and cardiovascular events: meta-analysis of observational studies. Int Dent J. 2009;59(4):197–209. [PubMed] [Google Scholar]
- 3.Holmlund A, Holm G, Lind L. Number of teeth as a predictor of cardiovascular mortality in a cohort of 7,674 subjects followed for 12 years. J Periodontol. 2010;81(6):870–876. doi: 10.1902/jop.2010.090680. [DOI] [PubMed] [Google Scholar]
- 4.Holmlund A, Hedin M, Pussinen PJ, Lerner UH, Lind L. Porphyromonas gingivalis (Pg) a possible link between impaired oral health and acute myocardial infarction. Int J Cardiol. 2011;148(2):148–153. doi: 10.1016/j.ijcard.2009.10.034. [DOI] [PubMed] [Google Scholar]
- 5.Fröhlich H, et al. Periodontitis in Chronic Heart Failure. Tex Heart Inst J. 2016;43(4):297–304. doi: 10.14503/THIJ-15-5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schulze-Späte U, et al. Periodontitis and bone metabolism in patients with advanced heart failure and after heart transplantation. ESC Heart Fail. 2017;4(2):169–177. doi: 10.1002/ehf2.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Humphrey LL, Fu R, Buckley DI, Freeman M, Helfand M. Periodontal disease and coronary heart disease incidence: a systematic review and meta-analysis. J Gen Intern Med. 2008;23(12):2079–2086. doi: 10.1007/s11606-008-0787-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cekici A, Kantarci A, Hasturk H, Van Dyke TE. Inflammatory and immune pathways in the pathogenesis of periodontal disease. Periodontol 2000. 2014;64(1):57–80. doi: 10.1111/prd.12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schenkein HA, Loos BG. Inflammatory mechanisms linking periodontal diseases to cardiovascular diseases. J Periodontol. 2013;84(4 Suppl):S51–S69. doi: 10.1902/jop.2013.134006. [DOI] [PubMed] [Google Scholar]
- 10.Shaddox LM, et al. Local inflammatory markers and systemic endotoxin in aggressive periodontitis. J Dent Res. 2011;90(9):1140–1144. doi: 10.1177/0022034511413928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeLeon-Pennell KY, et al. P. gingivalis lipopolysaccharide intensifies inflammation post-myocardial infarction through matrix metalloproteinase-9. J Mol Cell Cardiol. 2014;76:218–226. doi: 10.1016/j.yjmcc.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma Y, et al. Myofibroblasts and the extracellular matrix network in post-myocardial infarction cardiac remodeling. Pflugers Arch. 2014;466(6):1113–1127. doi: 10.1007/s00424-014-1463-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan X, et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol. 2013;62:24–35. doi: 10.1016/j.yjmcc.2013.04.023. [DOI] [PubMed] [Google Scholar]
- 15.Lee WW, et al. PET/MRI of inflammation in myocardial infarction. J Am Coll Cardiol. 2012;59(2):153–163. doi: 10.1016/j.jacc.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. 2012;110(1):159–173. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shiraishi M, et al. Alternatively activated macrophages determine repair of the infarcted adult murine heart. J Clin Invest. 2016;126(6):2151–2166. doi: 10.1172/JCI85782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moore BB, Murray L, Das A, Wilke CA, Herrygers AB, Toews GB. The role of CCL12 in the recruitment of fibrocytes and lung fibrosis. Am J Respir Cell Mol Biol. 2006;35(2):175–181. doi: 10.1165/rcmb.2005-0239OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Linthout S, Miteva K, Tschöpe C. Crosstalk between fibroblasts and inflammatory cells. Cardiovasc Res. 2014;102(2):258–269. doi: 10.1093/cvr/cvu062. [DOI] [PubMed] [Google Scholar]
- 20.Squires CE, et al. Altered fibroblast function following myocardial infarction. J Mol Cell Cardiol. 2005;39(4):699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006;324(3):475–488. doi: 10.1007/s00441-005-0144-6. [DOI] [PubMed] [Google Scholar]
- 22.López B, González A, Hermida N, Valencia F, de Teresa E, Díez J. Role of lysyl oxidase in myocardial fibrosis: from basic science to clinical aspects. Am J Physiol Heart Circ Physiol. 2010;299(1):H1–H9. doi: 10.1152/ajpheart.00335.2010. [DOI] [PubMed] [Google Scholar]
