

Abstract
The composition of the extracellular matrix (ECM) proteins and the expression of their cognate receptors dictate cell behavior and dynamics. In particular, the interactions of ECM proteins with integrin receptors are key mediators of these cellular processes, playing a crucial role in the progression of several diseases of the liver, including inflammation, fibrosis/cirrhosis and cancer. This study establishes a modeling approach combining computation and experiments to evaluate the kinetics of integrin receptor binding to hepatic ECM proteins. ECM ligand concentration was derived from LC-MS/MS quantification of the hepatic ECM from mice exposed to chronic carbon tetrachloride (CCl4); receptor density was derived from published literature. Mathematical models for ECM-integrin binding kinetics that were developed incorporate receptor divalence and an aggregation scheme to represent clustering. The computer simulations reproduced positive cooperativity in the receptor aggregation model when the aggregation equilibrium constant (Ka) was positive and greater than Keq for divalent complex formation. Importantly, the modeling projected an increase in integrin binding for several receptors for which signaling is known to be increased after CCl4 exposure in the liver. The proposed modeling approach may be of use to elucidate the kinetics of integrin receptor binding to ECM proteins for homeostatic and diseased livers.
Introduction
The extracellular matrix (ECM) consists of a broad range of components that interact bi-directionally with neighboring cells to create a dynamic and responsive microenvironment which regulates cell signaling, recruitment, and tissue function. The ECM not only provides structure and support for the cells in a tissue, but also acts as a reservoir for growth factors and cytokines and as a signaling mechanism by which cells can intercommunicate with their environment1. Quantitative and qualitative changes to the ECM structure and superstructure can impact overall health of the organ and the organism. In particular, the hepatic ECM changes predominantly described in published literature occur in the context of hepatic fibrosis, which is characterized by robust scarring of the liver with collagen fibrils. However, hepatic ECM is significantly more diverse than collagen ECM. Recent studies further indicate that hepatic ECM content changes dynamically in response to acute stress and injury2–4. Moreover, changes to the hepatic ECM may foster an environment that is conducive to cancer and metastasis. Although the concept that hepatic ECM changes drive hepatic dysfunction under several conditions is well understood, the mechanisms by which these effects are mediated are not.
Integrins comprise a family of heterodimeric transmembrane glycoprotein receptors that facilitate key interactions between cells and the ECM5–7. The binding of ECM ligands to integrins mediates critical processes, including cell adhesion, migration, proliferation, differentiation, inflammation and apoptosis. Indeed, several of the hallmarks of liver diseases and cancer (e.g., altered proliferation, angiogenesis and apoptosis) are hypothesized to be mediated via changes in ECM:integrin signaling8. Based on this assumption, integrins have become important therapeutic targets for diseases of dysregulation, including various cancers, fibrosis, and immune dysfunction. However, few integrin-based therapies have been effective to prevent and/or treat these diseases. This limitation is partly due, to an incomplete understanding of the complexity of the changes to integrin signaling under dysregulated conditions.
The kinetics of ECM:integrin interactions are highly intricate. Integrin receptor complexes are structured as non-covalently linked α and β subunits, the various combinations of which contribute to the diversity of receptor types9 (Fig. 1). The overall rate of binding is not driven simply by ligand binding to the receptor, but also by clustering at focal adhesion points and an increase in avidity for binding additional ligand (i.e., positive cooperativity). Masson-Gandais et al. described a two-step model wherein the α subunit binds ligand first, influencing ligand recognition and determinant of association kinetics10. The β subunit binds second, which creates bond stabilization and determines dissociation kinetics. Ligand binding to the extracellular domain activates the receptor and initiates its conformational changes to a high-affinity state11,12. This two-step process reflects a divalent kinetics model with the α subunit as the high affinity site, and the β subunit as the low affinity site13. In addition to binding processivity of individual receptors, ligand binding to distinct integrins favors subsequent binding by other receptors (i.e. focal adhesion clustering). Furthermore, integrin receptors bind promiscuously to various ECM ligands, creating redundancy, competition and diversity in biofunctionality5,9,14. These complex interdependent factors affect the kinetics of ECM-integrin interactions in the intact organism. Promiscuity among the repertoire of ECM ligands and integrin receptors, particularly those with RGD-binding motifs, implies a differential pattern of binding relative to the amounts of substrate available15,16.
Figure 1.

Repertoire of alpha and beta integrin subtype dimerization pairings. This diagram delineates the 24 possible integrin dimer species, classified by substrate type. For this study, of the integrins relevant to the CCl4 model, the collagen-binding β1 integrins and RGD-binding β1 and β3 receptors were evaluated. Binding here was treated as a two-step model with the α subunit binds ligand first, influencing ligand recognition and determinant of association kinetics. The β subunit binds second, creating bond stabilization and determining dissociation kinetics14, apropos to a divalent kinetics model with the α subunit being the high affinity site, and the β subunit as the low affinity site.
To explore these interactions as a system, several mathematical descriptions of integrin binding have been reported with outputs related to spatial clustering and signal transduction, liver fibrosis and haptotaxis17–19. Although these models recapitulate certain aspects of ECM-integrin interactions, they typically focus on one ligand (e.g. collagen or fibronectin) as the ECM substrate. In this study, modeling of integrin receptor binding kinetics is presented that considers divalent receptor characteristics and employs a simple model of integrin clustering. The kinetic indices of each integrin for each of its ligands were initially determined to establish a single-species integrin profile. Proteomic data were compiled that assess the liver ECM under homeostatic conditions as well as experimental fibrosis. These proteomic analyses provided information on relative abundance of hepatic ECM components to calibrate substrate concentrations for the kinetic simulations. Although data from human fibrotic livers has recently been analyzed20, an animal model was chosen here as it provides a more controlled environment for initial model calibration and testing. Longer term, by testing homeostatic conditions against the experimental treatment models, how the integrin binding phenotype changes in response to injury could be determined and used to predict the ECM-integrin binding within the context of transitional tissue remodeling.
Results
As expected, 4 weeks of CCl4 exposure caused robust fibrotic scarring of the liver in our mouse model. The resultant phenotype of injury and fibrosis has been previously described to include degradation of basement membrane-like ECM and replacement with fibrillar collagens and other integrin ligands (Fig. 2)21. The canonical change in ECM content during hepatic fibrosis is an increase in collagen 1 deposition. However, as has been previously described22,23, several other proteins increase in response to CCl4-induced fibrosis.
Figure 2.

