

Abstract
Antiphospholipid antibodies, present in one-third of lupus patients, increase the risk of thrombosis. We recently reported a key role for neutrophils — neutrophil extracellular traps (NETs), in particular — in the thrombotic events that define antiphospholipid syndrome (APS). To further elucidate the role of neutrophils in APS, we performed a comprehensive transcriptome analysis of neutrophils isolated from patients with primary APS. Moreover, APS-associated venous thrombosis was modeled by treating mice with IgG prepared from APS patients, followed by partial restriction of blood flow through the inferior vena cava. In patients, APS neutrophils demonstrated a proinflammatory signature with overexpression of genes relevant to IFN signaling, cellular defense, and intercellular adhesion. For in vivo studies, we focused on P-selectin glycoprotein ligand-1 (PSGL-1), a key adhesion molecule overexpressed in APS neutrophils. The introduction of APS IgG (as compared with control IgG) markedly potentiated thrombosis in WT mice, but not PSGL-1–KOs. PSGL-1 deficiency was also associated with reduced leukocyte vessel wall adhesion and NET formation. The thrombosis phenotype was restored in PSGL-1–deficient mice by infusion of WT neutrophils, while an anti–PSGL-1 monoclonal antibody inhibited APS IgG–mediated thrombosis in WT mice. PSGL-1 represents a potential therapeutic target in APS.
Keywords: Autoimmunity
Keywords: Autoimmune diseases
RNA sequencing in neutrophils from patients with primary antiphospholipid syndrome revealed a pro-inflammatory expression profile and overexpression of adhesion molecule P-selectin glycoprotein ligand-1.
Introduction
Vascular complications, including thrombotic events, are among the leading causes of morbidity and mortality in patients with lupus. Antiphospholipid antibodies, present in one-third of lupus patients, are a major driver of this thrombophilia and help define a complication coined antiphospholipid syndrome (APS). APS is diagnosed when circulating antiphospholipid antibodies are detected in patients with certain cardinal events, including deep vein thrombosis (DVT) and stroke (1). The diagnosis of APS is not limited to lupus patients and, about half the time, will be diagnosed as a standalone syndrome, primary APS (2). APS (with an estimated prevalence of at least 1 in 2,000) is the leading acquired cause of thrombosis in the US (3). APS also places patients at increased risk for pregnancy loss, cytopenias, cardiac valve lesions, seizure disorder, cognitive dysfunction, and nephropathy (4). Patients with APS are typically treated with anticoagulant drugs such as warfarin, which are not uniformly effective for antiphospholipid antibody–mediated thrombosis and offer little protection against the varied nonthrombotic manifestations of APS (5).
The pathophysiology of APS has yet to be fully defined. Despite the historical name “antiphospholipid”, most pathogenic antibodies in APS do not actually target phospholipids, but rather lipid-binding proteins. The best described antigen in APS is β-2 glycoprotein I (β2GPI). β2GPI, a cationic lipid–binding protein with unclear function, is made especially by the liver and circulates at high levels in plasma (50–200 μg/ml) (6, 7). It has been suggested that anti-β2GPI antibodies potentiate thrombosis by engaging β2GPI on cell surfaces, thereby promoting cell activation (8–10).
On this front, there has been a particular interest in endothelial cells. Given its constant interaction with whole blood, the endothelium necessarily has properties that potently counter coagulation/thrombosis (11). The endothelium is also the gateway by which inflammatory cells escape from blood to tissue, a tightly regulated process that involves rolling, firm adhesion, and extravasation. These critical events are regulated by selectin-mediated interactions that facilitate the initial rolling, and then stronger integrin-mediated engagement that promotes firm adhesion and the eventual escape of leukocytes through the vessel wall (12). In animal models of antiphospholipid antibody–mediated thrombosis, as well as in APS patients, there are signs suggesting increased endothelial activation. For example, tissue factor activity is increased in carotid homogenates from antiphospholipid antibody–treated mice (13). In parallel, such treatment also increases leukocyte-endothelium interplay in the microcirculation (14). Going further, antagonizing either E-selectin or P-selectin (the key selectins expressed by the endothelium) protects against thrombosis in mice (15, 16); the same is true for strategies blocking the integrin ligands VCAM-1 and ICAM-1 (16). Mechanistically, a study has suggested that downregulation of endothelial NOS (eNOS) by antiphospholipid antibodies may be another important factor in increased leukocyte-endothelium interplay (17). Mechanistically, NF-κB, p38 MAPK, and Krüppel-like factors (KLFs) have all been implicated in antiphospholipid antibody–mediated activation of endothelial cells (18–20), demonstrating how antiphospholipid antibodies may co-opt pathways normally associated with more “authentic” inflammatory stimuli.
Going beyond the endothelium, we have investigated the role of circulating cells, especially neutrophils, in antiphospholipid antibody–mediated thrombosis, with the idea that these cells may be hyperresponsive to subclinical endothelial activation (for example, as caused by the venous stasis that predisposes mice to DVT formation) (21). We have demonstrated in both human and murine systems that APS neutrophils are prone to exuberant release of neutrophil extracellular traps (NETs) — chromatin-derived extracellular “spider webs,” expelled from neutrophils in response to infectious or inflammatory stimuli (21, 22). While NETs aid the host by capturing and killing pathogens, they have also been reported to promote thrombosis by activating clotting factors and ensnaring platelets (23). When NETs do form in APS patients, they may be particularly resistant to degradation (24). Furthermore, APS patient blood appears to be enriched in a subset of low-density neutrophils called low-density granulocytes (LDGs), which are known to release NETs in an exaggerated fashion (25).
Here, we further explore pathways that may be primed in APS neutrophils, thereby leading to these exaggerated responses. We were particularly interested in identifying mediators of increased engagement between neutrophils and endothelial cells. While disruption of leukocyte-endothelial interactions has been explored as a potential therapeutic approach in inflammatory diseases such as sickle cell anemia and inflammatory bowel disease (26, 27), the concept has been relatively unstudied in APS.
Results
Analysis of the APS neutrophil transcriptome reveals an activated, proinflammatory signature.
Neutrophils were isolated from the peripheral blood of 9 patients with primary APS and 9 healthy controls matched for age, sex, and ethnicity (see Supplemental Table 1 for the details of all patients included in this study; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.93897DS1). The transcriptome was then comprehensively analyzed using RNA sequencing. We identified 1,363 differentially expressed genes in neutrophils from patients with primary APS as compared with controls, including 593 and 770 upregulated and downregulated genes in primary APS, respectively. Functional gene network analysis of upregulated genes revealed a clear proinflammatory expression signature in primary APS, highlighted by a number of significantly enriched inflammatory meta-groups including IFN-mediated signaling, TLR signaling, and cellular defense, among others (Figure 1).
Figure 1. Functional gene network analysis of genes upregulated in neutrophils from patients with primary APS as compared with healthy matched controls.

P values represent the significance of enrichment in the functional networks identified.
The IFN-mediated signaling pathway meta-group was the most significantly enriched group of upregulated genes in primary APS neutrophils as compared with controls (P = 4.0 × 10–27). Indeed, the type-I IFN–regulated gene IFIT1 was the most upregulated gene in primary APS neutrophils (8.5-fold compared with healthy controls) (Supplemental Table 2). Other upregulated IFN-regulated genes included IFIT3 (5.2-fold), IFI6 (4.9-fold), MX1 (4.8-fold), HERC5 (4.8-fold), and many others. Gene Ontology (GO) analysis showed the type I IFN signaling pathway as the most significant GO enriched in genes upregulated in primary APS (Supplemental Table 3). Other notably upregulated genes include TLRs TLR4, TLR5, and TLR8 (2-fold, 2.9-fold, and 2.9-fold, respectively), and a group of leukocyte immunoglobulin-like receptor (LILR) genes. These include all the activating LILR family members, LILRA1, LILRA2, LILRA3, LILRA4, LILRA5, and LILRA6, with the most overexpressed being LILRA4 (8.3-fold). Analysis of gene set enrichment in genes downregulated in primary APS revealed that downregulated genes are generally less cohesive as a functional group compared with upregulated genes, with only two functional categories reaching statistical significance (ATP binding and T cell receptor signaling pathway) (Supplemental Table 3). In summary, these data demonstrate an activated gene expression signature in the neutrophils of patients with primary APS as compared with matched healthy controls.
