

Abstract
Background
12N-benzyl matrinic acid analogues had been identified to be a novel scaffold of anti-HCV agents with a specific mechanism, and the representative compound 1 demonstrated a moderate anti-HCV activity. The intensive structure–activity relationship of this kind of compounds is explored so as to obtain anti-HCV candidates with good druglike nature.
Results
Taking compound 1 as the lead, 32 compounds (of which 27 were novel) with diverse structures on the 11-side chain, including methyl matrinate, matrinol, matrinic butane, (Z)-methyl Δβγ-matrinic crotonate derivatives were synthesized and evaluated for their anti-HCV activities. Among all the compounds, matrinol 7a demonstrated potential potency with a greatly improved SI value of 136. Pharmacokinetic studies of 7a showed the potential for oral administration that would allow further in vivo safety studies. The free hydroxyl arm in 7a made it possible to prepare pro-drugs for the potential in the treatment of HCV infection.
Conclusions
27 novel 12N-substituted matrinol derivatives were prepared. The SAR study indicated that the introduction of electron-donating substitutions on the benzene ring was helpful for the anti-HCV activity, and the unsaturated 11-side chain might not be favorable for the activity. This study provided powerful information on further strategic optimization and development of this kind of compounds into a novel family of anti-HCV agents.
Graphical abstract.
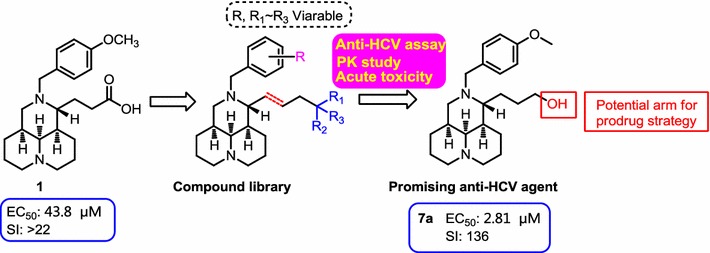
Matrinol derivatives as a class of novel anti-HCV agents
Keywords: Matrinol, Hepatitis C virus, Structure–activity relationship, Druglike
Background
Currently, at least 130–150 million people worldwide have been infected with hepatitis C virus (HCV) [1]. Each year, 3–4 million people are newly infected and HCV-related liver complications kill estimated 700,000 people annually [1, 2]. In recent years, new direct acting antivirals (DAAs) specifically targeting HCV proteins have made a great breakthrough to HCV treatment, and NS3/4A HCV protease inhibitors telaprevir, boceprevir and simeprevir, NS5A inhibitors asunaprevir and ledipasvir, NS5B polymerase inhibitors sofosbuvir and dasabuvir have been approved by FDA for the HCV treatment successively since 2011 [3]. To deal with the springing up of drug resistance challenges [4–6], multiple of DAA combinations have been developed [7–9]. Therefore, it is still imperative to develop new anti-HCV agents with novel structure skeleton or mechanism of action as a new component to DAA combination.
In our earlier studies, 12N-benzyl matrinic acid analogues had been successfully identified to be a novel class of anti-HCV agents from matrine, a natural product extracted from traditional Chinese herb. The representative compound, 12N-4-methoxylbenzyl matrinic acid (1, Fig. 1) was identified to be active against HCV with a novel mechanism targeting on host protein Hsc70 and demonstrated a moderate anti-HCV activity with SI over 22 [10, 11]. The special tricyclic flexible scaffold and appealing druglike of compound 1 strongly provoked our interesting to continuously explore the structure–activity relationship (SAR) of this kind of compounds, in an effort to discover novel anti-HCV candidates which could be used in the combination with current DAA.
Fig. 1.
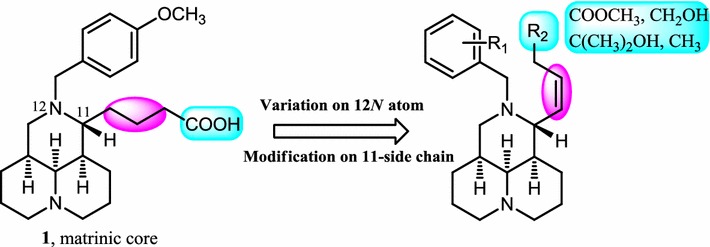
Modification sites based on compound 1
In the present study, as illustrated in Fig. 1, taking 1 as the lead, SAR studies were further conducted with the variations of the 11-side chain and diverse substituents on 12N-atom. Therefore, series of novel methyl matrinate, matrinol, matrinic butane, 1′, 1′-dialkyl matrinol, methyl (Z)-Δβγ-matrinic crotonate and (Z)-Δβγ-matrinic crotonl derivatives were designed, synthesized and evaluated for their in vitro anti-HCV activities as well as the in vivo pharmacokinetic (PK) and safety profile of the representative compounds.
Results and discussion
Chemistry
As displayed in Schemes 1 and 2, all the target compounds were synthesized using commercially available matrine or lehmannine with purity over 98% as the starting material. As shown in Scheme 1, following the procedure of preparing compound 2 [12], the rest methyl matrinates 6a–f were obtained from matrine through a three-step sequence including basic hydrolytic ring-opening, methyl esterification, 12N-substitution via substituted benzyl halides or benzaldehydes with good yields of 44–68% [13–17]. Similar to the preparation of 3a [12], the rest matrinols 7a–i were obtained by the LiAlH4 reduction of the corresponding methyl matrinate 6 as described in Scheme 1 with yields of 75–85%. The matrinic butane product 9 was achieved through hydroxyl sulfonylation, reductive-elimination of OTs by LiAlH4 from 7a in a yield of 56% and the alkylation of 6a–b and 6d–f with Grignard reagents afforded the 1′,1′-dialkyl substituted matrinols 10a–e in yields of 60–75% [12].
Scheme 1.
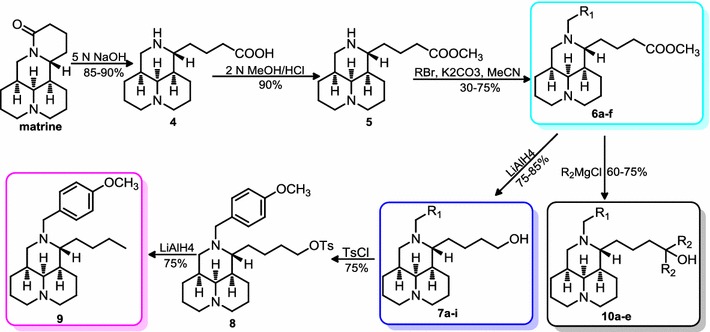
Synthetic procedures of methyl matrinate and matrinol derivatives. Reagents and conditions: (a) 5 N NaOH, reflux, 9 h, 6 N HCl, pH = 5–6; (b) 2 N MeOH/HCl, reflux, 2 h; (c) RBr, K2CO3, MeCN, r.t., overnight; (d) LiAlH4, THF, r.t., 30 min; (e) R2MgCl, THF, 0–25 °C, reflux, 2 h; (f) TsCl, CH2Cl2, TEA, 4-DMAP; (g) alkylmagnesium chloride, THF, reflux, 2 h
Scheme 2.
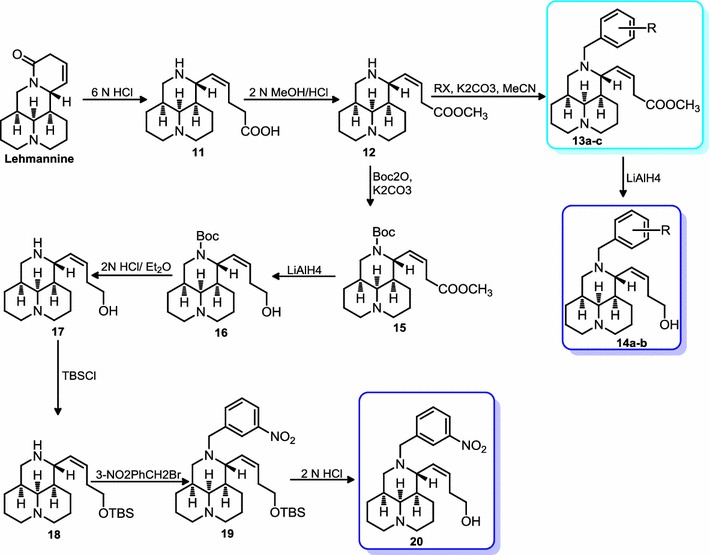
Synthetic procedures of methyl (Z)-Δβγ-matrinic crotonate and (Z)-Δβγ-matrinic crotonol derivatives. Reagents and conditions: (a) 6 N HCl, reflux, 9 h; (b) 2 N MeOH/HCl, reflux, 2 h; (c) RX, K2CO3, MeCN, r.t., overnight; (d) LiAlH4, THF, r.t., 30 min; (e) Boc2O, K2CO3, CH2Cl2, r.t., overnight; (f) 2 N HCl/Et2O, 30 min, (g) TBSCl, CH2Cl2, imidazole, r.t., overnight; (h) 3-NO2PhCH2Br, TEA, CH2Cl2, r.t., 4 h; (i) 2 N HCl
As depicted in Scheme 2, methyl (Z)-Δβγ-matrinic crotonate derivatives (13a–c) were obtained from lehmannine following the similar sequence including acidic hydrolytic ring-opening, methyl esterification, 12N-substitution with overall yields of 30–35% [18]. The targeted (Z)-Δβγ-matrinic crotonol derivatives (14a–b) were gained from a LiAlH4 reduction of 13a–b in 70–80% yields. Another nitro substituted crotonol derivative 20 was obtained from compound 12 via a six-step procedure, including 12N-tert-butoxycarbonyl (Boc) protection, ester reduction by LiAlH4, de-protection of Boc, silicane protection, 12N-substitution and deprotection with an overall yield of 30% [14, 15].