Schematic of aberrant ECM accumulation following CCl4 injury. Key extracellular matrix proteins (ECMPs) and cognate integrin receptors in CCl4 exposure mouse model of fibrosis. The phenomena include quiescent hepatic stellate cell (HSC) activation and their subsequent differentiation into myofibroblasts after which growth factor-induced proliferation leads to the aberrant ECM deposition that characterizes cirrhotic liver fibrosis. The chronic inflammatory response involves impaired matrix degradation which further contributes to dyshomeostasis of ECM proteins, and therefore tissue structure and errant signal transduction. Following exposure to CCl4, damaged hepatocytes release cellular and membrane components8, leading to recruitment of neutrophils and Kupffer cells. Profibrogenic and proinflammatory cytokines, reactive oxygen species (ROS), and proteases are released from resident immune cells, leading to stimulation and activation of quiescent HSCs, inducing their differentiation to myofibroblasts. Proliferation of activated myofibroblasts in response to fibrogenic factors results in excessive ECM deposition, leading to fibrotic scarring and end-stage liver disease. Integrin mediators known to be active in fibrotic pathology include β1, α1, α5, and α6 on hepatocytes, which correlate clinically with stage of fibrosis8. αvβ3 integrin signaling from HSCs/myofibroblasts is involved with regulating ECM-fibrolytic matrix metalloproteinases. De novo α8β1 expression in activated HSCs occurs in response to CCl4 injury; likewise, α1, α2, and α5 on HSCs is indicative of activation, enhancing attachment to basement membrane proteins8. Feed forward mechanism results from the fibrillar ECM itself enhancing HSC activation, implicating integrins α1β1, α2β1, and αVβ1 33.
Analysis of the proteomic data (Table 1) revealed ECM protein expression profiles, and a simple conversion for relating quantitative exponentially modified protein abundance index (emPAI) values to protein mass was employed as a proteomic ruler to estimate protein concentration under homeostatic and experimental treatment conditions24,25. Weighting the values with the concentration of extraction fractions, we estimated a relative protein concentration for ECM components. The composition of the liver ECM as quantitated via proteomic analysis has influence on integrin expression of cells that haptotactically migrate towards ECM protein gradients, and provides the pool of available ligands for subsequent binding.
Table 1.
Quantitative Differential Protein Expression of Liver ECM following 4 Weeks of CCl4 Exposure.
| Protein | GO Accession | MW (kDa) | Protein Abundance (µM) | Log2 FC | |
|---|---|---|---|---|---|
| Con | CCl4 | ||||
| Col 1α1 | CO1A1 | 138 | 3.60 | 6.42 | 0.83 |
| Col 1α2 | CO1A2 | 130 | 2.55 | 4.25 | 0.74 |
| Col 3α1 | CO3A1 | 139 | 0.75 | 1.64 | 1.13 |
| Col 4α1 | CO4A1 | 161 | 0.20 | 0.23 | −0.24 |
| Col 4α2 | CO4A2 | 167 | 0.46 | 0.28 | −0.71 |
| Col 5α1 | CO5A1 | 184 | 0 | 0.05 | 11.80 |
| Col 5α3 | Q9JLI2 | 172 | 0 | 0.06 | 11.90 |
| Col 18α1 | E9QPX1 | 182 | 0.07 | 0 | −11.81 |
| Dermatopontin | DERM | 24 | 0 | 7.11 | 16.06 |
| Fibronectin | FINC | 273 | 0.18 | 0.47 | 1.40 |
| Fibrinogen β chain | FIBB | 55 | 3.13 | 0.56 | −2.48 |
| Fibrinogen γ chain | FIBG | 49 | 5.94 | 1.94 | −0.45 |
| Galectin-1 | LEG1 | 15 | 8.45 | 21.18 | 1.33 |
| Galectin-3 | LG3BP | 64 | 0 | 0.63 | 13.97 |
| von Willebrand factor A | VMA5A | 87 | 0.33 | 0.84 | 1.33 |
Proteomic data for integrin-binding ECM proteins of interest (full dataset not shown). Zeroes were set to 0.00001 for calculation of Log2 fold change. The exponentially modified protein abundance index (emPAI) was used for estimation of absolute protein abundance25 and to approximate protein concentration of relevant integrin-binding proteins. Multidimensional protein identification technology (MudPIT) was used to artificially recombine fraction data from Mascot and SequestHT searches and produce quantitation that relates total protein signal in each treatment group46,47. To normalize for tissue fractionation, the dimensionless emPAI score was weighted with the concentration loaded for each fraction, i.e., 0.25 µg/µL, to calculate relative protein concentration as initial parameter values for the simulations.
Qualitatively, the majority of proteins identified were found in both the control and treatment groups; with seven proteins uniquely expressed in the CCl4 group and only one unique to the control group (Fig. 3). Collagens, glycoproteins and proteoglycans identified via proteomic analysis as ECM substrate were quantified and their relative concentration was determined (Table 1). Beta-1 and Beta-3 integrins selected for the simulations reflect those involved in hepatic events that relate to CCl4 fibrosis. Integrin-ECM binding microrates have been determined for various cell types and conditions.
Figure 3.
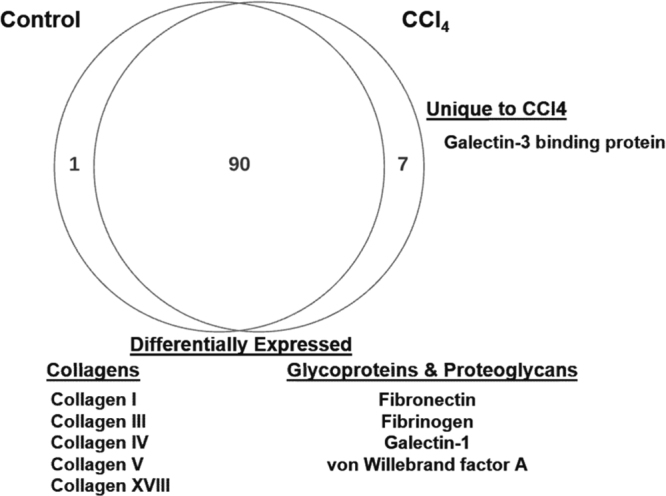
Qualitative Venn diagram of proteomic data. Differentially expressed proteins of interest for evaluation of ECMP-integrin bindings include collagens, fibrillar proteins, glycoproteins and proteoglycans. Of seven proteins uniquely expressed in the CCl4 experimental model, one ECMP protein, Galectin-3, was identified. One protein was unique to the control, and 90 were differentially expressed. ECMPs used for these simulations are listed in Table 1. Data for all relevant ECM proteins are given in Supplemental S1 Table.
Proteomic results were previously validated to confirm relative abundance of identified proteins qualitatively and quantitatively2. In particular, the amounts and distribution of collagens in the treatment group relative to the control were verified. Here, the presence of trace amounts of Col V in the CCl4 treatment group was validated to explore whether changes on the nanomolar scale would have pathological consequence. (Fig. 4).
Figure 4.

Immunofluorescent staining of hepatic cryosections. OCT sections were probed with Col V primary antibody and resolved with Alexa 488-tagged secondary antibody. The CCl4 treatment group shows a marked increase in Col V staining relative to the control.
Next, to provide for the capability of a system-level analysis, a computational framework was established using proteomic data for binding species to enable evaluation of integrin receptor binding kinetics (see modeling and experimental details in Methods). The model was developed taking into consideration sequential binding of subunits. In silico simulations of this model were parameterized using rate constants that correlate with published literature on binding, or otherwise estimated. The rates are listed in Tables 2 and 3.
Table 2.
