

Abstract
Pushing the frontier of fluorescence microscopy requires the design of enhanced fluorophores with finely tuned properties. We recently discovered that incorporation of four-membered azetidine rings into classic fluorophore structures elicits substantial increases in brightness and photostability, resulting in the ‘Janelia Fluor’ (JF) series of dyes. Here, we refine and extend this strategy, showing that incorporation of 3-substituted azetidine groups allows rational tuning of the spectral and chemical properties with unprecedented precision. This strategy yields a palette of new fluorescent and fluorogenic labels with excitation ranging from blue to the far-red with utility in cells, tissue, and animals.
Introduction
Small molecule fluorophores are essential tools for biochemical and biological imaging1,2. The development of new labeling strategies3 and innovative microscopy techniques4 is driving the need for new fluorophores with specific properties. A particularly useful class of dyes is the rhodamines, first reported in 18875, and now used extensively due to the superb brightness and excellent photostability of this fluorophore scaffold1,2,6. The photophysical and chemical properties of rhodamines can be modified through chemical substitution6–14, allowing the creation of fluorescent and fluorogenic labels, indicators, and stains in different colors7,11–20.
Despite a century of work on this dye class, the design and synthesis of new rhodamines remains severely limited by chemistry. The classic method of rhodamine synthesis—acid catalyzed condensation2,5,6—is incompatible with all but the simplest functional groups. To remedy this longstanding problem, our laboratory developed a method to synthesize rhodamine dyes using a Pd-catalyzed cross-coupling strategy starting from simple fluorescein derivatives21. This approach facilitated the discovery of a novel class of dyes containing four-membered azetidine rings, which exhibit substantial increases in the quantum yield relative to classic rhodamines containing N,N-dimethylamino groups13. The flagship member of this new dye class is ‘Janelia Fluor 549′ (JF549, 1, Fig. 1a). The enhanced brightness and photostability of this rhodamine dye has made it an exceptionally useful label for single-molecule experiments in living cells13,22–25; the recent development of a photoactivatable derivative has further extended its utility in advanced imaging experiments26.
Figure 1. Fine-tuning rhodamine dyes.
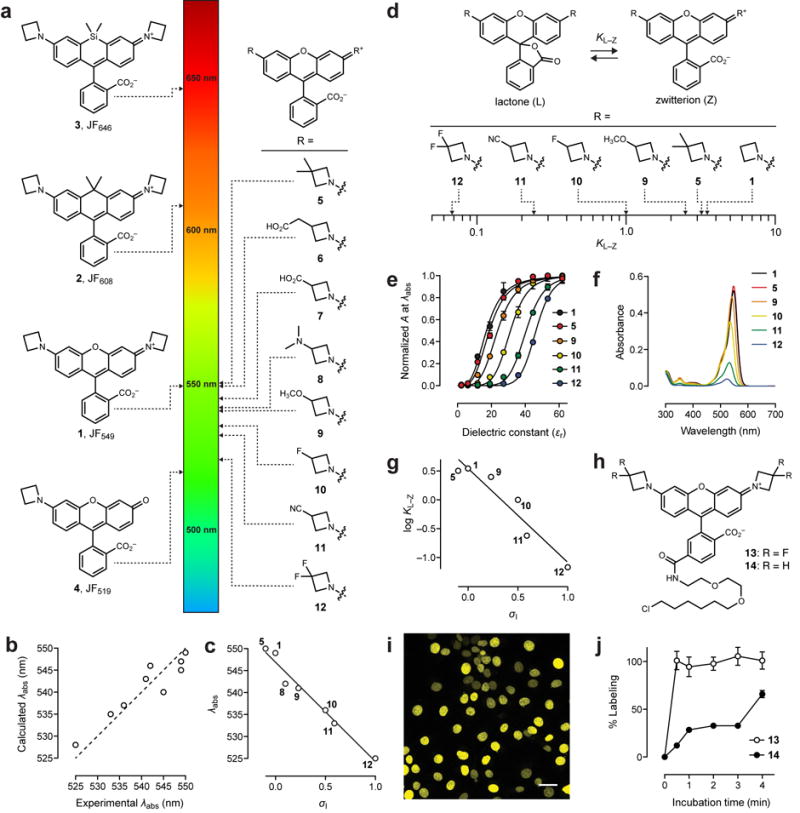
(a) Comparing coarse-tuning of λabs for dyes 1–4 and fine-tuning observed for azetidinyl rhodamines 5–12. (b) Correlation between calculated (DFT) and experimental λabs values for dyes 1, 5–12; dashed line shows ideal fit. (c) Correlation of experimental λabs vs. inductive Hammett constants (σI) for dyes 1, 5, 8–12. For the geminal disubstituted compounds 5 and 12 the σI of the substituent was doubled. Solid line shows linear regression (R2 = 0.97). (d) Fine-tuning of the lactone–zwitterion equilibrium constant (KL–Z) for dyes 1, 5, 9–12. (e) Normalized absorption vs. dielectric constant (εr) for dyes 1, 5, and 9–12; error bars show ± s.e.m; n = 4. (f) Absolute absorbance of 1, 5, 9–12 (5 μM) in 1:1 dioxane:H2O. (g) Correlation of KL–Z vs. inductive Hammett constants (σI) for dyes 1, 5, 9–12. For the geminal disubstituted compounds 5 and 12 the σI of the substituent was doubled. Solid line shows linear regression (R2 = 0.91). (h) Chemical structure of JF525–HaloTag ligand 13 and JF549–HaloTag ligand 14. (i) Image of live, washed COS7 cells expressing histone H2B–HaloTag fusions and labeled with ligand 13. Scale bar: 35 μm. (j) Plot of percent labeling of histone H2B–HaloTag fusions in live cells vs. incubation time for ligands 13 (100 nM) and 14 (100 nm); error bars show ± s.e.m; n = 113–248 (see Methods).
An important feature of rhodamine dyes is the ability to tune the spectral and chemical properties using chemistry13. JF549 (1, Fig. 1a, Table 1) absorbs green light (λabs/λem = 549 nm/571 nm, Φ = 0.88), making it an excellent match for light sources centered near 550 nm. Replacing the xanthene oxygen in JF549 (1) with a quaternary carbon yields carborhodamine Janelia Fluor 608 (JF608, 2) with an expected8 59-nm shift in spectral properties (λabs/λem = 608 nm/631 nm, Φ = 0.67). A larger red-shift can be achieved using the established Si-rhodamine strategy10,12 to afford Janelia Fluor 646 (JF646, 3, λabs/λem = 646 nm/664 nm, Φ = 0.54). Finally, a shift to shorter wavelengths can be imposed by replacing one azetidine group in JF549 (1) with an oxygen atom to yield rhodol27 Janelia Fluor 519 (JF519, 4, λabs/λem = 519 nm/546 nm, Φ = 0.85).
Table 1.
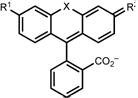
Properties of azetidine-containing fluorophores 1–12, 16, 21 and 25.