Anti-HCV activity and SAR analysis of matrinol derivatives
All the target compounds were evaluated for their anti-HCV activities (EC50) and cytotoxicities (CC50) in human Huh7.5 cells using specific real-time RT-PCR assay, as described earlier [11]. As an important indicator, the selectivity index (SI) was calculated as a ratio of CC50 to EC50. Anti-HCV ability of a given compound was estimated by combining its EC50 with SI values. Totally 32 compounds were gathered, and their structures and anti-HCV effects were shown in Table 1.
Table 1.
SAR of all the targeted compounds for anti-HCV activity in Huh7.5 cells
| |||||
|---|---|---|---|---|---|
| Code | R1 | R2 | CC50 (μM)a | EC50 (μM)b | SI |
| 1 | C6H4OCH3-4 | COOH | > 1000 | 43.8 ± 3.81 | > 22.8 |
| 2 | C6H4F-4 | COOCH3 | 274 ± 8.35 | 2.09 ± 1.65 | 131 |
| 3a | C6H4F-4 | CH2OH | 285 ± 7.45 | 2.99 ± 1.51 | 95.3 |
| 3b | CH2CH2CH3 | CH2OH | > 500 | 16.4 ± 10.9 | > 30.5 |
| 3c | CH2(CH2)5CH3 | CH2OH | 33.3 ± 6.49 | 1.01 ± 0.55 | 32.9 |
| 3d | CH2(CH2)6CH3 | CH2OH | 17.8 ± 2.44 | 0.80 ± 0.26 | 22.3 |
| 6a | C6H4OCH3-4 | COOCH3 | 414 ± 7.34 | 7.01 ± 1.51 | 59.1 |
| 6b | C6H4CH3-4 | COOCH3 | 132.9 ± 7.53 | 1.73 ± 1.36 | 76.8 |
| 6c | C6H4CH = CH2-4 | COOCH3 | 81.2 ± 12.9 | 1.61 ± 0.76 | 50.4 |
| 6d | C6H3F2-2,4 | COOCH3 | 261 ± 28.7 | 1.69 ± 1.47 | 154 |
| 6e | C6H4NO2-4 | COOCH3 | 235 ± 48.2 | 14.2 ± 3.13 | 16.5 |
| 6f | pyridyl-4 | COOCH3 | > 500 | 4.66 ± 0.35 | > 107 |
| 7a | C6H4OCH3-4 | CH2OH | 383 ± 30.2 | 2.81 ± 0.82 | 136 |
| 7b | C6H4CH3-4 | CH2OH | 252 ± 3.04 | < 2.06 | > 122 |
| 7c | C6H4CH = CH2-4 | CH2OH | 143 ± 32.3 | 3.16 ± 0.51 | 45.3 |
| 7d | C6H3F2-2,4 | CH2OH | 266 ± 9.47 | < 2.06 | > 129 |
| 7e | Pyrid-4-yl | CH2OH | > 500 | 9.03 ± 5.58 | > 55.4 |
| 7f | Pyrid-3-yl | CH2OH | > 500 | 4.39 ± 3.01 | > 114 |
| 7 g | pyrid-2-ylCl-5 | CH2OH | > 500 | 10.9 ± 5.59 | > 45.9 |
| 7 h | CONHC6H4 | CH2OH | > 500 | 46.7 ± 41.0 | > 10.7 |
| 7i | CONHC6H3CF3-4 | CH2OH | 86.2 ± 4.33 | 4.19 ± 1.44 | 20.6 |
| 9 | 41.9 ± 1.10 | 1.16 ± 0.25 | 36.1 | ||
| 10a | C6H4OCH3-4 | CH3 | > 140 ± 16.6 | 11.7 ± 0.06 | 12.0 |
| 10b | C6H4CH3-4 | CH3 | 55.7 ± 7.50 | 0.82 ± 0.26 | 67.9 |
| 10c | C6H4CH3-4 | C2H5 | 12.3 ± 4.23 | < 0.23 | > 53.5 |
| 10d | C6H3F2-2,4 | CH3 | 156 ± 37.9 | 6.55 ± 2.69 | 23.8 |
| 10e | Pyrid-4-yl | CH3 | > 500 | 80.4 ± 24.3 | 6.22 |
| 13a | 4-OCH3 | COOCH3 | 326 ± 21.0 | 16.7 ± 4.71 | 19.5 |
| 13b | 4-F | COOCH3 | 260 ± 84.8 | 18.1 ± 6.49 | 14.4 |
| 13c | 3-NO2 | COOCH3 | 402 ± 83.6 | 40.4 ± 3.64 | 10.0 |
| 14a | 4-OCH3 | CH2OH | 214 ± 95.0 | 8.80 ± 2.15 | 24.3 |
| 14b | 4-F | CH2OH | 156 ± 65.8 | 13.6 ± 2.63 | 11.5 |
| 20 | 3-NO2 | CH2OH | 312 ± 31.1 | 18.4 ± 1.64 | 17.0 |
| Tela | 47.6 ± 0.61 | 0.02 ± 0.02 | 1950 | ||
Tela telaprevir
aCytotoxic concentration required to inhibit Huh7.5 cell growth by 50%
bConcentration required to inhibit HCV growth by 50%
SAR investigation was initiated with the variation of carboxylic acid group, by which 7 methyl matrinates (2, 6a–f) and 13 matrinols (3a–d and 7a–i) were generated. As depicted in Table 1, except 4-nitrobenzyl derivative 6e, all methyl 12N-benzyl/pyridylmethyl substituted matrinates exerted higher activities than the lead 1 by showing lower EC50 values and higher SI values of over 50. In particular, 12N-4-fluorobenzyl 2, 4-methylbenzyl 6b, 4-vinylbenzyl 6c and 2,4-difluorobenzyl 6d displayed potent anti-HCV activities with EC50 values ranging from 1.61 to 2.09 µM, which were over 20 times more potent than that of 1. It appeared that the electron-donating substitutions on the benzene ring were more favorable than the electron-withdrawing groups in the methyl matrinate series.
Besides the substitutions mentioned above (2, 6a–d, 6f), 7 other substituents including long chain alkyl groups (3b–d), as well as pyridin-3-ylmethyl (7f), 5-chloropyridin-2-ylmethyl (7g), 2-oxo-2-(phenylamino)ethyl (7h), 2-oxo-2-((4- (trifluoromethyl) phenyl)amino)ethyl (7i) were also introduced on the 12N atom to generate the library of matrinols. As anticipated, most of the 12N-benzyl/pyridyl substituted matrinols (3a and 7a–g) gave inspiring anti-HCV activities with EC50 values in the range of 2.06–10.9 μM, and SI values in the range of 45–136. In particular, compounds 7a, 7b and 7d bearing electron-donating methoxy, methyl and 2,4-difluoro substitutions respectively gave excellent activities with EC50 values of less than 2.81 µM as well as SI values of over 122. However, alkyl (3b–d) or phenylamino carbonyl methyl compounds (7h–i) did not give favorable activities because of their either low activity or high cytotoxicity. It indicated again the favorability of electron-donating substitutions on the benzene ring to the anti-HCV activity.
Then, SAR investigation was focused on the influence of the structural type of the 11-side chain while the 12N-benzyl/pyridylmethyl substitution was retained. In the first round, matrinic butane (9), five 1′, 1′-dialkyl substituted matrinols (10a–e) were designed and synthesized. Among them, benzyl derived analogues (9, 10a–d) exhibited promising anti-HCV activities with low micro molar EC50 values ranging from 0.23 to 11.70 μM, as well as limited toxicity with CC50 between 12.3 and 155.8 µM, while the 12N-pyrid-4-ylmethyl derivative 10e showed a high EC50 value of 80.38 µM. The results indicated that 11-butane or 1′,1′-dialkyl butanol chain might not be helpful for the activity.
In the second round, to further examine the influence of saturation of 11-side chain on the activity, double bond was introduced to the β,γ position of the butyl acid chain, and the corresponding methyl Δβγ-matrinic crotonates (13a–c) and crotonyl alcohols (14a–b and 20) with 4-methoxyl, 4-fluoro, 4-nitrobenzyl substitution on the 12N atom were generated respectively. As described in Table 1, most compounds afforded very weak potencies with SI values between 10.0–24.3, inferring that the unsaturated side-chain might not be favorable for the HCV activity.
PK study
Based on above, methyl matrinates and matrinols exhibited the most potent anti-HCV activities, however, methyl matrinates might not possess favorable PK profiles in vivo owing to the exposed metabolically labile ester group. Therefore, two representative matrinols 7a and 7b were chosen to examine their PK parameters in SD rats at the single dosage of 25 mg kg−1 via oral route. As indicated in Table 2 and Fig. 2, both of them showed acceptable PK profiles with the areas under the curve (AUCs) of 1.58 and 2.36 μM·h and the half-times of 4.69 h and 3.39 h respectively, indicating reasonable stabilities in vivo. Meanwhile, the results demonstrated that the concentration of compounds 7a and 7b showed a significant difference at 2 h, owing to different dissolution rate at that time in vivo.
Table 2.
PK parameters of the key compoundsa
| Code | Tmax (h) | Cmax (μM) | AUC 0–t (μM h) | AUC 0–∞ (μM h) | MRT (h) | t1/2 (h) |
|---|---|---|---|---|---|---|
| 7a | 0.42 | 0.62 | 1.55 | 1.58 | 3.55 | 4.69 |
| 7b | 1.00 | 0.79 | 2.32 | 2.36 | 4.47 | 3.39 |
aPK parameters were calculated in rats after single oral dosing of 25 mg kg−1, (n = 3) by non-compartmental analysis using WinNonlin, version 5.3
Fig. 2.