Binding Rate Parameters.
| Integrin Species | Initial Value (nM) | Binding Kinetic Parameters (kon[s−1M−1];koff[s−1]) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Col I48,49 | Col IV49 | Fibronectin | von Willebrand factor A50 | ||||||
| kon | koff | kon | koff | kon | koff | kon | koff | ||
| α1β148,49,51 | 0.0001 | 5.6 × 104 | 1.3 × 10−3 | 8.0 × 105 | 5.0 × 10−3 | --- | --- | --- | --- |
| αVβ3 | 0.05 | --- | --- | --- | --- | 1.6 × 108 | 3.5 × 10−1 | 1.6 × 104 | 2.3 × 10−2 |
Initial conditions and reaction rate parameters of kinetic models – collagens, glycoproteins and proteoglycans. Rates for binding ECM components relevant to the CCl4 model are delineated per cognate integrin receptor. From published literature initial values of integrin receptor species are derived and given in nanomolar; similarly, binding rate parameters are given in s−1M−1 for kon and s−1 for koff.
Table 3.
Equilibrium binding constants for receptor aggregation model.
| Kd (nM) | Reaction | Microrates (on; nM−1s−1/off; s−1) | Manipulations |
|---|---|---|---|
| Ki | Integrin complex formation | k 2 /k 1 | Kp > Ki |
| increase in ligand affinity after aggregation | |||
| Kc | Filling divalent unpaired receptor | k 4 /k 3 | Kc = 0.01 Ki decreased unpaired receptor Keq for binding 2nd ligand |
| Ka | Empty receptor pairing with bound receptor | k 6 /k 5 | Ka > 0 |
| aggregation constant drives positive cooperativity | |||
| Kp | Population of empty paired receptors | k 10 /k 9 | Kp > Ki |
| increase in ligand affinity after aggregation | |||
| Kp = 100 Kx decreased aggregate receptor Keq for binding 2nd ligand | |||
| Kx | Receptor saturation | k 8 /k 7 | Kp = 100 Kx decreased aggregate receptor Keq for binding 2nd ligand |
Microrate parameters are derived from published values and set to implement positive cooperativity for sequential ligand binding and receptor aggregation. The rates for integrin complex formation (Ki) are set to simulate an increase in ligand affinity post-aggregation. Populating an empty unpaired receptor is set with a hundredth fold decrease in Keq for binding the second ligand. The aggregation equilibrium constant is set at ten times the equilibrium constant for initial complex formation to allow for aggregation to drive positive cooperativity. The population of empty paired receptors dictates an increase in ligand affinity after aggregation and is set to decrease aggregate receptor Keq for binding second ligand for receptor saturation. These parameters are adapted from Wanant et al.28, and applied here to simulate positive cooperativity in receptor aggregation pairing so that the model can be initialized and implemented with proteomic data to evaluate binding profiles.
The simulations were initialized using binding constants from published literature; where values were not available, parameters were estimated accordingly (see Methods). Collagen fragments for collagen I and IV were plotted together (Fig. 5) and assumed to have the same rates of binding for the purposes of these experiments. For the other fragmented protein, fibrinogen, only the gamma subunit was considered due to the binding motif located within this fragment26,27. The binding microrates were set to recapitulate positive cooperativity in divalent receptor saturation and in receptor aggregation pairs, as stipulated in Wanant et al.28, wherein the aggregation equilibrium constant, Ka, drove cooperativity in the aggregate model (Table 3).
Figure 5.

Kinetic simulations data. Model was initialized using ligand concentrations from proteomic analysis (Table 1) and kinetic rates listed in Table 2. The ECM:integrin binding pairs fibronectin:αvβ3, von Willebrand factor: αvβ3, and collagen I:α1β1 are shown, with binding curves and percent occupancy for fully occupied single divalent receptors (C d) and aggregated receptor pairs (A dd).
The simulation graphs in Fig. 5 show left-shifted curves with increased ECM ligand abundance, indicating increased affinity and avidity for ligand. This is reflected in both the curves for fully occupied divalent receptors (C d) and for fully occupied aggregate receptor pairs (A dd). Steady state values (SS) were recorded for each simulation (Table 4).
Table 4.
Steady state Values for Simulations of Binding.
| ECMP, Treatment | Integrin Receptor | Steady state [C d] |
|---|---|---|
| vWF, CCl4 | αvβ3 | 0.0471 |
| vWF, Control | αvβ3 | 0.0427 |
| Fibronectin, CCl4 | αvβ3 | 0.0448 |
| Fibronectin, Control | αvβ3 | 0.0369 |
| Col 1α1, CCl4 | α1β1 | 0.0001 |
| Col 1α2, CCl4 | α1β1 | 0.0001 |
| Col 1α1, Control | α1β1 | 9.99 × 10−5 |
| Col 1α2, Control | α1β1 | 9.99 × 10−5 |
| ECMP, Treatment | Integrin Receptor | Steady state [ A dd ] |
| vWF, CCl4 | αvβ3 | 0.0014 |
| vWF, Control | αvβ3 | 0.0035 |
| Fibronectin, CCl4 | αvβ3 | 0.002 |
| Fibronectin, Control | αvβ3 | 0.006 |
| Col 1α1, CCl4 | α1β1 | 7.79 × 10−10 |
| Col 1α2, CCl4 | α1β1 | 1.18 × 10−9 |
| Col 1α1, Control | α1β1 | 1.39 × 10−9 |
| Col 1α2, Control | α1β1 | 1.96 × 10−9 |
Steady state Values for Simulations of Binding. Steady state (SS) values for a fully occupied single divalent receptor (C d) and saturated aggregated receptor pairs (A dd). Top values for each parameter are given.
From these simulation data it appears that upregulated ECMPs reached steady state values in shorter time, and that aggregation of receptors produced positive cooperativity. Considering the single divalent receptor, C d, the ECM:integrin binding pairs that had the highest steady state values include both collagen 1 and fibrinogen γ chain in association with the αvβ3 integrin receptor. The combinations with the shortest time to SS were collagen 1 binding αvβ3 or α1β1 receptors. For the aggregated receptor pairs, A dd, the pairs with the highest SS values include von Willebrand factor, fibrinogen γ chain, and collagen 1 binding αvβ3, as well as fibronectin binding α5β1. The ECM:integrin pairings with the shortest time to SS for A dd pairs were collagen 1 binding αvβ3 and α1β1, and fibronectin binding α5β1. In nearly all cases, the CCl4 model ECM showed faster rise to SS compared to the control ECM; however, fibrinogen γ chain and Col 4α2 behaved in an opposite manner, owing to the fact that CCl4 actually downregulated these ECMPs in our dataset.