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Dye | X | R1 | R2 | λabs (nm) | ε (M−1cm−1) | εmax (M−1cm−1)a | λem (nm) | Φ | KL–Zb |
| 1 (JF549) |
|
|
|
549 | 101,000 | 134,000 | 571 | 0.88 | 3.5 |
| 5 |
|
|
|
550 | 110,000 | 143,000 | 572 | 0.83 | 3.2 |
| 6 |
|
|
|
549 | 111,000 | 138,000 | 572 | 0.87 | – |
| 7 |
|
|
|
545 | 108,000 | 130,000 | 568 | 0.87 | – |
| 8 |
|
|
|
542 | 111,000 | 127,000 | 565 | 0.57c | – |
| 9 |
|
|
|
541 | 109,000 | 137,000 | 564 | 0.88 | 2.5 |
| 10 |
|
|
|
536 | 113,000 | 141,000 | 560 | 0.87 | 1.0 |
| 11 |
|
|
|
533 | 108,000 | 133,000 | 557 | 0.89 | 0.24 |
| 12 (JF525) |
|
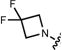
|
|
525 | 94,000 | 122,000 | 549 | 0.91 | 0.068 |
| 4 (JF519) |
|
|
|
519 | 59,000 | 69,000 | 546 | 0.85 | – |
| 16 (JF503) |
|
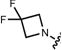
|
|
503 | 83,000 | 95,000 | 529 | 0.87 | – |
| 2 (JF608) |
|
|
|
608 | 99,000 | 121,000 | 631 | 0.67 | 0.091 |
| 21 (JF585) |
|
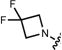
|
|
585 | 1,500 | 156,000 | 609 | 0.78 | <0.0001 |
| 3 (JF646) |
|
|
|
646 | 5,000 | 152,000 | 664 | 0.54 | 0.0012 |
| 25 (JF635) |
|
|
|
635 | ~400 | 167,000 | 652 | 0.56 | <0.0001 |
maximal extinction coefficient measured in EtOH or TFE with 0.1%TFA (rhodamines) or 0.01% Et3N (rhodols)
equilibrium constant measured in 1:1 v/v dioxane:water
cΦ = 0.89 in pH 5.0 buffer
In addition to large shifts in λabs and λem, these modifications modulate the equilibrium between the colorless, nonfluorescent ‘closed’ lactone (L) form and the colored, fluorescent, ‘open’ zwitterionic (Z) form. Rhodamine JF549 (1, ε = 1.01 × 105 M−1cm−1), carborhodamine JF608 (2, ε = 9.9 × 104 M−1cm−1) and rhodol JF519 (4, ε = 5.9 × 104 M−1cm−1) primarily adopt the open, zwitterionic form in water as evidenced by their large extinction coefficients (ε; Table 1). In contrast, JF646 (3) predominantly adopts the colorless, nonfluorescent, and lipophilic lactone form in aqueous solution (ε = 5.0 × 103 M−1cm−1). The shifted L–Z equilibrium of JF646 and other Si-rhodamines renders these dyes both highly cell permeable, chromogenic, and therefore fluorogenic, where the change in chemical environment upon binding of dye ligands to a variety of biomolecular targets can shift the equilibrium to the fluorescent zwitterionic form12–14,18.
Despite the flexibility of this fluorophore scaffold and the value in modulating rhodamine properties, the established strategies described above allow only ‘coarse’ tuning of spectral and chemical attributes: spectral shifts >30 nm and substantial changes in L–Z equilibrium and absorptivity. We sought, and now report, a general method to finely tune the spectral and chemical properties of rhodamine dyes with unprecedented precision. The use of 3-substituted azetidines on the Janelia Fluor 549 (1) scaffold allows modulation of λabs, λem, and the L–Z equilibrium without affecting fluorescence quantum yield. The shifts in chemical and spectral properties can be justified using physical organic chemistry principles and computational chemistry. The structure–activity relationships determined for rhodamine dyes are generalizable to rhodols, carborhodamines, and Si-rhodamine derivatives, allowing the rational design of improved fluorescent and fluorogenic labels across the visible spectrum.
Results
Fine-tuning rhodamines: Janelia Fluor 525
We reasoned we could finely tune the physicochemical properties of JF549 (1) by exploring different substitution patterns on the azetidine ring. Indeed, the azetidinyl-rhodamine system provides an ideal test case for N-substituent effects due to the following: (i) the high-yielding Pd-catalyzed cross-coupling synthesis21, (ii) the commercial availability of assorted 3-substituted azetidines, (iii) the short, three-bond separation between the substituent and the rhodamine aniline nitrogen, and (iv) the symmetry of the system. We hypothesized that electron withdrawing groups would decrease the λabs of the fluorophore and also shift the L–Z equilibrium towards the closed, colorless lactone form based on initial computational chemistry experiments (Fig. 1b, Methods) and reports of fluoroalkane-substituted rhodamine dyes19,28.
To test these predictions, we synthesized compounds 5–12 (Fig. 1a, Table 1) using our Pd-catalyzed cross-coupling approach13,21. We then evaluated the photophysical properties of compounds 5–12 in aqueous solution, comparing them to JF549 (1; Table 1). All of the substituted azetidinyl dyes showed high ε values above 1 × 105 M−1cm−1 except for the 3,3-difluoroazetidine compound 12, which exhibited a slightly lower absorptivity (ε = 9.4 × 104 M−1cm−1). Likewise, the quantum yield values of the azetidine dyes 5–12 were all >0.80 with the exception of the N,N-dimethyl-azetidin-3-amine compound 8, which showed Φ = 0.57 at pH 7.4. The quantum yield value for 8 is rescued at pH 5.0 (Φ = 0.89; Table 1), suggesting photoinduced electron transfer (PeT) quenching by the unprotonated dimethylamino groups29.
Although the ε and Φ of the different azetidinyl rhodamine dyes was largely immune to substitution at the 3-position, the λabs and λem values were strongly affected by the nature of the substituent (Table 1, Supplementary Fig. 1). Groups with greater electron-withdrawing character elicited larger hypsochromic shifts in λabs. This effect was additive. For example, the 3-fluoroazetidinyl compound 10 showed a 13-nm blue shift (λabs = 536 nm) relative to the parent dye 1 and the 3,3-difluoroazetidinyl-rhodamine (12) showed a further hypsochromic shift of 11 nm (λabs = 525 nm). We plotted λabs against the available Hammett inductive substituent constants (σI)30 for the azetidine substituents in dyes 1, 5, and 8–12 and observed an excellent correlation (Fig. 1c), suggesting that the inductive effect of the substituents was primarily responsible for the decrease in absorption and emission maxima. The experimental λabs values also showed excellent agreement with calculated λabs values (Fig. 1b).
We then analyzed how the azetidine substitutions can tune the lactone–zwitterion (L–Z) equilibrium (Fig. 1d), first examining the absorbance of fluorophores 1, 5, and 9–12 as a function of dielectric constant using dioxane–water titrations11,16 (Fig. 1e); compounds 6–8 were not examined due to the ionizable substituents on the azetidine ring. Based on these data, we determined the equilibrium constant (KL–Z)31 in 1:1 dioxane:water, which gave the largest distribution of absorbance measurements (Fig. 1f) and therefore KL–Z values (Fig. 1d, Table 1). We determined these equilibrium values from the maximal extinction coefficients (εmax) measured in acidic alcohol (Table 1, Methods). JF549 (1) and the 3,3-dimethylazetidinyl-rhodamine 5 showed KL–Z values >3, indicating that these dyes exist primarily in the open form. In contrast, the equilibrium constant of the 3,3-difluoroazetidinyl rhodamine (12) was substantially smaller (KL–Z = 0.068), showing that the electron-withdrawing fluorine substituents can shift the equilibrium toward the closed lactone form. The remainder of the dyes exhibited KL–Z values that were intermediate and correlated with σI (Fig. 1g). Collectively, these results yield rational and general rules for tuning both λabs and the L–Z equilibrium using different 3-substituted azetidines without compromising fluorophore brightness.
Rhodamine 12 exhibits λabs at 525 nm and a high quantum yield (Φ = 0.91), making it a useful label for imaging with blue-green excitation (514–532 nm). Based on the λabs, we named this fluorophore ‘Janelia Fluor 525′ (JF525) and prepared the JF525–HaloTag32 ligand (13, Fig. 1h), which showed excellent labeling in live cells expressing histone H2B–HaloTag fusions (Fig. 1i). We posited that the JF525–HaloTag ligand (13) would show improved cell permeability relative to the parent JF549–HaloTag ligand (14, Fig. 1h) based on its higher propensity to adopt the lactone form (Fig. 1d–f, Table 1). We therefore compared the labeling efficiency of 13 or JF549–HaloTag ligand 14 in live cells expressing HaloTag–histone H2B fusions (Methods). Compound 13 labeled intracellular proteins faster than JF549 ligand 14 (Fig. 1j). These results support the hypothesis that shifting the L–Z equilibrium towards the lactone form can improve cell permeability. We also synthesized the JF525–SNAP-tag ligand 15, which was useful for intracellular labeling (Supplementary Fig. 2a,b), validating JF525 as the first cell-permeable self-labeling tag ligand with an excitation maximum near 532 nm. None of the reported HaloTag or SNAP-tag ligands showed acute cellular toxicity at standard labeling concentrations and incubation times (Supplementary Fig. 2c)
Fine-tuning rhodols: Janelia Fluor 503
Since fluorine is the most electronegative atom, the difluoroazetidine-containing JF525 (12) represents the tuning limit of the azetidinyl-rhodamines towards the blue region of the spectrum. To access shorter wavelength dyes, we turned to the rhodol Janelia Fluor 519 (4, Fig. 1a, Fig. 2a). Based on the tuning rules determined for the rhodamine dyes (Fig. 1a) we surmised that replacement of the single azetidine substituent with a 3,3-difluoroazetidine could elicit a desirable ~15 nm blue-shift to yield a dye with maximal absorption closer to 488 nm. To test this hypothesis, we synthesized the 3,3-difluoroazetidinyl-rhodol 16, which showed the expected blue-shifted spectra with λabs/λem = 503 nm/529 nm, ε = 8.3 × 104 M−1cm−1, and Φ = 0.87 (Fig. 2a, Table 1, Supplementary Fig. 1); we named this compound ‘Janelia Fluor 503′ (JF503).
