

Abstract
Glioma is a malignant tumor for which new therapies are needed. Growing evidence has demonstrated that microRNAs (miRNAs) have a major effect on glioma development. Here, we aimed to characterize a novel anti-cancer miRNA, miR-625, by investigating its expression, function, and mechanism of action in glioma progression. The expression of miR-625 and its target mRNA in human glioma tissues and cell lines was assessed by real-time PCR, western blotting, and immunohistochemistry. Functional significance was assessed by examining cell cycle progression, proliferation, apoptosis, and chemosensitivity to temozolomide in vitro, and by examining growth of subcutaneous glioblastoma in a mouse model in vivo. We found that miR-625 expression was significantly lower in human glioma samples and cell lines than in normal brain tissue and human astrocytes. Furthermore, miR-625 overexpression not only suppressed glioma cell proliferation in culture and in the tumor xenograft model but also induced cell cycle arrest and apoptosis. AKT2 was identified as a direct miR-625 target in glioma cell lines, and AKT2 overexpression reversed the suppressive effects of miR-625 in the cell lines and the tumor xenograft model. Finally, we found that the sensitivity of glioma cells to temozolomide was increased by miR-625 overexpression, and this was reversed by concomitant AKT2 expression. In conclusion, our findings suggest that the miR-625-AKT2 axis could be a new prognostic marker and diagnostic target for gliomas.
Keywords: Glioma, miR-625, AKT2, proliferation
Introduction
Gliomas, the most common malignant brain tumors in adults, frequently harbor genetic abnormalities that make them refractory to treatment, leading to high morbidity and mortality [1]. The prognosis of glioma is highly dependent on the pathological grade, with the highest grades associated with worse prognosis [2]. The current standardized treatment for gliomas is comprehensive, involving surgical resection followed by chemotherapy and radiotherapy. Despite advances in modern therapy that have prolonged survival, the prognosis for patients with glioblastoma multiforme (GBM) remains grim, and the median survival time is only ~15 months after diagnosis [3]. Temozolomide (TMZ) is an antineoplastic DNA-alkylating drug that readily crosses the blood-brain barrier and is currently used to treat glioblastoma. TMZ can efficiently suppress the proliferation and promote apoptosis of glioma cell lines [4,5]. Unfortunately, GBM cells eventually develop resistance to this drug, and effective treatment remains challenging. Therefore, there is an urgent need for new therapeutic strategies and prognostic indicators to improve the survival rate of patients with glioma.
MicroRNAs (miRNAs) are a class of conserved endogenously expressed small noncoding RNAs (18-23 nucleotides) that post-transcriptionally regulate gene expression by binding to partially complimentary sequences in the 3’-untranslated regions (3’-UTRs) of mRNAs [6]. MiRNAs play a pivotal role in a variety of biological functions, including cell proliferation, invasion, apoptosis, differentiation, and development. Accordingly, aberrant expression of miRNAs is observed in many types of cancers, including gliomas [7-9]. Considerable evidence has shown that many miRNAs act as tumor promoters or suppressors, depending on their target mRNAs; consequently, miRNA function has been extensively investigated in various cancers [10,11]. A recent report examined the potential functions of miRNAs in the malignancy of human gliomas by comparing miRNA expression profiles in primary low-grade gliomas and secondary high-grade gliomas from individual patients [12]. To date, however, our understanding of how miRNAs affect glioma development and progression remains unclear.
AKT2, also known as protein kinase B, is a serine/threonine kinase [13] that plays a central role in signaling for cell survival, metastasis, metabolism, radioresistance, and drug resistance [14]. Aberrant AKT2 expression is known to contribute to the dysregulated differentiation, proliferation, metastasis, and invasion of various types of cancer cells, such as breast [15], hepatocellular [16], and ovarian [17] cancers. However, the expression, function, and molecular mechanisms of action of AKT2 in glioma are not known.