Sensitivity analysis was performed (Table 5) by evaluating the equilibrium constants listed in Table 3 for percent change in steady state after a ten-fold perturbation to the base values (Table 5). For simple integrin complex formation, i.e. when one ligand binds, increasing the Ki causes a significant increase in aggregated receptors, while a ten-fold decrease causes an approximate 100% decrease in aggregate pairing. Divalent receptors are moderately decreased when Ki increases, showing that decreased affinity suppresses the capacity for divalent receptor binding. Filling a single divalent receptor is negatively impacted by an increase in Kc, with aggregate receptor pairs decreasing ~83% for all three receptor:ligand pairings. A decrease in Kc positively increases the steady state for aggregate pairs, because a lower Keq for the second binding event increases affinity for receptors with one bound ligand. Kp, the constant for filling empty paired receptors, was nominally affected by perturbation, as were perturbations to filling aggregate pairs. Finally, perturbations to the aggregation constant, Ka, result in a decrease in steady sate for aggregated pairs when Ka is increased ten-fold, and an increase in steady state when Ka is decreased. This reflects the condition of a decreased equilibrium constant increasing the affinity for ligand binding, which is expected as the aggregation constant drives ligand affinity in this model.
Table 5.
Sensitivity Analysis.
| Parameter | Description | Baseline | 10 fold increase | 10 fold decrease | % Change in Steady State | |
|---|---|---|---|---|---|---|
| 10 fold increase | 10 fold decrease | |||||
| Ka | Aggregation Constant | |||||
| avb3 (vWF) | Aggregate Receptor Pair | 0.0014422 | 0.0001595 | 0.00731635 | −88.9417 | 4.07317 |
| Single Divalent Receptor | 0.047071 | 0.0496846 | 0.03206668 | 0.0555246 | −31.8759 | |
| avb3 (Fn) | Aggregate Receptor Pair | 0.0025167 | 0.0003024 | 0.00916465 | −87.9828 | 2.64149 |
| Single Divalent Receptor | 0.0448266 | 0.0494085 | 0.0250326 | 0.102215 | −44.1568 | |
| a1b1 (Col I) | Aggregate Receptor Pair | 7.787 × 10−10 | 7.788 × 10−11 | 7.7855 × 10−9 | −89.9997 | 899.743 |
| Single Divalent Receptor | 0.0001 | 0.0001 | 9.9988 × 10−5 | 1.4009 × 10−5 | −0.0001401 | |
| Kp | Population of Empty Paired Receptors | |||||
| avb3 (vWF) | Aggregate Receptor Pair | 0.0014422 | 0.0012618 | 0.00144284 | −12.5058 | 0.0466698 |
| Single Divalent Receptor | 0.047071 | 0.0470725 | 0.04706991 | 0.00319051 | −0.0022229 | |
| avb3 (Fn) | Aggregate Receptor Pair | 0.0025167 | 0.0022089 | 0.00252032 | −12.2318 | 0.142716 |
| Single Divalent Receptor | 0.0448266 | 0.0448329 | 0.04482091 | 0.0140776 | −0.0126299 | |
| a1b1 (Col I) | Aggregate Receptor Pair | 7.787 × 10−10 | 7.489 × 10−10 | 7.7875 × 10−10 | −3.83664 | −4.768 × 10−5 |
| Single Divalent Receptor | 0.0001 | 0.0001 | 0.0001 | −2.454 × 10−8 | −1.34 × 10−10 | |
| Kx | Aggregate Receptor Saturation | |||||
| avb3 (vWF) | Aggregate Receptor Pair | 0.0014422 | 0.0010941 | 0.00144273 | −24.1374 | 0.03937633 |
| Single Divalent Receptor | 0.047071 | 0.0470717 | 0.04707014 | 0.00154618 | −0.0017387 | |
| avb3 (Fn) | Aggregate Receptor Pair | 0.0025167 | 0.0019215 | 0.00251985 | −23.650441 | 0.12406835 |
| Single Divalent Receptor | 0.0448266 | 0.0448308 | 0.04482188 | 0.00949427 | −0.0104646 | |
| a1b1 (Col I) | Aggregate Receptor Pair | 7.787 × 10−10 | 7.06 × 10−10 | 7.7875 × 10−10 | −9.3388012 | −7.855 × 10−5 |
| Single Divalent Receptor | 0.0001 | 0.0001 | 0.0001 | −4.37 × 10−9 | −2.26 × 10−10 | |
| Ki | Integrin Complex Formation | |||||
| avb3 (vWF) | Aggregate Receptor Pair | 0.0014422 | 0.0142873 | 2.8307 × 10−5 | 890.686233 | −98.03722 |
| Single Divalent Receptor | 0.047071 | 0.0306523 | 0.04969114 | −34.880649 | 5.56644996 | |
| avb3 (Fn) | Aggregate Receptor Pair | 0.0025167 | 0.0206362 | 5.2505 × 10−5 | 719.962077 | −97.91377 |
| Single Divalent Receptor | 0.0448266 | 0.0236912 | 0.04943082 | −47.14931 | 10.2712401 | |
| a1b1 (Col I) | Aggregate Receptor Pair | 7.787 × 10−10 | 9.496 × 10−9 | 1.3021 × 10−11 | 1119.42405 | −98.327913 |
| Single Divalent Receptor | 0.0001 | 9.694 × 10−5 | 1 × 10−4 | −3.0666187 | −0.0017795 | |
| Kc | Single Divalent Receptor Saturation | |||||
| avb3 (vWF) | Aggregate Receptor Pair | 0.0014422 | 0.0002512 | 0.00269898 | −82.584496 | 87.1478824 |
| Single Divalent Receptor | 0.047071 | 0.0426039 | 0.04702548 | −9.4901208 | −0.0966041 | |
| avb3 (Fn) | Aggregate Receptor Pair | 0.0025167 | 0.0004241 | 0.00482601 | −83.148576 | 91.7572123 |
| Single Divalent Receptor | 0.0448266 | 0.0410334 | 0.04467362 | −8.4619004 | −0.3412082 | |
| a1b1 (Col I) | Aggregate Receptor Pair | 7.787 × 10−10 | 1.279 × 10−10 | 1.2887 × 10−9 | −83.571112 | 65.4777672 |
| Single Divalent Receptor | 0.0001 | 9.695 × 10−5 | 9.9998 × 10−5 | −3.0532152 | −0.003242 | |
Sensitivity Analysis for Integrin Receptor Concentration. Sensitivity analysis was performed by modulating equilibrium binding constants, calculating the percent change in steady state value attained by the system. Values for CCl4 treatment groups used for analysis.
Discussion
Integrin binding to ECM is a vital mechanism for cell migration, invasion, proliferation, and signal transduction between cells and their microenvironment. Diseases of chronic inflammation and injury, including fibroses and cancer, involve persistent dysregulation of ECM-integrin processes and induce remodeling of the ECM. In addition to their intrinsic utility in cellular processes, association between immune cells and the ECM is regulated via the β1 & β3 integrin receptor subfamilies29. Elucidating these complex cell-ECM-driven pathological conditions could lead to improved prognostics and clinical outcomes via more precise therapeutic management of the tissue microenvironment. Several mathematical models of integrin binding have been reported with outputs relating to spatial clustering and signal transduction, liver fibrosis, and cell migration17,19,30,31. These models recapitulated certain aspects of integrin interactions; however, these previous studies typically modeled only one ligand, mainly fibronectin or collagen, and utilized generic cognate receptor.