Cell adhesion genes are upregulated in APS patient neutrophils.
A number of upregulated genes within the cellular defense meta-group are relevant to neutrophil recruitment and adhesion (Figure 1). Moreover, the GO “leukocyte migration” was significantly represented by upregulated genes (Supplemental Table 3). To identify all upregulated genes in APS neutrophils that are relevant to cell adhesion, we performed a literature mining analysis to determine relationships between these upregulated genes and “cell adhesion” in the published literature. Among the 593 upregulated genes, 548 were present in MEDLINE (https://www.nlm.nih.gov/pubs/factsheets/medline.html). Of these 548 genes, 209 were linked to “cell adhesion” in MEDLINE, thereby demonstrating significant enrichment for this relationship (P < 0.0001) (Figure 2). Furthermore, a number of these genes were specifically linked to “neutrophil adhesion,” such as SELPLG (PSGL1) and SELL (encoding for L-selectin) (Figure 2). We took particular note of SELPLG, which encodes P-selectin glycoprotein ligand-1 (PSGL-1) and which was upregulated 2.5-fold in patients with primary APS as compared with controls. PSGL-1 engages with endothelial selectins to mediate neutrophil rolling (step-wise transit along the endothelial surface) (12). Rolling may be followed by arrest and (depending on the vascular bed) transmigration, with these subsequent steps dependent upon the interaction of neutrophil β-2 integrins with endothelial partners such as ICAM-1 (12). PSGL-1, as a key initiator of cell adhesion, is an attractive therapeutic target in APS. Humanized monoclonal antibodies targeting PSGL-1 have been developed (28), and inhibiting PSGL-1 has been shown to be effective in models of other immune-mediated diseases such as inflammatory bowel disease (27, 29). P-selectin/PSGL-1 inhibitors have demonstrated efficacy in sickle cell disease and certain models of thrombosis (26, 30, 31). Moreover, PSGL-1 polymorphisms have been shown to be associated with thrombosis risk in APS patients (32). Therefore, we selected PSGL-1 for further study as a potential target in APS.
Figure 2. Genes upregulated in APS neutrophils that had been associated with “cell adhesion” within MEDLINE abstracts.

Genes that had also been associated with “neutrophil adhesion” are shown in blue. The size of each node is proportional to the number of connections within this network, and the thickness of the edges is proportional to the number of times the terms were co-mentioned in the literature. Relationships with a strength of 5 or greater (see Methods) and genes with at least 4 connections within the network were included in the figure. This was done to reduce clutter and emphasize which genes were highly interconnected within the cellular adhesion network.
PSGL-1 is upregulated in the neutrophils of patients with primary APS.
Having demonstrated overexpression of the gene encoding for PSGL-1 by RNA sequencing analysis (Supplemental Figure 1), we turned our attention to an independent group of 15 patients with primary APS and 15 healthy controls. As compared with controls, the neutrophils of patients with primary APS demonstrated a median 3.4-fold increase in expression of the PSGL-1 gene (Figure 3A). In a parallel experiment, we conditioned control neutrophils with 24 unique APS patient serum samples for 4 hours and then analyzed gene expression. This experiment demonstrated a median 2.1-fold increase in the expression of the gene encoding for PSGL-1 upon exposure to APS serum, as compared with control serum (which was always heterologous to the control neutrophils) (Figure 3B). Importantly, we also extended these findings to the protein level, demonstrating that, as compared with control serum, APS serum increases PSGL-1 expression on the neutrophil surface (Supplemental Figure 2).
Figure 3. PSGL-1 is upregulated in APS patient neutrophils.
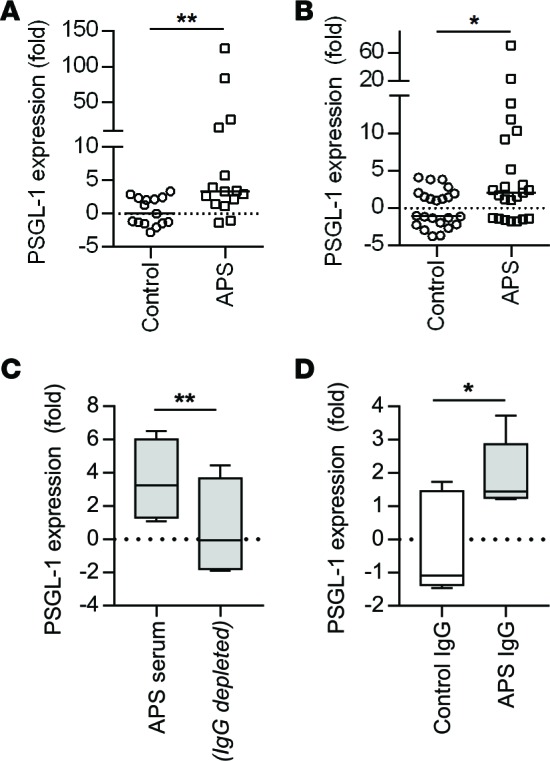
(A) RNA was prepared from patients with primary APS, none of whom were a part of the RNA-sequencing analysis. Expression of the gene encoding for PSGL-1 was determined by quantitative PCR, with normalization to the β-actin gene. The median for each group is denoted by a solid horizontal line, while each data point represents a unique control/patient; **P < 0.01 by Mann-Whitney test. (B) Neutrophils were isolated from healthy controls and then incubated with heterologous control serum or APS serum for 4 hours. RNA was prepared and subjected to quantitative PCR as in panel A. The median for each group is denoted by a solid horizontal line, while each data point represents stimulation with a unique control/patient serum sample; *P < 0.05 by Mann-Whitney test. (C) Control neutrophils were incubated with either APS serum (n = 5) or the same APS serum samples depleted of total IgG. After 4 hours, RNA was prepared and subjected to quantitative PCR. Box-and-whisker plots denote minimum, 25th percentile, median, 75th percentile, and maximum; **P < 0.01 by paired t test. (D) Control neutrophils were incubated with IgG purified from controls or APS patients (n = 5). After 4 hours, RNA was prepared and subjected to quantitative PCR. *P < 0.05 by two-tailed t test.
To understand whether increased PSGL-1 expression in APS was attributable to antiphospholipid antibodies themselves or other factors in serum, we depleted total IgG from the serum of 5 APS patients. Interestingly, IgG depletion significantly neutralized the ability of APS serum to increase expression of the PSGL-1 gene in control neutrophils (Figure 3C). In a parallel experiment, we conditioned control neutrophils with purified IgG from the same 5 APS patients. As compared with IgG from control patients, APS IgG significantly increased PSGL-1 gene expression (Figure 3D). In summary, these data demonstrate upregulation of the gene encoding for PSGL-1 in the neutrophils of patients with primary APS, with that upregulation likely attributable to a direct interaction between antiphospholipid antibodies and neutrophils.
PSGL-1 deficiency protects against venous thrombosis in a mouse model of APS.
We recently developed a new model of APS thrombosis (Figure 4A), in which the endothelium is not explicitly damaged but rather physiologically activated by flow restriction (21). This is accompanied by injection of APS IgG (purified from the blood of patients with high-titer anti-β2GPI antibodies) 24 or 48 hours before the flow restriction (21). Our work to date in this model has revealed that both neutrophils and NETs are required for APS IgG to accelerate the thrombosis phenotype (21), as evidenced by the ability of either neutrophil depletion or DNase administration (which dissolves NETs) to preferentially mitigate thrombosis in APS mice as compared with control mice (21). Furthermore, we have demonstrated the direct engagement of APS IgG with mouse neutrophils in vivo, as injection of APS IgG (but not control IgG) significantly increases the percentage of IgG-bound neutrophils in circulation (21). Here, injection of APS IgG significantly raised anti-β2GPI levels in mouse blood, with peak levels achieved by 24 hours after injection and maintained through 48 hours (Supplemental Figure 3). At this same 24-hour time point, we detected more PSGL-1 on the neutrophil surface (Supplemental Figure 4).