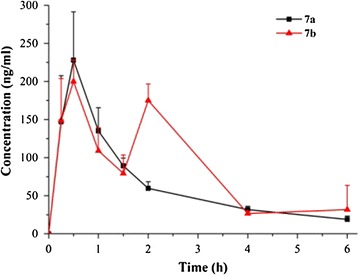
Mean plasma concentration-time profiles of the key compounds (25 mg kg−1, orally)
Acute toxicity study
The acute toxicity tests of 7a and 7b were performed in Kunming mice. Each compound was given orally in a single-dosing experiment at 250, 500, 750 or 1000 mg kg−1, respectively. The mice were closely monitored for 7 days. As indicated in Table 3, the LD50 values for 7a and 7b were 708 and 392 mg kg−1, respectively, therefore, 7a seemed to be more promising as a parent drug from a safety prospective.
Table 3.
Acute toxicity of the key compounds
| Code | 7a | 7b |
|---|---|---|
| LD50 (mg kg−1) | 708 | 392 |
Experimental
Instruments
Unless otherwise noted, all commercial reagents and solvents were obtained from the commercial provider and used without further purification. Melting points (mp) were obtained with CXM-300 melting point apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance 400 (400/101 MHz for 1H/13C) spectrometer or Bruker Avance III 500 (500/126 MHz for 1H/13C) spectrometer (Varian, San Francisco, USA) respectively, in DMSO-d6 with Me4Si as the internal standard. ESI high-resolution mass spectra (HRMS) were recorded on an AutospecUitima-TOF spectrometer (Micromass UK Ltd., Manchester, U.K.). Flash chromatography was performed on Combiflash Rf 200 (Teledyne, Nebraska, USA).
General procedures for methyl 12N-substituted matrinate derivatives 6a–f
Matrine (5.0 g, 20.0 mmol) was added to 5 N NaOH in water (30 mL), and the reaction mixture was refluxed for 9 h, cooled in an ice bath and then acidified with HCl (2 N) to pH 6–7. The solvent was removed in vacuo and the residue was dissolved with 2 N HCl in methanol and then heated at refluxing for 2 h. The solvent methanol was removed under reduced pressure to give crude 5 (5.5 g, yield 77%), which was applied directly in the next step without further purification.
To a stirred solution of 5 (10.0 mmol) and K2CO3 (35.0 mmol) in chloroethane (50 mL), the substituted benzyl halide (10 mmol) was added. The reaction mixture was stirred at room temperature for 5–8 h until TLC analysis showed completion of the reaction. Water (20 mL) was added to the mixture and the organic phase was separated and dried with anhydrous Na2SO4, concentrated, and the gained residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluent to afford the title compounds.
Methyl 12N-(4-methoxybenzyl)matrinate dihydrochloride (6a)
The title compound was prepared from 5 and 4-methoxybenzyl bromide in the same manner as described above followed by an acidification with 2 N hydrochloride/ether (10 mL). Yield: 61%; white solid; mp 208–209 °C; 1H NMR (500 MHz) δ 11.42 (br, 1H), 11.06 (br, 1H), 7.53 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 4.93–4.89 (m, 1H), 4.22–4.18 (m, 1H), 4.00–3.88 (m, 2H), 3.79 (s, 3H), 3.61 (s, 3H), 3.58 (d, J = 10.4 Hz, 1H), 3.30–3.24 (m, 2H), 2.99–2.87 (m, 2H), 2.68–2.65 (m, 1H), 2.60–2.54 (m, 1H), 2.49–2.46 (m, 3H), 2.05–1.98 (m, 2H), 1.94–1.88 (m, 1H), 1.82–1.58 (m, 8H), 1.47 (d, J = 13.7 Hz, 1H); 13C NMR (126 MHz) δ 173.7, 160.3, 133.4 (2), 122.0, 114.6 (2), 60.7, 60.6, 57.2, 55.7, 54.7, 54.6, 51.8, 48.8, 36.3, 32.9, 30.4, 28.0, 24.5, 23.9, 21.8, 18.3 (2). HRMS: calcd for C24H37O3N2·2HCl [M−2HCl+H]+: 401.2799, found: 401.2790.
Methyl 12N-(4-methylbenzyl)matrinate (6b)
The title compound was prepared from 5 and 4-methylbenzyl bromide in the same manner as described in the general procedures. Yield: 67%; white solid; mp 89–91 °C; 1H NMR (500 MHz) δ 7.17 (d, J = 7.4 Hz, 2H), 7.10 (d, J = 7.4 Hz, 2H), 3.96 (d, J = 13.0 Hz, 1H), 3.55 (s, 3H), 3.01 (d, J = 12.9 Hz, 1H), 2.79 (s, 1H), 2.72 (d, J = 8.6 Hz, 1H), 2.65 (d, J = 9.1 Hz, 1H), 2.55 (d, J = 11.5 Hz, 1H), 2.30 (d, J = 6.3 Hz, 1H), 2.27 (s, 3H), 2.18 (d, J = 8.5 Hz, 1H), 1.96 (s, 1H), 1.85–1.24 (m, 17H); 13C NMR (126 MHz) δ 174.0, 137.6, 135.8, 129.2 (2), 128.9 (2), 64.3, 57.4, 57.1, 56.9, 55.4, 52.0, 51.6 (2), 37.6, 33.8, 28.4, 27.8, 27.4, 21.5, 21.2 (2), 19.0. HRMS: calcd for C24H37O2N2 [M+H]+: 385.2850, found: 385.2844.
Methyl 12N-(4-vinylbenzyl)matrinate (6c)
The title compound was prepared from 5 and 4-vinylbenzyl chloride in the same manner as 6b. Yield: 70%; white solid; mp 75–77 °C. 1H NMR (500 MHz) δ 7.40 (d, J = 7.9 Hz, 2H), 7.27 (d, J = 7.9 Hz, 2H), 6.80–6.68 (m, 1H), 5.79 (d, J = 17.7 Hz, 1H), 5.21 (d, J = 11.1 Hz, 1H), 3.99 (d, J = 13.7 Hz, 1H), 3.54 (s, 3H), 3.10–3.01 (m, 1H), 2.88–2.78 (m, 1H), 2.78–2.67 (m, 1H), 2.70–2.60 (m, 1H), 2.63–2.56 (m, 1H), 2.29 (t, J = 6.7 Hz, 2H), 2.18 (d, J = 8.5 Hz, 1H), 1.96 (s, 1H), 1.85–1.52 (m, 11H), 1.36–1.24 (m, 5H); 13C NMR (126 MHz) δ 174.0, 140.7, 137.0, 135.9, 129.1 (2), 126.4 (2), 114.0, 64.3, 57.4, 57.1, 56.9, 55.4, 52.2, 51.6 (2), 37.6, 33.7, 28.3, 27.9, 27.4, 21.5, 21.2, 19.0. HRMS: calcd for C25H37O2N2 [M+H]+: 397.2850, found: 397.2838.
Methyl 12N-(2,4-difluorobenzyl)matrinate (6d)
The title compound was prepared from 5 and 2,4-difluorobenzyl bromide in the same manner as 6b. Yield: 73%; white solid; mp 68–70 °C; 1H NMR (500 MHz) δ 7.48–7.43 (m, 1H), 7.18–7.14 (m, 1H), 7.08–7.04 (m, 1H), 3.95 (d, J = 13.8 Hz, 1H), 3.55 (s, 3H), 3.15 (d, J = 13.7 Hz, 1H), 2.85–2.83 (m, 1H), 2.72 (d, J = 10.7 Hz, 1H), 2.67–2.61 (m, 2H), 2.31–2.28 (m, 2H), 2.23–2.05 (m, 1H), 1.96 (s, 1H), 1.85–1.73 (m, 4H), 1.67–1.47 (m, 7H), 1.37–1.25 (m, 5H); 13C NMR (126 MHz) δ 173.9, 162.3, 160.3, 132.4, 123.4, 111.7, 103.9, 64.2, 57.3, 57.1 (2), 52.1, 51.6, 47.9, 37.6, 33.7, 33.6, 28.3, 27.9, 27.3, 21.5, 21.2, 19.1. HRMS: calcd for C23H33O2N2 F [M+H]+: 407.2505, found: 407.2488.
Methyl 12N-(4-nitrobenzyl)matrinate dihydrochloride (6e)
The title compound was prepared from 5 and 4-nitrobenzyl bromide in the same manner as 6a. Yield: 75%; white solid; mp 215–217 °C; 1H NMR (500 MHz) δ 11.87 (br, 1H), 11.07 (br, 1H), 8.52–8.52 (m, 1H), 8.32–8.30 (m, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.77 (t, J = 8.0 Hz, 1H), 5.10 (d, J = 11.7 Hz, 1H), 4.27–4.23 (m, 1H), 4.22–4.16 (m, 1H), 4.00–3.93 (m, 1H), 3.61 (s, 3H), 3.60–3.56 (m, 1H), 3.35–3.10 (m, 2H), 3.00–2.87 (m, 2H), 2.82–2.77 (m, 1H), 2.61–2.57 (m, 1H), 2.53–2.37 (m, 2H), 2.12–1.51 (m, 13H); 13C NMR (126 MHz) δ 173.7, 148.3, 138.7, 132.2, 130.7, 126.9, 124.8, 60.8, 60.6, 56.6, 54.6, 51.8 (2), 49.2, 36.4, 32.9, 30.5, 28.0, 24.3, 23.9, 21.9, 18.3, 18.3. HRMS: calcd for C23H34O4N3·2HCl [M−2HCl+H]+: 416.2544, found: 416.2539.
