

Abstract
Myasthenia gravis (MG) is a B cell–mediated autoimmune disorder of neuromuscular transmission. Pathogenic autoantibodies to muscle-specific tyrosine kinase (MuSK) can be found in patients with MG who do not have detectable antibodies to the acetylcholine receptor (AChR). MuSK MG includes immunological and clinical features that are generally distinct from AChR MG, particularly regarding responsiveness to therapy. B cell depletion has been shown to affect a decline in serum autoantibodies and to induce sustained clinical improvement in the majority of MuSK MG patients. However, the duration of this benefit may be limited, as we observed disease relapse in MuSK MG patients who had achieved rituximab-induced remission. We investigated the mechanisms of such relapses by exploring autoantibody production in the reemerging B cell compartment. Autoantibody-expressing CD27+ B cells were observed within the reconstituted repertoire during relapse but not during remission or in controls. Using two complementary approaches, which included production of 108 unique human monoclonal recombinant immunoglobulins, we demonstrated that antibody-secreting CD27hiCD38hi B cells (plasmablasts) contribute to the production of MuSK autoantibodies during relapse. The autoantibodies displayed hallmarks of antigen-driven affinity maturation. These collective findings introduce potential mechanisms for understanding both MuSK autoantibody production and disease relapse following B cell depletion.
Keywords: Autoimmunity, Immunology
Keywords: Autoimmune diseases, B cells, Immunotherapy
Myasthenia gravis autoantibody–expressing memory B cells and plasmablasts emerge during disease relapse in patients previously treated with rituximab.
Introduction
Acetylcholine receptor (AChR) autoantibody–negative myasthenia gravis (MG) patients can harbor pathogenic autoantibodies to muscle-specific tyrosine kinase (MuSK) (1, 2). Distinguishing features of MuSK MG include a predominantly IgG4 subclass-driven immunopathology, frequent bulbar symptoms, and responsiveness to immunotherapy (3, 4). Rituximab, an anti-CD20 biologic, has gained support in managing MuSK MG, as its application affects sustained clinical improvement along with a marked decline in MuSK autoantibody titer in the majority of patients (5, 6). In comparison with AChR MG, fewer relapses have been reported to date in MuSK MG patients who achieved stable clinical remission after rituximab (7–12).
Although broad immune dysregulation (13–15) in MuSK MG is recognized, the mechanistic details of autoantibody production remain undefined. Given that MuSK MG autoantibodies are required for pathogenesis (2) and that they are, consequently, the fundamental target of immunotherapy, it is important to establish how they are generated. The study of patients treated with rituximab presents an excellent opportunity to investigate such mechanisms (16). Due to the pronounced clinical improvement commonly observed with CD20 depletion in MuSK MG, immunosuppressive agents such as steroids, mycophenolate mofetil, azathioprine, and other broad-acting agents can be tapered and often completely withdrawn (5, 6, 10). Once the confounding effects of such therapeutics are absent, the reemergence and evolution of the B cell compartment can be investigated and associated with clinical status.
It is recognized that B cell populations that do not express CD20, such as long-lived plasma cells, produce a considerable portion of circulating Ig (17). Given the absence of CD20 on their surface, they are not directly affected by rituximab. This is supported by unchanged immunization-generated and plasma cell–dependent antibody titers to microbial antigens, such as tetanus, varicella, and pneumococcus, following B cell depletion (5, 18, 19). Accordingly, the markedly diminished MuSK autoantibody titer, as early as 3 months after rituximab-mediated CD20+ B cell depletion (5), suggests that long-lived plasma cells are unlikely candidates for MuSK autoantibody production. Rather, short-lived antibody-secreting cells such as plasmablasts are more viable candidates. As only a small fraction of these cells express CD20, the effectiveness of rituximab in MuSK MG may depend upon depletion of a pool of plasmablast-progenitor CD20+ memory B cells (20, 21).
We recently had the opportunity to observe 3 previously rituximab-treated MuSK MG patients who were experiencing relapses after having achieved remission, indicating that the disease was not extinguished but dormant. To investigate which cells produce the autoantibodies, and thereby further define the mechanism of immunopathology in MuSK MG, we tested whether autoantibody-expressing plasmablasts and memory B cells emerge during disease relapse in these patients.
Results
Study subject characteristics.
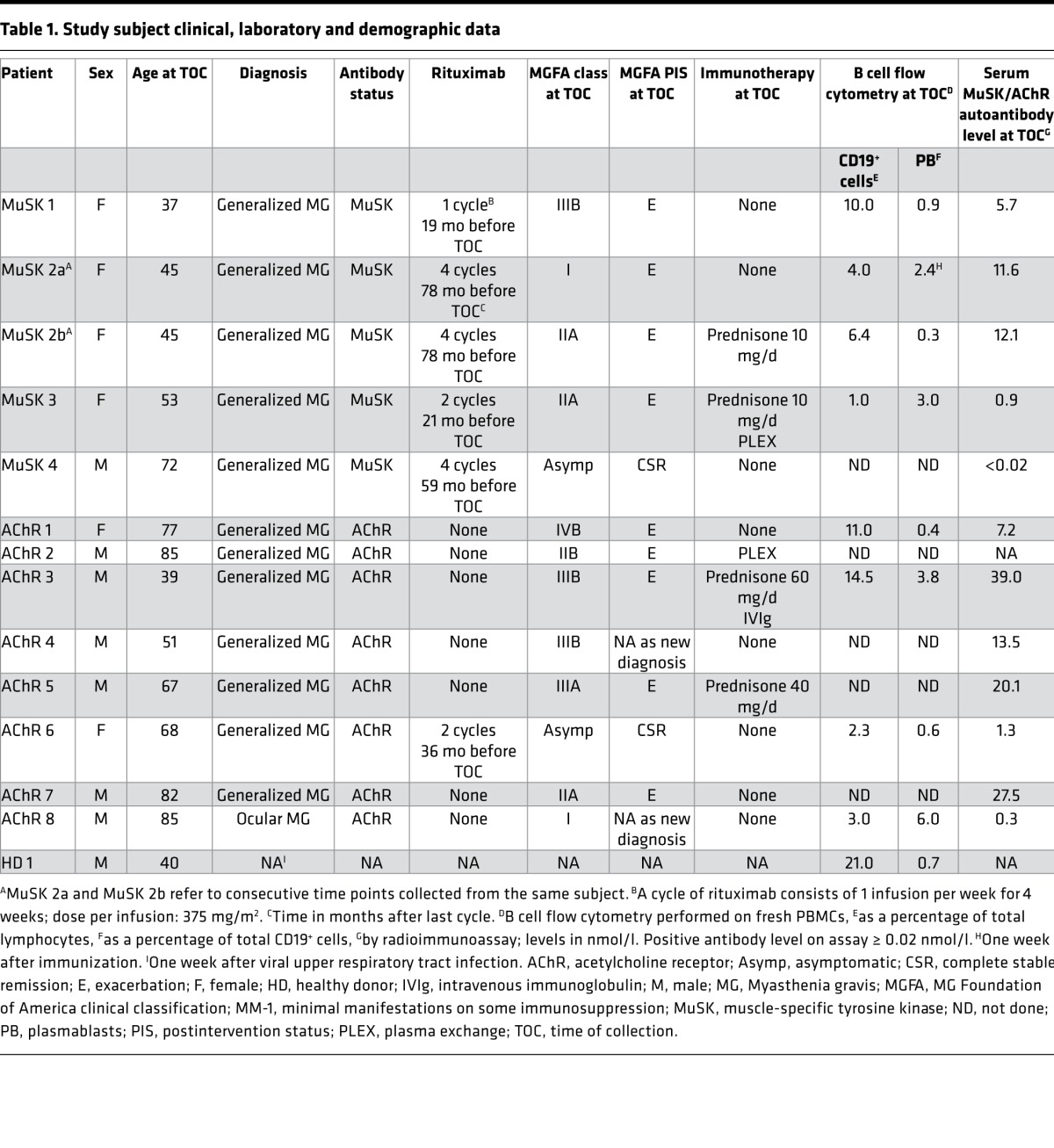
Four rituximab-treated MuSK MG subjects (3 female, 1 male) were included and had a mean age of 51.8 years (range 37–72). Table 1 summarizes their demographic and clinical data. At the time of sampling, 3 of the subjects met Myasthenia Gravis Foundation of America (MGFA) postintervention status (PIS) criteria for “exacerbation” (E) and had positive MuSK autoantibody titers, while the fourth met criteria for complete stable remission (CSR) and had undetectable MuSK autoantibodies. The severity of relapses/exacerbations as measured by an MGFA clinical classification assignment ranged from mild ocular symptoms to moderate generalized disease with bulbar features (MGFA grades, I–IIIb). B cell and memory B cell fractions of the MuSK MG patients were monitored during treatment (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.94263DS1). In one (MuSK 2a) of the 3 relapse patients, an inflammatory event (immunization) preceded clinical relapse by 1 week. The B cell compartment increasingly reconstituted after rituximab in all 4 subjects. An elevated proportion of memory B cells was observed in the relapse patients compared with the subject that remained in remission. Controls included 8 AChR MG patients and 1 healthy donor (HD 1). Table 1 summarizes their demographic and clinical data. The 8 AChR MG subjects (2 female, 6 male) had a mean age of 69 years (range 39–85) and ranged from asymptomatic to severe generalized disease (MGFA grades 0–IVb) with typically variable AChR autoantibody levels. HD 1 (male, aged 40 years) had no history of autoimmune disease and no recent inflammatory events, and was negative for AChR and MuSK autoantibodies.