Figure 2. Rational fine-tuning of other dyes.
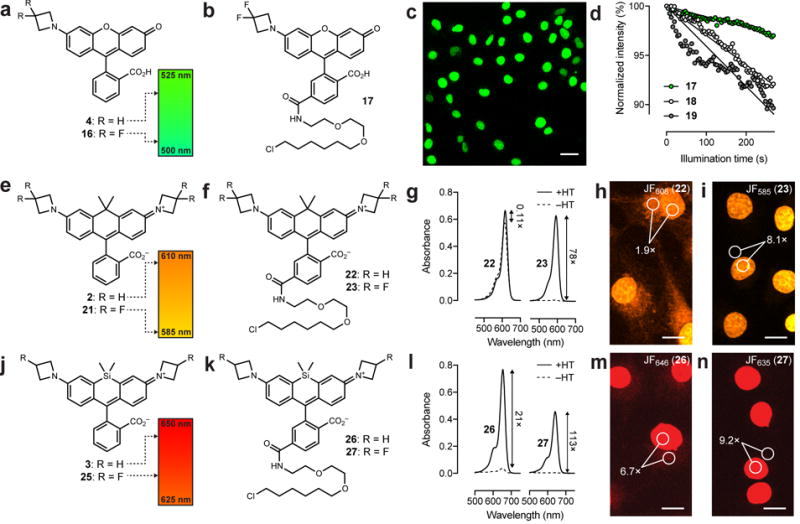
(a) Tuning of JF519 (4) to yield JF503 (16). (b) Structure of JF503–HaloTag ligand 17. (c) Image of live, washed COS7 cells expressing histone H2B–HaloTag fusions and labeled with ligand 17. Scale bar: 35 μm. (d) Comparison of the photostability of cells labeled with 17 and cells labeled with 488 nm-excited dyes 18 and 19 (Supplementary Fig. 2d); the initial photobleaching measurements are fitted to a linear regression. (e) Tuning of JF608 (2) to yield JF585 (21). (f) Structure of HaloTag ligands derived from JF608 (22) and JF585 (23). (g) Absorbance of HaloTag ligands 22 and 23 in the presence (+HT) or absence (–HT) of excess HaloTag protein; n = 2. (h,i) Representative images of COS7 cells expressing HaloTag–histone H2B fusion and labeled with 250 nM of HaloTag ligands 22 and 23 for 1 h and imaged directly without washing. The image for each dye pair was taken with identical microscope settings, λex = 594 nm. Numbers indicate mean signal (nuclear) to background (cytosol) ratio (S/B) in three fields of view. (h) JF608 ligand 22 (S/B from n = 224 areas). (i) JF585 ligand 23 (S/B from n = 235 areas). (j) Tuning of JF646 (3) to yield JF635 (25). (k) Structure of HaloTag ligands derived from JF646 (26) and JF635 (27) (l) Absorbance of HaloTag ligands 26 and 27 in the presence (+HT) or absence (−HT) of excess HaloTag protein (n = 2). (m,n) Representative images of COS7 cells expressing HaloTag–histone H2B fusion and labeled with 250 nM of HaloTag ligands 26 and 27 for 1 h and imaged directly without washing. The image for each dye pair was taken with identical microscope settings, λex = 647 nm. Numbers indicate mean signal (nuclear) to background (cytosol) ratio (S/B) in three fields of view. (m) JF646 ligand 26 (S/B from n = 175 areas). (n) JF635 ligand 27 (S/B from n = 278 areas). Scale bars for h,i,m,n: 15 μm.
We then synthesized the JF503–HaloTag ligand (17, Fig. 2b), which was an excellent label for histone H2B–HaloTag fusions in live cells (Fig. 2c). We compared this novel label to two other 488 nm-excited HaloTag ligands based on the classic rhodamine 110 (λabs/λem = 497 nm/520 nm; 18) and the recently described N,N´-bis(2,2,2-trifluoroethyl)rhodamine (λabs/λem = 501 nm/525 nm; 19, Supplementary Fig. 2d)19. The cell-loading time course for these structurally distinct and relatively polar dyes was similar (Supplementary Fig. 2e) but the JF503 ligand showed higher photostability than the other two dyes in live cells (Fig. 2d), consistent with previous reports comparing the photostability of rhodols to rhodamines27. JF503 could be extended to the SNAP-tag labeling system with JF503–SNAP-tag ligand (20; Supplementary Fig. 2f,g).
Fine-tuning carborhodamines: Janelia Fluor 585
In previous work, we extended our Pd-catalyzed cross-coupling approach to carborhodamines11, resulting in the synthesis of JF608 (2, Fig. 1a, Fig. 2e, Table 1)13. We also discovered that carborhodamines generally exhibit a higher propensity to adopt the colorless lactone form compared to rhodamines (KL–Z for 2 = 0.091). Nevertheless, this shift in the L–Z equilibrium is not sufficient to achieve fluorogenic ligands and its relatively long λabs value makes JF608 suboptimal for multicolor cellular imaging experiments using orange (e.g., 589 nm) and red (e.g., 640 nm) excitation. Based on the rhodamine tuning (Fig. 1a) we expected that incorporation of a 3,3-difluoroazetidine would elicit a blue-shift of approximately 24 nm, bringing the λabs closer to the desired excitation wavelengths. We were also curious if this modification would create a fluorogenic label since, in the rhodamine series, the 3,3-difluoroazetidine motif decreased the KL–Z by nearly two log units (Fig. 1d). Given the linear relationship between log KL–Z and σI (Fig. 1g) we reasoned that this substitution should tune the equilibrium of JF608 (2, KL–Z = 0.091) closer to the fluorogenic JF646 (3, KL–Z = 0.0012; Table 1). We note previous efforts to shift the L–Z equilibrium of carborhodamines using direct fluorination produced a HaloTag ligand with modest, nine-fold fluorogenicity, but this modification severely decreased quantum yield19. Based on the general trend to higher Φ values upon incorporation of electron-withdrawing substituents in both rhodamines and rhodols (Table 1), we expected that substitution with fluorine atoms on the 3-position of azetidine would increase quantum yield.
To test these predictions, we synthesized the 3,3-difluoroazetidinyl carborhodamine (21), which showed the expected blue-shift in spectra (λabs/λem = 585 nm/609 nm) and increase in quantum yield (Φ = 0.78; Fig. 2e, Table 1, Supplementary Fig. 1). Based on these properties the dye was named ‘Janelia Fluor 585′ (JF585). As predicted, JF585 (21) also exhibited low visible absorption in water (ε = 1.5 × 103 M−1cm−1) and a KL–Z near zero (Table 1). We then evaluated these carborhodamines as biomolecule labels, preparing JF608–HaloTag ligand (22) and JF585–HaloTag ligand (23, Fig. 2f)11,13,21. We first determined the absorbance of these ligands in the absence and presence of excess HaloTag protein. JF608–HaloTag ligand (22) showed only an 11% increase in absorption upon reaction with the HaloTag protein, but JF585 ligand 23 showed a substantially higher absorbance increase of 80-fold (Fig. 2g). We then evaluated these dyes in ‘no wash’ cellular imaging experiments. Incubation of JF608–HaloTag ligand (22, 250 nM) with cells expressing histone H2B–HaloTag showed excellent nuclear labeling but high background due to the free ligand staining internal membrane structures (Fig. 2h). In contrast, cells that were incubated with 250 nM of JF585–HaloTag ligand 23 and imaged directly showed bright nuclei with low fluorescence background (Fig. 2i). The JF585–SNAP-tag ligand (24) also functioned as a cellular label (Supplementary Fig. 2h,i), and the orange JF585–HaloTag ligand (23) could be used in three-color experiments with the green JF503–SNAP-tag ligand (20) and our previously described red JF646–Hoechst stain33 (Supplementary Fig. 2j).