- 23.Tao ZY, Cavasin MA, Yang F, Liu YH, Yang XP. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci. 2004;74(12):1561–1572. doi: 10.1016/j.lfs.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 24.Gupta KB, Ratcliffe MB, Fallert MA, Edmunds LH, Bogen DK. Changes in passive mechanical stiffness of myocardial tissue with aneurysm formation. Circulation. 1994;89(5):2315–2326. doi: 10.1161/01.CIR.89.5.2315. [DOI] [PubMed] [Google Scholar]
- 25.Krishnasamy R, et al. Left Ventricular Global Longitudinal Strain (GLS) Is a Superior Predictor of All-Cause and Cardiovascular Mortality When Compared to Ejection Fraction in Advanced Chronic Kidney Disease. PLoS One. 2015;10(5):e0127044. doi: 10.1371/journal.pone.0127044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chakravortty D, Nanda Kumar KS. Bacterial lipopolysaccharide induces cytoskeletal rearrangement in small intestinal lamina propria fibroblasts: actin assembly is essential for lipopolysaccharide signaling. Biochim Biophys Acta. 2000;1500(1):125–136. doi: 10.1016/s0925-4439(99)00098-8. [DOI] [PubMed] [Google Scholar]
- 27.Gutiérrez-Venegas G, Contreras-Marmolejo LA, Román-Alvárez P, Barajas-Torres C. Aggregatibacter actinomycetemcomitans lipopolysaccharide affects human gingival fibroblast cytoskeletal organization. Cell Biol Int. 2008;32(4):417–426. doi: 10.1016/j.cellbi.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Wu L, Qu JM. Inhibited proliferation of human lung fibroblasts by LPS is through IL-6 and IL-8 release. Cytokine. 2011;54(3):289–295. doi: 10.1016/j.cyto.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 29.Sarafi MN, Garcia-Zepeda EA, MacLean JA, Charo IF, Luster AD. Murine monocyte chemoattractant protein (MCP)-5: a novel CC chemokine that is a structural and functional homologue of human MCP-1. J Exp Med. 1997;185(1):99–109. doi: 10.1084/jem.185.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majmudar MD, et al. Monocyte-directed RNAi targeting CCR2 improves infarct healing in atherosclerosis-prone mice. Circulation. 2013;127(20):2038–2046. doi: 10.1161/CIRCULATIONAHA.112.000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kalderén C, et al. CCL2 mediates anti-fibrotic effects in human fibroblasts independently of CCR2. Int Immunopharmacol. 2014;20(1):66–73. doi: 10.1016/j.intimp.2014.02.020. [DOI] [PubMed] [Google Scholar]
- 32.Hegefeld WA, Kuczera K, Jas GS. Structural dynamics of neuropeptide hPYY. Biopolymers. 2011;95(7):487–502. doi: 10.1002/bip.21608. [DOI] [PubMed] [Google Scholar]
- 33.Deleon KY, Patel AP, Kuczera K, Johnson CK, Jas GS. Structure and reorientational dynamics of angiotensin I and II: a microscopic physical insight. J Biomol Struct Dyn. 2012;29(6):671–690. doi: 10.1080/07391102.2011.672631. [DOI] [PubMed] [Google Scholar]
- 34.Cardenas AE, Jas GS, DeLeon KY, Hegefeld WA, Kuczera K, Elber R. Unassisted transport of N-acetyl-L-tryptophanamide through membrane: experiment and simulation of kinetics. J Phys Chem B. 2012;116(9):2739–2750. doi: 10.1021/jp2102447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Lemos JA, et al. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndromes. Circulation. 2003;107(5):690–695. doi: 10.1161/01.CIR.0000049742.68848.99. [DOI] [PubMed] [Google Scholar]
- 36.Anderson G, Horvath J. The growing burden of chronic disease in America. Public Health Rep. 2004;119(3):263–270. doi: 10.1016/j.phr.2004.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gifford AH, Matsuoka M, Ghoda LY, Homer RJ, Enelow RI. Chronic inflammation and lung fibrosis: pleotropic syndromes but limited distinct phenotypes. Mucosal Immunol. 2012;5(5):480–484. doi: 10.1038/mi.2012.68. [DOI] [PubMed] [Google Scholar]
- 38.Rieder F, Fiocchi C. Intestinal fibrosis in inflammatory bowel disease - Current knowledge and future perspectives. J Crohns Colitis. 2008;2(4):279–290. doi: 10.1016/j.crohns.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Shamhart PE, Luther DJ, Hodson BR, Koshy JC, Ohanyan V, Meszaros JG. Impact of type 1 diabetes on cardiac fibroblast activation: enhanced cell cycle progression and reduced myofibroblast content in diabetic myocardium. Am J Physiol Endocrinol Metab. 2009;297(5):E1147–E1153. doi: 10.1152/ajpendo.00327.2009. [DOI] [PubMed] [Google Scholar]