In this study, the relative abundance of ECM components that are canonical substrates of integrin receptors was developed for the proposed modeling framework based on experimentally-obtained liver ECM data. With binding parameters from published literature, an integrin binding pattern was established for each integrin involved in hepatic processes that are involved in fibrosis. The model from Wanant et al. was adapted to implement the basic model for divalent binding28. Specifically, this model aptly describes initial integrin binding leading to a conformational switch of the receptor complex from low- to high-affinity. A model of receptor aggregation, which can describe integrin clustering upon attachment to ECM via adhesions5, was also implemented. The simulations include how each integrin binds with cognate ECM ligands and incorporates the varying affinities that drive this interaction. From these calculations, the kinetic indices of each integrin for each of its binding partners were determined separately. The impact of changes to the ECM (e.g., in response to CCl4-induced fibrosis) on integrin binding was modeled by calibrating the substrate concentration based on the proteomic analyses. The extracellular matrix proteome was consistent with the known disease phenotype of the mouse model, with upregulation of specific ECMPs involved in fulminant fibrosis. The computational results show that in simulations using these ECMPs as substrate for key integrin receptors, interactions involving profibrotic integrins were predominant.
The CCl4 mouse model of liver fibrosis was chosen here due to its robustly characterized pathology and ECM/integrin phenotype (Fig. 2). This model is imperfect in its recapitulation of human liver fibrosis, but it is the current research standard and therefore has well-defined pathology and changes to the ECM32. Using proteomic data from CCl4-exposed mouse livers, integrin binding can be explored within the context of fulminant fibrosis. Collagen type Iα1, type III and type IV are excessively deposited due to activated hepatic stellate cells (HSCs) in response to myofibroblastic transformation induced by activated Kupffer cells and damaged hepatocytes33,34. In agreement with these established phenomena, collagens I, III, and V were upregulated in the CCl4 cohort in the current study (Table 1). Collagen I is aberrantly produced in this mouse model, and collagen V, a potent nucleating effector for the co-upregulated fibronectin, exhibited a slight increase from trace levels. In contrast, collagen IV and XVIII levels were decreased relative to the control. Interestingly, collagen XVIII was identified at relatively minimal levels in the controls, and absent in the CCl4 treated animals (Table 1). This is contrary to an expected increase in collagen XVIII following CCl4 treatment22. Nevertheless, interactions simulated with this ECMP are still based on experimental proteomic analysis. Integrin receptors were not able to be resolved with this particular method of proteomic analysis, so further proteomic analysis of integrin adhesion complexes in culture is a key component of the future directions for this project.
Owing to their involvement in several critical functions that drive homeostasis and dyshomeostasis, integrins have been identified as key druggable targets in several diseases. For example, integrin inhibitors have been evaluated to suppress liver fibrogenesis, disrupt attachment and invasion of cancer cells, and to mediate immune response35–38. Regrettably, many of these drugs fail in early trials and rarely reach clinical use, perhaps due to an incomplete understanding of integrin binding kinetics, which are traditionally based on single-species models and assumptions; indeed, even antibodies and small peptide mimetics with specificities for multiple integrins have limited clinical application39,40. Though necessary to target multiple integrins to maximize efficacy in vivo, perhaps the missing link is knowing which targeted doses are most effective for each anti-integrin molecule. In attempting to begin to develop a predictive tool for effective dosing, the primary goal of this work was to create a framework to simulate simple receptor aggregation and reproduce positive cooperativity induced by aggregate pairing. The simulations were parameterized to analyze for positive cooperativity of binding in the divalent and aggregation cases. The steady state values and time to steady state for each pairing correlated to upregulation of key ECMPs in CCl4 liver injury (Fig. 5; Table 4). The integrin receptors that predominated simulations of occupancy were consistent with those known to be at play in the disease model (Fig. 2).
This study offers a first step in which the proposed modeling framework has been initially evaluated using data from a model of fulminant fibrosis and by which other liver pathologies and how the transitional remodeling of the ECM affects ECM-integrin interactions could be explored. We acknowledge that a more comprehensive test of the model and its assumptions would require further experiments, which will be pursued in follow-up work. By testing homeostatic conditions against experimental treatment models, this platform could be broadly employed to predict or confirm changes in integrin binding (and by extension, signaling) caused by remodeling of the hepatic ECM in response to insult or injury. Longer term, a more complex stochastic model for concurrent integrin binding building upon the results of this study could be developed that considers competitive binding of multiple species. This would lay the foundation for a more detailed and nuanced analysis of ECM:integrin interactions.
Methods
All experiments were performed in accordance with the guidelines and regulations of the University of Louisville Office of Research Integrity and Institutional Review Board and Biosafety Committee.
Animals and treatments
Male C57BL/6J mice (4–6 w) were purchased from Jackson Laboratory (Bar Harbor, ME). Mice were housed in a pathogen-free barrier facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and procedures were approved by the University of Louisville’s Institutional Animal Care and Use Committee. Food and tap water were provided ad libitum. Mice were administered CCl4 (1 ml/kg i.p.; diluted 1:4 in olive oil; Sigma-Aldrich, St. Louis, MO) 2×/wk for 4 wk. Twenty-four h after the last CCl4 administration, mice were anesthetized by injection of a ketamine HCl/xylazine solution (100/15 mg/kg i.m.; Sigma-Aldrich, St. Louis, MO). Other animals received the same dose of CCl4, but only once, and were sacrificed 12–72 h after intoxication. Blood was collected from the vena cava just prior to sacrifice by exsanguination and citrated plasma was stored at −80 °C for further analysis. Portions of liver tissue were frozen immediately in liquid nitrogen, while others were fixed in 10% neutral buffered formalin or embedded in frozen specimen medium (Tissue-Tek OCT compound, Sakura Finetek, Torrance, CA) for subsequent sectioning and mounting on microscope slides.
3-step ECM extraction
Sample preparation and wash
Snap-frozen liver tissue (75–100 mg) was immediately added to ice-cold phosphate-buffered saline (pH 7.4) wash buffer containing commercially available protease and phosphatase inhibitors (Sigma Aldrich) and 25 mM EDTA to inhibit proteinase and metalloproteinase activity, respectively. While immersed in wash buffer, liver tissue was diced into small fragments and washed five times to remove contaminants. Between washes, samples were pelleted by centrifugation at 10,000 × g for 5 min and wash buffer was decanted.
NaCl extraction
Diced samples were incubated in 10 volumes of 0.5 M NaCl buffer, containing 10 mM Tris HCl (pH 7.5), proteinase/phosphatase inhibitors, and 25 mM EDTA. The samples were gently mixed on a plate shaker (800 rpm) overnight at room temperature. The following day, the remaining tissue pieces were pelleted by centrifugation at 10,000 × g for 10 min. The supernatant was saved and labeled as the NaCl fraction.
SDS extraction
The pellet from the NaCl extraction was subsequently incubated in 10 volumes (based on original weight) of a 1% SDS solution, containing proteinase/phosphatase inhibitors and 25 mM EDTA. The samples were gently mixed on a plate shaker (800 rpm) overnight at room temperature. The following day, the remaining tissue pieces were pelleted by centrifugation at 10,000 × g for 10 min. The supernatant was saved and labeled as the SDS extract.