Tuning of Si-rhodamines: Janelia Fluor 635
Having success in applying these tuning rules to rhodol and carborhodamine dyes, we then turned to the Si-rhodamine JF646 (3, Fig. 1a, Fig. 2j)13. We anticipated that addition of a single fluorine atom on each azetidine ring would elicit a ~13 nm hypsochromic shift and further decrease KL–Z, yielding a dye with a λabs near 633 nm and higher degree of fluorogenicity. True to this prediction, the synthesis of the desired fluorinated derivative 25 (Fig. 2j) afforded a dye that showed λabs/λem = 635 nm/652 nm and a slightly higher Φ = 0.56 relative to JF646 (3). Compound 25 also exhibited an extremely low absorbance in water with an extinction coefficient value of approximately 400 M−1cm−1, which gave a KL–Z near zero (Table 1, Supplementary Fig. 1). Based on these data we gave this dye the moniker ‘Janelia Fluor 635′ (JF635).
Analogous to the experiments with JF608 and JF585, we synthesized the JF635–HaloTag ligand (27) and compared it to the JF646 ligand 26 (Fig. 2k). As reported previously13, ligand 26 shows a 21-fold increase in absorbance upon binding to the HaloTag protein (Fig. 2l). The shifted L–Z equilibrium of JF635 causes HaloTag ligand 27 to show exceptionally low background and a 113-fold increase in absorbance upon conjugation (Fig. 2l). Both of these absorbance increases are substantially larger than the previously published SiTMR ligand 28 (Supplementary Fig. 2k), which shows a 6.7-fold increase in absorption upon reaction with the HaloTag protein12,13. These in vitro results were mirrored in no wash cellular imaging experiments, where we incubated cells expressing histone H2B–HaloTag fusions with 250 nM ligands 26–28. JF646 ligand 26 (Fig. 2m) and JF635 ligand 27 (Fig. 2n) exhibited substantially lower nonspecific extranuclear fluorescence than the SiTMR compound 28 (Supplementary Fig. 2l) with JF635 showing the highest contrast. The SNAP-tag ligand of JF635 (29) effectively labels SNAP-tag fusions in cells (Supplementary Fig. 2m,n) and the JF635–HaloTag ligand (27) could be used in a two-color experiment with JF525–SNAP-tag ligand (15; Supplementary Fig. 2o).
Applications in tissue and in vivo
The HaloTag ligands of tuned fluorophores Janelia Fluor 585 (23) and Janelia Fluor 635 (27) are small, cell permeable, and exhibit high fluorogenicity upon reactyion with the HaloTag protein. We were curious if these properties would make them useful for labeling in more complex biological environments such as tissue or whole animals. We first attempted labeling in living brain tissue from Drosophila larvae using the JF635–HaloTag ligand (27, Fig. 2k) due to its far-red excitation (Fig. 2j, Table 1) and high on:off ratio (Fig. 2l,n). We used a Drosophila GAL4 line expressing myristoylated HaloTag protein in ‘Basin’ neurons, which project basin-shaped arbors into the ventral nerve cord (VNC) of the larval fly34. Explants from Drosophila third instar larvae were dissected, incubated briefly with 27 (1 μM, 10 min), and imaged using the SiMView light-sheet microscope35. As shown in the projection of the SiMView 3D-reconstruction, the JF635 label exhibited consistent labeling throughout the living tissue and low nonspecific background staining (Fig. 3a,b, Supplementary Fig. 3a–c), demonstrating its utility beyond simple cell culture.
Figure 3. Labeling in tissue and in vivo.
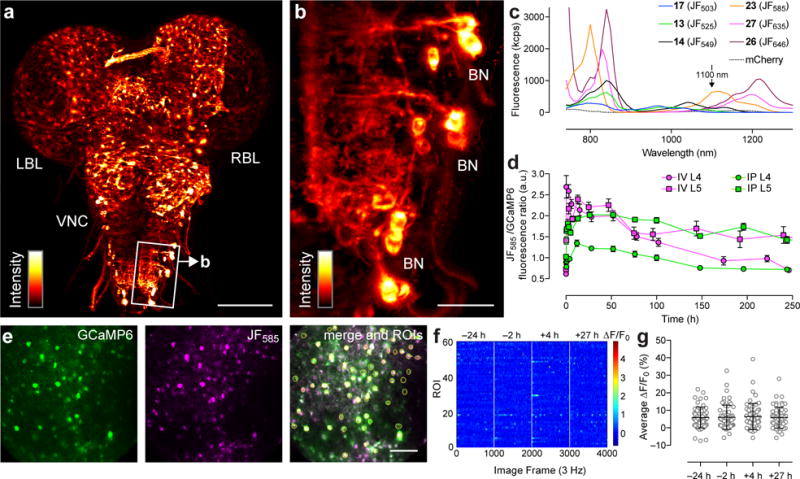
(a) SiMView light-sheet microscopy image (3D projection) of the central nervous system of a third instar Drosophila larva expressing myristoylated HaloTag protein in ‘Basin’ neurons (BNs) and stained with JF635–HaloTag ligand (27); LBL: left brain lobe; VNC: ventral nerve cord; RBL: right brain lobe. Scale bar: 100 μm. (b) Zoom in of boxed area in panel a showing individual BN cell bodies. Scale bar: 20 μm. (c) Two-photon fluorescence excitation spectra of HaloTag conjugates (1 μM) from HaloTag ligands 13, 14, 17, 23, 26, and 27 in 10 mM HEPES buffer (pH 7.3). The two-photon excitation spectra for mCherry is shown for reference. (d) Ratio of JF585 fluorescence to GCaMP6s epifluorescence at different time points after a single injection of JF585–HaloTag ligand (23, 100 nmol) either intravenous (IV) or intraperitoneal (IP) into mice expressing HaloTag protein in either layer 4 (L4) or layer 5 (L5) cortical neurons; n = 3 fields of view; error bars show ± s.e.m. (e) Two-photon microscopy images of neurons in layer 5 of the visual cortex coexpressing GCaMP6s (green) and JF585-labeled HaloTag (magenta) after IV injection of ligand 23 (t = 5 h). Scale bar: 100 μm. Yellow circles in the merged image indicate individual neurons as regions of interest (ROIs). (f) Raster plot of spontaneous neuronal activity in different ROIs (n = 61) before and after labeling with JF585–HaloTag ligand (23). (g) Plot of average spontaneous neural activity in each ROI before and after labeling with JF585–HaloTag ligand (23); central line shows mean; error bars show ± s.d.; no significant difference is observed between time points (one-way ANOVA: p = 0.95).
We next evaluated the JF dyes in the brains of living mice. The JF585–HaloTag ligand 23 was chosen based on its high fluorogenicity (Fig. 2g,i) and superior 2-photon fluorescence at 1100 nm excitation (Fig. 3c), which is sufficiently separated from GFP-based indicators such as GCaMP6 (2-photon λex = 940 nm)36 to allow multicolor imaging. Cytosolic HaloTag protein was co-expressed in layer 4 or layer 5 visual cortex (V1) neurons with GCaMP6s via viral transduction and the mice were fitted with a chronic cranial window (Methods). Injection of 100 nmol of HaloTag ligand 23 into the tail vein (intravenous, IV) showed the JF585 ligand was blood–brain barrier-permeable and gave measureable labeling in the brain within 5 minutes, peaking around 6 hours and lasting for nearly two weeks as measured by epifluorescence (Fig. 3d, Supplementary Fig. 3d). Subsequent intraperitoneal injection (IP) into the same set of mice also showed effective delivery to the brain, although with different pharmacokinetics in the early time points (<7 days; Fig. 3d). Under 2-photon imaging, we observed that the GCaMP6s and JF585 signals co-localized (Fig. 3e, Supplementary Fig. 3e, Supplementary Video 1), and the labeling showed no significant effect on spontaneous neuronal activity (Fig. 3f,g), establishing the utility of this fluorophore in vivo.