Methyl 12N-(pyridin-4-ylmethyl)matrinate (6f)
The title compound was prepared from 5 and 4-(chloromethyl)pyridine in the same manner as 6b. Yield: 45%; white solid; mp 84–86 °C; 1H NMR (500 MHz) δ 8.49–8.48 (m, 2H), 7.32 (d, J = 5.9 Hz, 2H), 4.02 (d, J = 14.7 Hz, 1H), 3.53 (s, 3H), 3.15 (d, J = 14.7 Hz, 1H), 2.90–2.87 (m, 1H), 2.74–2.64 (m, 3H), 2.29–2.26 (m, 2H), 2.25–2.08 (m, 1H), 1.98 (s, 1H), 1.85–1.76 (m, 4H), 1.65–1.25 (m, 12H); 13C NMR (126 MHz) δ 173.9, 150.3, 149.9 (2), 123.9 (2), 64.2, 57.3, 57.1, 56.7, 54.3, 52.6, 51.6, 37.6, 33.6, 33.5, 28.2, 27.8, 27.4, 21.5, 21.2, 19.0. HRMS: calcd for C22H34O2N3 [M+H]+: 372.2646, found: 372.2635.
General procedures for 12N-substituted matrinol derivativess 7a–e
A solution of LiAlH4 (12 mmol) in anhydrous THF (20 mL) was added to the solution of compound 6 (10 mmol) in anhydrous THF (3 mL) in an ice bath, the mixture solution was then stirred at room temperature for 30 min before the reaction was quenched with acetone. Saturated ammonium chloride (2 mL) was then added and the mixture was stirred for 30 min, and the precipitation was filtered off. The solvent was evaporated, and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluent or followed by an acidification with 2 N hydrochloride/ether (10 mL) to afford target compounds.
12N-(4-Methoxybenzyl)matrinol dihydrochloride (7a)
The title compound was prepared from 6a as described above. Yield: 82%; white solid; mp 241–243 °C; 1H NMR (400 MHz) δ 11.04 (br, 1H), 10.99 (br, 1H), 7.52 (d, J = 8.7 Hz, 2H), 7.02 (d, J = 8.7 Hz, 2H), 4.82 (d, J = 11.2 Hz, 1H), 4.36 (s, 4H), 4.21–4.11 (m, 1H), 4.03–3.87 (m, 2H), 3.79 (s, 3H), 3.55 (d, J = 10.2 Hz, 1H), 3.27 (t, J = 13.0 Hz, 2H), 3.00–2.84 (m, 2H), 2.73–2.63 (m, 1H), 2.41 (d, J = 11.2 Hz, 1H), 1.92 (d, J = 9.3 Hz, 2H), 1.87–1.75 (m, 3H), 1.75–1.64 (m, 3H), 1.65–1.56 (m, 2H), 1.52 (s, 4H); 13C NMR (101 MHz) δ 159.9, 132.9 (2), 121.6 (2), 114.2, 60.4, 60.3, 60.1, 57.0, 55.2, 54.2, 54.2, 48.4, 36.1, 31.8, 30.0, 28.2, 24.1, 23.6, 22.7, 17.9, 17.8. HRMS: calcd for C23H37O2N2 ·2HCl [M−2HCl+H]+: 373.2850, found: 373.2848.
12N-(4-Methylbenzyl)matrinol dihydrochloride (7b)
The title compound was prepared from 6b as described above. Yield: 85%; white solid; mp 111–113 °C; 1H NMR (400 MHz) δ 11.20 (s, 1H), 11.06 (s, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.26 (d, J = 8.0 Hz, 2H), 4.85–4.80 (m 1H), 4.21–4.14 (m, 1H), 3.99–3.89 (m, 2H), 3.55 (d, J = 10.0 Hz, 1H), 3.26 (t, J = 13.6 Hz, 2H), 3.16 (s, 1H), 2.95 (m, 2H), 2.67–2.62 (m, 1H), 2.55 (m, 1H), 2.47–2.42 (m, 1H), 2.33 (s, 3H), 1.92–1.41 (m, 16H); 13C NMR (101 MHz) δ 138.8, 131.4 (2), 129.4 (2), 126.8, 60.6, 60.2, 60.1, 57.2, 54.2, 54.2, 48.6, 36.1, 31.8, 30.0, 28.2, 24.1, 23.6, 22.7, 20.8, 17.9, 17.8. HRMS: calcd for C23H37ON2·2HCl [M−2HCl+H]+: 357.2900, found: 357.2898.
12N-(4-Vinylbenzyl)matrinol dihydrochloride (7c)
The title compound was prepared from 6c as described above. Yield: 75%; white solid; mp 121–123 °C; 1H NMR (400 MHz) δ 11.02 (br, 2H), 7.92–7.50 (m, 4H), 6.78 (dd, J = 17.6, 10.8 Hz, 1H), 5.93 (d, J = 17.6 Hz, 1H), 5.34 (d, J = 11.2 Hz, 1H), 4.87 (d, J = 11.6 Hz, 1H), 3.54 (d, J = 10.0 Hz, 1H), 3.45 (m, 2H), 3.33–3.20 (m, 2H), 3.16 (s, 1H), 3.08–2.81 (m, 3H), 2.78–2.57 (m, 2H), 2.45–2.40 (m, 1H), 2.11–1.33 (m, 16H); 13C NMR (101 MHz) δ 138.0, 136.0, 131.7 (2), 129.3, 126.4 (2), 115.6, 60.6, 60.2, 60.1, 57.2, 54.2, 48.7, 36.1, 31.8, 30.0, 28.2, 24.0, 23.6, 22.7, 18.6, 17.9, 17.8. HRMS: calcd for C24H37ON2·2HCl [M−2HCl+H]+: 369.2900, found: 369.2900.
12N-(2,4-Difluorobenzyl)matrinol dihydrochloride (7d)
The title compound was prepared from 6d as described above. Yield: 80%; white solid, mp 124–126 °C; 1H NMR (400 MHz) δ 11.13 (br, 1H), 10.87 (br, 1H), 7.92–7.80 (m, 1H),7.45–7.38 (m, 1H), 7.28–7.18 (m, 1H), 4.77 (d, J = 13.0 Hz, 1H), 4.28–4.08 (m, 2H), 4.08–3.92 (m, 2H), 3.54 (d, J = 10.2 Hz, 2H), 3.28 (t, J = 12.0 Hz, 2H), 2.99–2.87 (m, 4H), 2.42–2.38 (m, 1H), 2.02–1.87 (m, 3H), 1.87–1.83 (m, 2H), 1.79–1.68 (m, 3H), 1.68–1.58 (m, 3H), 1.52 (s, 4H); 13C NMR (101 MHz) δ 135.5, 132.6, 113.4, 112.1, 111.9, 104.4, 60.4, 60.1, 60.1, 54.2, 54.2, 49.8, 48.7, 36.0, 31.8, 30.0, 28.1, 23.9, 23.7, 22.4, 17.9, 17.8. HRMS: calcd for C22H33ON2F2·2HCl [M−2HCl+H]+: 379.2556, found: 379.2551.
12N-(Pyridin-4-ylmethyl)matrinol dihydrochloride (7e)
The title compound was prepared from 6f as described above. Yield: 77%; white solid; mp 205–206 °C; 1H NMR (400 MHz) δ 12.40 (br, 1H), 11.08 (br, 1H), 9.00 (d, J = 5.5 Hz, 2H), 8.34 (d, J = 5.5 Hz, 2H), 5.17 (s, 1H), 4.42–4.16 (m, 2H), 3.99–3.95 (m, 1H), 3.62 (d, J = 9.7 Hz, 1H), 3.43 (t, J = 5.6 Hz, 2H), 3.30–3.17 (m, 3H), 2.95–2.89 (m, 3H), 2.71 (d, J = 9.8 Hz, 1H), 2.07 (s, 1H), 1.95–1.39 (m, 14H); 13C NMR (101 MHz) δ 148.3 (2), 143.5, 129.4 (2), 61.4, 60.6, 56.1, 54.6, 49.9,49.0, 39.6 (2), 36.5, 32.3, 30.5, 28.8, 24.2, 24.1, 23.3, 18.3. HRMS: calcd for C21H34ON3·2HCl [M−2HCl+H]+: 344.2696, found: 344.2694.
General procedures for 12N-substituted matrinol derivatives 7f–i
To a stirred solution of 5 (5.0 mmol) and K2CO3 (17.0 mmol) in dichloroethane (50 mL), substituted pyridylmethyl halide or phenylcarbamic chloride (5 mmol) was added. The reaction mixture was stirred at room temperature for 8 h until TLC analysis showed completion of the reaction. Water (20 mL) was added to the mixture and the organic phase was separated and dried with anhydrous Na2SO4, concentrated. To a solution of the gained residue in anhydrous THF (3 mL) in an ice bath, a solution of LiAlH4 (6 mmol) in anhydrous THF (10 mL) was added, the mixture solution was stirred at room temperature for 30 min before the reaction was quenched with acetone. The saturated ammonium chloride (2 mL) was then added and the mixture was stirred for 30 min, and the precipitation was filtered off. Then the solvent was evaporated, and the residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluent to afford the target compounds.