Table 1. Study subject clinical, laboratory and demographic data.
MuSK autoantibody–producing B cells emerge during relapse.
Given that clinical relapse and remission were respectively associated with elevated or undetectable autoantibody titers, we sought to explore whether circulating autoantibody-producing B cells emerge during relapse. To that end, antigen-experienced CD27+ B cells were isolated and then cultured for 7 days in a cocktail of stimuli (CD40L, CpG, IL-21, and IL-6) that led to an induction and maintenance of terminally differentiated Ig-producing cells (22). The resulting culture supernatants were subsequently tested for the presence of MuSK autoantibody.
A live cell-based antibody assay (CBA) was used for measurement of MuSK autoantibodies (4, 23). This assay format presented human MuSK on the human embryonic kidney (HEK) cell surface such that native conformation and posttranslational modifications were maintained (24). The sensitivity of the CBA was first validated by testing sera from all subjects (MuSK 1–4, AChR 1–8, HD 1). Representative MuSK CBA flow cytometry plots are shown in Figure 1, A and B; a summary of the sera CBA data is shown in Figure 2A; and CBA numerical values are shown in Supplemental Table 2. Positivity was measured as the difference in the percentage (Δ%) of positive cells, i.e. the fraction of MuSK-transfected cells that was bound by an Ig-containing sample minus the fraction of GFP-transfected cells that was bound by the same sample. Postrituximab relapse (MuSK 1, -2a, -2b, and -3) sera were positive for MuSK binding (Δ% positive cells of 95, 94, 97, and 73, respectively). The remaining sera were negative, remaining below the cutoff. Further validation was achieved by testing 2 monoclonal recombinant Igs (rIg), each respectively specific for MuSK (4A3) or AChR (637); 4A3 was positive for MuSK binding, whereas 637 was negative (Δ% positive cells of 96 and 2, respectively) (Figure 1, G and H, and Supplemental Table 2). Cell culture medium or CBA buffer (PBS 1% BSA) was consistently negative for MuSK binding (Δ% positive cells of 1) (Supplemental Table 2).
Figure 1. Representative MuSK cell-based assay (CBA) flow cytometry plots.

Control sera, cell culture supernatants, and monoclonal rIgs were tested for surface binding to MuSK on MuSK-GFP–transfected HEK cells. The x axis represents GFP fluorescence intensity and, consequently, the fraction of HEK cells transfected with MuSK. The y axis represents Alexa Fluor 647 fluorescence intensity, which corresponds to secondary anti–human IgG Fc antibody binding and, consequently, primary antibody binding to MuSK. Hence, transfected cells are located in the right quadrants and cells with MuSK autoantibody binding in the upper quadrants. The upper right quadrant shows cells that are both transfected with MuSK-GFP and that bind MuSK autoantibodies, whereas the upper left quadrant represents nonspecific antibody binding to HEK cell antigens. All results shown were reproduced in duplicate experiments. (A–F) Serum and B cell culture supernatants; (G–L) monoclonal rIg. (A) Post–rituximab relapse (MuSK 2b) serum; (B) post–rituximab remission (MuSK 4) serum; (C) post–rituximab relapse (MuSK 2a) CD27+ B cell culture supernatant; (D) post–rituximab remission (MuSK 4) CD27+ B cell culture supernatant; (E) post–rituximab relapse (MuSK 2b) plasmablast culture supernatant; (F) post–viral URI (HD 1) plasmablast culture supernatant; (G) 4A3, a humanized murine MuSK–specific monoclonal rIg; (H) 637, a human AChR–specific monoclonal rIg; (I) post–rituximab relapse (MuSK 1) PB–derived rIg 1-1; (J) post–rituximab relapse (MuSK 3) PB–derived rIg 3-29; (K) post–rituximab relapse (MuSK 3) PB–derived rIg 3-33; (L) AChR MG (AChR 7) PB–derived rIg 7-3. AChR, acetylcholine receptor; HD, healthy donor; HEK, human embryonic kidney; MuSK, muscle-specific tyrosine kinase; rIg, recombinant Ig; URI, upper respiratory tract infection.
Figure 2. Summary of MuSK CBA data performed with sera, B cell culture supernatants, and recombinant immunoglobulin (rIg).

Results are presented as Δ% positive cells on the y axis. Δ% positive cells = (%frequency of positive MuSK-GFP–transfected cells/%frequency of MuSK-GFP–transfected cells) – (%frequency of positive GFP-transfected cells/%frequency of GFP-transfected cells). Testing of all samples was performed in duplicate. (A–C) Bars represent means, dots represent individual values, and error bars represent range of values; (D) lines represent means, and dots represent individual rIg values. (A) Sera of MuSK 1–4, AChR 1–8, and HD 1; (B) CD27+ B cell culture supernatants of MuSK 1–4, AChR 1–6 and HD 1; (C) Plasmablast culture supernatants from MuSK 1, -2b, -3, -4, AChR 1–3 and HD 1; (D) Plasmablast-derived rIg from MuSK 1 (n = 4), 2b (n = 33), 3 (n = 45) and AChR 7 (n = 15), 8 (n = 11). AChR, acetylcholine receptor; HD, healthy donor; MuSK, muscle-specific tyrosine kinase.
The application of this validated assay to the CD27+ cell culture supernatants from post–rituximab relapse subjects (MuSK 1, -2a, -2b, -3) demonstrated that they were positive for MuSK autoantibodies (Δ% positive cells of 77, 84, 64, and 32, respectively). By contrast, CD27+ cell culture supernatants prepared from a post–rituximab MuSK MG subject in CSR (MuSK 4), from 6 AChR MG subjects (AChR 1–6), and from HD 1 were all negative for MuSK autoantibodies, remaining below the cutoff. Representative MuSK CBA flow cytometry plots are shown in Figure 1, C and D; a summary of the CD27+ cell culture supernatant CBA data is shown in Figure 2B; and CBA numerical results are shown in Supplemental Table 2. These findings indicate that MuSK-specific B cells are present in the CD27+ compartment during disease relapse.
MuSK autoantibodies utilize the IgG1 and IgG4 subclasses during relapse.
In most patients with MuSK MG, the autoantibodies are primarily of the IgG4 subclass, but the IgG1 subclass is also represented (4, 25). Moreover, the IgG1 subclass can supersede the IgG4 as the dominant subclass after rituximab treatment (5). To investigate how these IgG subclasses were represented in our MuSK MG patient cohort, we used IgG1- and IgG4-specific secondary antibodies in the MuSK CBA to examine the autoantibodies in the serum and the CD27+ cell culture supernatants (Table 2). Postrituximab relapse serum samples (MuSK 1, -2a, -2b, -3) were positive for IgG1 MuSK autoantibodies (percent of positive cells of 50, 95, 97, and 68, respectively) and IgG4 MuSK autoantibodies (% positive cells of 89, 97, 98, and 55, respectively). Serum samples from the patient in remission (MuSK 4) and the control cohort (AChR 1–6 and HD 1) were negative (below the cutoff) for MuSK autoantibodies of either subclass (Table 2).