- 40.Frangogiannis NG, Entman ML. Chemokines in myocardial ischemia. Trends Cardiovasc Med. 2005;15(5):163–169. doi: 10.1016/j.tcm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Rollins BJ. Monocyte chemoattractant protein 1: a potential regulator of monocyte recruitment in inflammatory disease. Mol Med Today. 1996;2(5):198–204. doi: 10.1016/1357-4310(96)88772-7. [DOI] [PubMed] [Google Scholar]
- 42.Gu L, Tseng SC, Rollins BJ. Monocyte chemoattractant protein-1. Chem Immunol. 1999;72:7–29. doi: 10.1159/000058723. [DOI] [PubMed] [Google Scholar]
- 43.Gupta M, Chaturvedi R, Jain A. Role of monocyte chemoattractant protein-1 (MCP-1) as an immune-diagnostic biomarker in the pathogenesis of chronic periodontal disease. Cytokine. 2013;61(3):892–897. doi: 10.1016/j.cyto.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 44.Dewald O, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96(8):881–889. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 45.Frangogiannis NG, et al. Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation. 2007;115(5):584–592. doi: 10.1161/CIRCULATIONAHA.106.646091. [DOI] [PubMed] [Google Scholar]
- 46.Kaikita K, Hayasaki T, Okuma T, Kuziel WA, Ogawa H, Takeya M. Targeted deletion of CC chemokine receptor 2 attenuates left ventricular remodeling after experimental myocardial infarction. Am J Pathol. 2004;165(2):439–447. doi: 10.1016/S0002-9440(10)63309-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nahrendorf M, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Jesus NM, et al. Atherosclerosis exacerbates arrhythmia following myocardial infarction: Role of myocardial inflammation. Heart Rhythm. 2015;12(1):169–178. doi: 10.1016/j.hrthm.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panizzi P, et al. Impaired infarct healing in atherosclerotic mice with Ly-6C(hi) monocytosis. J Am Coll Cardiol. 2010;55(15):1629–1638. doi: 10.1016/j.jacc.2009.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lew WY, Bayna E, Dalle Molle E, Contu R, Condorelli G, Tang T. Myocardial fibrosis induced by exposure to subclinical lipopolysaccharide is associated with decreased miR-29c and enhanced NOX2 expression in mice. PLoS ONE. 2014;9(9):e107556. doi: 10.1371/journal.pone.0107556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res. 2014;114(2):266–282. doi: 10.1161/CIRCRESAHA.113.301720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deleon-Pennell KY, de Castro Brás LE, Lindsey ML. Circulating Porphyromonas gingivalis lipopolysaccharide resets cardiac homeostasis in mice through a matrix metalloproteinase-9-dependent mechanism. Physiol Rep. 2013;1(5):e00079. doi: 10.1002/phy2.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zamilpa R, et al. Cardiac wound healing post-myocardial infarction: a novel method to target extracellular matrix remodeling in the left ventricle. Methods Mol Biol. 2013;1037:313–324. doi: 10.1007/978-1-62703-505-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Altschul SF, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Altschul SF, et al. Protein database searches using compositionally adjusted substitution matrices. FEBS J. 2005;272(20):5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Papadopoulos JS, Agarwala R. COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007;23(9):1073–1079. doi: 10.1093/bioinformatics/btm076. [DOI] [PubMed] [Google Scholar]
- 57.Biasini M, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014;42(Web Server issue):W252–W258. doi: 10.1093/nar/gku340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 59.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4(1):1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 60.Vu TH, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93(3):411–422. doi: 10.1016/S0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia J, Sinelnikov, Han B, Wishart DS. MetaboAnalyst 3.0--making metabolomics more meaningful. Nucleic Acids Res. 2015;43(W1):W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 63.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8(6):e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.