Guanidine HCl extraction
The pellet from the SDS extraction was incubated with five volumes (based on original weight) of a denaturing guanidine buffer containing 4 M guanidine HCl (pH 5.8), 50 mM sodium acetate, 25 mM EDTA, and proteinase/phosphatase inhibitors. The samples were vigorously mixed on a plate shaker at 1200 rpm for 48 h at room temperature; vigorous shaking is necessary at this step to aid in the mechanical disruption of ECM components. The remaining insoluble components were pelleted by centrifugation at 10,000 × g for 10 minutes. This insoluble pellet was retained and solubilized as described below. The supernatant was saved and labeled as the GnHCl fraction.
Deglycosylation and solubilization
The supernatants from each extraction were desalted using Zeba Spin columns (Pierce) according to manufacturer’s instructions. The desalted extracts were then mixed with five volumes of 100% acetone and stored at −20 °C overnight to precipitate proteins. The precipitated proteins were pelleted by centrifugation at 16,000× g for 45 min. Acetone was evaporated by vacuum drying in a RotoVap for one hour. Dried protein pellets were resuspended in 500 µL deglycosylation buffer (150 mM NaCl, 50 mM sodium acetate, pH 6.8, 10 mM EDTA, and proteinase/phosphatase inhibitors) that contained chondroitinase ABC (P. vulgaris; 0.025 U/sample), endo-beta-galactosidase (B. fragilis; 0.01 U/sample) and heparitinase II (F. heparinum; 0.025 U/sample). Samples were incubated overnight at 37 °C; those containing the pellet remaining after the guanidine HCl step received 20 µL DMSO for solubilization. Protein concentrations were estimated by absorbance at 280 nm using bovine serum albumin (BSA) in deglycosylation buffer for reference standards.
LC-MS/MS analysis of samples
Sample cleanup and preparation for liquid chromatography
Pooled samples in deglycosylation buffer were thawed to room temperature and clarified by centrifugation at 5,000 × g for 5 min at 4 °C. Samples were reduced by adding 1 M DTT to 50 µL (25 µg) of each sample and then incubating at 60 °C for 30 min before addition of 8 M urea in 0.1 M Tris-HCl (pH 8.5) was added to each sample. Each reduced and diluted sample was digested with a modified Filter-Aided Sample Preparation (FASP) method. Recovered material was dried in a SpeedVac and redissolved in 200 µL of 2% v/v acetonitrile (ACN)/0.4% formic acid (FA). The samples were then trap-cleaned with a C18 PROTOTM 300 Å Ultra MicroSpin Column (The Nest Group). The sample eluates were incubated at −80 °C for 30 min, dried in a SpeedVac, and stored at −80 °C. Before liquid chromatography, dried samples were warmed to room temperature and dissolved in 2%v/v ACN/0.1% FA to a final concentration of 0.25 µg/µL. A volume of 16 µL (4 µg) of sample was injected into the Orbitrap Elite.
Liquid Chromatography
Dionex Acclaim PepMap 100, 75 µM × 2 cm nanoViper (C18, 3 µm, 100 Å) trap and Dionex Acclaim PepMap RSLC, 50 µM × 15 cm nanoViper (C18, 2 µm, 100 Å) separating column were used. An EASY n-LC (Thermo) UHPLC system was used with mobile phase buffer A (2% v/v acetonitrile/0.1% v/v formic acid), and buffer B (80% v/v acetonitrile/0.1% v/v formic acid). Following injection of the sample onto the trap, separation was accomplished with a 140 min linear gradient from 0% B to 50% B, followed by a 30 min linear gradient from 50% B to 95% B, and lastly a 10 min wash with 95% B. A 40-mm stainless-steel emitter (Thermo P/N ES542) was coupled to the outlet of the separating column. A Nanospray Flex source (Thermo) was used to position the end of the emitter near the ion transfer capillary of the mass spectrometer. The ion transfer capillary temperature of the mass spectrometer was set at 225 °C, and the spray voltage was set at 1.6 kV.
Mass Spectroscopy
An Orbitrap Elite – ETD mass spectrometer (Thermo) was used to collect data from the LC eluate. An Nth Order Double Play with ETD Decision Tree method was created in Xcalibur v2.2. Scan event one of the method obtained an FTMS MS1 scan for the range 300–2000 m/z. Scan event two obtained ITMS MS2 scans on up to ten peaks that had a minimum signal threshold of 10,000 counts from scan event one. A decision tree was used to determine whether collision induced dissociation (CID) or electron transfer dissociation (ETD) activation was used. An ETD scan was triggered if any of the following held: an ion had charge state 3 and m/z less than 650, an ion had charge state 4 and m/z less than 900, an ion had charge state 5 and m/z less than 950, or an ion had charge state greater than 5; a CID scan was triggered in all other cases. The lock mass option was enabled (0% lock mass abundance) using the 371.101236 m/z polysiloxane peak as an internal calibrant.
Proteome Data Analysis
Proteome Discoverer v1.4.0.288 was used to analyze the data collected by the mass spectrometer. The database used in Mascot v2.4 and SequestHT searches was the 6/2/2014 version of the UniprotKB Mus musculus reference proteome canonical and isoform sequences. In order to estimate the false discovery rate, a Target Decoy PSM Validator node was included in the Proteome Discoverer workflow. The Proteome Discoverer analysis workflow allows for extraction of MS2 scan data from the Xcalibur RAW file, separate searches of CID and ETD MS2 scans in Mascot and Sequest, and collection of the results into a single file (.msf extension). The resulting.msf files from Proteome Discoverer were loaded into Scaffold Q + S v4.3.2. Scaffold was used to calculate the false discovery rate using the Peptide and Protein Prophet algorithms. The results were annotated with mouse gene ontology information from the Gene Ontology Annotations Database.
Computational Modeling
First is considered the divalent receptor model that corresponds to ECM ligand binding of the α subunit occurring prior to the β subunit7,28,41, where k 1 is the first-order association rate constant and k 2 is the dissociation constant for singly occupied receptors (C m). C d indicates a fully occupied integrin receptor with two bound ECM ligands, and k 3 and k 4 define the rate constants for association and dissociation, respectively, of the doubly bound integrin receptor. Differential equations for this model are:
| 1 |
| 2 |
| 3 |
| 4 |
The scheme for receptor aggregation and ligand binding is shown in Fig. 6. In the model of receptor aggregation, we utilized the same scheme as Wanant et al.28, wherein receptors pair in a manner such that either singly- or doubly-bound receptors can aggregate only with an unbound receptor with the aggregation equilibrium constant KA (where KA = k 5 /k 6), and disaggregation equilibrium constant KA’ (KA’ = k 6 /k 5). Binding constants for an additional ECM ligand binding to the unbound portion of an aggregate pair are the same regardless of whether the bound portion has one or two ligands, where the equilibrium association constant is KC = k 9 /k 10. The equilibrium constant for adding a second ECM ligand to a singly bound receptor in any pair-configuration is KF = k 7 /k 8. The differential equations describing receptor aggregation are listed below:
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
| 11 |
| 12 |
| 13 |
where A im indicates an aggregate pair comprised of one unbound integrin receptor coupled with a singly-bound receptor; A id is the same combination, except featuring a doubly-bound receptor. A pair with two singly bound receptors is defined as A mm, with two doubly bound receptors is A dd, and A md indicates a singly bound receptor paired with a doubly bound one (Fig. 6).