Table 2. IgG subclass analysis of MuSK autoantibodies.
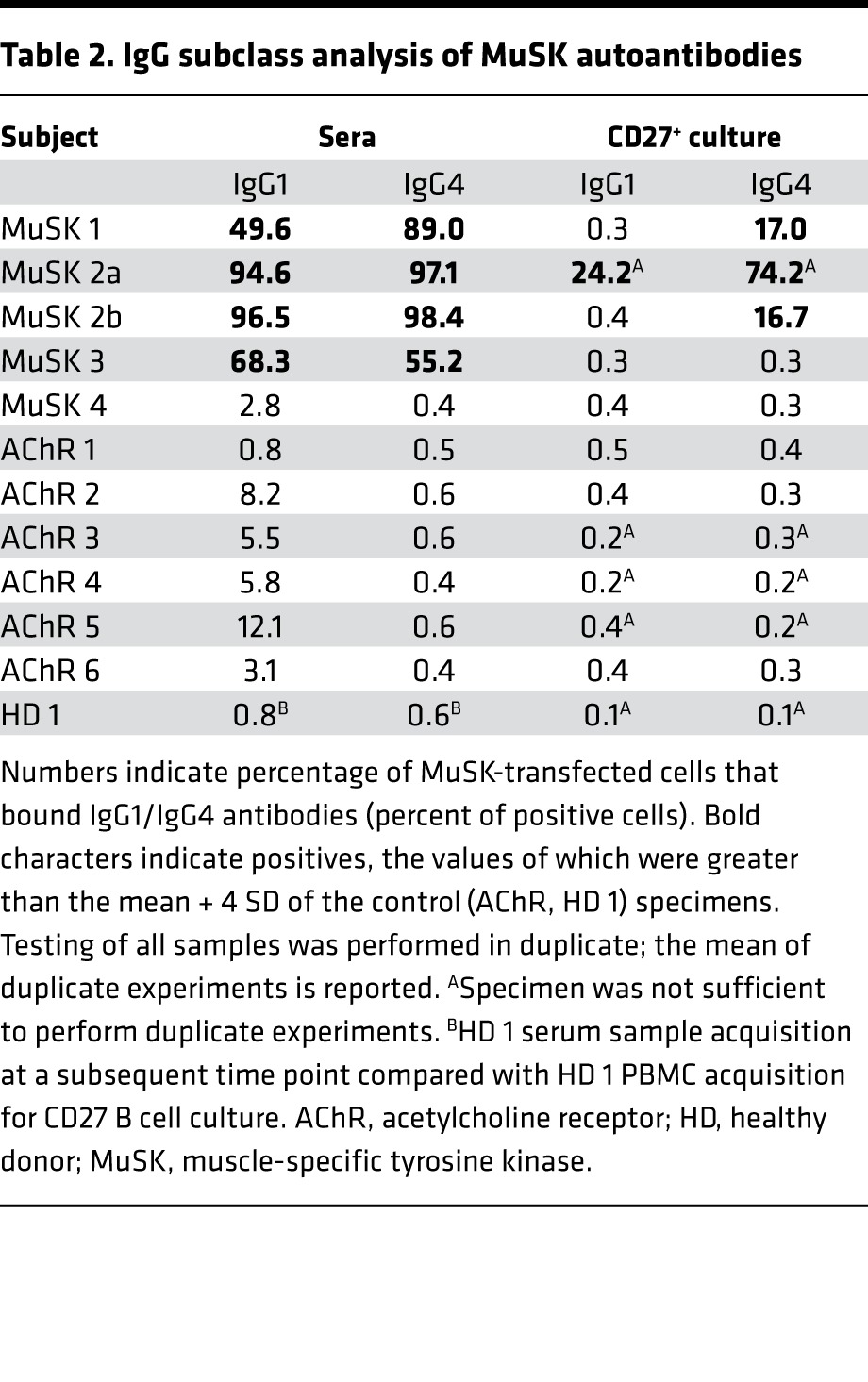
Testing of the CD27+ culture supernatants (Table 2) showed that the supernatant from post–rituximab relapse patient MuSK 1 included detectable IgG4 autoantibodies (percent of positive cells of 17). Relapse patient MuSK 2a included an IgG1 and IgG4 response (percent of positive cells of 24 and 74, respectively), while the second time point from the same patient (MuSK 2b) only included detectable IgG4 autoantibodies (percent of positive cells of 17). The CD27+ culture supernatants prepared from the post–rituximab relapse patient MuSK 3, the patient in remission (MuSK 4), and the control cohort (AChR 1–6 and HD 1) did not have detectable MuSK autoantibodies of either the IgG1 or IgG4 subclass (percent of positive cells below cutoff).
Antibody-secreting plasmablasts contribute to MuSK autoantibody production.
The cultured CD27+ B cells mostly include memory cells, which were specifically stimulated to produce antibodies. However, the cultures may also have included a fraction of plasmablasts that would be expected to spontaneously secrete antibody. To further explore whether such circulating plasmablasts are contributors to MuSK autoantibody production, we isolated plasmablasts using cell sorting. The plasmablast gating strategy we adopted was first validated by sorting the cell populations, followed by morphologic examination of the B cell subsets. This analysis confirmed that a high level of enrichment for plasmablasts among sorted cells was achieved (Figure 3). Using this strategy, we also performed immunophenotyping of B cell lineages to determine whether our MuSK MG subjects included an expanded compartment of plasmablasts, as previously observed in a small subset of MG patients (13, 26). Plasmablast frequency was marginally elevated in the post–rituximab relapse MuSK MG subjects (Table 1), relative to the low levels we and others have observed in HDs (27, 28).
Figure 3. Plasmablast FACS gating strategy and validation.

A representative example of the plasmablast population sorting strategy is shown. PBMCs were initially gated in the SSC/FSC graph (upper left). The SSC/FSC gate was determined based on plasmablast back-gating (not shown). After doublet and dead cell exclusion, CD19+CD3–CD14– cells were gated as B cells (lower left). Plasmablasts were subsequently gated from the B cell gate as CD27hiCD38hi cells (upper right). Naive B cells were defined as IgD+CD27– B cells (lower right). Morphological analysis (400× magnification) is shown for plasmablasts (upper picture) and naive B cells (lower picture). Plasmablasts display an eccentric nucleus, perinuclear hof region, prominent nucleoli, and abundant cytoplasm. In contrast, naive B cells display a large nucleus with a thin rim of cytoplasm. These results were replicated in a repeat experiment. FSC, forward scatter; PBMC, peripheral blood mononuclear cells; SSC, side scatter.
To test for MuSK autoantibody production by plasmablasts in post–rituximab relapse MuSK MG patients, these cells were directly sorted into culture media supplemented with IL-6, and supernatants were harvested after 7 days. They were subsequently tested for the presence of MuSK autoantibodies with the MuSK CBA. Representative MuSK CBA flow cytometry plots are shown in Figure 1, E and F; a summary of the CBA data is shown in Figure 2C; and CBA numerical results are shown in Supplemental Table 2. Post–rituximab relapse MuSK 1, -2b, and -3 plasmablast culture supernatants were positive for MuSK binding (Δ% positive cells of 47, 30, and 11, respectively). By contrast, post–rituximab remission (MuSK 4) and the control (AChR 1–3, HD 1) plasmablasts culture supernatants did not include detectable MuSK autoantibodies (Δ% positive cells below the cutoff).