Figure 4. PSGL-1–KO mice are protected from antiphospholipid antibody–mediated thrombosis.
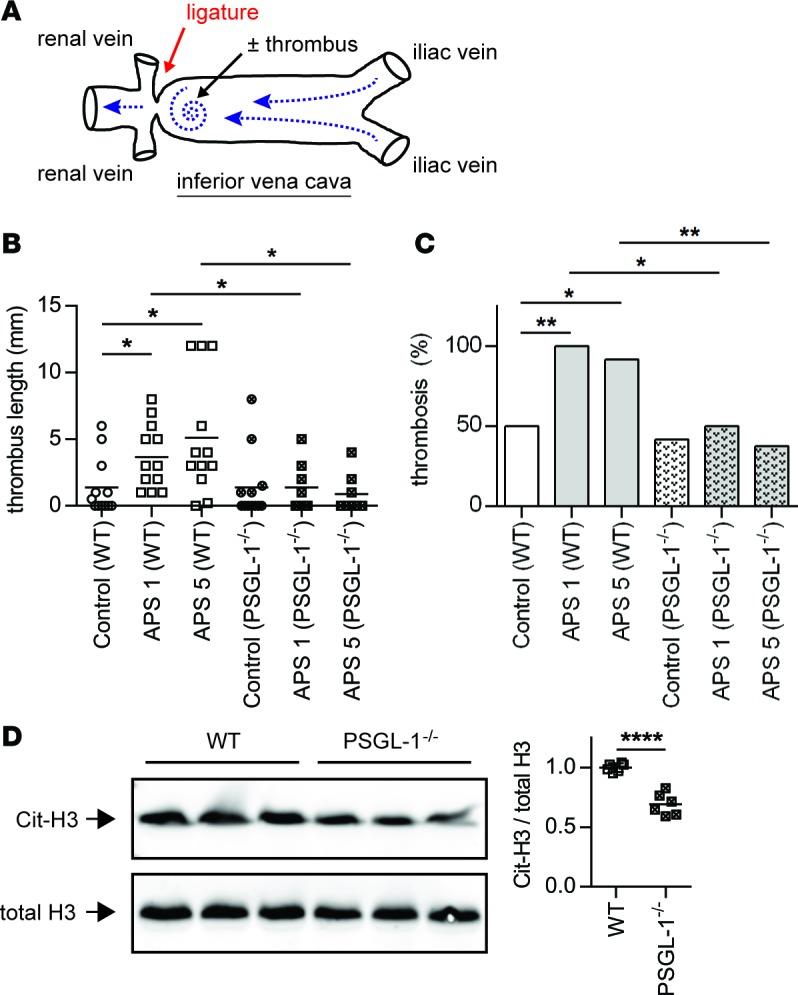
(A) Schematic of flow-restriction model of venous thrombosis. Mice were treated with IgG isolated from either healthy controls or patients with primary APS as described in Methods. The inferior vena cava (IVC) was then narrowed by fixing a ligature around the IVC (using a spacer to ensure uniformity). When a thrombus formed, it was just distal to the stenosis, in the area indicated by the spiral. (B) Thrombus length was measured 6 hours after flow restriction in WT or PSGL-1–/– mice. APS 1 and APS 5 denote IgG from two different patients. The mean for each group is denoted by a horizontal line, while each data point represents an individual mouse; *P < 0.05 adjusted using Kruskal-Wallis test followed by Dunn’s multiple comparisons test. (C) This is a contingency analysis of the data presented in panel B. *P < 0.05 and **P < 0.01 by χ2 test. (D) NET content of APS thrombi (presented in panel B) was assessed by Western blotting for citrullinated histone H3 (Cit-H3) and total histone H3 (the two blots were run in parallel/contemporaneously). Quantification is based on n = 6 per group (3 each from APS 1 and APS 5); ****P < 0.0001 by two-tailed t test.
As seen in our previous work, administration of APS IgG to WT mice resulted in larger (Figure 4B) and more frequent (Figure 4C) thrombi. In comparison, APS IgG–mediated thrombosis was markedly blunted in PSGL-1–/– mice (Figure 4, B and C). In WT mice, we have previously demonstrated by both IHC and Western blotting that NETs are quantitatively increased in the thrombi of APS mice, as compared with controls (21). For this quantification, we and others have relied on the only known biochemical marker for NETs, citrullinated histones (33). Citrullination of core histones, mediated by peptidylarginine deiminase 4, appears to be required for the massive chromatin decondensation of neutrophil extracellular trap (NET) release and has been used regularly in the literature as a specific marker of NETs (34–37). Here, we found that NETs were less abundant in APS-PSGL-1–/– thrombi as compared with APS-WT thrombi (Figure 4D), providing a potential mechanism to explain the reduced thrombosis in APS-PSGL-1–/– mice. In summary, these data demonstrate that PSGL-1 deficiency can protect against antiphospholipid antibody–mediated thrombosis, with a potential mechanism involving neutrophils and NETs.
Leukocyte adhesion is reduced in the IVC of PSGL-1–/– mice.
We hypothesized that the results of Figure 4 could be explained by a reduction in leukocyte adhesion to the wall of the inferior vena cava (IVC) in the absence of PSGL-1. Indeed, as compared with controls, mice treated with APS IgG demonstrated a marked increase in IVC leukocyte adhesion 1 hour after flow restriction (Figure 5A and Supplemental Video 1). Importantly, this increase was absent in PSGL-1–/– mice (Figure 5B and Supplemental Video 2). In summary, these data reveal potentiation of leukocyte adhesion by antiphospholipid antibodies, which is mitigated by PSGL-1 deficiency (Figure 5C).
Figure 5. Antiphospholipid antibodies promote increased leukocyte adhesion to the inferior vena cava following flow restriction.
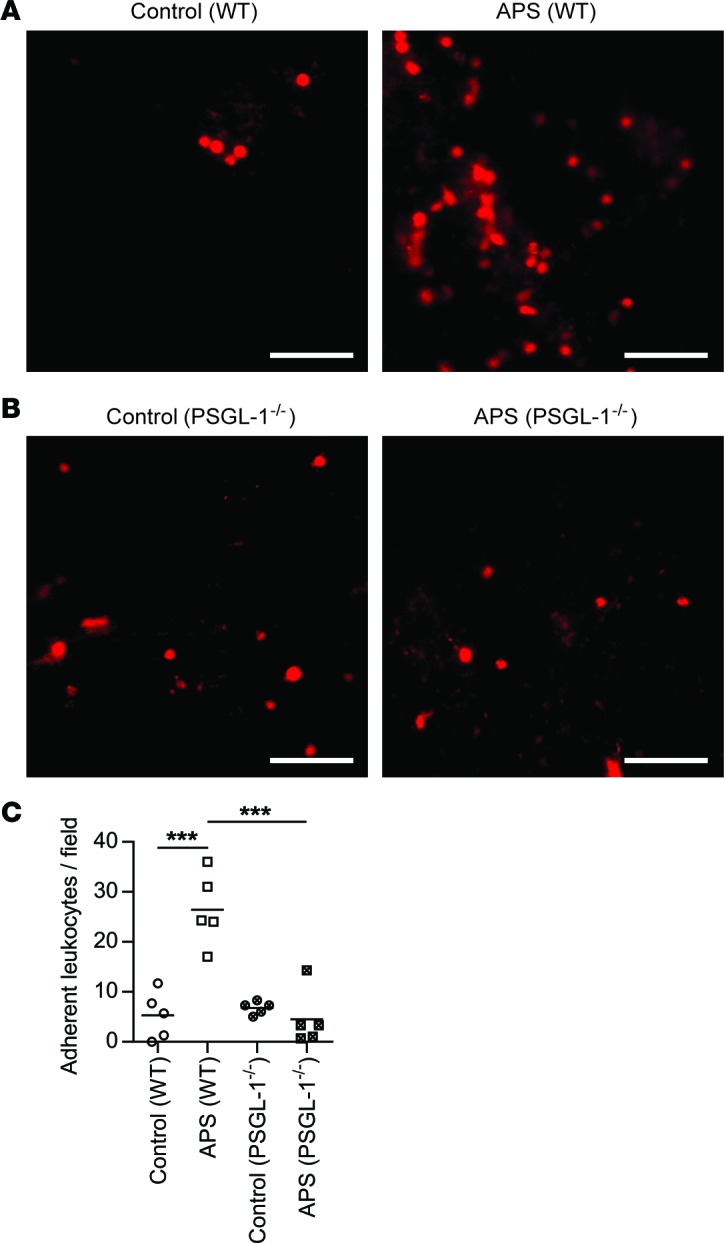
Mice were treated with control or APS IgG (specifically APS 1 as in Figure 4). (A) WT mice. Representative images of the inferior vena cava, 1 hour after flow restriction. Leukocytes are stained red by Rhodamine 6G. Scale bars: 50 μm. (B) PSGL-1–/– mice. Representative images and quantification of adherent leukocytes as described in panel A. Scale bars: 50 μm. (C) Quantification of adherent leukocytes per 40× field. The mean for each group is denoted by a horizontal line, while each data point represents a unique mouse; ***P < 0.001 adjusted using one-way ANOVA followed by Tukey’s multiple comparisons test.