12N-(Pyridin-3-ylmethyl)matrinol (7f)
The title compound was prepared from 5 and 3-chloromethylpyridine as described above. Yield: 43%; yellow oil; 1H NMR δ (500 MHz) 9.13 (s, 1H), 8.96 (d, J = 5.5 Hz, 1H), 8.78 (d, J = 5.5 Hz, 1H), 8.07 (t, J = 5.5 Hz, 1H), 5.04–4.97(m, 1H), 4.37–4.17 (m, 2H), 3.95–3.92 (m, 1H), 3.64–3.62 (m, 1H), 3.45 (t, J = 5.9 Hz, 2H), 3.29–3.18 (m, 3H), 3.07–2.86 (m, 4H), 2.68–2.66 (m, 1H), 2.09–2.08 (m, 1H), 1.93–1.91 (m, 2H), 1.86–1.47 (m, 11H); 13C NMR (126 MHz) δ 148.1, 145.8, 143.8, 129.5, 127.1, 61.2, 60.8, 60.6, 54.6, 53.8, 49.8, 49.6, 36.6, 32.3 (2), 30.6, 28.5, 24.3, 24.1, 22.9, 18.3. HRMS: calcd for C21H34ON3 [M+H]+: 344.2696, found: 344.2694.
12N-(5-Chloropyridin-2-ylmethyl)matrinol dihydrochloride (7g)
The title compound was prepared from 5 and 5-chloro-2-(chloromethyl)pyridine in the same manner as 7f followed by an acidification with 2 N hydrochloride/ether (3 mL). Yield: 48%; light yellow solid; mp: 91–92 °C; 1H NMR (500 MHz) δ 11.98 (br, 1H), 11.06 (br, 1H), 8.61 (d, J = 2.4 Hz, 1H), 8.19 (dd, J = 8.2, 2.4 Hz, 1H), 7.64 (d, J = 8.2 Hz, 1H), 5.01–4.95 (m, 1H), 4.33–4.22 (m, 2H), 3.99–3.95 (m, 1H), 3.64–3.62 (m, 1H), 3.43–3.41 (m, 2H), 3.30–3.17 (m, 3H), 2.95–2.89 (m, 3H), 2.71 (d, J = 9.8 Hz, 1H), 2.07 (s, 1H), 1.97–1.35 (m, 14H); 13C NMR (126 MHz) δ 152.9, 151.7, 143.3, 125.8, 124.8, 60.7, 60.3, 54.7, 53.9, 51.8, 49.2, 39.5, 36.5, 33.2, 32.9, 30.5, 28.0, 24.3, 23.9, 21.5, 18.4. HRMS: calcd for C21H33ON3Cl·2HCl [M−2HCl+H]+: 378.2307, found: 378.2304.
12N-(2-Oxo-2-(phenylamino)ethyl)matrinol (7h)
The title compound was prepared from 5 and phenylcarbamic chloride in the same manner as 7f. Yield: 46%; white solid; mp: 136–137 °C; 1H NMR (400 MHz) δ 9.64 (br, 1H), 7.64–7.57 (m, 2H), 7.31 (t, J = 7.9 Hz, 2H), 7.06 (t, J = 7.4 Hz, 1H), 4.41–4.25 (m, 1H), 3.41–3.37 (m, 2H), 3.03 (s, 2H), 2.75–2.72 (m, 2H), 2.44–2.31 (m, 1H), 2.00–1.93 (m, 2H), 1.85–1.76 (m, 3H), 1.70–1.48 (m, 4H), 1.47–1.20 (m, 12H); 13C NMR (101 MHz) δ 170.1, 138.9, 131.1 (2), 123.9, 119.8 (2), 64.2, 61.2, 61.1, 57.3, 56.4, 55.7, 53.9, 37.9, 33.2, 29.7 (2), 28.7, 28.1, 27.4, 21.5, 20.8. HRMS: calcd for C23H36O2N3 [M+H]+: 386.2802, found: 386.2800.
12N-(2-Oxo-2-((4-(trifluoromethyl)phenyl)amino)ethyl)matrinol dihydrochloride (7i)
The title compound was prepared from 5 and 4-(trifluoromethyl)phenylcarbamic chloride in the same manner as 7 g. Yield: 62%; white solid; mp: 185–187 °C; 1H NMR (400 MHz) δ 11.20 (br, 1H), 10.48 (br, 1H), 10.08 (br, 1H), 8.44 (d, J = 1.6 Hz, 1H), 7.56–7.46 (m, 1H), 7.30 (d, J = 8.6 Hz, 1H), 4.65–4.55 (m, 1H), 4.32–4.16 (m, 2H), 4.09 (d, J = 9.4 Hz, 1H), 3.46–3.30 (m, 2H), 3.26 (t, J = 9.5 Hz, 2H), 3.03–2.87 (m, 2H), 2.60–2.56 (m, 1H), 2.46–2.42 (m, 1H), 1.95–1.60 (m, 12H), 1.50–1.30 (m, 6H); 13C NMR (101 MHz) δ 164.5, 152.8, 127.2 (2), 126.2, 118.9 (2), 61.2, 60.7, 60.4, 56.8, 54.7, 54.6, 52.3, 36.6, 32.3, 30.7, 29.7, 29.2, 24.3, 24.1, 23.7, 18.4, 18.3. HRMS: calcd for C24H35O2N3F3·2HCl [M−2HCl+H]+: 454.2676, found: 454.2679.
Synthesis of 12N-4-methoxybenzyl matrinic butane 9
To a solution of 7a (5 mmol) in anhydrous CH2Cl2 (20 mL), TsCl (5 mmol), TEA (10 mmol) and dimethylamino pyridine (0.5 mmol) were added and stirred at room temperature until the TLC showed completion of the reaction. The solution was washed successively by water (10 mL), saturated ammonium chloride solution (10 mL) and brine (10 mL), dried over anhydrous sodium sulfate, and concentrated to obtain crude 8. To a solution of the crude 8 in anhydrous THF, a solution of LiAlH4 (6 mmol) in anhydrous THF was added in an ice bath, then the mixture was stirred at room temperature for 30 min, the reaction was then quenched with acetone, 2 ml saturated ammonium chloride was added and stirred for 30 min, and the precipitation was filtrated. The gained residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluent to afford the title compound 9 as a yellow solid. Yield: 56%; mp: 73–74 °C; 1H NMR (400 MHz) δ 7.22 (d, J = 8.8 Hz, 2H), 6.88 (d, J = 8.8 Hz, 2H), 3.93–3.88 (m, 1H), 3.74 (s, 3H), 3.43–3.21 (m, 1H), 3.00 (d, J = 10.3 Hz, 1H), 2.88–2.61 (m, 3H), 2.53 (d, J = 11.7 Hz, 1H), 2.22-2.15 (m, 1H), 1.96 (s, 1H), 1.90–1.73 (m, 3H), 1.64 (s, 2H), 1.50-1.37 (m, 12H), 0.87 (s, 3H); 13C NMR (101 MHz) δ 158.4, 129.9, 128.4, 114.1 (2), 113.9, 64.3, 63.0, 57.1, 55.5, 55.4, 55.1, 52.0, 37.8, 37.5, 33.7, 28.4, 27.5, 25.9, 23.0, 21.6, 21.2, 14.5. HRMS: calcd for C23H37ON2 [M+H]+: 357.2900, found: 357.2899.
General procedures for 1′,1′-dialkyl-12N-substituted matrinol derivatives 10a–e
To a solution of compound 6 (5 mmol) in anhydrous THF (10 mL), a solution of 2 N alkylmagnesium chloride in THF (25 mmol) was added in an ice bath, and the mixture solution was heated at refluxing for 2 h. After reaction completed, the reaction was quenched with a solution of saturated aqueous ammonium chloride (2 mL). The residue was purified by flash column chromatography on silica gel with CH2Cl2/CH3OH as the eluent followed by the acidification with 2 N hydrochloride/ether (3 mL) to afford the title compounds.
1′,1′-Dimethyl-12N-(4-methoxybenzyl)matrinol dihydrochloride (10a)
The title compound was prepared from 6a and methylmagnesium chloride using the same method as described above. Yield: 67%; white solid; mp: 125–127 °C; 1H NMR (400 MHz) δ 11.35 (br, 1H), 11.05 (br, 1H), 7.53 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 4.78 (d, J = 11.2 Hz, 1H), 4.22–4.12 (m, 1H), 3.94–3.89 (m, 2H), 3.77 (s, 3H), 3.59 (d, J = 10.0 Hz, 1H), 3.25 (t, J = 13.6 Hz, 2H), 3.00–2.87 (m, 2H), 2.68–2.57 (m, 2H), 2.46 (d, J = 12.4 Hz, 1H), 2.02–1.51 (m, 12H), 1.46–1.39 (m, 4H), 1.11-1.07 (m, 5H); 13C NMR (126 MHz) δ 159.8, 132.9 (2), 121.6, 114.1 (2), 72.4, 68.7, 57.0, 55.2, 54.2, 54.1, 44.8, 42.9, 35.7, 32.3, 32.0, 29.9, 29.6, 29.2, 27.6, 25.5, 24.1, 23.6, 21.3. HRMS: calcd for C25H41O2N2·2HCl [M−2HCl+H]+: 401.3163, found: 401.3163.
1′,1′-Dimethyl-12N-(4-methylbenzyl)matrinol dihydrochloride (10b)
The title compound was prepared from 6b and methylmagnesium chloride using the same method as described above. Yield 63%; white solid; mp: 120–122 °C; 1H NMR (400 MHz) δ 11.12 (br, 1H), 10.98 (br, 1H), 7.48 (dd, J = 8.0, 5.6 Hz, 2H), 7.27 (d, J = 7.6 Hz, 2H), 4.91–4.74 (m, 1H), 4.33–4.14 (m, 2H), 4.04 (s, 5H), 3.62–3.54 (m, 1H), 3.29–3.24 (m, 2H), 2.99–2.88 (m, 2H), 2.73–2.68 (m, 1H), 2.34 (s, 3H), 2.04–1.56 (m, 15H), 1.49–1.42 (m, 2H), 1.10 (d, J = 3.2 Hz, 2H); 13C NMR (126 MHz) δ 132.1, 123.1 (2), 121.5 (2), 117.9, 62.3, 61.7, 53.3, 53.0, 50.1, 46.9, 41.5, 36.8, 34.4, 28.4, 23.4, 23.3, 22.9 (2), 19.9 (2), 15.9, 15.6, 11.8. HRMS: calcd for C25H41ON2·2HCl [M−2HCl+H]+: 385.3213, found: 385.3214.