Figure 6.

Model description: (a), mass-action kinetics scheme of species variables. Divalent receptors bind ligand sequentially to α, β subunits, with Ki = k 2 /k 1 for equilibrium of initial binding event (C m) and Kc = k 4 /k 3 for fully occupied ECMP-Integrin receptor complex (C d). The receptor aggregation scheme incorporates divalent binding and receptor pairing such that half/fully occupied receptors can aggregate only with an unbound receptor [aggregation equilibrium constant Ka = k 6 /k 5]. Binding constants for an additional ECM ligand binding to unbound receptor in an aggregate pair are the same regardless of occupancy status of its paired receptor [population equilibrium constant is Kp = k 10 /k 9]. The equilibrium constant for adding a second ECM ligand to a singly bound receptor in any pair-configuration is Kx = k 8 /k 7. Adapted from Wanant and Quon (2000)28. (b), model species description.
The affinity of integrin receptors for ECM proteins fibronectin and laminin are generally in the micromolar range. The Kd measured for ECM:integrin and, in particular, fibronectin binding, ranges between approximately 10−7–10−6 M42; Takagi et al. report nanomolar Kd values for fibronectin binding43. Mallet et al. utilized a Kd of 2 × 10−4 M for tethered RGD peptides in their model of integrin binding17.
Simulations
Computer simulations were run using Spyder for Tellurium software version 2.3.5.2; Python version 2.744. Binding curves were plotted using SigmaPlot 13.0. The model was initialized using ligand concentrations from proteomic analysis (Table 1) and initial integrin concentrations were derived from published values. Ligand concentration was developed by collapsing the fractionated sample data using MudPIT functionality in Scaffold. Rappsilber et al. defined protein abundance index (PAI) for estimation of absolute protein abundance45, and Ishihama et al. report that the emPAI, i.e. exponentially modified PAI, is approximately proportional to protein abundance25. Using the emPAI quantitative method, proteomic output was normalized by the tissue loading concentration of 0.25 µg/µL; these values for concentration were then divided by the molecular weight of the protein to convert to molar concentration. Kinetic rates listed in Table 2 were used to calculate microrate parameters relative to the established binding rates from literature; where exact microrates were unavailable, rates were estimated from various published literature sources. Table 3 relates the equilibrium constants of the system relative to initial integrin complex formation, such that subsequent binding and clustering steps produce cooperativity when simulated in these proportions. Sensitivity analysis was performed by varying levels of integrin receptor concentration in 10-fold increments, to explore binding when surface membrane integrin receptor expression is upregulated or downregulated as a consequence of disease state or in response to microenvironmental fluctuations.
Data Availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
Electronic supplementary material
Acknowledgements
We are grateful to William L. Dean (Biochemistry & Molecular Biology, University of Louisville) for useful discussions and advice.
Author Contributions
Conceived and organized the study (S.V.H., H.B.F., G.E.A.); directed proteomics analysis (M.L.M.); directed animal study for proteomics samples (V.L.M., J.I.B.); prepared proteomic samples (L.G.P.); performed proteomic analysis (C.E.D., D.W.); implemented modeling and simulations (S.V.H.); analyzed the results (S.V.H., H.B.F.); wrote initial manuscript (S.V.H.); revised and approved final manuscript (S.V.H., G.E.A., H.B.F.).
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Hermann B. Frieboes and Gavin E. Arteel jointly supervised this work.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-017-12691-y.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hynes RO. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Massey VL, et al. The hepatic “matrisome” responds dynamically to injury: characterization of transitional changes to the extracellular matrix. Hepatology. 2017;65:969–982. doi: 10.1002/hep.28918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox TR, Erler JT. Remodeling and homeostasis of the extracellular matrix: implications for fibrotic diseases and cancer. Dis Model Mech. 2011;4:165–178. doi: 10.1242/dmm.004077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poole, L. G. & Arteel, G. E. Transitional remodeling of the hepatic extracellular matrix in alcohol-induced liver injury. BioMed Res Intl2016 (2016). [DOI] [PMC free article] [PubMed]
- 5.Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci. 2006;119:3901–3903. doi: 10.1242/jcs.03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hynes RO. Integrins: a family of cell surface receptors. Cell. 1987;48:549–554. doi: 10.1016/0092-8674(87)90233-9. [DOI] [PubMed] [Google Scholar]
- 7.Hynes RO. Integrins. Cell. 2002;110:p673–p687. doi: 10.1016/S0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 8.Patsenker E, Stickel F. Role of integrins in fibrosing liver diseases. Am J Physiol Gastrointest Liver Physiol. 2011;301:G425–G434. doi: 10.1152/ajpgi.00050.2011. [DOI] [PubMed] [Google Scholar]
- 9.Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011;3:a004994. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Masson-Gadais B, Pierres A, Benoliel AM, Bongrand P, Lissitzky JC. Integrin (alpha) and beta subunit contribution to the kinetic properties of (alpha)2beta1 collagen receptors on human keratinocytes analyzed under hydrodynamic conditions. J Cell Sci. 1999;112:2335–2345. doi: 10.1242/jcs.112.14.2335. [DOI] [PubMed] [Google Scholar]
- 11.Legate KR, Wickstrom SA, Fassler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes & Dev. 2009;23:397–418. doi: 10.1101/gad.1758709. [DOI] [PubMed] [Google Scholar]
- 12.Pouwels J, Nevo J, Pellinen T, Ylänne J, Ivaska J. Negative regulators of integrin activity. J Cell Sci. 2012;125:3271–3280. doi: 10.1242/jcs.093641. [DOI] [PubMed] [Google Scholar]
- 13.Zhao T, Li Y, Dinner AR. How focal adhesion size depends on integrin affinity. Langmuir. 2009;25:1540–1546. doi: 10.1021/la8026804. [DOI] [PubMed] [Google Scholar]
- 14.Plow EF, Haas TA, Zhang L, Loftus J, Smith JW. Ligand binding to integrins. J Biological Chem. 2000;275:21785–21788. doi: 10.1074/jbc.R000003200. [DOI] [PubMed] [Google Scholar]
- 15.Sánchez-Cortés J, Mrksich M. The platelet integrin aIIbb3 binds to the RGD and AGD motifs in fibrinogen. Chem Biol. 2009;16:990–1000. doi: 10.1016/j.chembiol.2009.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garratt AN, Humphries MJ. Recent insights into ligand binding, activation and signalling by integrin adhesion receptors. Acta Anatomica. 1995;154:34–45. doi: 10.1159/000147750. [DOI] [PubMed] [Google Scholar]
- 17.Mallet DG, Pettet GJ. A mathematical model of integrin-mediated haptotactic cell migration . Bull Math Biol. 2006;68:231–253. doi: 10.1007/s11538-005-9032-1. [DOI] [PubMed] [Google Scholar]