Antiphospholipid antibody–mediated thrombosis is dependent upon neutrophil PSGL-1.
As discussed above, we have previously demonstrated that neutrophils are required for antiphospholipid antibody–mediated venous thrombosis (21). To confirm that the phenotype is specifically dependent upon neutrophil PSGL-1, we attempted to rescue thrombosis in PSGL-1–/– mice with WT neutrophils. Indeed, WT neutrophils triggered thrombosis in PSGL-1–/– mice that had been conditioned with APS IgG (Figure 6A). This was importantly in contrast to (i) mice treated with WT neutrophils but conditioned with control IgG, and (ii) mice conditioned with APS IgG but treated with PSGL-1–/– neutrophils (Figure 6A). In summary, these data confirm that PSGL-1 expression by neutrophils is required for antiphospholipid antibodies to accelerate venous thrombosis.
Figure 6. Antiphospholipid antibody–mediated thrombosis is dependent upon neutrophil PSGL-1 and can be mitigated by anti-PSGL-1 therapy.
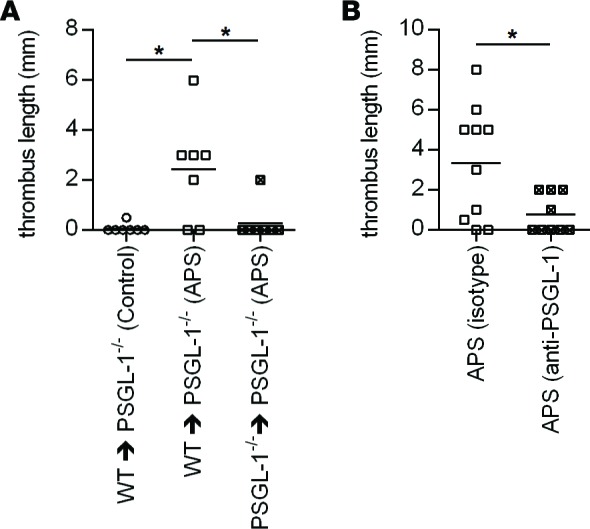
(A) PSGL-1–/– mice were treated with control or APS IgG (specifically APS 1 as in Figure 4). At the time of flow restriction in the inferior vena cava, mice were administered WT or PSGL-1–/– neutrophils by i.v. injection. Thrombus length was measured 6 hours later. The mean for each group is denoted by a horizontal line, while each data point represents an individual mouse; *P < 0.05 adjusted using Kruskal-Wallis test followed by Dunn’s multiple comparisons test. (B) WT mice were treated with APS IgG. Twenty-four hours before flow restriction, mice were also administered a single dose of either anti–PSGL-1 or isotype control antibody. Thrombus length was measured 6 hours after flow restriction. The mean for each group is denoted by a horizontal line, while each data point represents an individual mouse; *P < 0.05 by Mann-Whitney test.
Antiphospholipid antibody–mediated thrombosis can be mitigated by anti-PSGL-1 therapy.
Finally, we turned our attention to the potential role of PSGL-1 antagonism as a therapeutic strategy. Indeed, there is much interest in related drug concepts in other inflammatory conditions such as sickle cell anemia and inflammatory bowel disease (26, 27). Here, pretreatment with anti–PSGL-1 was effective in reducing the size of APS thrombi (Figure 6B). These data demonstrate the potential of PSGL-1 as a therapeutic target in APS.
Discussion
Historically, three cell types — endothelial cells, platelets, and monocytes — have received the majority of attention as drivers of thrombosis in APS (38). This work has demonstrated that so-called antiphospholipid antibodies actually engage proteins such as β2GPI, which themselves associate with cell-surface phospholipids (39). This engagement may lead to cell activation and upregulation of prothrombotic molecules, such as tissue factor. More recently, our group and others have suggested a role for neutrophils, the most abundant leukocytes in circulation, in the prothrombotic diathesis inherent to APS (21, 22, 24, 25, 40). This work was spurred by a new emphasis in the thrombosis literature regarding the importance of neutrophils and NETs in pathologic clotting and, in particular, venous thrombosis (41). As examples, the DNA component of NETs activates the intrinsic coagulation cascade (42, 43), histones stimulate platelets (44), and NET-derived proteases inactivate certain anticoagulant factors (42). In some contexts, NETs may even be an important source of tissue factor (45).
It should be emphasized that APS is an autoimmune disorder that is still primarily treated with anticoagulation, rather than immunomodulation. While anticoagulation clearly has some efficacy against the thrombotic and pregnancy complications of APS, the approach comes with inherent and sometimes life-threatening complications, including bleeding. Furthermore, anticoagulation has less proven efficacy in other APS manifestations such as thrombocytopenia, lower extremity ulcerations, nephropathy, seizures, and cognitive decline. While current clinical efforts are rightfully focused on testing novel oral anticoagulants (46), we would argue that a continued emphasis must be placed on understanding the inflammatory pathophysiology of APS, with the goal of establishing therapeutics that are more targeted and more effective.
To further understand the inflammatory aspects of APS, we used RNA sequencing to perform a comprehensive transcriptome analysis of neutrophils isolated from patients with primary APS. As compared with matched controls, APS neutrophils demonstrated an unequivocally activated signature. Interestingly, a number of the upregulated genes have already been defined as playing a role in APS, such as TLR4 (13), the complement component 5a receptor (47), protease-activated receptor 2 (48), and Fms-related tyrosine kinase 1 (FLT1, a receptor for vascular endothelial growth factor) (49). Furthermore, TLR and IFN-mediated signaling stood out in the meta-group analysis — both are receiving active attention in APS (50–53).
Here, we especially focused on another upregulated pathway revealed by the RNA sequencing data — intercellular adhesion. There were several rationales for doing so. First, there are certainly hints in the literature that disrupting these pathways may protect against antiphospholipid antibody–accelerated thrombosis in vivo, albeit with a focus on endothelial factors more so than the role of circulating cells (15, 16). Second, leukocyte adhesion is a potential therapeutic target, with active drug development programs already underway in other fields, including the study of sickle cell disease (26, 31). Finally, we felt that our recently described animal model was particularly amenable to assessing the role of neutrophil adhesion in antiphospholipid antibody–mediated thrombosis (21). Notably, the majority of models used to study APS in mice have relied on explicit vessel wall damage such as femoral vein pinch injury (13, 54), laser injury to the cremaster microcirculation (55, 56), and ferric chloride application (57). The flow restriction model used by our group and others leads to what is likely more physiologically relevant activation of the endothelium (21, 42, 58), as might be seen in a patient with stagnant blood flow in the deep veins of the lower extremities.