Discussion
Despite the broad utility of rhodamines, the extant methods to modulate the physicochemical properties of this dye class are relatively coarse and empirical. Here, we describe a new method that allows rational fine-tuning of fluorophore properties for specific biological applications. We first determined the tuning rules in the rhodamine system by synthesizing a panel of rhodamine variants using the bright and photostable JF549 scaffold (Fig. 1a). This resulted in the development of JF525 (12) and its derivatives 13 and 15, which constitute the first ligands for self-labeling tags with absorption maxima near 532 nm. The tuning rules discovered are generalizable to other fluorophore classes—rhodols, carborhodamines, and Si-rhodamines—allowing the rational design of finely-tuned fluorophores such as JF503 (16, Fig. 2a), JF585 (21, Fig. 2e), and JF635 (25, Fig. 2j). Together with JF549 (1) and JF646 (3, Fig. 1a)13, we have now described six dyes that span the visible region of the spectrum and match common excitation wavelengths for fluorescence microscopy. These bright fluorophores can be used immediately for structured illumination (SIM) and stimulated emission depletion (STED) imaging and could be converted to photoactivatable derivatives26 for single-molecule localization microscopy (SMLM) experiments. Our general rules should allow fine-tuning of a variety of fluorescent reagents including classic fluorophores13, emerging red-shifted rhodamine variants37, and fluorogenic ligands12,14,18,19,33 to further extend the range of bright fluorophores useful for fluorescence microscopy. Importantly, the new HaloTag ligands derived from JF585 and JF635 show a high degree of chromogenicity and fluorogenicity (Fig. 2g,i,l,n), a critical parameter in advanced imaging experiments38. Of particular interest is the ability to deliver these dyes to neural tissue in explants (Fig. 3a,b) or whole animals (Fig. 3d–g), which could allow the imaging of deeper structures in the brain or the in vivo assembly of semisynthetic indicators for monitoring cellular activity3.
Although we focused on the fluorine-substituted azetidines in this paper, the other substitutions (Fig. 1a) could be exploited to prepare fluorophores for specific applications. For example, the carboxy groups in compounds 6 and 7 could serve as attachment sites for a variety of chemical modifiers to improve solubility7, quench unwanted triplet states39, or allow the molecule to serve as a multivalent fluorescent cross-linker. The modest pH sensitivity and presence of the basic amine in compound 8 could allow it to function as a pH sensor or stain for lysosomes. The methoxy group on compound 9 could be elaborated to a polyethylene glycol (PEG) or other solubilizing group. Finally, the cyano group in compound 11 could be used in multimodal imaging regimes where both fluorescence and Raman40 modalities are used for imaging. In all, this general method to rationally tune photophysical and chemical properties against a backdrop of high quantum yield will allow the precise design of many new fluorophores for specific, sophisticated biological imaging experiments in increasingly complex systems.
Online Methods
Chemical synthesis
Methods for chemical synthesis and full characterization of all novel compounds can be found in the Supplementary Note.
UV–vis and fluorescence spectroscopy
Fluorescent and fluorogenic molecules for spectroscopy were prepared as stock solutions in DMSO and diluted such that the DMSO concentration did not exceed 1% v/v. Spectroscopy was performed using 1-cm path length, 3.5-mL quartz cuvettes or 1-cm path length, 1.0-mL quartz microcuvettes from Starna Cells. All measurements were taken at ambient temperature (22 ± 2 °C). Absorption spectra were recorded on a Cary Model 100 spectrometer (Agilent). Fluorescence spectra were recorded on a Cary Eclipse fluorometer (Varian). Maximum absorption wavelength (λabs), extinction coefficient (ε), and maximum emission wavelength (λem) were taken in 10 mM HEPES, pH 7.3 buffer unless otherwise noted; reported values for ε are averages (n = 3). Normalized spectra are shown for clarity.
Determination KL–Z and εmax
To determine KL–Z we first performed dioxane–H2O titrations in spectral grade dioxane (Aldrich) and milliQ H2O (Fig. 1e). The solvent mixtures contained 0.01% v/v triethylamine to ensure the rhodamine dyes were in the zwitterionic form. The absorbance values at λabs were measured on 5 μM samples (n = 4) using a quartz 96-well microplate (Hellma) and a FlexStation3 microplate reader (Molecular Devices). Values of dielectric constant (εr) were as previously reported41. We then calculated KL–Z using the following equation31: KL–Z = (εdw/εmax)/(1 – εdw/εmax). εdw is the extinction coefficient of the dyes in a 1:1 v/v dioxane:water solvent mixture (Fig. 1f); this dioxane–water mixture was chosen to give the maximum spread of KL–Z values (Fig. 1e). εmax is the maximal extinction coefficients measured in different solvent mixtures depending on dye type: 0.1% v/v trifluoroacetic acid (TFA) in 2,2,2-trifluoroethanol (TFE) for the rhodamines (1, 5–12) and carborhodamines (2, 21); 0.1% v/v TFA in ethanol for the Si-rhodamines (3, 25); 0.01% v/v Et3N in TFE for the rhodols (4, 16).
Quantum yield determination
All reported absolute fluorescence quantum yield values (Φ) were measured in our laboratory under identical conditions using a Quantaurus-QY spectrometer (model C11374, Hamamatsu). This instrument uses an integrating sphere to determine photons absorbed and emitted by a sample. Measurements were carried out using dilute samples (A < 0.1) and self-absorption corrections42 were performed using the instrument software. Reported values are averages (n = 3). The quantum yield for compound 8 at pH 5.0 was taken in 10 mM sodium citrate buffer containing 150 mM NaCl.
Computational chemistry
Computational experiments were performed using Gaussian 09.43 DFT and TD-DFT methods were used to calculate the spectral properties of the azetidinyl rhodamine compounds (Fig. 1b). Calculations were performed at the B3LYP/6-31+G(d,p)/IEFPCM and TD-B3LYP/6-31+G(d,p)/IEFPCM theory levels for the ground states and excited states respectively. Frequency calculations confirmed that an energy minimum was found in geometry optimizations. Linear response solvation with the IEFPCM model was sufficient to study the excited state energies. Evaluations of TD-DFT theory have discussed the overestimation of excitation energies44,45, and previous studies of rhodamine excited states have reported using ~0.4 eV correction to account for this overestimation46,47. We applied a consistent –0.4 eV correction to the calculated excited state energies, which gave good agreement with spectroscopy experiments (Fig. 1b).
Measurement of increase in absorbance of HaloTag ligands 22, 23, 26–28 upon attachment with HaloTag protein
HaloTag protein used as a 100 μM solution in 75 mM NaCl, 50 mM TRIS·HCl, pH 7.4 with 50% v/v glycerol (TBS–glycerol). Absorbance measurements were performed in 1 mL quartz cuvettes. HaloTag ligands 22, 23, 26–28 (5 μM) were dissolved in 10 mM HEPES, pH 7.3 containing 0.1 mg·mL−1 CHAPS. An aliquot of HaloTag protein (1.5 equiv) or an equivalent volume of TBS–glycerol blank was added and the resulting mixture was incubated until consistent absorbance signal was observed (~60 min). Absorbance scans are averages (n = 2).
Multiphoton spectroscopy
HaloTag ligands 13, 14, 17, 23, 26, and 27 (5 μM) were incubated with excess purified HaloTag protein (1.5 equiv) in 10 mM HEPES, pH 7.3 containing 0.1 mg·mL−1 CHAPS as above and incubated for 24 h at 4 °C. These solutions were then diluted to 1 μM in 10 mM HEPES buffer, pH 7.3 and the two-photon excitation spectra were measured as previously described48,49. Briefly, measurements were taken on an inverted microscope (IX81, Olympus) equipped with a 60×, 1.2NA water objective (Olympus). Dye–protein samples were excited with pulses from an 80 MHz Ti-Sapphire laser (Chameleon Ultra II, Coherent) for 710–1080 nm and with an OPO (Chameleon Compact OPO, Coherent) for 1000–1300 nm. Fluorescence collected by the objective was passed through a dichroic filter (675DCSXR, Omega) and a short pass filter (720SP, Semrock) and detected by a fiber-coupled Avalanche Photodiode (SPCM_AQRH-14, Perkin Elmer). For reference, a two-photon excitation spectrum was also obtained for the red fluorescent protein mCherry (1 μM), in the same HEPES buffer. All excitation spectra are corrected for the wavelength-dependent transmission of the dichroic and band-pass filters, and quantum efficiency of the detector.