The aim of this study was to further our understanding of miR-625 by studying its function and mechanisms of action in human glioma. We show that miR-625 is downregulated in human glioma compared with normal human brain tissues (NBTs), and its expression regulates the metastatic capacity and tumorigenic properties of glioma cells, including tumor initiation, colony formation, and drug resistance phenotypes. Moreover, miR-625 acts as a novel tumor suppressor by directly targeting AKT2 mRNA. Our results not only provide new insights into the molecular mechanisms of glioma pathogenesis but also highlight new therapeutic opportunities for the development of glioma treatments.
Materials and methods
Human tissue samples
Gene expression data were obtained from The Cancer Genome Atlas (TCGA) database (http://tcga-data.nci.nih.gov). NBTs (n = 5) and glioma specimens (n = 26) were obtained postoperatively from the Department of Neurosurgery, the First Affiliated Hospital of Nanjing Medical University, China. The pathologic features of all tissues samples were classified and graded by pathologists according to the World Health Organization pathologic classification. None of the patients had received radiotherapy or chemotherapy before tumor resection. The study protocol was approved by the Ethics Committee of Nanjing Medical University, China, and written informed consent was obtained from all participants. All samples were frozen immediately after collection and stored in liquid nitrogen until analysis.
Cell culture and antibodies
Human glioma cell lines U87, LN229, U251, A172, and U118 were obtained from the Chinese Academy of Sciences Cell Bank (Shanghai, China). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 ng/ml streptomycin, and were maintained at 37°C in a humidified 5% CO2 atmosphere. Normal human astrocytes (NHAs) were purchased from Lonza (Walkersville, MD, USA) and cultured according to the supplier’s instructions. Antibodies against AKT2 and O6-methylguanine-DNA methyltransferase (MGMT) were purchased from Cell Signaling Technology. An antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was obtained from Beyotime Biotechnology. Antibodies against Cyclin D1, Cyclin E1, and cyclin-dependent kinase 4 (CDK4) were obtained from Open Biosystems. TMZ was obtained from Sigma-Aldrich.
Oligonucleotides, plasmid construction, and transfection
Lentiviruses carrying miR-625 or a miRNA negative control (miR-NC) were produced by Ribobio (Guangzhou, China). Small interfering (si) RNA targeting AKT2 and noncoding control siRNA (si-Ctrl) oligonucleotides were obtained from GenePharma (Shanghai, China). A plasmid containing the human AKT2 coding sequence was constructed according to the manufacturer’s instructions (Genechem; Shanghai, China). A human AKT2 cDNA was inserted into the vector pGL3 to generate pGL3-AKT2. Oligonucleotides and plasmids were transfected into cells using Lipofectamine 2000 Transfection Reagent (Invitrogen Corporation) according to the manufacturer’s instructions.
Lentiviral packaging and establishment of stably transfected cell lines
A lentiviral packaging kit was purchased from Genechem. Lentiviruses carrying miR-625 or miR-NC were packaged in HEK293T cells and harvested from the culture supernatant according to the kit manufacturer’s instructions. Stable cell lines were established by selection of infected U87 cells and U251 cells with puromycin.
RNA Isolation and quantitative reverse-transcriptase PCR
RNA was extracted from harvested human glioma samples, NBTs, glioma cells, or NHAs using TRIzol reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. Primers for quantitative RT-(qRT)-PCR of AKT2 and miR-625 were purchased from Ribobio. Quantitative real-time PCR was conducted using an ABI StepOne Plus system (Applied Biosystems, CA, USA) with the Bulge-loop miRNA qRT-PCR Primer Kit (Ribobio) to detect miR-625. Expression of U6 was measured as an endogenous control. All reactions were performed in triplicate and repeated three times. The fold change in expression was calculated by the 2-ΔΔCt method [18].