While the in vitro culture of plasmablasts provides a direct means for evaluating expressed Ig, the approach provides a very limited amount of material on which to perform tests. As a means to both produce an inexhaustible source of human MuSK autoantibodies and to validate plasmablast culture findings, we applied a complementary approach: we cloned and expressed fully human monoclonal rIg from single cell–sorted plasmablasts. A total of 108 rIgs were produced; 82 from 3 post–rituximab relapse MuSK MG subjects (MuSK 1, -2b, and -3) and 26 from 2 AChR MG subjects (AChR 7 and -8), which were used as controls. These were tested for MuSK binding with the CBA. Representative MuSK CBA flow cytometry plots are shown in Figure 1, I–L; a summary of the CBA data is shown in Figure 2D; and CBA numerical results are shown in Supplemental Table 2. Seven plasmablast-derived rIgs from post–rituximab relapse patients were positive for MuSK binding: MuSK 1–derived MuSK 1-1 (Δ% positive cells of 53) and MuSK 3–derived MuSK 3-1, –3-20, –3-28, –3-29, –3-30, and –3-33 (Δ% positive cells of 22, 32, 99, 86, 82, and 87, respectively). The 26 AChR MG–derived rIgs were all negative for MuSK binding, remaining below the cutoff; they produced values that were marginally above background (mean Δ% positive cells of 5 [SD = 4]). Furthermore, group analysis showed that MuSK 1– and -3–derived rIg MuSK binding was significantly different from that of AChR-derived rIg (Mann-Whitney U test, P = 0.048). In addition, Δ mean fluorescence intensity (MFI) values of MuSK 3-28, –3-29, –3-30, and –3-33 were particularly high, exceeding values of 2,000. The ΔMFI of MuSK 3-28 was 114,666, a value directly comparable with that of 4A3 (ΔMFI of 118,554), while the ΔMFI of MuSK 3-33 was 27,952. In sharp contrast, the mean ΔMFI of the 26 AChR MG–derived rIg was 41 (SD = 20). These combined data, from plasmablast cultures and rIg, demonstrate that MuSK-specific autoantibodies can be produced by circulating plasmablasts.
Specificity of each positive MuSK MG plasmablast–derived rIg was further tested by CBA using HEK cells transfected with MuSK, AChR, and GFP. The 7 rIg that were positive for MuSK binding, (MuSK 1-1, –3-1, –3-20, –3-28, –3-29, –3-30, and –3-33) were tested, and results are graphically summarized in Figure 4. The mean percent of positive cells in the CBAs for the 7 rIg were 70, 5, and 3 using MuSK-transfected, AChR-transfected, and GFP-transfected cells, respectively. Group analysis showed a significant difference between these 3 groups (Kruskal-Wallis, P < 0.001). These data indicate that the MuSK MG plasmablast–derived rIgs do not crossreact with the 4 subunits of the AChR or with antigens present on the surface of HEK cells.
Figure 4. Specificity of positive MuSK MG relapse plasmablast-derived rIg.

Seven monoclonal rIg, MuSK 1-1, 3-1, 3-20, 3-28, 3-29, 3-30, and 3-33, were tested for binding on HEK cells transfected with MuSK (upper panel), AChR (middle panel), and GFP (lower panel). Results are shown as percentage of transfected cells that bound secondary antibody (percent of positive cells). Testing of all samples was performed in duplicate. Bars represent means, dots represent individual values, and error bars represent range of values. AChR, acetylcholine receptor; HD, healthy donor; HEK, human embryonic kidney; MuSK, muscle-specific tyrosine kinase; rIg, recombinant Ig.
MuSK autoantibodies are clonally diverse and the product of affinity maturation.
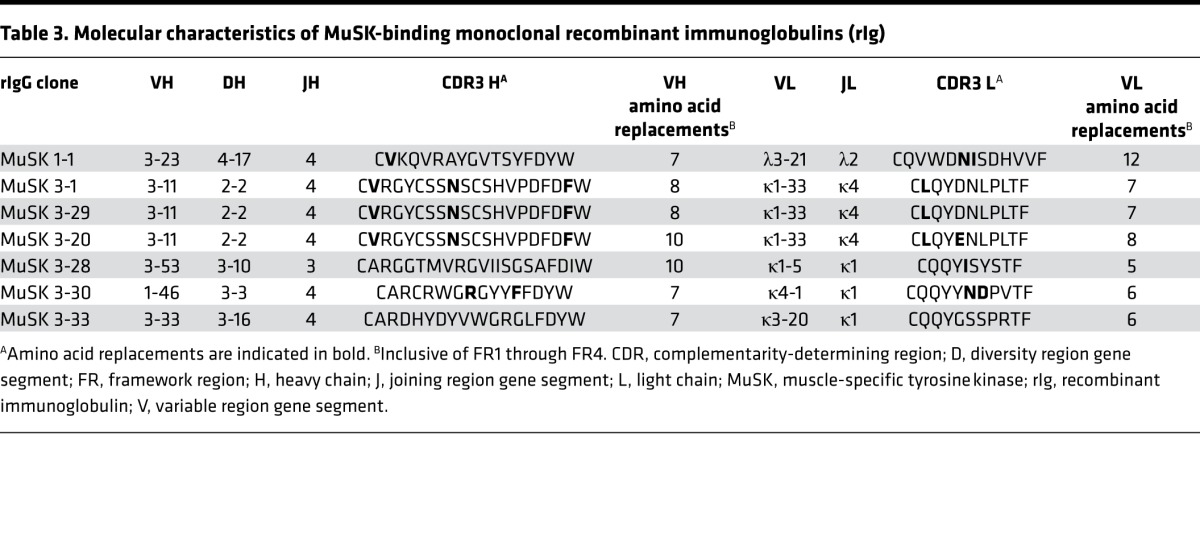
To explore the B cell receptor diversity among the MuSK-specific autoantibodies, we examined the characteristics of the sequences. Sequence analysis of the 7 rIgs that bound to MuSK revealed that these autoantibodies are represented by diverse clones that utilize many variable region gene segments. Table 3 summarizes heavy and light chain variable region gene usage and CDR3 amino acid sequences for the 7 positive monoclonal rIgs. A number of somatic mutations, which are a hallmark of affinity maturation, were concentrated in the variable heavy (VH) and variable light (VL) CDR regions of all autoantibodies, strongly suggesting that antigenic selection had occurred. Clonal expansion represents another hallmark of antigen-driven maturation of B cells. Three of the MuSK-specific plasmablast clones (MuSK 3-1, –3-20, and –3-29) had identical VH-D-JH and VL-JL gene rearrangements in the heavy and light chains, respectively. Of those, 2 (MuSK 3-1 and –3-29) showed identical heavy and light chain sequences while the third (MuSK 3-20), although sharing somatic mutations with the two first clones, also showed unique mutations in both the heavy and light chain variable region. Thus, it was identified as a clonal variant, which is a unique descendant of a shared parental clone. These sequencing data suggest that the development of MuSK autoantibodies requires processes of clonal selection and affinity maturation, perhaps mediated by T cell help.
Table 3. Molecular characteristics of MuSK-binding monoclonal recombinant immunoglobulins (rIg).
Discussion
The precise cellular origins of autoantibodies are seldom easy to study, largely because such cells are very rare and most patients are on immunotherapies that modify the number and circulation of the responsible cells. We were able to study autoantibody-producing cells during disease relapse of 3 MuSK MG subjects that had previously achieved CSR/minimal manifestation (MM) status following rituximab treatment. From a clinical standpoint, these are among the few reported post–rituximab MuSK MG relapses, which collectively indicate that the disease is not extinguished but rather remains dormant, in at least some individuals, and consequently, relapses can occur. Our investigation into the mechanisms of immunopathology during relapse revealed that circulating MuSK-specific cells are present in the antigen-experienced CD27+ compartment of repopulated B cells. Their occurrence qualitatively associates with post–rituximab serum MuSK autoantibodies, as well as clinical relapse. Circulating ex vivo plasmablasts were identified as an autoantibody-secreting population. Specific autoantibody producers in this cell population were of low frequency. The autoantibodies that recognized MuSK appeared to be the product of antigen-stimulated affinity maturation, as they all included replacement mutations in the CDRs, and 3 of the 7 positive clones were related. The diversity among the positive clones also suggests that MuSK autoantibodies present in the serum are heterogeneous.
Based on the findings reported here and on previously reported observations concerning autoantibody production and B cell depletion, we propose a model for the cellular immunopathology of MuSK MG in which memory B cells and plasmablasts are key players. We suggest that MuSK-specific memory cells continuously supply a population of short-lived, autoantibody-secreting plasmablasts (29). Rituximab depletes CD20+ memory cells and, thus, indirectly diminishes the short-lived, mostly CD20– plasmablast population (21, 30), thereby affecting serological and clinical remission within a few months (5, 6, 31). One alternative version of this model acknowledges that a fraction of plasmablasts may be CD20+ (20); their direct depletion by rituximab could contribute to clinical response.