General cell culture and fluorescence microscopy
COS7 and U2OS cells (ATCC) were cultured in Dulbecco’s modified Eagle medium (DMEM, phenol red-free; Life Technologies) supplemented with 10% (v/v) fetal bovine serum (Life Technologies), 1 mM GlutaMAX (Life Technologies) and maintained at 37 °C in a humidified 5% (v/v) CO2 environment. The COS7 cells have integrated a histone H2B–HaloTag expressing plasmid via the piggyback transposase (i.e., ‘H2B–Halo’ cells), and the U2OS cells have integrated a Sec61β–HaloTag expressing plasmid via the piggyback transposase. Both cells were kept under the selection of 500 μg/mL Geneticin (Life Technologies). Cell lines undergoes regular mycoplasma testing by the Janelia Cell Culture Facility. Cells were imaged on confocal microscopes in the Janelia Imaging Facility (Zeiss LSM 710, W Plan APO 20×/1.8 D -or- Zeiss LSM 880, C-APO 40×/1.2 W Corr FCS M27) using the indicated filter sets.
Comparison of JF549 and JF525
For the dye loading comparison (Fig 1j), H2B-Halo COS7 cells were stained for varying amounts of time with 100 nM of either JF525–HaloTag ligand 13 or JF549–HaloTag ligand 14. The dye was washed from the cells and subsequently labeled with JF646–HaloTag ligand 26 at 1 μM for 30 min. Fluorescence of JF646–HaloTag ligand was quantified from the nuclear signals in summed confocal image stacks collected with 633 nm Ex/638–759 nm Em and analysed using Fiji.50 The integrated density of the nuclear signal was corrected by subtracting the integrated density of adjacent background regions. Labeling is expressed as the percent of the JF646–HaloTag fluorescence displaced by the JF525– and JF549–HaloTag ligands. The nuclear staining of these cells by the JF525–HaloTag ligand (Fig. 1i) is displayed as a maximum intensity projection of confocal image stacks, 514 nm Ex/530–657 Em.
Comparison of JF503 and other 488 nm-excited dyes
H2B–Halo COS7 cells were labeled with 200 nM of JF503–HaloTag ligand 17, HaloTag® R110Direct™ ligand (18, Promega), or HaloTag ligand 1928, over a time course of 0–2 h. Cells were washed 2× with PBS and fixed with 4% w/v paraformaldehyde in 0.1 M phosphate for 30 min, followed by two more washes with PBS. Cells were imaged using confocal microscopy with 488 nm Ex/515–565 nm Em. The nuclear staining of these cells by the JF503–HaloTag ligand (17; Fig. 2c) is displayed as a maximum intensity projection of confocal image stacks. Corrected nuclear fluorescence was calculated as above to determine the cell loading profile (Supplementary Fig. 2e). To test the relative bleaching rates of these three dyes under imaging conditions (Fig. 2d), cells were stained and fixed as previously described for 2 h and then bleached with 488 nm at twice the typical power and imaged after each of 70 cycles. Bleached fluorescence data are normalized to the initial fluorescence levels.
Comparison of HaloTag ligands 22, 23, and 26–28 in cells
H2B–Halo COS7 cells were labeled with 250 nM of JF608–HaloTag ligand (22), JF585–HaloTag ligand (23), JF646-HaloTag ligand (26), JF635-HaloTag ligand (27), or SiTMR–HaloTag ligand (28; Supplementary Fig. 2k) and imaged by confocal microscopy using 594 nm Ex/599–734 nm Em (JF608 and JF585) or 633 nm Ex/638–759 nm Em (JF646 JF635, or SiTMR). All five samples were imaged via confocal microscopy without washing out the dyes. Signal to noise ratios were determined using the mean fluorescence of the nuclei relative to a region adjacent to each nuclei using Fiji50 (n = 152–275 areas as noted; Fig. 2h,i,m,n, Supplementary Fig. 2l).
Staining with SNAP-tag ligands
COS7 cells were transfected with histone H2B–pSNAP-tag (New England Biolabs) and stable integration of this plasmid was selected for using 600 μg/mL Geneticin® (Life Technologies). This cell line expresses the histone H2B protein fused to the 26m version of the SNAP-tag protein. Cells were stained with four different dyes as follows; JF503–SNAP-tag ligand (20, 2 μM for 90 min), JF525–SNAP-tag ligand (15, 3 μM for 30 min), JF585–SNAP-tag ligand (24, 2 μM for 3 hr with 0.2% w/v Pluronic F-127), JF635–SNAP-tag ligand (29, 2 μM for 2 h with 0.2% w/v Pluronic F-127). After staining, cells were washed three times with complete media, followed by a 20-min incubation in a 37 °C, 5% CO2, humidified incubator. The media was replaced again immediately prior to imaging.
Multiplexed imaging using HaloTag and SNAP-tag
U2OS cells expressing Sec61β–HaloTag fusion were transfected with either histone H2B–SNAP-tag piggybac orTOMM20–pSNAPf plasmids using Lipofectamine 2000 (ThermoFisher). Sec61β encodes an endoplasmic reticulum membrane protein translocator protein, and TOMM20 encodes an outer mitochondrial membrane protein as part of a protein translocase complex. Live cells were simultaneously stained with combinations of JF503–SNAP-tag ligand (20, 1 μM) and JF585–HaloTag ligand (23, 100 nM; Supplementary Fig. 2j) or JF525–SNAP-tag ligand (15, 1 μM) and JF635–HaloTag ligand (27, 100 nM, Supplementary Fig. 2o) for 60 min. Cells were fixed with 4% paraformaldehyde (20 min), and subsequently stained with JF646–Hoechst33 (5 μM, 30 min), as indicated, prior to imaging.
Cell viability assays
The effects of various Janelia Fluor ligand compounds on cell viability were tested using the tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Supplementary Fig. 2c). This colorimetric cell viability assay relies on NAD(P)H-dependent oxidoreductase enzymes that reduce the dye to an insoluble and highly absorbing formazan51. COS7 cells were plated at 1.5 × 104 cells per well of a 96-well plate 24 h prior to the assay. Dyes were applied to cells at different concentrations to span standard labeling conditions in live cells (n = 3). HaloTag ligands 13, 14, 17, 23, 26, and 27 were applied for 1 hour and SNAP-tag ligands 15, 20, 24, and 29 were applied for 3 hours prior to addition of MTT to reflect typical maximum staining times for these dyes.
Staining of Drosophila larvae
The central nervous system of third instar Drosophila melanogaster larvae were dissected in physiological saline. For the JF635–HaloTag ligand staining (Fig. 3a,b, Supplementary Fig. 3a,c), we used larva expressing a previously described52 HaloTag containing a myristoylation sequence in the ‘Basin’ neurons under control of the enhancer fragment R72F11.34 The isolated nervous system was incubated in physiological saline containing 1 μM JF635–HaloTag ligand (27) for 10 min at room temperature. For the pan-neuronal comparison (Supplementary Fig. 3b), this animal expressed GCaMP6s via the Gal4/UAS system using a 57C10-Gal4 driver line. The specimens were then embedded in agarose and imaged with the SiMView light-sheet microscope.35 The VNC zoomed images for direct comparison (Supplementary Fig. 3a,b) show the raw image data after fusion and deconvolution, whereas the full image of JF635–HaloTag ligand labeling (Fig. 3a,b, Supplementary Fig. 3c) additionally uses filtering and gamma correction to show neuronal morphologies more clearly.
General information for mouse in vivo experiments
Male mice, 3–8 months old, were used for viral infection, dye injection, and in vivo imaging of neurons in the visual cortex (V1): The Scnn1a-Tg3-Cre (Jax no. 009613) line was used for imaging in layer 4 cortical neurons (L4); and Rbp4-Cre mice (MMRRC no. 031125-UCD) were used for imaging in layer 4 cortical neurons (L5) neurons. All experimental protocols were conducted according to the National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee at the Janelia Research Campus, HHMI.