Protein extraction and western blotting
Protein extraction and western blot analysis were performed as described previously [19]. Briefly, cells or tissues were lysed on ice for 30 min in RIPA buffer (150 mM NaCl, 100 mM Tris, pH 8.0, 0.1% sodium dodecyl sulfate [SDS], 1% Triton X-100, 1% sodium deoxycholate, 5 mM EDTA, and 10 mM sodium fluoride) supplemented with 1 mM sodium vanadate, 2 mM leupeptin, 2 mM aprotinin, 1 mM phenylmethylsulfonyl fluoride, 1 mM dithiothreitol, and 2 mM pepstatin A. The lysates were centrifuged at 12,000 rpm at 4°C for 15 min, and the supernatants were collected and assayed for protein concentration using the bicinchoninic acid assay (KenGEN, Jiangsu, China). Proteins were separated by SDS-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes (Thermo Fisher Scientific, MA, USA) in transfer buffer (20 mM Tris, 150 mM glycine, and 20% methanol). Membranes were blocked with 5% nonfat milk for 2 h and then incubated with primary and secondary antibodies. An electrochemiluminescence detection system (Thermo Fisher Scientific) was used for signal detection. GAPDH was probed as an internal control.
Cell proliferation and colony-forming assays
To assess cell proliferation, cells were seeded at 2 × 103 per well in 96-well plates, incubated for 24, 48, or 96 h, Cell growth was measured using a CCK-8 kit (Dojindo Laboratories) following the manufacturer’s protocol. Assays were performed in triplicate. To assess colony-forming ability, 5 × 102 cells were seeded in a 6-well plate and cultured for 2 weeks. Cells were fixed with 100% methanol and stained with 0.1% crystal violet for 30 min. Colony-forming efficiency was calculated as the cell activity after transfection. Experiments were performed three times, each in triplicate. To quantify DNA synthesis, the 5-ethynyl-2-deoxyuridine (EDU) assay was performed using a Cell-Light EDU In Vitro Imaging Detection Kit (Guangzhou Ribobio) according to the manufacturer’s instructions. Cells were visualized by fluorescence microscopy. Data were recorded from three separate experiments, each with four replicates.
Fluorescence in situ hybridization (FISH)
MiR-625 expression in glioma specimens and NBTs was also detected by FISH. The 5’-FAM-labeled miR-625 probe sequence (5’-GGACTATAGAACTTTCCCCCT-3’) was synthesized by GoodBio (Wuhan, China). The FISH procedure followed by the BioSense manufacturer’s instructions. The frozen tissues were fixed with 4% paraformaldehyde for 25 min and then washed three times with phosphate-buffered saline (PBS). The sections were digested with proteinase K at 37°C for 5 min and dehydrated in 70%, 85%, and 100% ethanol for 10 min. The probe was added to the sections, which were denatured at 78°C for 5 min and hybridized overnight at 42°C in a humidified chamber. The sections were washed sequentially with pre-warmed 2× saline sodium citrate (SSC) at 37°C for 10 min, 1× SSC at 37°C for 10 min, and 0.5× SSC at 37°C for 10 min. Finally, sections were counterstained with 4’,6-diamidino-2-phenylindole (DAPI; Sigma) for 10 min and examined with a Zeiss LSM 700 Meta confocal microscope (Oberkochen, Germany).
Immunohistochemistry (IHC)
IHC detection of AKT2 (antibody from Abcam), cleaved caspase-3, and Ki-67 (antibodies from Cell Signaling Technology) in subcutaneous tumor tissues from nude mice was performed as described previously [20].
Flow cytometric analysis of cell cycle progression and apoptosis
For the cell cycle distribution assay, cells transfected with lentiviruses and/or plasmids were harvested and fixed with 70% ethanol at -20°C for 24 h. Cells were resuspended in Cell Cycle Staining Kit reagent (Multi Sciences, Hangzhou, China), incubated for 30 min in the dark, and then analyzed by flow cytometry. The apoptosis assay was performed using an Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, CA, USA) according to the manufacturer’s instructions. For both assays, cells were analyzed using a Gallios flow cytometer (Beckman Coulter, CA, USA).