1′,1′-Diethyl-12N-(4-methylbenzyl)matrinol dihydrochloride (10c)
The title compound was prepared from 6b and ethylmagnesium chloride using the same method as described above. Yield 72%; yellow oil; 1H NMR (400 MHz) δ 10.91 (br, 1H), 10.54 (br, 1H), 7.45 (d, J = 8.0 Hz, 2H), 7.29 (d, J = 7.6 Hz, 2H), 4.85–4.80 (m, 1H), 4.56–4.14 (m, 3H), 4.06–3.82 (m, 3H), 3.61–3.53 (m, 3H), 3.35–3.24 (m, 2H), 3.12–2.85 (m, 3H), 2.78 (d, J = 6.8 Hz, 1H), 2.34 (s, 3H), 2.09–1.99 (m, 3H), 1.95–1.69 (m, 9H), 1.69–1.53 (m, 3H), 1.53–1.33 (m, 2H), 0.96 –0.92 (m, 4H); 13C NMR (126 MHz) δ 138.9, 131.3 (2), 129.3 (2), 126.8, 80.6, 60.3, 60.2, 57.3, 54.2 (2), 48.9, 35.6, 32.9, 32.8, 30.0, 24.0, 23.6, 20.8 (2), 19.9, 17.8, 13.2, 12.6, 8.6 (2). HRMS: calcd for C27H45ON2·2HCl [M−2HCl+H]+: 413.3526, found: 413.3524.
1′,1′-Dimethyl-12N-(2,4-difluorobenzyl)matrinol dihydrochloride (10d)
The title compound was prepared from 6d and methylmagnesium chloride using the same method as described above. Yield: 75%; white solid; mp: 237–238 °C; 1H NMR (400 MHz) δ 11.02 (s, 1H), 10.40 (s, 1H), 7.93–7.81 (m, 1H),7.47–7.40 (m, 1H), 7.29–7.19 (m, 1H), 4.78 (d, J = 13.2 Hz, 1H), 4.30–4.20 (m, 1H), 4.20–4.05 (m, 1H), 4.10–3.85 (m, 1H), 3.53 (d, J = 8.4 Hz, 3H), 3.29 (t, J = 12.0 Hz, 2H), 3.06–2.82 (m, 4H), 2.00–1.51 (m, 11H), 1.47–1.44 (m, 3H), 1.11 (d, J = 3.6 Hz, 6H); 13C NMR (126 MHz) δ 164.2, 162.2, 135.6, 113.5, 112.0, 104.3, 68.6, 60.5, 60.1, 54.2 (2), 49.8, 48.8, 42.9, 35.9, 29.9, 29.5, 29.2, 28.7, 23.9, 23.7, 20.4, 17.8 (2). HRMS: calcd for C24H37ON2F2·2HCl [M−2HCl+H]+: 407.2868, found: 407.2863.
1′,1′-Dimethyl-12N-(4-pyridylmethyl)matrinol (10e)
The title compound was prepared from 6f and methylmagnesium chloride using the same method as described above without acidification. Yield: 68%; yellow solid; mp: 243–245 °C; 1H NMR (400 MHz) δ 8.47 (d, J = 5.6 Hz, 2H), 7.33 (d, J = 5.6 Hz, 2H), 4.02 (s, 1H), 3.94 (d, J = 15.2 Hz, 1H), 3.32 (s, 1H), 3.19 (d, J = 14.4 Hz, 1H), 2.89 (d, J = 8.4 Hz, 1H), 2.72 (t, J = 11.2 Hz, 3H), 2.17 (d, J = 8.8 Hz, 1H), 2.00 (s, 1H), 1.81 (d, J = 12.0 Hz, 3H), 1.64–1.54 (m, 4H), 1.42–1.24 (m, 10H), 1.01 (d, J = 2.8 Hz, 6H); 13C NMR (126 MHz) δ 149.7, 149.4 (2), 123.3 (2), 68.7, 63.8, 56.5, 56.4, 53.7, 51.9, 43.9, 37.0, 34.7, 32.7, 29.4, 29.1, 29.0, 27.4, 26.7, 20.8, 20.5, 18.1. HRMS: calcd for C23H38ON3 [M+H]+: 372.3009, found: 372.3008.
General procedures for methyl (Z)-12N-substituted Δβγ-matrinic crotonate derivatives 13a–c
Lehmannine (3.0 g, 12.2 mmol) was added to a solution of 5 N HCl (30 mL). The reaction mixture was heated at reflux for 9 h. The solvent was then removed in vacuo, and the residue was recrystallized by methanol and ethyl acetate to afford the intermediate 11 (2.5 g, 60%) as white solid. mp: 191–193 °C. 1H NMR (400 MHz) δ 12.39 (s, 1H), 11.21 (d, J = 8.0 Hz, 1H), 10.27 (d, J = 9.3 Hz, 1H), 9.30 (d, J = 9.0 Hz, 1H), 6.01 (dt, J = 10.8, 7.3 Hz, 1H), 5.49 (t, J = 10.4 Hz, 1H), 5.04–4.92 (m, 1H), 3.99–3.764 (m, 1H), 3.65 (d, J = 10.1 Hz, 1H), 3.44–3.33 (m, 2H), 3.25–3.20 (m, 2H), 3.20–3.02 (m, 1H), 2.97–2.89 (m, 2H), 2.55–2.51 (m, 1H), 2.40–2.23 (m, 1H), 1.89–1.56 (m, 8H); 13C NMR (101 MHz) δ 172.4, 132.4, 125.7, 60.4, 54.8, 54.7, 49.8, 41.5, 35.5, 33.8, 30.8, 24.6, 23.6, 18.5(2); HRMS: calcd for C15H25N2O2·2HCl [M−2HCl+H]+: 265.1911, found: 265.1909.
Compound 11 (1.0 g, 3.0 mmol) was dissolved in 2 N MeOH/HCl (30 mL), and the reaction mixture was refluxed for 2 h. Compound 12 was obtained by evaporation and used in the next reaction without further purification. Anhydrous K2CO3 (3.5 equiv) and substituted benzyl bromide (1.5 equiv) were added to a solution of compound 12 in acetonitrile (30 mL), and the reaction solution was then stirred at room temperature until TLC analysis showed completion of the reaction. The reaction mixture was filtered, and the filtrate was washed by water and brine, dried with anhydrous Na2SO4, filtrated, and concentrated to afford crude compound 13. The title compounds were obtained by purifying with flash column chromatography on silica gel with dichloromethane and methanol as the eluent.
(Z)-Methyl 12N-(4-methoxybenzyl)-Δβγ-matrinic crotonate (13a)
The title compound was prepared from 12 and 4-methoxybenzyl bromide using the same method as described above. Yield: 62%; white solid; mp: 98–100 °C. 1H NMR (400 MHz) δ 7.14 (d, J = 8.4 Hz, 2H), 6.84 (d, J = 8.4 Hz, 2H), 5.81–5.75 (m, 1H), 5.33 (t, J = 10.4 Hz, 1H), 3.92 (d, J = 13.2 Hz, 1H), 3.73 (s, 3H), 3.61 (s, 3H), 3.34–3.15 (m, 3H), 2.89–2.86 (m, 1H), 2.72–2.75(m, 2H), 2.51–2.44 (m, 1H), 2.21–2.18 (m, 1H), 1.98 (s, 1H),1.84–1.75 (m, 2H), 1.65–1.24 (m, 10H); 13C NMR (126 MHz) δ 171.3, 158.0, 136.0, 131.5, 129.7 (2), 124.8, 113.4 (2), 62.5, 58.2, 56.8, 56.7, 54.9 (2), 51.6 (2), 50.8, 34.7, 33.3, 28.1, 26.8, 21.4, 21.2. HRMS: calcd for C24H35N2O3 [M+H]+: 399.2642, found: 399.2642.
(Z)-Methyl 12N-(4-fluorobenzyl)-Δβγ-matrinic crotonate dihydrochloride (13b)
The title compound was prepared from 12 and 4-fluorobenzyl bromide using the same method as described above. Yield: 68%; white solid; mp: 151–153 °C. MS–ESI m/s: 387; 1H NMR (400 MHz) δ 11.93 (d, J = 8.0 Hz, 1H), 11.08 (d, J = 7.6 Hz, 1H), 7.64–7.61 (m, 2H), 7.32–7.27 (m, 2H), 6.27–6.20 (m, 1H), 5.88–5.78 (m, 1H), 5.28 (t, J = 11.2 Hz, 1H), 4.63 (d, J = 12.0 Hz, 1H), 4.00–3.85 (m, 2H), 3.68–3.17 (m, 9H), 3.00–2.79 (m, 3H), 2.63 (s, 1H), 1.83–1.56 (m, 8H); 13C NMR (126 MHz) δ 170.7, 161.7, 133.7, 133.6, 133.2, 126.0, 124.9, 115.9, 115.8, 59.6, 58.6, 56.7, 54.2, 54.1, 51.9, 47.3, 35.1, 33.3, 30.2, 24.0, 23.9, 18.0, 17.9. HRMS: calcd for C23H32FN2O2·2HCl [M−2HCl+H]+: 387.2442, found: 387.2446.