While the PSGL-1–/– mice used here are global KOs, we added specificity to the model with adoptive transfer experiments. Transfer of WT neutrophils into APS–PSGL-1–/– mice rescued the thrombosis phenotype, pointing to PSGL-1 contained within the neutrophil compartment as necessary for thrombosis in this model. As neutrophils transition from rolling to firm adhesion, PSGL-1 — working in concert with other surface molecules such as L-selectin — mediates the activation of β-2 integrins (59). Interestingly, we have previously shown that disruption of β-2 integrin signaling reduces the efficiency of antiphospholipid antibody–mediated NET release (22), and we therefore speculate that neutrophil adhesion plays a dual role in triggering thrombosis. The first, and obvious, role would be to localize the thrombotic event to a particular vascular location. Second, it is well accepted that signaling through both selectin ligands and integrins can further activate neutrophils (12). We therefore propose that activation through these pathways is required for efficient NET release. Our Western blotting data demonstrating increased NET content in APS-WT thrombi as compared with APS-PSGL-1–/– thrombi support this hypothesis. Future studies should interrogate whether these concepts might also extend to other conditions prone to inflammatory thrombosis, such as cancer and sepsis (60, 61).
Our work is not the first to link PSGL-1 and APS. In 2007, Diz-Kucukkaya and colleagues demonstrated that a particular pattern of PSGL-1 alleles, as defined by a variable-number-of-tandem-repeats (VNTR) polymorphism in the mucin-like region of PSGL-1, was a risk factor for thrombosis in antiphospholipid antibody–positive patients (32). The group’s prior work had demonstrated that these alleles, by defining PSGL-1 length, could regulate the efficiency by which PSGL-1 binds to P-selectin and thereby modulate the risk of events such as strokes (62). Their APS study posited that dimerization of PSGL-1 molecules of different lengths (the mixed AB genotype is predictive of APS) is actually the highest-risk situation for thrombosis (32). It would be of interest to connect that work with our study by asking whether the polymorphic PSGL-1 alleles may be expressed at different levels, as defined by quantitative mRNA analysis.
Going forward, we would now argue that the therapeutic potential of PSGL-1 inhibitors and other cell adhesion antagonists should be further pursued. Indeed, antagonism of PSGL-1 is not only an effective therapeutic approach in these experiments, but has also been shown to protect against stroke in a lupus model (63) and against microvascular occlusions in sickle cell anemia (26). The next step will be to dissect these pathways in other established APS models that target arteries and the microvasculature. In conclusion, our transcriptome profiling has confirmed the recent interest in APS neutrophils and the need to continue their study. Our proof-of-principle data have demonstrated that antagonism of neutrophil adhesion can protect against thrombosis. Future studies should focus on identifying other thrombosis-relevant adhesion pathway targets and on elucidating the role of adhesive intercellular interactions in triggering NET release. Both concepts will surely have implications that extend beyond APS to other disease states.
Methods
Human subjects.
Patients were recruited from rheumatology and hematology clinics at the University of Michigan. All patients with APS fulfilled the laboratory and clinical requirements for APS established by the Sydney classification criteria (1). None of the patients met American College of Rheumatology (ACR) criteria for SLE (64). Healthy volunteers were recruited through a posted flyer; exclusion criteria included history of a systemic autoimmune disease, active infection, and pregnancy. Blood was collected by phlebotomist venipuncture, and serum was prepared by standard methods and stored at –80ºC until ready for use. IgG, IgM, and IgA anti-β2GPI, as well as IgG and IgM anticardiolipin, were determined by multiplex assay on a BioPlex 2200 System (Bio-Rad). Lupus anticoagulant (LAC) was tested according to published guidelines (65).
Neutrophil isolation and culture.
Citrated blood was fractionated by density gradient centrifugation using Ficoll-Paque Plus (GE Healthcare). Neutrophils were then further purified by dextran sedimentation of the rbc layer, before lysing residual rbcs with 0.2% sodium chloride. Neutrophil preparations were at least 95% pure, as confirmed by both flow cytometry and nuclear morphology. In some instances, neutrophils were cultured in RPMI media (Gibco) supplemented with 2% BSA (Sigma-Aldrich) and 10 mM HEPES buffer (Gibco) for 4 hours. In some experiments, the media was additionally supplemented with 10% human serum, 10% human serum depleted of total IgG, or purified human IgG (at a concentration of 10 μg/ml). IgG depletion was accomplished with Protein G Agarose (Thermo Fisher Scientific) as described previously (22).
RNA preparation.
Purified neutrophils were resuspended in TriPure Isolation Reagent (Roche) and stored at –80°C. RNA was later prepared by the Direct-zol RNA MiniPrep kit (Zymo Research) according to manufacturer’s instructions. RNA integrity number (RIN) was > 7 for all included samples.
RNA sequencing.
Sequencing libraries were created using the TruSeq Stranded mRNA library kit (Illumina), and 100 bp single-end mRNA reads were generated on an Immunina HiSeq 2500 instrument. Sequenced reads were cleaned using the Trimmomatic (v. 0.32) preprocessing workflow and then mapped to the human genome (hg19) with SHRiMP (version 2.2.3) (66, 67). Differential expression analysis of mapped reads at the gene level was accomplished using the edgeR analysis package in the R programming environment. Only genes with a fold difference of > 2.0, a counts per million (CPM) of > 1 in at least 50% of samples (9 samples), and a FDR-adjusted P value of < 0.01 were considered for further analysis. RNA sequencing data were deposited in Gene Expression Omnibus (GEO), under GEO accession number GSE102215.
Bioinformatics analysis.
Bioinformatics analysis was conducted using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (68, 69). Gene set enrichment was calculated for biological process and molecular function GO terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. A Benjamini-Hochberg corrected P value of < 0.05 was considered significant. Additional bioinformatics analyses were also performed using both FGNet and GeneTerm Linker Bioconductor packages for the R statistical programming language (70, 71). Specifically, GeneTerm Linker was used to perform an initial gene enrichment analysis, which produced gene-term sets based on gene ontologies for biological processes and molecular functions. These gene-term sets were then used for a subsequent analysis by FGNet to predict functional gene networks. Significantly enriched functional networks were those with an adjusted P value < 0.05 and a silhouette width > 0, representing a greater functional coherence of genes and gene-term sets.
A program called IRIDESCENT was used to create the literature-based network of associations between concepts and genes (72, 73). Briefly, IRIDESCENT processes MEDLINE records (titles and abstracts) using a thesaurus of names and synonyms obtained from public databases of genes, diseases, phenotypes, chemical compounds, and GO categories. It searches for cooccurring entities and weights the cooccurrence by whether the entities were found in the same sentence (+0.8 weight) or abstract (+0.5 weight). Cumulatively, this creates a weighted network of published relationships. The upregulated genes in primary APS were analyzed for how they intersected with the concepts “cell adhesion” (which includes synonyms like “cellular adhesion”) and “neutrophil adhesion.”
Quantitative PCR.
cDNA was synthesized using MMLV RT (Invitrogen) and 200 ng of RNA in a MyCycler thermocyler (Bio-Rad). Quantitative PCR was performed with the SYBR Green PCR Master Mix (Applied Biosystems) according to manufacturer’s instructions and was carried out using an ABI PRISM 7900HT (Applied Biosystems). Primers for the human PSGL-1 and β-actin genes were purchased from Qiagen (QuantiTect Primer Assays, which have proprietary primer sequences). Ct values were normalized to the β-actin housekeeping gene to determine ΔCt. ΔΔCt values were then determined by comparing each ΔCt to a reference value within the control group (typically a value near the control mean). Data was presented as relative fold change by the formula 2ΔΔCt.
Preparation of human IgG.
IgG was purified from human serum with Protein G Agarose according to the manufacturer’s instructions (Pierce Biotechnology). Briefly, serum was diluted in IgG binding buffer and passed through a Protein G Agarose column (at least 5 times). Elution of IgG was done with 0.1 M glycine. The solution was neutralized with 1 M Tris, followed by overnight dialysis against PBS at 4ºC. After passing through a 0.2-μm filter, IgG purity was verified by SDS-PAGE. IgG was quantified by BCA protein assay (Pierce Biotechnology). IgG preparations were free of endotoxin contamination as determined by a chromogenic endotoxin quantification kit (Pierce Biotechnology). IgG samples used for injection into mice (see below) were prepared from APS patients who had high-titer IgG anti-β2GPI, high-titer IgG anticardiolipin, and positive testing for lupus anticoagulant (so-called triple-positive patients).