Dual luciferase reporter assay
The wild-type (WT) and mutated putative (mut) miR-625 target sequence in the AKT2 3’-UTR were amplified from human AKT2 cDNA by PCR and then cloned into the SacI and HindIII sites of the pmiRNA-Report firefly luciferase vector (Genechem). U87 and U251 cells were seeded in a 24-well plate and co-transfected with the WT or mut reporter plasmid, a Renilla luciferase plasmid, and miR-625 mimic or miR-NC. Luciferase activities were measured 24 h after transfection using a dual luciferase assay kit (Promega, Madison, WI, USA). Each experiment was performed in triplicate and repeated at least once.
In vitro chemosensitivity assay
Cells were seeded overnight at a density of 3 × 103 cells per well in a 96-well plate. Freshly prepared TMZ solution was added to the cells at final concentrations ranging from 25 μM to 400 μM. Cell survival was assessed using the CCK-8 assay 48 h later. Percentage cell survival was normalized to cells incubated without TMZ. U87 and U251 cells transfected with miR-NC, miR-625, or miR-625+AKT2 were incubated with 100 μM TMZ, and cell survival was assessed every 24 h. The percentage cell survival was normalized to that on day 0.
Nude mouse model of subcutaneous glioma
Eight male BALB/c nude mice (6 weeks old) were purchased from Shanghai Laboratory Animal Experimental Animal Center of the Chinese Academy of Sciences. Animal experiments were approved by the Animal Management Rule of the Chinese Ministry of Health (document 55, 2001) and approved by the Animal Experimental Ethics Committee of Nanjing Medical University. Mice were randomly divided into two groups of four and injected subcutaneously with viable U87 cells transfected with either miR-625 or miR-NC (5 × 105 cells/injection). Once tumors became visible, the sizes were measured with a vernier caliper every 3 days. Tumor volume was calculated as: volume = 0.5 × length × width2. Mice were euthanized 30 days after injection, and the tumors were excised, trimmed, and weighed. Protein and RNA extracts were prepared for western blotting and qRT-PCR analysis.
Statistical analysis
All experiments were performed in triplicate, and data are presented as the mean ± standard deviation. Differences between groups were analyzed using Student’s t test. Correlations between miR-625 expression and AKT2 levels in glioma tissues were analyzed using Spearman’s rank test. P < 0.05 was considered statistically significant.
Results
MiR-625 is downregulated in human glioma tissues and cell lines
To assess the expression of miR-625, we performed real-time PCR analysis of five NBT samples and 26 glioma tissue samples. The results show that miR-625 was expressed at much lower levels in glioma tissues compared with NBTs (Figure 1A). Moreover, glioma samples classified as WHO grades II (n = 7), III (n = 7), and IV (n = 12) expressed significantly lower levels of miR-625 compared with NBTs (P < 0.001, Figure 1B), and significantly lower levels were detected in grade III-IV tumors than in grade II tumors (P < 0.05, Figure 1B). These results were confirmed by FISH analysis of miR-625 expression in representative grades II-IV glioma samples (Figure 1C). We also assessed miR-625 levels in the human glioma cell lines U87, U251, LN229, A172, and U118, and found that expression was lower in glioma cells, particularly U87 and U251 cells, than in NHAs (Figure 1D). Thus, miR-625 expression levels are lower in glioma tumor tissues and cell lines than in NBTs and NHAs, and the expression levels inversely correlate with glioma grade.
Figure 1.