Support for this model includes studies that point toward autoantibody production by plasmablasts in several autoimmune, rituximab-responsive disorders. In neuromyelitis optica (NMO) patients, most of whom respond well to rituximab (32, 33), an enlarged peripheral plasmablast compartment is associated with disease activity and produces aquaporin-4 autoantibodies (34, 35). Similarly, circulating plasmablasts produce citrullinated protein autoantibodies in rheumatoid arthritis (36–38). In addition, rituximab treatment of patients with diseases that include IgG4 autoantibodies, such as pemphigus and bullous pemphigoid, leads to a decrease in autoantibody titer within 3 months, correlating with clinical improvement (19, 31, 39, 40).
Interestingly, an autoreactive long-lived plasma cell compartment does not appear to be established in MuSK MG, which exhibits a distinction between this autoimmune disease and immunization/infection-based serological memory. Long-lived, CD20– plasma cells contribute to the majority of circulating Ig, yet they are unlikely candidates for MuSK autoantibody production because they are not directly targeted by rituximab. This is substantiated by stable serum IgG titers against vaccine antigens such as tetanus, varicella, and pneumococcus (5, 18, 19) in post–rituximab patients, while MuSK serum autoantibodies decline remarkably during such treatment (5, 6). Moreover, in AChR MG, where long-lived plasma cells are implicated in autoantibody production (41–44), the rituximab response appears to be considerably more delayed and the autoantibody titer decline less pronounced when compared with MuSK MG (5, 7).
Although our study identifies a role for plasmablasts in MuSK MG autoantibody production, further study is required to determine whether B cell depletion directly or indirectly targets autoantibody-specific plasmablasts. Ex vivo characterization and measurement of autoantibody producers during treatment using novel technologies that allow for direct isolation of such cells (45, 46) may facilitate these studies. It is also of interest to determine whether the clones that emerge in the reconstituting B cell compartment are newly produced or existed prior to treatment. It is unlikely that rituximab affects absolute eradication of the CD20+ B cell compartment. Consequently, there are B cells that escape the depletion process, especially in the tissue. A recent study demonstrated that such tissue-resident memory cells are refractory to depletion and mount robust recall responses (47). In our study, all 7 MuSK-specific plasmablasts identified during postrituximab disease relapse had the hallmarks of the affinity maturation process: class switching, accumulation of somatic mutations, and clonal expansion (44, 48–50). These data may point to a predepletion antigen-driven selection process and a recall response. Thus, the MuSK-specific clones we identified could be the product of B cells that were present prior to treatment. However, the presence of a clonal variant pair, representing different steps in affinity maturation, could also suggest a newly formed, postdepletion pathogenic process including, but not limited to, de novo oligoclonal expansions that emerge after antigen reexperience in the reconstituted repertoire. Similar models of nascent B cell responses have been proposed for IgG4-related disease (51). Large-scale B cell repertoire sequencing studies applied both prior to and after B cell depletion therapy will be required to identify whether historical and/or de novo clonal production contributes to posttherapy relapse.
We noted that plasmablast culture–derived autoantibodies bound to MuSK less efficiently than those of other populations. This could be attributed to the low plasmablast frequency in peripheral blood, which led to a limitation in the number of sorted cells (approximately 80-fold fewer sorted cells than in the CD27+ culture) and, consequently, to a reduced concentration of MuSK-specific Ig in the culture. This interpretation is further supported by our quantitative assessment of single plasmablast–derived rIg MuSK binding. Among the rIg produced from single plasmablasts of the patients experiencing a relapse, a small fraction bound MuSK, which suggests that cells secreting MuSK autoantibody may be very scarce within this population of cells. While this observation stands in contrast to the high frequency of vaccine-specific plasmablasts demonstrated after immunization (52), it is more consistent with the low frequency of autoantibody-producing plasmablasts described in both systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) (36, 53).
The MuSK-specific, monoclonal rIgs displayed a range of binding that included high, intermediate, and lower binding capacities when tested in the CBA. For example, rIg MuSK 1-1 consistently displayed a lower binding capacity indicated by fractions of MuSK-transfected cells that were both rIg-bound (shifted upward on the FACS y axis) and rIg-unbound (not shifted upward). We noted, however, that none of the control rIgs derived from non–MuSK MG subjects displayed this binding pattern. Accordingly, we viewed rIgs such as MuSK 1-1 as positive but characterized them as weaker binders. On the other hand, several rIgs such as MuSK 3-28 and MuSK 3-33 displayed very strong binding, as they consistently bound nearly 100% of the transfected cells, which emulated the strong binding observed with the murine hybridoma-derived control (4A3). MuSK 3-28, in particular, displayed a ΔMFI value that was similar to that of 4A3; these two rIgs both displayed ΔMFIs that were 3,000-fold higher than the mean ΔMFI of control rIgs. This range of binding strengths is in agreement with other studies of human monoclonal rIg derived from patients with autoantibody-mediated diseases. This includes pemphigus (54, 55), NMO (56), and AChR MG (44), all of which include rIg reported to exhibit a range of binding capacities. It is important to highlight that it is not known how the strength of binding correlates with pathogenic potential of MuSK-specific rIg. It would be presumptuous to assume that those with lower binding capacity may not be pathogenic, given that human-derived MuSK rIg have not been studied. However, the human-derived MuSK-specific rIgs we isolated will provide excellent tools for future studies investigating the molecular mechanisms of MuSK autoantibody pathogenicity. Such studies may elucidate the divergent pathogenesis between IgG4 (MuSK MG, pemphigus) and IgG1 (AChR MG, NMO) autoantibody-related diseases and further the understanding of their distinct response to B cell depletion therapy.
Given the substantial rarity of MuSK MG, our study included a narrow cohort of subjects. The procurement of research material from MuSK MG subjects who have not received aggressive, broad-acting immunomodulating therapy reflects an ongoing constraint in our field. Thus, the investigation of this disease following B cell depletion presents a unique and valuable opportunity for study, since additional immunotherapy is often withdrawn with improving clinical status. The postdepletion period leading up to relapse may reflect mechanisms transpiring in the early stages of the disease. The limited specimens in the present study notwithstanding, our results are consistent in all 3 relapsing MuSK MG subjects and provide new insight into MuSK autoantibody production and mechanisms of relapse.
MuSK serum autoantibody titer associates with clinical presentation (25, 57) and may be predictive of relapse following rituximab treatment. We speculate that our study presents findings that may also have potential clinical utility. The frequency of B cell subsets, such as disease-associated plasmablasts, can be useful clinical biomarkers to monitor rituximab treatment and effectiveness (51, 58). Given their modest elevation in MuSK MG, plasmablast frequency distributions do not appear to be reliable markers of disease activity. Furthermore, they can be elevated following immunizations and infections that are possibly unrelated to the disease. In their place, B cell–derived cell culture approaches that include measurements of autoantibody production by memory B cells or plasmablasts, such as those described here, could provide a useful putative biomarker to monitor disease activity following rituximab and other B cell targeting agents. Larger longitudinal studies are critical to further explore such candidate biomarkers and establish their possible role in the management paradigm. The need is considerably important: biomarker-assisted detection of subclinical activity may be able to predict potentially life-threatening relapse and guide retreatment decisions.
Methods
Patients and controls.
Peripheral blood was collected from MG and HD subjects (Yale Myasthenia Gravis Clinic, New Haven, Connecticut, USA). We identified a MuSK MG cohort (n = 3) for purposes of investigation of postrituximab relapse that shared the following characteristics: (i) a rituximab-induced CSR/MM clinical status (>1 year), (ii) repopulation of the B cell compartment after rituximab, and (iii) disease relapse following sustained CSR/MM after rituximab. These 3 relapse patients correspond to samples MuSK 1, -2a, -2b, and -3; MuSK 2a and MuSK 2b refer to the same subject. This subject experienced a clinical relapse 1 week following influenza vaccination and was seen urgently in clinic a week after symptom onset. Sample MuSK 2a was collected at this time, followed by initiation of prednisone 10 mg/day. Within 2 weeks, symptoms resolved and dose was reduced to 5 mg/day. Following this, clinical worsening reoccurred, at which time the prednisone dose was increased to 10 mg/day. While the patient’s clinical condition improved, active signs and symptoms of disease were present at the time of sample MuSK 2b collection (8 weeks from MuSK 2a collection). Controls for the immunopathology-based investigations included a post–rituximab MuSK MG subject in CSR (59 months since last rituximab treatment) with a repopulated B cell compartment, a HD one-week after viral upper respiratory tract infection, and 8 AChR MG subjects. All MG subjects included met definitive diagnostic criteria for MG, including positive serology.