Flow cytometry.
For in vitro studies with human neutrophils, cells were washed and stained with anti-CD15 (W6D3, BioLegend), anti-CD16 (3G8, BioLegend), and anti-PSGL-1 (KPL-1, BD Biosciences). Staining was for 30 minutes at 4°C. After washing, cells were fixed in 2% paraformaldehyde. For in vivo studies, mouse whole blood was stained using the following antibodies: anti-Ly6G (1A8, BD Biosciences), anti–PSGL-1 (2PH1, BD Biosciences), anti-Ly6C (HK1.4, BioLegend), and anti-CD11b (M1/70, eBioscience). Cells were stained for 30 minutes at 4°C. After washing, cells were lysed and fixed using eBioscience 1-step Fix/Lyse Solution. Flow cytometry was with a LSRFortessa Cell Analyzer (BD Biosciences). Further data analysis was done in FlowJo.
Animal housing, surgery, and treatments.
Mice were housed in a specific pathogen-free barrier facility and fed standard chow. C57BL/6 mice were purchased from The Jackson Laboratory (000664) and used at approximately 10 weeks of age. PSGL-1–/– mice were originally purchased from The Jackson Laboratory and backcrossed to the C57BL6/J strain > 16 generations before use in these experiments (63).
To model antiphospholipid antibody–mediated thrombosis, mice were administered 2 doses (500 μg each) of either control or APS IgG by i.p. injection, 48 hours apart. Just prior to the second IgG treatment, a laparotomy was performed under anesthesia, and a ligature was fastened around the IVC over a blunted 30-gauge needle (which served as a spacer). After removal of the spacer, the abdomen was closed, and the mouse was allowed to recover. Mice were humanely euthanized, and thrombus formation was assessed 6 hours after laparotomy. In some experiments, mice were pretreated with anti–PSGL-1 antibody or isotype (4RA10, BD Pharmingen, 100 μg by i.p. injection) 24 hours before the laparotomy.
For neutrophil adoptive transfer, neutrophils were prepared from the BM of 10-week-old mice with the Mouse Neutrophil Enrichment Kit (Stem Cell Technologies, catalog 19762A) according to manufacturer’s instructions. To model antiphospholipid antibody–mediated thrombosis, mice were administered a single dose of either control or APS IgG (2,000 μg) by i.p. injection 24 hours before the laparotomy. Immediately following the laparotomy (and IVC flow restriction), 2.5 million neutrophils were infused by retroorbital injection. Mice were humanely euthanized, and thrombus formation was assessed 6 hours later.
ELISA for anti-β2GPI IgG.
Serum was tested according to manufacturer’s instructions (Inova Diagnostics).
Intravital microscopy.
Mice were administered a single dose of either control or APS IgG (2,000 μg) by i.p. injection. Twenty-four hours later, a laparotomy was performed under anesthesia and IVC flow was restricted (stenosis procedure, as described above for thrombosis experiments). One hour after flow restriction, rhodamine-6G (200 μl of a 0.067 mg/ml solution) was injected i.v. via the retroorbital sinus. The abdomen was opened, and the IVC was again exposed. Labeled leukocytes were visualized with a Nikon FN1 fixed-stage microscopy system with X-cite for epi-fluorescence. Videos were recorded with a Photometrics Coolsnap Cascade 512B color digital camera system (1,500 frames over 2 minutes). Analysis was with the MetaMorph Premier software package (Molecular Devices). The area of attention was 3–5 mm distal (i.e., in the direction of the tail) to the suture/stenosis. Adherent cells (stable for the duration of the 2-minute video) were quantified in at least three 40× fields per mouse. Finally, fields were averaged to generate a single value for each mouse.
Western blotting.
After separation from the vessel wall, thrombi were prepared for SDS-PAGE by sonication in RIPA buffer (50 mM Tris pH 8.0, 200 mM NaCl, 0.5% Nonidet P-40, and a Roche protease inhibitor cocktail pellet). Lysates were cleared by centrifugation at 14,000 g for 10 minutes at 4°C. Protein concentration was determined by the BCA Protein Assay Kit (Pierce Biotechnology) according to manufacturer’s instructions. Samples were resolved by 15% SDS-PAGE and transferred to a 0.45-μm nitrocellulose membrane. Primary antibodies were against citrullinated histone H3 (5103, Abcam) and histone H3 (1791, Abcam). Detection was with IRDye-labeled secondary antibodies (Rockland Immunochemicals). Images were captured and analyzed with an Odyssey imaging system (LI-COR Biosciences).
Statistics.
Data analysis was performed with GraphPad Prism software version 6. Normally distributed data were analyzed by two-tailed t test, and skewed data were assessed by Mann-Whitney test. When multiple comparisons on a single data set were made, these comparisons were analyzed using one-way ANOVA followed by Tukey’s test or by using Kruskal-Wallis test followed by Dunn’s test for normally distributed and nonnormally distributed data, respectively. For assessment of dichotomous variables (thrombosis frequency), the analysis was by χ2 test. Statistical significance was defined as P < 0.05.
Study approval.
These human and animal studies were reviewed and approved by the University of Michigan IRB and University of Michigan IACUC, respectively. All protocols complied with the standards of the National Research Council.
Author contributions
HM, PC, SY, GS, AAG, RCG, LFM, RAA, PR, JDW, and HW conducted experiments and analyzed data. JSK, PLB, DTE, and AHS designed the study and analyzed data. All authors participated in writing the manuscript and gave approval before submission.
Supplementary Material
Acknowledgments
JSK was supported by NIH K08AR066569 and career development awards from the Burroughs Wellcome Fund and Rheumatology Research Foundation. The work was supported by a grant from the Arthritis National Research Foundation to JSK and the National Institute of Allergy and Infectious Diseases under award number R01 AI097134 to AHS.
Version 1. 09/21/2017
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information: JCI Insight. 2017;2(18):e93897. https://doi.org/10.1172/jci.insight.93897.
Contributor Information
He Meng, Email: hemeng@med.umich.edu.
Patrick Coit, Email: pscoit@med.umich.edu.
Srilakshmi Yalavarthi, Email: syalavar@med.umich.edu.
Gautam Sule, Email: gsule@umich.edu.
Paul Renauer, Email: paul.renauer@yale.edu.
Hui Wang, Email: wanghui74@gmail.com.
Daniel T. Eitzman, Email: deitzman@umich.edu.