(Z)-Methyl 12N-(3-nitrobenzyl)-Δβγ-matrinic crotonate dihydrochloride (13c)
The title compound was prepared from 12 and 3-nitrobenzyl bromide using the same method as described above. Yield: 70%; white solid; mp: 185–187 °C; 1H NMR (400 MHz) δ 12.33 (s, 1H), 11.07 (s, 1H), 8.41 (s, 1H), 8.26 (d, J = 8.4 Hz, 1H), 8.02 (d, J = 7.5 Hz, 1H), 7.71 (t, J = 8.0 Hz, 1H), 6.22 (dt, J = 15.1, 7.5 Hz, 1H), 5.84 (t, J = 10.6 Hz, 1H), 5.39–5.21 (m, 1H), 4.70 (d, J = 12.9 Hz, 1H), 4.15–3.78 (m, 3H), 3.64 (s, 3H), 3.47–3.41 (m, 1H), 3.26 (d, J = 11.7 Hz, 2H), 2.99–2.83 (m, 3H), 2.61 (s, 1H), 2.56–2.48 (m, 1H), 1.80–1.76 (m, 2H), 1.70–1.49 (m, 7H); 13C NMR (126 MHz) δ 170.7, 147.8, 138.1, 133.4, 131.6, 130.4, 126.3, 124.9, 124.4, 59.6, 58.7, 56.5, 54.3, 54.1, 51.9, 47.7, 35.2, 33.3, 30.3, 23.9, 23.9, 18.0, 17.9. HRMS: calcd for C23H32N3O4·2HCl [M−2HCl+H]+: 414.2387, found: 414.2391.
General procedures for (Z)-12N-substituted Δβγ-matrinic crotonol derivatives 14a–b
A solution of the LiAlH4 in THF (2.4 N, 1.2 equiv) was added to the solution of compound 13 in anhydrous THF in ice bath, then the mixture solution was stirred at room temperature for 30 min, the reaction was then quenched with acetone, 2 mL saturated ammonium chloride solution was added and stirred for 30 min, and the precipitation was filtrated off. The filtrate was concentrated, and the residue was purified by flash column chromatography on silica gel with dichloromethane and methanol as the eluent followed by the acidification by 2 N hydrochloride/ether to afford compounds.
(Z)-12N-(4-Methoxybenzyl)-Δβγ-matrinic crotonol dihydrochloride (14a)
The title compound was prepared from 13a using the same method as described above. Yield: 86%; white solid; mp: 175–177 °C; 1H NMR (400 MHz) δ 11.46 (br, 1H), 11.15 (br, 1H), 7.48–7.45 (d, J = 8.8 Hz, 2H), 7.01–6.99 (d, J = 8.8 Hz, 2H), 6.17–6.11 (m, 1H), 5.68 (t, J = 10.8 Hz, 1H), 5.16 (dd, J = 10.4 Hz, 1H), 4.66 (d, J = 11.6 Hz, 1H), 4.02–3.93 (m, 1H), 3.89–3.78 (m, 1H), 3.74 (s, 3H), 3.69–3.43 (m, 5H), 3.39–3.28 (m, 3H), 2.99–2.88 (m, 2H), 2.78–2.75 (m, 1H), 2.55–2.49 (m, 1H), 2.37–2.31 (m, 1H), 1.89–1.53 (m, 8H); 13C NMR (126 MHz) δ 159.9, 139.2, 132.8 (2), 123.5, 121.6, 114.2 (2), 59.8, 59.7, 58.6, 57.1, 55.2 (2), 54.2, 47.1, 35.3, 31.8, 30.2, 24.2, 24.0, 18.0, 17.9. HRMS: calcd for C23H35N2O2·2HCl [M−2HCl+H]+: 371.2693, found: 371.2698.
(Z)-12N-4-(Fluorobenzyl)-Δβγ-matrinic crotonol dihydrochloride (14b)
The title compound was prepared from 13b using the same method as described above. Yield: 87%; white solid; mp: 194–196 °C; 1H NMR (400 MHz) δ 11.77 (br, 1H), 11.15 (br, 1H), 7.64 (dd, J = 5.6 Hz, 2H), 7.28 (t, J = 8.8 Hz, 2H), 6.18–6.11 (m, 1H), 5.71 (t, J = 10.8 Hz, 1H), 5.18 (dd, J = 10.8 Hz, 1H), 4.70 (d, J = 12.4 Hz, 1H), 4.04–3.87 (m, 2H), 3.77–3.41 (m, 5H), 3.41–3.28 (m, 2H), 3.00–2.76 (m, 3H), 2.58–2.51 (m, 2H), 2.37–2.29 (m, 1H), 1.90–1.56 (m, 8H); 13C NMR (126 MHz) δ 163.6, 139.4, 133.7, 133.6, 126.2, 123.4, 115.9, 115.7, 59.8, 59.6, 58.8, 56.7, 54.2 (2), 47.3, 35.3, 31.8, 30.2, 24.1, 24.0, 18.0, 17.9. HRMS: calcd for C22H32FN2O·2HCl [M−2HCl+H]+: 359.2493, found: 359.2492.
Synthesis of (Z)-12N-(3-nitrobenzyl)-Δβγ-matrinic crotonol dihydrochloride 20
The compound 12 (1.0 g, 4.0 mmol) was dissolved in 2 N HCl/MeOH (30 mL). The reaction mixture was refluxed for 2 h, then anhydrous K2CO3 (3.5 equiv) and Boc2O (1.5 equiv) were added to the reaction solution, and the mixture solution was stirred at room temperature until TLC analysis showed completion of the reaction. The reaction mixture was filtered, and the filtrate was washed by water and brine, dried with anhydrous Na2SO4, filtrated and concentrated to afford the crude 15.
A solution of the LiAlH4 in THF (2.4 N, 1.2 equiv) was added to the solution of compound 15 in anhydrous THF in ice-bath, then the mixture solution was stirred at room temperature for 30 min, the reaction was then quenched with acetone, 2 mL saturated ammonium chloride solution was added and stirred for 30 min, and the precipitation was filtrated off. The filtrate was concentrated, and the residue of compound 16 was dissolved in ethyl acetate, and washed with water and brine, dried with anhydrous Na2SO4, filtrated and concentrated. The residue was stirred in 2 N HCl/Et2O (20 mL) to remove the Boc protection group, then the mixute was filtrated to give the crude 17.
The crude 17 (1.0 equiv), TBSCl (1.2 equiv) and imidazole (1.5 equiv) were used to synthesize compound 18 in CH2Cl2, after reaction was complete, 3-nitrobenzyl bromide (3.0 equiv) and TEA (3.0 equiv) were added to the reaction solution, which was stirred at room temperature until TLC analysis showed completion of the reaction. The reaction solution was washed by water and brine, dried over anhydrous Na2SO4, filtrated and concentrated to afford the crude compound 19.
The crude 19 was dissolved in 2 N HCl (15 mL), and the mixture was stirred until TLC analysis showed completion of the reaction. The pH of the reaction solution was then adjusted to 7–8 by addition of ammonium hydroxide. The solvent was removed under reduced pressure, and the residue was dissolved in MeOH and filtered to remove the organic salts. The solution was concentrated, and the residue was purified by flash column chromatography on silica gel with dichloromethane and methanol as the eluent to afford compound 20 as white solid. Yield: 30%; mp: 143–145 °C; 1H NMR (400 MHz) δ 12.8 (br, 1H), 11.17 (br, 1H), 8.44 (s, 1H), 8.30 (d, J = 8.0 Hz, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.75 (t, J = 8.0 Hz, 1H), 6.20–6.14 (m, 1H), 5.73 (t, J = 10.4 Hz, 1H), 5.27–5.20 (m,1H), 4.85(d, J = 12.8 Hz, 1H), 4.10–4.02 (m, 3H), 3.65 (d, J = 10.4 Hz, 1H), 3.57–3.49 (m, 2H), 3.29 (d, J = 12.0 Hz, 2H), 3.00–2.88 (m, 3H), 2.60–2.53 (m, 3H), 2.37–2.32 (m, 1H), 1.91–1.56 (m, 8H); 13C NMR (126 MHz) δ 147.8, 139.7, 138.0, 131.9, 130.4, 126.3, 124.3, 123.3, 59.8, 59.6, 58.9, 56.5, 54.2, 47.6, 35.3, 31.8, 30.3, 29.2, 24.1, 23.9, 18.0, 17.9. HRMS: calcd for C22H32N3O3·2HCl [M−2HCl+H]+: 386.2438, found: 386.2440.
Biology assay
Cell culture
Human liver cell line Huh7.5 cells (kindly provided by Vertex Pharmaceuticals, Inc., Boston, MA) were cultured in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% inactivated fetal bovine serum and 1% penicillin–streptomycin (invitrogen). Cells were digested with 0.05% trypsin-ethylene diamine tetraacetic acid (EDTA) and split twice a week.
Anti-HCV effect in vitro
Huh7.5 cells were seeded into 96-well or 6-well plates (Costar) at a density of 3 × 104 cells cm−2. After 24 h incubation, the cells were infected with HCV viral stock (45 IU cell−1) and simultaneously treated with the test compounds at various concentrations or solvent as control. The culture medium was removed after 72 h inoculation, the intracellular total RNA (in 96-well plates) was extracted with RNeasy Mini Kit (Qiagen), and total intracellular proteins (in 6-well plates) were extracted with Cyto-Buster Protein Extraction Reagent added with 1 mM protease inhibitor cocktail. The intracellular HCV RNA was quantified with a real time one-step reverse-transcription polymerase chain reaction (RT-PCR).