Multi-chromatic flow cytometry and cell sorting.
PBMCs were isolated and were either used fresh or cryopreserved (14). They were incubated with live/dead stain and fluorescently labeled antibodies against CD3 (Invitrogen, Pacific orange; UCTH1), CD14 (Invitrogen, Pacific orange; TUK4), CD19 (BD Biosciences, PE Cy7; SJ25C1), CD27 (BD Biosciences, PE; M-T271), and CD38 (BD Biosciences, V450; HB7) before analysis on an LSRII cytometer or sorting with a FACSaria (BD Biosciences) cell sorter. Plasmablasts were defined as live, single CD3–CD14–CD19+CD27hiCD38hi cells. To validate the plasmablast gating strategy, this population was sorted for morphological phenotyping with Cytospin (ThermoFisher Scientific) and subsequent Giemsa staining. For standard B cell immunophenotyping, B cells were defined as live, single, CD3–CD14–CD19+ cells and memory B cells as live, single, CD3–CD14–CD19+CD27+IgD– cells.
Cell culture and stimulation.
Fresh or cryopreserved PBMCs were enriched for CD27+ B cells in a two-step magnetic bead separation (Stemcell Technologies and Miltenyi Biotec), which invariably resulted in a > 95% (by flow cytometry) pure fraction of CD27+ cells. Separated cells were immediately cultured in conditions driving antibody secretion (1.5 × 105 to 2.0 × 105 cells per culture) in culture medium (RPMI with FBS, L-glutamine, penicillin/streptomycin; Gibco) supplemented with IL-21 (eBiosciences), IL-6 (R&D Systems), CpG (Invitrogen), and CD40L (R&D Systems) (22, 34). Alternatively, plasmablasts were bulk-sorted from fresh PBMCs directly into culture medium (an average of 2,000 cells per culture) supplemented with IL-6; for MuSK 2a, the plasmablast cell number requirement (2,000 per culture) was not fulfilled. Supernatants were harvested after 7 days. Ig was quantified by ELISA (14).
MuSK and AChR monoclonal Ig engineering.
Hybridomas producing monoclonal Igs to MuSK were prepared from lymph nodes of immunized mice using standard protocols (59). Mice were obtained from the Jackson Laboratory. The hybridoma cells were produced off-site at a core facility (The Hybridoma Facility-Laboratory of Vijay Kuchroo, Harvard Medical School, Boston, Massachusetts, USA) from mice immunized with the extracellular domain of recombinant human MuSK. The aforementioned MuSK extracellular domain was produced in S2 insect cells (transfected cells were provided by Patrick Waters, of the University of Oxford, Oxford, United Kingdom). A single monoclonal Ig (4A3) recognizing human MuSK was used for the production of a partially humanized rIg. Briefly, the VH and VL domains of the murine IgG were amplified using degenerate primers (60). These regions were then subcloned into expression vectors containing the human IgG heavy and κ constant regions (61) so anti-human secondary antibodies could be used for detection in antibody assays. The variable regions (VH/VL) of a human AChR-specific autoantibody (637) were similarly subcloned (44). These monoclonal rIgs were expressed and purified using protocols we have previously described (14, 61).
rIg production.
Reverse transcription of fresh or frozen single-cell–sorted plasmablast RNA, nested PCR reactions, cloning into expression vectors, antibody expression, and purification were performed as previously described (14).
MuSK autoantibody CBA.
Binding to MuSK was tested using 293T HEK cells (purchased from ATCC) transiently transfected with DNA encoding full-length human recombinant MuSK intracellularly tagged to GFP or GFP alone as previously described (plasmids were provided by David Beeson and Patrick Waters, of the University of Oxford) (4, 62). Live cells were incubated with serum (1:20 dilution in PBS 1% BSA), cell culture supernatant, or rIg such that Ig was normalized to 10 μg/ml for 1 hour at 4°C. After washing, binding to MuSK was detected by a 45-minute and 4°C incubation with an Alexa Fluor 647–labeled, rabbit anti–human IgG Fc secondary antibody with no IgM binding (Jackson ImmunoResearch, 309-605-008) that was used at 1:1,000 dilution in PBS 1% BSA. Each sample was tested on MuSK-GFP–transfected cells and GFP-transfected cells. For every experiment, the Alexa Fluor 647 cutoff was set on a sample of PBS 1% BSA incubated with MuSK-GFP–transfected cells so as to control for batch effect. Results were calculated as ΔMFI and Δ% of transfected cells that bound secondary antibody (termed positive cells) as follows: (i) ΔMFI = Alexa Fluor 647 MFI in MuSK-GFP–transfected cells minus Alexa Fluor 647 MFI in GFP-transfected cells, (ii) Δ% positive cells = (%frequency of positive MuSK-GFP–transfected cells/%frequency of MuSK-GFP–transfected cells) – (%frequency of positive GFP-transfected cells/%frequency of GFP-transfected cells). Controls included a humanized murine MuSK monoclonal rIg (4A3) and a human thymus–derived AChR monoclonal rIg (637) (44). Δ% positive cell values greater than 4 SD above the mean of the non–MuSK MG (AChR, HD) subjects were considered positive. Δ% positive cell cutoffs were as follows: serum (AChR 1–8, HD 1); mean + 4 SD = 36.0%. CD27+ cultures (AChR 1–6, HD 1); mean + 4 SD = 31.3%. PB cultures (AChR 1–3, HD 1); mean + 4 SD = 6.6%. Recombinant Ig (n = 26; AChR 7, -8); mean + 4 SD = 21.9%. CBA testing of all samples was performed in duplicate.
IgG1/IgG4 subclass-specific MuSK autoantibody CBA.
MuSK MG patient and control sera and CD27+ culture supernatants were tested for IgG1 and IgG4 binding to MuSK, using the CBA as described above, with substitution of the human IgG Fc–specific secondary antibody with either a PE-labeled, mouse anti–human IgG1 Fc (clone HP6001) or a PE-labeled, mouse anti–human IgG4 Fc secondary antibody (clone HP6025), both of which were from Southern Biotech. Both the secondary antibodies were used at a 1:500 dilution in PBS 1% BSA. Each sample was tested on MuSK-GFP–transfected cells with either the IgG1 or the IgG4 secondary antibody. For every experiment, the PE cutoff was set on a sample of PBS 1% BSA incubated with MuSK-GFP–transfected cells. Results were calculated as percentage of transfected cells that bound secondary antibody (percent of positive cells). Percent of positive cell values greater than 4 SD above the mean of the non–MuSK MG (AChR 1–6, HD 1) subjects were considered positive. Percent of positive cell cutoffs were as follows: Serum IgG1 mean + 4 SD = 21.6%; Serum IgG4 mean + 4 SD = 0.9%; CD27+ IgG1 mean + 4 SD = 0.9%; CD27+ IgG4 mean + 4 SD = 6.0%. Testing of all samples was performed in duplicate; the mean of duplicate experiments is reported.
MuSK/AChR/GFP reactivity CBA.
rIgs that tested positive for MuSK binding were also tested for reactivity to AChR. Binding was tested using 293T HEK cells transiently transfected with DNA encoding the full-length human recombinant MuSK tagged to GFP, adult AChR with rapsyn-GFP, or GFP alone as previous described (plasmids were provided by David Beeson and Patrick Waters, of the University of Oxford) (62). Live cells were incubated with rIg such that Ig was normalized to 10 μg/ml. Binding to MuSK/AChR was detected as described in the CBA above. For every experiment, the Alexa Fluor 647 cutoff was set on a sample of PBS 1% BSA incubated with MuSK-GFP–transfected cells. Results were calculated as percentage of transfected cells that bound secondary antibody (percent of positive cells). Testing of all samples was performed in duplicate.
Ig sequence analyses.
The heavy- and light-chain variable region germline gene segments were assigned with IMGT/HighV-QUEST (http://imgt.org) (63) using the July 7, 2015, version of the IMGT gene database. Somatic mutations resulting in replacement amino acids were evaluated through the alignment to germline genes provided by the IMGT V-base algorithm.
Statistics.