References
- 1.Miyakis S, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J Thromb Haemost. 2006;4(2):295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 2.Bertolaccini ML, et al. 14th International Congress on Antiphospholipid Antibodies Task Force. Report on antiphospholipid syndrome laboratory diagnostics and trends. Autoimmun Rev. 2014;13(9):917–930. doi: 10.1016/j.autrev.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Gómez-Puerta JA, Cervera R. Diagnosis and classification of the antiphospholipid syndrome. J Autoimmun. 2014;48-49:20–25. doi: 10.1016/j.jaut.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 4.Abreu MM, et al. The relevance of “non-criteria” clinical manifestations of antiphospholipid syndrome: 14th International Congress on Antiphospholipid Antibodies Technical Task Force Report on Antiphospholipid Syndrome Clinical Features. Autoimmun Rev. 2015;14(5):401–414. doi: 10.1016/j.autrev.2015.01.002. [DOI] [PubMed] [Google Scholar]
- 5.Erkan D, et al. 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun Rev. 2014;13(6):685–696. doi: 10.1016/j.autrev.2014.01.053. [DOI] [PubMed] [Google Scholar]
- 6.Conti F, et al. Beta-2-glycoprotein I expression on monocytes is increased in anti-phospholipid antibody syndrome and correlates with tissue factor expression. Clin Exp Immunol. 2003;132(3):509–516. doi: 10.1046/j.1365-2249.2003.02180.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caronti B, et al. Beta2-glycoprotein I (beta2-GPI) mRNA is expressed by several cell types involved in anti-phospholipid syndrome-related tissue damage. Clin Exp Immunol. 1999;115(1):214–219. doi: 10.1046/j.1365-2249.1999.00770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma K, Simantov R, Zhang JC, Silverstein R, Hajjar KA, McCrae KR. High affinity binding of beta 2-glycoprotein I to human endothelial cells is mediated by annexin II. J Biol Chem. 2000;275(20):15541–15548. doi: 10.1074/jbc.275.20.15541. [DOI] [PubMed] [Google Scholar]
- 9.Allen KL, Fonseca FV, Betapudi V, Willard B, Zhang J, McCrae KR. A novel pathway for human endothelial cell activation by antiphospholipid/anti-β2 glycoprotein I antibodies. Blood. 2012;119(3):884–893. doi: 10.1182/blood-2011-03-344671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sorice M, et al. Anti-beta2-glycoprotein I antibodies induce monocyte release of tumor necrosis factor alpha and tissue factor by signal transduction pathways involving lipid rafts. Arthritis Rheum. 2007;56(8):2687–2697. doi: 10.1002/art.22802. [DOI] [PubMed] [Google Scholar]
- 11.de Groot PG, Urbanus RT. Antiphospholipid Syndrome--Not a Noninflammatory Disease. Semin Thromb Hemost. 2015;41(6):607–614. doi: 10.1055/s-0035-1556725. [DOI] [PubMed] [Google Scholar]
- 12.McEver RP. Selectins: initiators of leucocyte adhesion and signalling at the vascular wall. Cardiovasc Res. 2015;107(3):331–339. doi: 10.1093/cvr/cvv154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierangeli SS, et al. Toll-like receptor and antiphospholipid mediated thrombosis: in vivo studies. Ann Rheum Dis. 2007;66(10):1327–1333. doi: 10.1136/ard.2006.065037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pierangeli SS, Colden-Stanfield M, Liu X, Barker JH, Anderson GL, Harris EN. Antiphospholipid antibodies from antiphospholipid syndrome patients activate endothelial cells in vitro and in vivo. Circulation. 1999;99(15):1997–2002. doi: 10.1161/01.CIR.99.15.1997. [DOI] [PubMed] [Google Scholar]
- 15.Espinola RG, Liu X, Colden-Stanfield M, Hall J, Harris EN, Pierangeli SS. E-Selectin mediates pathogenic effects of antiphospholipid antibodies. J Thromb Haemost. 2003;1(4):843–848. doi: 10.1046/j.1538-7836.2003.00119.x. [DOI] [PubMed] [Google Scholar]
- 16.Pierangeli SS, Espinola RG, Liu X, Harris EN. Thrombogenic effects of antiphospholipid antibodies are mediated by intercellular cell adhesion molecule-1, vascular cell adhesion molecule-1, and P-selectin. Circ Res. 2001;88(2):245–250. doi: 10.1161/01.RES.88.2.245. [DOI] [PubMed] [Google Scholar]
- 17.Ramesh S, et al. Antiphospholipid antibodies promote leukocyte-endothelial cell adhesion and thrombosis in mice by antagonizing eNOS via β2GPI and apoER2. J Clin Invest. 2011;121(1):120–131. doi: 10.1172/JCI39828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunoyer-Geindre S, de Moerloose P, Galve-de Rochemonteix B, Reber G, Kruithof EK. NFkappaB is an essential intermediate in the activation of endothelial cells by anti-beta(2)-glycoprotein 1 antibodies. Thromb Haemost. 2002;88(5):851–857. [PubMed] [Google Scholar]
- 19.Vega-Ostertag M, Casper K, Swerlick R, Ferrara D, Harris EN, Pierangeli SS. Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum. 2005;52(5):1545–1554. doi: 10.1002/art.21009. [DOI] [PubMed] [Google Scholar]
- 20.Allen KL, Hamik A, Jain MK, McCrae KR. Endothelial cell activation by antiphospholipid antibodies is modulated by Kruppel-like transcription factors. Blood. 2011;117(23):6383–6391. doi: 10.1182/blood-2010-10-313072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meng H, et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. 2017;69(3):655–667. doi: 10.1002/art.39938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yalavarthi S, et al. Release of Neutrophil Extracellular Traps by Neutrophils Stimulated With Antiphospholipid Antibodies: A Newly Identified Mechanism of Thrombosis in the Antiphospholipid Syndrome. Arthritis Rheumatol. 2015;67(11):2990–3003. doi: 10.1002/art.39247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rao AN, Kazzaz NM, Knight JS. Do neutrophil extracellular traps contribute to the heightened risk of thrombosis in inflammatory diseases? World J Cardiol. 2015;7(12):829–842. doi: 10.4330/wjc.v7.i12.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leffler J, Stojanovich L, Shoenfeld Y, Bogdanovic G, Hesselstrand R, Blom AM. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin Exp Rheumatol. 2014;32(1):66–70. [PubMed] [Google Scholar]
- 25.van den Hoogen LL, Fritsch-Stork RD, van Roon JA, Radstake TR. Low-Density Granulocytes Are Increased in Antiphospholipid Syndrome and Are Associated With Anti-beta2 -Glycoprotein I Antibodies: Comment on the Article by Yalavarthi et al. Arthritis Rheumatol. 2016;68(5):1320–1321. doi: 10.1002/art.39576. [DOI] [PubMed] [Google Scholar]
- 26.Telen MJ. Cellular adhesion and the endothelium: E-selectin, L-selectin, and pan-selectin inhibitors. Hematol Oncol Clin North Am. 2014;28(2):341–354. doi: 10.1016/j.hoc.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 27.Yacyshyn BR. Adhesion molecule therapeutics in IBD. Inflamm Bowel Dis. 2008;14 Suppl 2:S279–S280. doi: 10.1002/ibd.20710. [DOI] [PubMed] [Google Scholar]
- 28.Muz B, et al. Inhibition of P-Selectin and PSGL-1 Using Humanized Monoclonal Antibodies Increases the Sensitivity of Multiple Myeloma Cells to Bortezomib. Biomed Res Int. 2015;2015:417586. doi: 10.1155/2015/417586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Inoue T, et al. Blockade of PSGL-1 attenuates CD14+ monocytic cell recruitment in intestinal mucosa and ameliorates ileitis in SAMP1/Yit mice. J Leukoc Biol. 2005;77(3):287–295. doi: 10.1189/jlb.0204104. [DOI] [PubMed] [Google Scholar]
- 30.Ramacciotti E, et al. P-selectin/ PSGL-1 inhibitors versus enoxaparin in the resolution of venous thrombosis: a meta-analysis. Thromb Res. 2010;125(4):e138–e142. doi: 10.1016/j.thromres.2009.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ataga KI, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med. 2017;376(5):429–439. doi: 10.1056/NEJMoa1611770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Diz-Kucukkaya R, Inanc M, Afshar-Kharghan V, Zhang QE, López JA, Pekcelen Y. P-selectin glycoprotein ligand-1 VNTR polymorphisms and risk of thrombosis in the antiphospholipid syndrome. Ann Rheum Dis. 2007;66(10):1378–1380. doi: 10.1136/ard.2007.075945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neeli I, Dwivedi N, Khan S, Radic M. Regulation of extracellular chromatin release from neutrophils. J Innate Immun. 2009;1(3):194–201. doi: 10.1159/000206974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong SL, et al. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat Med. 2015;21(7):815–819. doi: 10.1038/nm.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laridan E, et al. Neutrophil extracellular traps in ischemic stroke thrombi. Ann Neurol. doi: 10.1002/ana. [published online ahead of print July 11, 2017]. https://doi.org/10.1002/ana.24993. [DOI] [PubMed] [Google Scholar]