- 18.Dutta-Moscato J, et al. A multiscale agent-based in silico model of liver fibrosis progression. Front Bioeng Biotechnol. 2014;2:18. doi: 10.3389/fbioe.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Welf ES, Naik UP, Ogunnaike BA. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophys J. 2012;103:1379–1389. doi: 10.1016/j.bpj.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baiocchini A, et al. Extracellular matrix molecular remodeling in human liver fibrosis evolution. PLoS One. 2016;11:e0151736. doi: 10.1371/journal.pone.0151736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iredale JP, Thompson A, Henderson NC. Extracellular matrix degradationin liver fibrosis: Biochemistry and regulation. Biochimica et Biophysica Acta Mol Basis Disease. 2013;1832:876–883. doi: 10.1016/j.bbadis.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Duncan MB, et al. Type XVIII collagen is essential for survival during acute liver injury in mice. Dis Model Mech. 2013;6:942–951. doi: 10.1242/dmm.011577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Y, Liu J, Waalkes M, Kang YJ. Changes in the gene expression associated with carbon tetrachloride-induced liver fibrosis persist after cessation of dosing in mice. Toxicological Sci. 2004;79:404–410. doi: 10.1093/toxsci/kfh120. [DOI] [PubMed] [Google Scholar]
- 24.Wisniewski JR, Hein MY, Cox J, Mann M. A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol Cell Proteomics. 2014;13:3497–3506. doi: 10.1074/mcp.M113.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishihama Y, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by th enumber of sequeced peptides per protein. Mol & Cellular Proteomics. 2005;4:1265–1272. doi: 10.1074/mcp.M500061-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Peerschke EI, Francis CW, Marder VJ. Fibrinogen binding to human blood platelets: effect of gamma chain carboxyterminal structure and length. Blood. 1986;67:385–390. [PubMed] [Google Scholar]
- 27.Ware S, Donahue JP, Hawiger J, Anderson WF. Structure of the fibrinogen gamma-chain integrin binding and factor XIIIa cross-linking sites obtained through carrier protein driven crystallization. Protein Sci. 1999;8:2663–2671. doi: 10.1110/ps.8.12.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wanant S, Quon MJ. Insulin receptor binding kinetics: Modeling and simulation studies. J Theor Biol. 2000;205:355–364. doi: 10.1006/jtbi.2000.2069. [DOI] [PubMed] [Google Scholar]
- 29.Bruck R, et al. The use of synthetic analogues of Arg-Gly-Asp (RGD) and soluble receptor of tumor necrosis factor to prevent acute and chronic experimental liver injury. Yale J Biol Med. 1997;70:391–402. [PMC free article] [PubMed] [Google Scholar]
- 30.Caré BR, Soula HA. Impact of receptor clustering on ligand binding. BMC Systems Biol. 2011;5:48. doi: 10.1186/1752-0509-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nieto N, Lutolf MP. Extracellular matrix bioengineering and systems biology approaches in liver disease. Systems Synthetic Biol. 2011;5:11. doi: 10.1007/s11693-011-9085-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delire B, Stärkel P, Leclercq I. Animal models for fibrotic liver diseases: What we have, what we need, and what is under development. J Clin Translational Hepatol. 2015;3:53–66. doi: 10.14218/JCTH.2014.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman SL. Molecular Regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biological Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 34.Liedtke C, et al. Experimental liver fibrosis research: update on animal models, legal issues and translational aspects. Fibrogenesis Tissue Repair. 2013;6:19. doi: 10.1186/1755-1536-6-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosenow F, et al. Integrins as antimetastatic targets of RGD-independent snake venom components in liver metastasis. Neoplasia. 2008;10:168–176. doi: 10.1593/neo.07898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimaoka M, Springer TA. Therapeutic antagonists and conformational regulation of integrin function. Nature Rev Drug Discovery. 2003;2:703–716. doi: 10.1038/nrd1174. [DOI] [PubMed] [Google Scholar]
- 37.Henderson NC, et al. Targeting of av integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nature Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agarwal SK. Integrins and cadherins as therapeutic targets in fibrosis. Front Pharmacol. 2014;5:131. doi: 10.3389/fphar.2014.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Millard M, Odde S, Neamati N. Integrin targeted therapeutics. Theranostics. 2011;1:154–188. doi: 10.7150/thno/v01p0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodman SL, Picard M. Integrins as therapeutic targets. Trends Pharmacol Sci. 2012;33:405–412. doi: 10.1016/j.tips.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 41.Sauro, H. M. Enzyme Kinetics for Systems Biology, 2nd Ed.(Ambrosius Publishing and Future Skill Software, Seattle, 2012).
- 42.Buck CA, Horwitz AF. Cell surface receptors for extracellular matrix molecules. Ann Rev Cell Biol. 1987;3:179–205. doi: 10.1146/annurev.cb.03.110187.001143. [DOI] [PubMed] [Google Scholar]
- 43.Takagi J, Strokovich K, Springer TA, Walz T. Structure of integrin a5b1 in complex with fibronectin. EMBO J. 2003;22:4607–4615. doi: 10.1093/emboj/cdg445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi, K., Medley, J. K., König, M., Stocking, K. & Cannistra, C. Tellurium, version 2.3.5.2. Computer Program (2014).
- 45.Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Mol & Cellular Proteomics. 2002;4:1265–1272. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kislinger T, Gramolini AO, MacLennan DH, Emili A. Multidimensional protein identification technology (MudPIT): Technical overview of a profiling method optimized for the comprehensive proteomic investigation of normal and diseased heart tissue. Proteomics and Disease. 2005;16:1207. doi: 10.1016/j.jasms.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 47.Schirmer EC, Yates IJR, Gerace L. MudPIT: A powerful proteomics tool for discovery. Discov Med. 2003;3:38–39. [PubMed] [Google Scholar]
- 48.Kim JK, et al. A novel binding site in collagen type III for integrins a1b1 and a2b1. J Biological Chem. 2005;280:32512–32520. doi: 10.1074/jbc.M502431200. [DOI] [PubMed] [Google Scholar]
- 49.Kern A, Eble J, Golbik R, Kühn K. Interaction of type IV collagen with the isolated integrins a1b1. Eur J Biochem. 1993;215:151–159. doi: 10.1111/j.1432-1033.1993.tb18017.x. [DOI] [PubMed] [Google Scholar]
- 50.Denis C, Williams JA, Lu X, Meyer D, Baruch D. Solid-phase von Willebrand factor contains a conformationally active RGD motif that mediates endothelial cell adhesion through the alpha v beta 3 receptor. Blood. 1993;82:3622–3630. [PubMed] [Google Scholar]
- 51.Xu Y, et al. Multiple binding sites in collagen type I for the integrins a1b1 and a2b1. J Biological Chem. 2000;275:38981–38989. doi: 10.1074/jbc.M007668200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.