Positivity on the CBA was determined by the application of a cutoff value, which was determined by calculating the mean plus 4 SD of non–MuSK MG subject samples. In parallel, a comparison of rIg CBA results was applied with usage of the nonparametric Mann-Whitney U and Kruskal-Wallis tests. P values below 0.05 were considered significant. Statistics were performed on Graphpad Prism software.
Study Approval.
The study was approved by the Human Investigation Committee at the Yale School of Medicine and after informed consent was obtained from all subjects.
Author contributions
The immunological model and the experimental approach was conceived and designed by KCO with input from PS, AK, and RJN. Experiments were conducted and data was acquired and analyzed by all authors. RJN oversaw all clinical aspects of the study, including subject selection, collection of clinical specimens, associated clinical data, and interpretation of these data. The manuscript was written by all authors.
Supplementary Material
Acknowledgments
The authors thank Walker Marsh for assisting with the production of the 4A3 anti–MuSK monoclonal rIg and Vijay Kuchroo and Nasim Kassam of Harvard Medical School for generation of the hybridomas that yielded 4A3. The authors also thank colleagues from The Weatherall Institute of Molecular Medicine at the University of Oxford: Angela Vincent for critical reading of the manuscript and Nick Willcox for valuable discussions. The project described was supported by the National Institute of Allergy and Infectious Diseases of the NIH through a grant to KCO, under award number R01AI114780. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. This work was also supported (in part) by a pilot research award from Conquer Myasthenia Gravis.
Version 1. 08/29/2017
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(17):e94263. https://doi.org/10.1172/jci.insight.94263.
Contributor Information
Panos Stathopoulos, Email: pmstathopoulos@gmail.com.
Aditya Kumar, Email: aditya.kumar@uth.tmc.edu.
References
- 1.Hoch W, McConville J, Helms S, Newsom-Davis J, Melms A, Vincent A. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7(3):365–368. doi: 10.1038/85520. [DOI] [PubMed] [Google Scholar]
- 2.Viegas S, et al. Passive and active immunization models of MuSK-Ab positive myasthenia: electrophysiological evidence for pre and postsynaptic defects. Exp Neurol. 2012;234(2):506–512. doi: 10.1016/j.expneurol.2012.01.025. [DOI] [PubMed] [Google Scholar]
- 3.Suh J, Goldstein JM, Nowak RJ. Clinical characteristics of refractory myasthenia gravis patients. Yale J Biol Med. 2013;86(2):255–260. [PMC free article] [PubMed] [Google Scholar]
- 4.McConville J, et al. Detection and characterization of MuSK antibodies in seronegative myasthenia gravis. Ann Neurol. 2004;55(4):580–584. doi: 10.1002/ana.20061. [DOI] [PubMed] [Google Scholar]
- 5.Díaz-Manera J, et al. Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology. 2012;78(3):189–193. doi: 10.1212/WNL.0b013e3182407982. [DOI] [PubMed] [Google Scholar]
- 6.Keung B, et al. Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatr. 2013;84(12):1407–1409. doi: 10.1136/jnnp-2012-303664. [DOI] [PubMed] [Google Scholar]
- 7.Robeson KR, et al. Durability of the Rituximab Response in Acetylcholine Receptor Autoantibody-Positive Myasthenia Gravis. JAMA Neurol. 2017;74(1):60–66. doi: 10.1001/jamaneurol.2016.4190. [DOI] [PubMed] [Google Scholar]
- 8.Evoli A, Alboini PE, Bisonni A, Mastrorosa A, Bartoccioni E, Bartocccioni E. Management challenges in muscle-specific tyrosine kinase myasthenia gravis. Ann N Y Acad Sci. 2012;1274:86–91. doi: 10.1111/j.1749-6632.2012.06781.x. [DOI] [PubMed] [Google Scholar]
- 9.Guptill JT, Sanders DB, Evoli A. Anti-MuSK antibody myasthenia gravis: clinical findings and response to treatment in two large cohorts. Muscle Nerve. 2011;44(1):36–40. doi: 10.1002/mus.22006. [DOI] [PubMed] [Google Scholar]
- 10.Nowak RJ, Dicapua DB, Zebardast N, Goldstein JM. Response of patients with refractory myasthenia gravis to rituximab: a retrospective study. Ther Adv Neurol Disord. 2011;4(5):259–266. doi: 10.1177/1756285611411503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blum S, et al. Use and monitoring of low dose rituximab in myasthenia gravis. J Neurol Neurosurg Psychiatr. 2011;82(6):659–663. doi: 10.1136/jnnp.2010.220475. [DOI] [PubMed] [Google Scholar]
- 12.Tandan R, Hehir MK, Waheed W, Howard DB. Rituximab treatment of myasthenia gravis: A systematic review. Muscle Nerve. 2017;56(2):185–196. doi: 10.1002/mus.25597. [DOI] [PubMed] [Google Scholar]
- 13.Guptill JT, et al. Characterization of B cells in muscle-specific kinase antibody myasthenia gravis. Neurol Neuroimmunol Neuroinflamm. 2015;2(2):e77. doi: 10.1212/NXI.0000000000000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JY, et al. Compromised fidelity of B-cell tolerance checkpoints in AChR and MuSK myasthenia gravis. Ann Clin Transl Neurol. 2016;3(6):443–454. doi: 10.1002/acn3.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vander Heiden JA, et al. Dysregulation of B Cell Repertoire Formation in Myasthenia Gravis Patients Revealed through Deep Sequencing. J Immunol. 2017;198(4):1460–1473. doi: 10.4049/jimmunol.1601415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Colliou N, et al. Long-term remissions of severe pemphigus after rituximab therapy are associated with prolonged failure of desmoglein B cell response. Sci Transl Med. 2013;5(175):175ra30. doi: 10.1126/scitranslmed.3005166. [DOI] [PubMed] [Google Scholar]
- 17.Manz RA, Hauser AE, Hiepe F, Radbruch A. Maintenance of serum antibody levels. Annu Rev Immunol. 2005;23:367–386. doi: 10.1146/annurev.immunol.23.021704.115723. [DOI] [PubMed] [Google Scholar]
- 18.Cambridge G, et al. Serologic changes following B lymphocyte depletion therapy for rheumatoid arthritis. Arthritis Rheum. 2003;48(8):2146–2154. doi: 10.1002/art.11181. [DOI] [PubMed] [Google Scholar]
- 19.Hall RP, et al. Association of serum B-cell activating factor level and proportion of memory and transitional B cells with clinical response after rituximab treatment of bullous pemphigoid patients. J Invest Dermatol. 2013;133(12):2786–2788. doi: 10.1038/jid.2013.236. [DOI] [PubMed] [Google Scholar]
- 20.Quách TD, et al. Distinctions among Circulating Antibody-Secreting Cell Populations, Including B-1 Cells, in Human Adult Peripheral Blood. J Immunol. 2016;196(3):1060–1069. doi: 10.4049/jimmunol.1501843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lanzillotta M, Della-Torre E, Stone JH. Roles of Plasmablasts and B Cells in IgG4-Related Disease: Implications for Therapy and Early Treatment Outcomes. Curr Top Microbiol Immunol. 2017;401:85–92. doi: 10.1007/82_2016_58. [DOI] [PubMed] [Google Scholar]
- 22.Cao Y, et al. An optimized assay for the enumeration of antigen-specific memory B cells in different compartments of the human body. J Immunol Methods. 2010;358(1-2):56–65. doi: 10.1016/j.jim.2010.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Tsonis AI, et al. MuSK autoantibodies in myasthenia gravis detected by cell based assay--A multinational study. J Neuroimmunol. 2015;284:10–17. doi: 10.1016/j.jneuroim.2015.04.015. [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez Cruz PM, Huda S, López-Ruiz P, Vincent A. Use of cell-based assays in myasthenia gravis and other antibody-mediated diseases. Exp Neurol. 2015;270:66–71. doi: 10.1016/j.expneurol.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 25.Niks EH, et al. Clinical fluctuations in MuSK myasthenia gravis are related to antigen-specific IgG4 instead of IgG1. J Neuroimmunol. 2008;195(1-2):151–156. doi: 10.1016/j.jneuroim.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 26.Kohler S, et al. Disturbed B cell subpopulations and increased plasma cells in myasthenia gravis patients. J Neuroimmunol. 2013;264(1-2):114–119. doi: 10.1016/j.jneuroim.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 27.Perez-Andres M, et al. Human peripheral blood B-cell compartments: a crossroad in B-cell traffic. Cytometry B Clin Cytom. 2010;78 Suppl 1:S47–S60. doi: 10.1002/cyto.b.20547. [DOI] [PubMed] [Google Scholar]