- 36.Martinod K, et al. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc Natl Acad Sci USA. 2013;110(21):8674–8679. doi: 10.1073/pnas.1301059110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–213. doi: 10.1083/jcb.200806072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. 2010;376(9751):1498–1509. doi: 10.1016/S0140-6736(10)60709-X. [DOI] [PubMed] [Google Scholar]
- 39.Giannakopoulos B, Krilis SA. The pathogenesis of the antiphospholipid syndrome. N Engl J Med. 2013;368(11):1033–1044. doi: 10.1056/NEJMra1112830. [DOI] [PubMed] [Google Scholar]
- 40.Pérez-Sánchez C, et al. Ubiquinol Effects on Antiphospholipid Syndrome Prothrombotic Profile: A Randomized. Arteriosclerosis, thrombosis, vascular biology. Arterioscler Thromb Vasc Biol. doi: 10.1161/ATVBAHA.117. [published online ahead of print July 6, 2017]. https://doi.org/10.1161/ATVBAHA.117.309225. [DOI] [PubMed] [Google Scholar]
- 41.Martinod K, Wagner DD. Thrombosis: tangled up in NETs. Blood. 2014;123(18):2768–2776. doi: 10.1182/blood-2013-10-463646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.von Brühl ML, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–835. doi: 10.1084/jem.20112322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gould TJ, et al. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler Thromb Vasc Biol. 2014;34(9):1977–1984. doi: 10.1161/ATVBAHA.114.304114. [DOI] [PubMed] [Google Scholar]
- 44.Fuchs TA, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107(36):15880–15885. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kambas K, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One. 2012;7(9):e45427. doi: 10.1371/journal.pone.0045427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen H, et al. Rivaroxaban versus warfarin to treat patients with thrombotic antiphospholipid syndrome, with or without systemic lupus erythematosus (RAPS): a randomised, controlled, open-label, phase 2/3, non-inferiority trial. Lancet Haematol. 2016;3(9):e426–e436. doi: 10.1016/S2352-3026(16)30079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Romay-Penabad Z, Liu XX, Montiel-Manzano G, Papalardo De Martínez E, Pierangeli SS. C5a receptor-deficient mice are protected from thrombophilia and endothelial cell activation induced by some antiphospholipid antibodies. Ann N Y Acad Sci. 2007;1108:554–566. doi: 10.1196/annals.1422.058. [DOI] [PubMed] [Google Scholar]
- 48.Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G. Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest. 2008;118(10):3453–3461. doi: 10.1172/JCI36089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cuadrado MJ, et al. Vascular endothelial growth factor expression in monocytes from patients with primary antiphospholipid syndrome. J Thromb Haemost. 2006;4(11):2461–2469. doi: 10.1111/j.1538-7836.2006.02193.x. [DOI] [PubMed] [Google Scholar]
- 50.Grenn RC, et al. Endothelial progenitor dysfunction associates with a type I interferon signature in primary antiphospholipid syndrome. Ann Rheum Dis. 2017;76(2):450–457. doi: 10.1136/annrheumdis-2016-209442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van den Hoogen LL, Fritsch-Stork RD, Versnel MA, Derksen RH, van Roon JA, Radstake TR. Monocyte type I interferon signature in antiphospholipid syndrome is related to proinflammatory monocyte subsets, hydroxychloroquine and statin use. Ann Rheum Dis. 2016;75(12):e81. doi: 10.1136/annrheumdis-2016-210485. [DOI] [PubMed] [Google Scholar]
- 52.Laplante P, et al. Antiphospholipid antibody-mediated effects in an arterial model of thrombosis are dependent on Toll-like receptor 4. Lupus. 2016;25(2):162–176. doi: 10.1177/0961203315603146. [DOI] [PubMed] [Google Scholar]
- 53.Wu M, Barnard J, Kundu S, McCrae KR. A novel pathway of cellular activation mediated by antiphospholipid antibody-induced extracellular vesicles. J Thromb Haemost. 2015;13(10):1928–1940. doi: 10.1111/jth.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pericleous C, et al. Proof-of-concept study demonstrating the pathogenicity of affinity-purified IgG antibodies directed to domain I of β2-glycoprotein I in a mouse model of anti-phospholipid antibody-induced thrombosis. Rheumatology (Oxford) 2015;54(4):722–727. doi: 10.1093/rheumatology/keu360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arad A, Proulle V, Furie RA, Furie BC, Furie B. β2-Glycoprotein-1 autoantibodies from patients with antiphospholipid syndrome are sufficient to potentiate arterial thrombus formation in a mouse model. Blood. 2011;117(12):3453–3459. doi: 10.1182/blood-2010-08-300715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Proulle V, Furie RA, Merrill-Skoloff G, Furie BC, Furie B. Platelets are required for enhanced activation of the endothelium and fibrinogen in a mouse thrombosis model of APS. Blood. 2014;124(4):611–622. doi: 10.1182/blood-2014-02-554980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laplante P, et al. Antiphospholipid antibody-mediated effects in an arterial model of thrombosis are dependent on Toll-like receptor 4. Lupus. 2016;25(2):162–176. doi: 10.1177/0961203315603146. [DOI] [PubMed] [Google Scholar]
- 58.Manukyan D, et al. Cofactor-independent human antiphospholipid antibodies induce venous thrombosis in mice. J Thromb Haemost. 2016;14(5):1011–1020. doi: 10.1111/jth.13263. [DOI] [PubMed] [Google Scholar]
- 59.Stadtmann A, et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J Exp Med. 2013;210(11):2171–2180. doi: 10.1084/jem.20130664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Demers M, Wagner DD. NETosis: a new factor in tumor progression and cancer-associated thrombosis. Semin Thromb Hemost. 2014;40(3):277–283. doi: 10.1055/s-0034-1370765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kimball AS, Obi AT, Diaz JA, Henke PK. The Emerging Role of NETs in Venous Thrombosis and Immunothrombosis. Front Immunol. 2016;7:236. doi: 10.3389/fimmu.2016.00236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lozano ML, et al. Polymorphisms of P-selectin glycoprotein ligand-1 are associated with neutrophil-platelet adhesion and with ischaemic cerebrovascular disease. Br J Haematol. 2001;115(4):969–976. doi: 10.1046/j.1365-2141.2001.03151.x. [DOI] [PubMed] [Google Scholar]
- 63.Wang H, et al. Psgl-1 Deficiency is Protective against Stroke in a Murine Model of Lupus. Sci Rep. 2016;6:28997. doi: 10.1038/srep28997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hochberg MC. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1997;40(9):1725. doi: 10.1002/art.1780400928. [DOI] [PubMed] [Google Scholar]
- 65.Pengo V, et al. Update of the guidelines for lupus anticoagulant detection. Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibody of the Scientific and Standardisation Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2009;7(10):1737–1740. doi: 10.1111/j.1538-7836.2009.03555.x. [DOI] [PubMed] [Google Scholar]
- 66.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rumble SM, Lacroute P, Dalca AV, Fiume M, Sidow A, Brudno M. SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol. 2009;5(5):e1000386. doi: 10.1371/journal.pcbi.1000386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 69.Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Aibar S, Fontanillo C, Droste C, De Las Rivas J. Functional Gene Networks: R/Bioc package to generate and analyse gene networks derived from functional enrichment and clustering. Bioinformatics. 2015;31(10):1686–1688. doi: 10.1093/bioinformatics/btu864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fontanillo C, Nogales-Cadenas R, Pascual-Montano A, De las Rivas J. Functional analysis beyond enrichment: non-redundant reciprocal linkage of genes and biological terms. PLoS ONE. 2011;6(9):e24289. doi: 10.1371/journal.pone.0024289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wren JD, Bekeredjian R, Stewart JA, Shohet RV, Garner HR. Knowledge discovery by automated identification and ranking of implicit relationships. Bioinformatics. 2004;20(3):389–398. doi: 10.1093/bioinformatics/btg421. [DOI] [PubMed] [Google Scholar]
- 73.Wren JD, Garner HR. Shared relationship analysis: ranking set cohesion and commonalities within a literature-derived relationship network. Bioinformatics. 2004;20(2):191–198. doi: 10.1093/bioinformatics/btg390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.