Cranial window implant and virus injection
A craniotomy was carried out at the same time as the virus injection to provide optical access for in vivo imaging experiments. Mice were anesthetized with isoflurane (1–2% v/v in O2) and given the analgesic buprenorphine (SC, 0.3 mg/kg). Using aseptic technique, a 3.5 mm-diameter craniotomy was made over the left V1 region of the brain of anaesthetized mouse (center: 3.4 mm posterior to Bregma; 2.7 mm lateral from midline). The dura was left intact. HaloTag and GCaMP6s was cotranduced using the viral vector: AAV2/1.synapsin.FLEX.GCaMP6s.P2A.HaloTag.WPRE (~5 × 1012 infectious units per ml, 30 nl per site). The virus was injected using a glass pipette beveled at 45° with a 15–20-μm opening and back-filled with mineral oil. A fitted plunger controlled by a hydraulic manipulator (Narashige, MO10) was inserted into the pipette and used to load and inject the solution into 6 sites of left V1 (3.4–4.4 mm posterior to Bregma; 2.2–2.8 mm lateral from midline; ~0.5 mm distance between each injection site, 0.5 mm below pia). A cranial window made of a single glass coverslip (Fisher Scientific no. 1.5) was embedded in the craniotomy and sealed in place with dental acrylic. A titanium head-post was attached to the skull with cyanoacrylate glue and dental acrylic.
Dye administration in vivo
JF585–HaloTag ligand (23) was administered to mice 3–4 weeks after the cranial window installation and viral injection. Dye solution was prepared by first dissolving 100 nmol (76 μg) of 23 in 20 μL DMSO. After vortexing, 20 μL of a Pluronic F-127 solution (20% w/w in DMSO) was added and this stock solution was diluted into 100 μL or 200 μL sterile saline for IV (tail vein) or IP injection, respectively.
In vivo wide-field imaging and analysis
Mice were head-fixed and awake during the imaging period and were therefor habituated to experimental handling and head fixation starting 1-week post-surgery. During each habituation session, mice were head-fixed onto the sample stage with body restrained under a half-cylindrical cover. The habituation procedure was repeated 3–4 times for each animal for a duration of 15–60 minutes. For in vivo wide-field imaging, an external fluorescence light source (Leica EL6000, Leica) was used for excitation of GCaMP6s (green channel) and JF585–HaloTag ligand (red channel). Images were acquired via Leica Application Suite 4.5 (Leica). Wide-field images in green (1 second exposure) and red (4 second exposure) channels were acquired at multiple time intervals over two weeks under the same imaging conditions and the images were aligned with the Stackreg plugin in ImageJ. The mean values in the same area of red and green channels were plotted to track the labeling kinetics and turnover of JF585–HaloTag in vivo.
In vivo two-photon imaging and analysis
For in vivo two-photon imaging, GCaMP6s and JF585–HaloTag were excited at 940 nm and 1100 nm, respectively, using a femtosecond laser source (InSight DeepSee, Spectra-Physics), and imaged using an Olympus 25× 1.05 NA objective and a homebuilt two-photon microscope53. Images were acquired from 200 to 550 μm below the pia with post-objective power ranging between 20 and 60 mW. No photobleaching or photodamage of tissue was observed. Typical imaging settings were composed of 256 × 256 pixels, with 1.2 μm per pixel, and a ~3 Hz frame rate. The time-lapse calcium images of spontaneous neuronal activity in awake, head fixed mice were recorded and analyzed with custom programs written in MATLAB (Mathworks). Lateral motion present in head-fixed awake mice was corrected using a cross-correlation-based registration algorithm54, where cross-correlation was calculated to determine frame shift in x and y directions. Cortical neurons were outlined by hand as regions of interest (ROIs). The fluorescence time course of each ROI was used to calculate its calcium transient as ΔF/F (%) = (F−F0)/F0 × 100, with the baseline fluorescence F0 being the mode of the fluorescence intensity histogram of this ROI. For the Pearson correlation coefficient calculation, the JF585 (red channel) and GCaMP6s (green channel) fluorescence signals in each ROI were averages from 1000 imaging frames (3 Hz).
Statistics
For spectroscopy measurements (Fig. 1e,f, Fig. 2g,l, Table 1, and Supplementary Fig. 1) reported n values for absorption spectra, extinction coefficient (ε) and quantum yield (Φ) represent measurements of different samples prepared from the same dye DMSO stock solution. For the cell loading experiment (Fig. 1j) the following reported n values represent the number of intensity values measured from three fields of view for the time points at 30 s, 1 min, 2 min, 3 min, and 4 minutes, respectively: JF525–HaloTag ligand 13: n= 112, 120, 130, 114, 128; JF549– HaloTag ligand 14: n = 135, 129, 135, 158, 161. For the reference stain using JF646–HaloTag ligand n = 248. For cellular toxicity (Supplementary Fig. 2c) assays, reported n values represent different cell culture samples in separate microplate wells. For the contrast measurements (Fig. 2h,i,m,n, Supplementary Fig. 2l) reported n values represent intensity values measured from three fields of view for each dye type. For the IV and IP experiments (Fig. 3d), reported n = 3 values represent different fields of view taken via wide-field imaging. The one-way ANOVA analysis of spontaneous neuronal activity before and after dye administration (Fig. 3g) gave F (3, 236) = 0.1204.
Data availability
The data that support the findings of this study are provided in the Source Data files or available from the corresponding author upon request.
Supplementary Material
Acknowledgments
We thank A. Berro and E. Schreiter (Janelia) for purified HaloTag protein, and H. Choi (Janelia) for the Sec61β–HaloTag plasmid, contributive discussions, and a critical reading of the manuscript. This work was supported by the Howard Hughes Medical Institute.
Footnotes
Author Contributions
L.D.L. and J.B.G. conceived the project. J.B.G. contributed organic synthesis and 1-photon spectroscopy measurements. A.K.M. contributed organic synthesis and computational chemistry experiments. Y.J., R.L. and N.J. contributed mouse imaging experiments. T.A.B. contributed cultured cell imaging experiments. W.C.L. and P.J.K. contributed larval explant imaging experiments. R.P. and J.J.M contributed 2-photon spectroscopy measurements. L.D.L. contributed 1-photon spectroscopy measurements and wrote the paper with input from the other authors.
Competing Financial Interests Statement
The authors declare competing interests: J.B.G. and L.D.L. have filed patent applications whose value may be affected by this publication.