- 28.Morbach H, Eichhorn EM, Liese JG, Girschick HJ. Reference values for B cell subpopulations from infancy to adulthood. Clin Exp Immunol. 2010;162(2):271–279. doi: 10.1111/j.1365-2249.2010.04206.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herrera D, Rojas OL, Duarte-Rey C, Mantilla RD, Angel J, Franco MA. Simultaneous assessment of rotavirus-specific memory B cells and serological memory after B cell depletion therapy with rituximab. PLoS ONE. 2014;9(5):e97087. doi: 10.1371/journal.pone.0097087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallace ZS, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74(1):190–195. doi: 10.1136/annrheumdis-2014-205233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmed AR, Spigelman Z, Cavacini LA, Posner MR. Treatment of pemphigus vulgaris with rituximab and intravenous immune globulin. N Engl J Med. 2006;355(17):1772–1779. doi: 10.1056/NEJMoa062930. [DOI] [PubMed] [Google Scholar]
- 32.Lindsey JW, Meulmester KM, Brod SA, Nelson F, Wolinsky JS. Variable results after rituximab in neuromyelitis optica. J Neurol Sci. 2012;317(1-2):103–105. doi: 10.1016/j.jns.2012.02.017. [DOI] [PubMed] [Google Scholar]
- 33.Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70(9):1110–1117. doi: 10.1001/jamaneurol.2013.3071. [DOI] [PubMed] [Google Scholar]
- 34.Chihara N, et al. Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci USA. 2011;108(9):3701–3706. doi: 10.1073/pnas.1017385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chihara N, et al. Plasmablasts as migratory IgG-producing cells in the pathogenesis of neuromyelitis optica. PLoS ONE. 2013;8(12):e83036. doi: 10.1371/journal.pone.0083036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li S, et al. Autoantibodies From Single Circulating Plasmablasts React With Citrullinated Antigens and Porphyromonas gingivalis in Rheumatoid Arthritis. Arthritis Rheumatol. 2016;68(3):614–626. doi: 10.1002/art.39455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerkman PF, et al. Circulating plasmablasts/plasmacells as a source of anticitrullinated protein antibodies in patients with rheumatoid arthritis. Ann Rheum Dis. 2013;72(7):1259–1263. doi: 10.1136/annrheumdis-2012-202893. [DOI] [PubMed] [Google Scholar]
- 38.Tan YC, et al. Barcode-enabled sequencing of plasmablast antibody repertoires in rheumatoid arthritis. Arthritis Rheumatol. 2014;66(10):2706–2715. doi: 10.1002/art.38754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joly P, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357(6):545–552. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
- 40.Ahmed AR, Kaveri S, Spigelman Z. Long-Term Remissions in Recalcitrant Pemphigus Vulgaris. N Engl J Med. 2015;373(27):2693–2694. doi: 10.1056/NEJMc1508234. [DOI] [PubMed] [Google Scholar]
- 41.Fujii Y, Monden Y, Hashimoto J, Nakahara K, Kawashima Y. Acetylcholine receptor antibody production by bone marrow cells in a patient with myasthenia gravis. Neurology. 1985;35(4):577–579. doi: 10.1212/WNL.35.4.577. [DOI] [PubMed] [Google Scholar]
- 42.Willcox HN, Newsom-Davis J, Calder LR. Cell types required for anti-acetylcholine receptor antibody synthesis by cultured thymocytes and blood lymphocytes in myasthenia gravis. Clin Exp Immunol. 1984;58(1):97–106. [PMC free article] [PubMed] [Google Scholar]
- 43.Gomez AM, et al. Proteasome inhibition with bortezomib depletes plasma cells and specific autoantibody production in primary thymic cell cultures from early-onset myasthenia gravis patients. J Immunol. 2014;193(3):1055–1063. doi: 10.4049/jimmunol.1301555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graus YF, et al. Human anti-nicotinic acetylcholine receptor recombinant Fab fragments isolated from thymus-derived phage display libraries from myasthenia gravis patients reflect predominant specificities in serum and block the action of pathogenic serum antibodies. J Immunol. 1997;158(4):1919–1929. [PubMed] [Google Scholar]
- 45.Franz B, May KF, Dranoff G, Wucherpfennig K. Ex vivo characterization and isolation of rare memory B cells with antigen tetramers. Blood. 2011;118(2):348–357. doi: 10.1182/blood-2011-03-341917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Love JC, Ronan JL, Grotenbreg GM, van der Veen AG, Ploegh HL. A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat Biotechnol. 2006;24(6):703–707. doi: 10.1038/nbt1210. [DOI] [PubMed] [Google Scholar]
- 47.Cho A, et al. Robust memory responses against influenza vaccination in pemphigus patients previously treated with rituximab. JCI Insight. 2017;2(12):93222. doi: 10.1172/jci.insight.93222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardona A, Pritsch O, Dumas G, Bach JF, Dighiero G. Evidence for an antigen-driven selection process in human autoantibodies against acetylcholine receptor. Mol Immunol. 1995;32(16):1215–1223. doi: 10.1016/0161-5890(95)00101-8. [DOI] [PubMed] [Google Scholar]
- 49.Farrar J, et al. Diverse Fab specific for acetylcholine receptor epitopes from a myasthenia gravis thymus combinatorial library. Int Immunol. 1997;9(9):1311–1318. doi: 10.1093/intimm/9.9.1311. [DOI] [PubMed] [Google Scholar]
- 50.Sims GP, Shiono H, Willcox N, Stott DI. Somatic hypermutation and selection of B cells in thymic germinal centers responding to acetylcholine receptor in myasthenia gravis. J Immunol. 2001;167(4):1935–1944. doi: 10.4049/jimmunol.167.4.1935. [DOI] [PubMed] [Google Scholar]
- 51.Mattoo H, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134(3):679–687. doi: 10.1016/j.jaci.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wrammert J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453(7195):667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tipton CM, et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol. 2015;16(7):755–765. doi: 10.1038/ni.3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. 2008;128(4):939–948. doi: 10.1038/sj.jid.5701132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Payne AS, et al. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115(4):888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bennett JL, et al. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66(5):617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bartoccioni E, Scuderi F, Minicuci GM, Marino M, Ciaraffa F, Evoli A. Anti-MuSK antibodies: correlation with myasthenia gravis severity. Neurology. 2006;67(3):505–507. doi: 10.1212/01.wnl.0000228225.23349.5d. [DOI] [PubMed] [Google Scholar]
- 58.Owczarczyk K, et al. A plasmablast biomarker for nonresponse to antibody therapy to CD20 in rheumatoid arthritis. Sci Transl Med. 2011;3(101):101ra92. doi: 10.1126/scitranslmed.3002432. [DOI] [PubMed] [Google Scholar]
- 59.Greenfield EA, et al. Monoclonal antibodies to distinct regions of human myelin proteolipid protein simultaneously recognize central nervous system myelin and neurons of many vertebrate species. J Neurosci Res. 2006;83(3):415–431. doi: 10.1002/jnr.20748. [DOI] [PubMed] [Google Scholar]
- 60.Wang Z, et al. Universal PCR amplification of mouse immunoglobulin gene variable regions: the design of degenerate primers and an assessment of the effect of DNA polymerase 3’ to 5’ exonuclease activity. J Immunol Methods. 2000;233(1-2):167–177. doi: 10.1016/S0022-1759(99)00184-2. [DOI] [PubMed] [Google Scholar]
- 61.Ray A, et al. Autoantibodies produced at the site of tissue damage provide evidence of humoral autoimmunity in inclusion body myositis. PLoS ONE. 2012;7(10):e46709. doi: 10.1371/journal.pone.0046709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leite MI, et al. IgG1 antibodies to acetylcholine receptors in ‘seronegative’ myasthenia gravis. Brain. 2008;131(Pt 7):1940–1952. doi: 10.1093/brain/awn092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569–604. doi: 10.1007/978-1-61779-842-9_32. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.