Cytotoxicity assay
Huh7.5 cells were seeded into 96-well plates (Costar) at a density of 3 × 104 cells cm−2. After 24 h incubation, fresh culture medium containing test compounds at various concentrations were added. 72 h later, cytotoxicity was evaluated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT).
PK studies
Three male SD mice were used in each study. Each of them was dosed with a tested compound at 25 mg kg−1 via oral administration. Eight blood samples were respectively collected at 0, 0.25, 0.5, 1.0, 2.0, 4.0, and 6.0 h and were immediately centrifuged to separate the plasma fractions. The separated plasma samples were stored at −20 °C for analysis. Concentration-versus-time profiles were obtained for each analyte, and standard non-compartmental analysis was performed on the data using WinNonlin software, version 5.3, to recover the AUC and other non-compartmental parameters.
Acute toxicity
Female Kunming mice with weight of 20.0 ± 1.0 g were fed with regular rodent chow and housed in an air conditioned room. The mice were randomly divided into different groups with six mice each. Each compound was given orally in a single-dosing experiment at 0, 250, 500, 750 or 1000 mg kg−1 (ddH2O as control), respectively. The mice were closely monitored for 7 days. Body weight as well as survival was monitored.
Conclusion
In conclusion, 32 compounds (of which 27 were novel) with diverse structures, including methyl matrinate, matrinols, matrinic butane, 1′, 1′-dialkylmatrinols, (Z)-methyl Δβγ-matrinic crotonates, (Z)-Δβγ-matrinic crotonols were synthesized and evaluated for their anti-HCV activities, taking compound 1 as the lead. The SAR study indicated that the introduction of electron-donating substitutions on the benzene ring was helpful for the anti-HCV activity, and the unsaturated 11-side chain might not be favorable for the activity. Out of the gathered compounds, matrinol 7a demonstrated a potential anti-HCV effect with the SI value of 136. Further study showed that compound 7a possessed reasonable PK and safety profiles in vivo, indicating a fair druggability nature. Besides, the free hydroxyl arm in 7a would make it possible to be a parent structure to make pro-drug candidates for their potential in the treatment of HCV infection. This study provided powerful information on further strategic optimization and development of this kind of compounds into a novel family of anti-HCV agents.
Authors’ contributions
The current study is an outcome of the constructive discussion with DQS and YXW, who offered necessary guidance to ST, YHL and XZ to carry out their synthesis and characterization experiments. ZGP and TS performed the biological assay against HCV, XYC carried out the 1H NMR and 13C NMR spectral analyses and HRMS analysis. JDJ provided theoretical guidance. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21472246), the Beijing Natural Science Foundation (7152097) and CAMS Innovation Fund for Medical Sciences (CIFMS, 2016-12 M-3-009).
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Sheng Tang and Zong-Gen Peng equally contributed to this work
Contributor Information
Sheng Tang, Email: tang13874204108@163.com.
Zong-Gen Peng, Email: pumcpzg@126.com.
Ying-Hong Li, Email: yinghongli523@aliyun.com.
Xin Zhang, Email: yaxuzhangxin@163.com.
Tian-Yun Fan, Email: fty1668@163.com.
Jian-Dong Jiang, Email: jiang.jdong@163.com.
Yan-Xiang Wang, Phone: +86 10 63033012, Email: wangyanxiang@imb.pumc.edu.cn.
Dan-Qing Song, Phone: +86 10 6316 5268, Email: songdanqingsdq@hotmail.com.
References
- 1.World Health Organization Fact sheet No. 164. http://www.who.int/mediacentre/factsheets/fs164/en/. Accessed 28 Nov 2016
- 2.Mohd HK, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age-specific antibody to HCV seroprevalence. Hepatology. 2013;57:1333–1342. doi: 10.1002/hep.26141. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Grande R, Jimenez-Perez M, Gonzalez AC, Mostazo TJ. New approaches in the treatment of hepatitis C. World J Gastroenterol. 2016;22:1421–1432. doi: 10.3748/wjg.v22.i4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esposito I, Trinks J, Soriano V. Hepatitis C virus resistance to the new direct-acting antivirals. Expert Opin Drug Metab Toxicol. 2016;12:1197–1209. doi: 10.1080/17425255.2016.1209484. [DOI] [PubMed] [Google Scholar]
- 5.Kan H, Imamura M, Uchida T, Hiraga N, Hayes CN, Tsuge M, Abe H, Aikata H, Makokha GN, Chowdhury S, et al. Protease inhibitor resistance remains even after mutant strains become undetectable using deep sequencing. J Infect Dis. 2016;214:1687–1694. doi: 10.1093/infdis/jiw437. [DOI] [PubMed] [Google Scholar]
- 6.Ramirez S, Mikkelsen LS, Gottwein JM, Bukh J. Robust HCV genotype 3a infectious cell culture system permits identification of escape variants with resistance to sofosbuvir. Gastroenterol. 2016 doi: 10.1053/j.gastro.2016.07.013. [DOI] [PubMed] [Google Scholar]
- 7.Mizokami M, Dvory-Sobol H, Izumi N, Nishiguchi S, Doehle B, Svarovskaia ES, De-Oertel S, Knox S, Brainard DM, Miller MD, et al. Resistance analyses of Japanese hepatitis C-infected patients receiving sofosbuvir or ledipasvir/sofosbuvir containing regimens in phase 3 studies. J Viral Hepat. 2016;23:780–788. doi: 10.1111/jvh.12549. [DOI] [PubMed] [Google Scholar]
- 8.Werner CR, Schwarz JM, Egetemeyr DP, Beck R, Malek NP, Lauer UM, Berg CP. Second-generation direct-acting-antiviral hepatitis C virus treatment: efficacy, safety, and predictors of SVR12. World J Gastroenterol. 2016;22:8050–8059. doi: 10.3748/wjg.v22.i35.8050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alavian SM, Hajarizadeh B, Bagheri LK, Sharafi H, Ebrahimi DN, Merat S, Mohraz M, Mardani M, Fattahi MR, Poustchi H, et al. Recommendations for the clinical management of hepatitis C in Iran: a consensus-based national guideline. Hepat Mon. 2016;16:e40959. doi: 10.5812/hepatmon.guideline. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Du N-N, Peng Z-G, Bi C-W, Tang S, Li Y-H, Li J-R, Zhu Y-P, Zhang J-P, Wang Y-X, Jiang J-D, et al. N-substituted benzyl matrinic acid derivatives inhibit hepatitis C virus (HCV) replication through down-regulating host heat-stress cognate 70 (Hsc70) expression. PLoS ONE. 2013;8:e58675. doi: 10.1371/journal.pone.0058675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng Z-G, Fan B, Du N-N, Wang Y-P, Gao L-M, Li Y-H, Li Y-H, Liu F, You X-F, Han Y-X, et al. Small molecular compounds that inhibit hepatitis C virus replication through destabilizing heat shock cognate 70 messenger RNA. Hepatology. 2010;52:845–853. doi: 10.1002/hep.23766. [DOI] [PubMed] [Google Scholar]
- 12.Tang S, Li Y-H, Cheng X-Y, Li Y-H, Wang H-Q, Kong L-Y, Zhang X, Jiang J-D, Song D-Q. SAR evolution and discovery of benzenesulfonyl matrinanes as a novel class of potential coxsakievirus inhibitors. Future Med Chem. 2016;8:495–508. doi: 10.4155/fmc-2015-0019. [DOI] [PubMed] [Google Scholar]
- 13.Fu HG, Tang S, Li YH, Song DQ, Wang YX. Synthesis and antitubercular activities of novel 12-n-substituted matrinic acid derivatives. Chin J Synth Chem. 2014;6:739–743. [Google Scholar]
- 14.Tang S, Kong L-Y, Li Y-H, Jiang J-D, Gao L-M, Cheng X-Y, Ma L-L, Zhang X, Li Y-H, Song D-Q. Novel N-benzenesulfonyl sophocarpinol derivatives as coxsackie B virus inhibitors. ACS Med Chem Lett. 2015;6:183–186. doi: 10.1021/ml500525s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bi C-W, Zhang C-X, Li Y-H, Tang S, Wang S, Shao R-G, Fu H-G, Su F, Song D-Q. Synthesis and biological evaluation of sophoridinol derivatives as a novel family of potential anticancer agents. ACS Med Chem Lett. 2014;5:1225–1229. doi: 10.1021/ml500289h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li Y-H, Peng Z-G, Gao L-M, Song D-Q. Synthesis and biological evaluation of sophocarpinic acid derivatives as anti-HCV agents. Acta Pharm Sin B. 2014;4:307–312. doi: 10.1016/j.apsb.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du N-N, Li X, Wang Y-P, Liu F, Liu Y-X, Li C-X, Peng Z-G, Gao L-M, Jiang J-D, Song D-Q. Synthesis, structure-activity relationship and biological evaluation of novel N-substituted matrinic acid derivatives as host heat-stress cognate 70 (Hsc70) down-regulators. Bioorg Med Chem Lett. 2011;21:4732–4735. doi: 10.1016/j.bmcl.2011.06.071. [DOI] [PubMed] [Google Scholar]
- 18.Gao L-M, Tang S, Wang Y-X, Gao R-M, Zhang X, Peng Z-G, Li J-R, Jiang J-D, Li Y-H, Song D-Q. Synthesis and biological evaluation of N-substituted sophocarpinic acid derivatives as coxsackievirus B3 inhibitors. ChemMedChem. 2013;8:1545–1553. doi: 10.1002/cmdc.201300224. [DOI] [PubMed] [Google Scholar]