References
- 1.Lavis LD, Raines RT. Bright ideas for chemical biology. ACS Chem Biol. 2008;3:142–155. doi: 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lavis LD, Raines RT. Bright building blocks for chemical biology. ACS Chem Biol. 2014;9:855–866. doi: 10.1021/cb500078u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xue L, Karpenko IA, Hiblot J, Johnsson K. Imaging and manipulating proteins in live cells through covalent labeling. Nat Chem Biol. 2015;11:917–923. doi: 10.1038/nchembio.1959. [DOI] [PubMed] [Google Scholar]
- 4.Liu Z, Lavis LD, Betzig E. Imaging live-cell dynamics and structure at the single-molecule level. Mol Cell. 2015;58:644–659. doi: 10.1016/j.molcel.2015.02.033. [DOI] [PubMed] [Google Scholar]
- 5.Ceresole M. Verfahren zur Darstellung von Farbstoffen aus der Gruppe des Meta-amidophenolphtaleïns. 44002. Germany Patent. 1887
- 6.Beija M, Afonso CAM, Martinho JMG. Synthesis and applications of rhodamine derivatives as fluorescent probes. Chem Soc Rev. 2009;38:2410–2433. doi: 10.1039/b901612k. [DOI] [PubMed] [Google Scholar]
- 7.Panchuk-Voloshina N, et al. Alexa Dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J Histochem Cytochem. 1999;47:1179–1188. doi: 10.1177/002215549904700910. [DOI] [PubMed] [Google Scholar]
- 8.Arden-Jacob J, Frantzeskos J, Kemnitzer NU, Zilles A, Drexhage KH. New fluorescent markers for the red region. Spectrochim Acta, Part A. 2001;57:2271–2283. doi: 10.1016/s1386-1425(01)00476-0. [DOI] [PubMed] [Google Scholar]
- 9.Liu JX, et al. Rational design and synthesis of a novel class of highly fluorescent rhodamine dyes that have strong absorption at long wavelengths. Tetrahedron Lett. 2003;44:4355–4359. [Google Scholar]
- 10.Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T. Evolution of Group 14 rhodamines as platforms for near-infrared fluorescence probes utilizing photoinduced electron transfer. ACS Chem Biol. 2011;6:600–608. doi: 10.1021/cb1002416. [DOI] [PubMed] [Google Scholar]
- 11.Grimm JB, et al. Carbofluoresceins and carborhodamines as scaffolds for high-contrast fluorogenic probes. ACS Chem Biol. 2013;8:1303–1310. doi: 10.1021/cb4000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lukinavičius G, et al. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nature Chem. 2013;5:132–139. doi: 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- 13.Grimm JB, et al. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods. 2015;12:244–250. doi: 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lukinavicius G, et al. Fluorogenic probes for multicolor imaging in living cells. J Am Chem Soc. 2016;138:9365–9368. doi: 10.1021/jacs.6b04782. [DOI] [PubMed] [Google Scholar]
- 15.Lavis LD, Chao TY, Raines RT. Fluorogenic label for biomolecular imaging. ACS Chem Biol. 2006;1:252–260. doi: 10.1021/cb600132m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watkins RW, Lavis LD, Kung VM, Los GV, Raines RT. Fluorogenic affinity label for the facile, rapid imaging of proteins in live cells. Org Biomol Chem. 2009;7:3969–3975. doi: 10.1039/b907664f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wysocki LM, et al. Facile and general synthesis of photoactivatable xanthene dyes. Angew Chem, Int Ed. 2011;50:11206–11209. doi: 10.1002/anie.201104571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukinavicius G, et al. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat Methods. 2014;11:731–733. doi: 10.1038/nmeth.2972. [DOI] [PubMed] [Google Scholar]
- 19.Butkevich AN, et al. Fluorescent rhodamines and fluorogenic carbopyronines for super-resolution STED microscopy in living cells. Angew Chem Int Ed. 2016;55:3290–3294. doi: 10.1002/anie.201511018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grimm JB, et al. Synthesis of a far-red photoactivatable Si-rhodamine for super resolution microscopy. Angew Chem Int Ed. 2016;55:1723–1727. doi: 10.1002/anie.201509649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grimm JB, Lavis LD. Synthesis of rhodamines from fluoresceins using Pd-catalyzed C–N cross-coupling. Org Lett. 2011;13:6354–6357. doi: 10.1021/ol202618t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z, et al. 3D imaging of Sox2 enhancer clusters in embryonic stem cells. Elife. 2014;3:e04236. doi: 10.7554/eLife.04236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knight SC, et al. Dynamics of CRISPR-Cas9 genome interrogation in living cells. Science. 2015;350:823–826. doi: 10.1126/science.aac6572. [DOI] [PubMed] [Google Scholar]
- 24.Swinstead EE, et al. Steroid receptors reprogram FoxA1 occupancy through dynamic chromatin transitions. Cell. 2016;165:593–605. doi: 10.1016/j.cell.2016.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bisson-Filho AW, et al. Treadmilling by FtsZ filaments drives peptidoglycan synthesis and bacterial cell division. Science. 2017;355:739–743. doi: 10.1126/science.aak9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grimm JB, et al. Bright photoactivatable fluorophores for single-molecule imaging. Nat Methods. 2016;13:985–988. doi: 10.1038/nmeth.4034. [DOI] [PubMed] [Google Scholar]
- 27.Whitaker JE, et al. Fluorescent rhodol derivatives: Versatile, photostable labels and tracers. Anal Biochem. 1992;207:267–279. doi: 10.1016/0003-2697(92)90011-u. [DOI] [PubMed] [Google Scholar]
- 28.Mitronova GY, et al. New fluorinated rhodamines for optical microscopy and nanoscopy. Chem Eur J. 2010;16:4477–4488. doi: 10.1002/chem.200903272. [DOI] [PubMed] [Google Scholar]
- 29.Asanuma D, et al. Acidic-pH-activatable fluorescence probes for visualizing exocytosis dynamics. Angew Chem Int Ed. 2014;126:6199–6203. doi: 10.1002/anie.201402030. [DOI] [PubMed] [Google Scholar]
- 30.Hansch C, Leo A, Taft RW. A survey of Hammett substituent constants and resonance and field parameters. Chem Rev. 1991;91:165–195. [Google Scholar]
- 31.Hinckley DA, Seybold PG. A spectroscopic/thermodynamic study of the rhodamine B lactone–zwitterion equilibrium. Spectrochim Acta, Part A. 1988;44:1053–1059. [Google Scholar]
- 32.Los GV, et al. HaloTag: A novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 33.Legant WR, et al. High-density three-dimensional localization microscopy across large volumes. Nat Methods. 2016;13:359–365. doi: 10.1038/nmeth.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohyama T, et al. A multilevel multimodal circuit enhances action selection in Drosophila. Nature. 2015;520:633–639. doi: 10.1038/nature14297. [DOI] [PubMed] [Google Scholar]
- 35.Lemon WC, et al. Whole-central nervous system functional imaging in larval Drosophila. Nat Commun. 2015;6:7924. doi: 10.1038/ncomms8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou X, Lai R, Beck JR, Li H, Stains CI. Nebraska Red: A phosphinate-based near-infrared fluorophore scaffold for chemical biology applications. Chem Commun. 2016;52:12290–12293. doi: 10.1039/c6cc05717a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bruchez MP. Dark dyes–bright complexes: Fluorogenic protein labeling. Curr Opin Chem Biol. 2015;27:18–23. doi: 10.1016/j.cbpa.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Altman RB, et al. Cyanine fluorophore derivatives with enhanced photostability. Nat Methods. 2012;9:68–71. doi: 10.1038/nmeth.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palonpon AF, Sodeoka M, Fujita K. Molecular imaging of live cells by Raman microscopy. Curr Op Chem Biol. 2013;17:708–715. doi: 10.1016/j.cbpa.2013.05.021. [DOI] [PubMed] [Google Scholar]
Methods-Only References
- 41.Critchfield FE, Gibson JA, Jr, Hall JL. Dielectric constant for the dioxane–water system from 20 to 35°. J Am Chem Soc. 1953;75:1991–1992. [Google Scholar]
- 42.Suzuki K, et al. Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector. Phys Chem Chem Phys. 2009;11:9850–9860. doi: 10.1039/b912178a. [DOI] [PubMed] [Google Scholar]
- 43.Frisch MJ, et al. Gaussian 09, revision D. 01. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 44.Dreuw A, Weisman JL, Head-Gordon M. Long-range charge-transfer excited states in time-dependent density functional theory require non-local exchange. J Chem Phys. 2003;119:2943–2946. [Google Scholar]
- 45.Jacquemin D, et al. Assessment of the efficiency of long-range corrected functionals for some properties of large compounds. J Chem Phys. 2007;126:144105. doi: 10.1063/1.2715573. [DOI] [PubMed] [Google Scholar]
- 46.Guthmuller J, Champagne B. Resonance Raman scattering of rhodamine 6G as calculated by time-dependent density functional theory: vibronic and solvent effects. J Phys Chem A. 2008;112:3215–3223. doi: 10.1021/jp7112279. [DOI] [PubMed] [Google Scholar]
- 47.Setiawan D, Kazaryan A, Martoprawiro MA, Filatov M. A first principles study of fluorescence quenching in rhodamine B dimers: How can quenching occur in dimeric species? Phys Chem Chem Phys. 2010;12:11238–11244. doi: 10.1039/c004573j. [DOI] [PubMed] [Google Scholar]
- 48.Mütze J, et al. Excitation spectra and brightness optimization of two-photon excited probes. Biophys J. 2012;102:934–944. doi: 10.1016/j.bpj.2011.12.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akerboom J, et al. Optimization of a GCaMP calcium indicator for neural activity imaging. J Neurosci. 2012;32:13819–13840. doi: 10.1523/JNEUROSCI.2601-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schindelin J, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 52.Kohl J, et al. Ultrafast tissue staining with chemical tags. Proc Natl Acad Sci U S A. 2014;111:E3805–3814. doi: 10.1073/pnas.1411087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ji N, Milkie DE, Betzig E. Adaptive optics via pupil segmentation for high-resolution imaging in biological tissues. Nat Methods. 2010;7:141–147. doi: 10.1038/nmeth.1411. [DOI] [PubMed] [Google Scholar]
- 54.Sun W, Tan Z, Mensh BD, Ji N. Thalamus provides layer 4 of primary visual cortex with orientation- and direction-tuned inputs. Nat Neurosci. 2016;19:308–315. doi: 10.1038/nn.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are provided in the Source Data files or available from the corresponding author upon request.