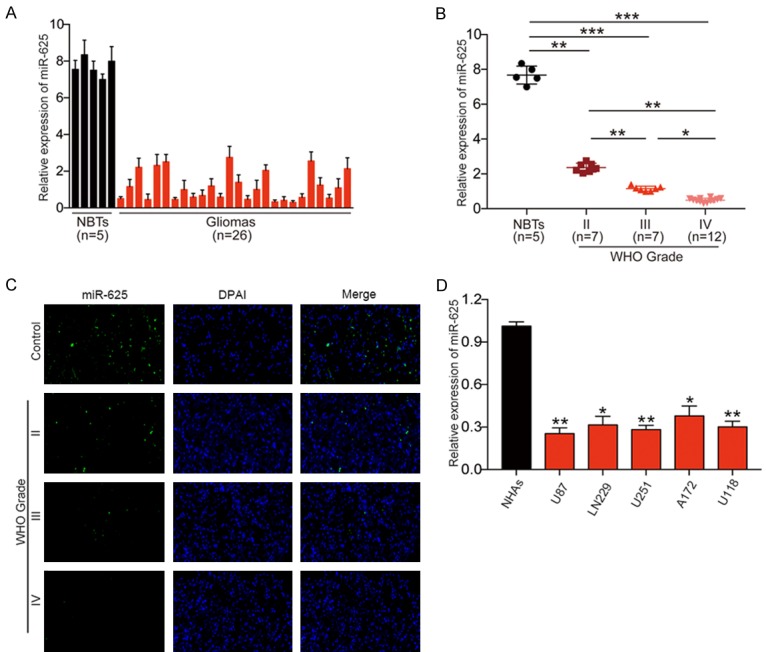
Downregulation of miR-625 in glioma tissues and cell lines. A. qRT-PCR analysis of miR-625 expression in normal brain tissues (NBTs, n = 5) and glioma tissues (n = 26). B. qRT-PCR analysis of miR-625 expression in NBTs (n = 5) and glioma specimens (n = 26) divided according to WHO pathological classification criteria into grade II (n = 7), grade III (n = 7), and grade IV (n = 12). C. FISH analysis of miR-625 expression shows positive correlation with WHO grade in glioma specimens. D. qRT-PCR analysis of miR-625 expression in normal human astrocytes (NHAs) and five glioma cell lines (U87, LN229, U251, A172, U118). *P < 0.05, **P < 0.01, ***P < 0.001.
MiR-625 overexpression suppresses cell proliferation and colony formation and induces G0/G1 arrest in glioma cell lines
To investigate the biological functions of miR-625 in glioma cells, we examined the effects of miR-625 overexpression on the proliferation and colony-forming ability of U87 and U251 cells. Lentiviral-mediated overexpression of miR-625 significantly decreased the proliferative capacity (Figure 2A) and the colony-forming ability (Figure 2B) of both cell lines compared with cells expressing miR-NC or mock-transduced cells. Similarly, EDU assays showed that DNA synthesis was significantly suppressed in miR-625-transduced cells compared with miR-NC- or mock-transduced cells.
Figure 2.

MiR-625 overexpression induces cell cycle arrest and inhibits glioma cell growth in vitro. A. CCK-8 assay of proliferation of U87 and U251 glioma cell lines transfected with miR-NC or miR-625. B. Colony-forming assays of U87 and U251 cells transfected with miR-NC or miR-625. C. Representative single or merged images of DAPI- and EDU-stained U87 and U251 cells transfected with miR-NC or miR-625. D. Flow cytometric analysis of cell cycle phase of U251 and U87 cells transfected with miR-NC or miR-625. E. Western blot analysis of Cyclin D1, CDK4, and Cyclin E1 in U87 and U251 cells 48 h after transfection with miR-NC or miR-625. GAPDH served as a loading control. **P < 0.01.
Because inhibition of cell proliferation often involves changes in cell cycle progression, we next analyzed the cell cycle distribution of cells using a flow cytometric assay. We found that miR-625 overexpression significantly increased the percentage of cells in G0/G1 phase and decreased the percentage of cells in S phase compared with the mock-transduced and miR-NC-expressing cells (Figure 2D). Consistent with this, expression of the cell cycle-related protein Cyclin D1 was clearly reduced in the miR-625-overexpressing group compared with control groups (Figure 2E). However, miR-625 had no effects on Cyclin E1 or CDK4 expression. The cyclin and CDK proteins have previously been reported to be important regulators of entry into the G1 phase [21,22]. Therefore, these results suggest that miR-625 suppresses glioma cell cycle progression.
AKT2 is a direct target of miR-625 in glioma cells
To understand the molecular mechanisms by which miR-625 influences glioma biology, we used the bioinformatics algorithms miRNAWalk 2.0 and TargetScan to identify potential miR-625 target genes. Among the candidates identified was AKT2, which contained a 3’-UTR sequence complementary to the seed sequence of miR-625 (Figure 3A). To determine whether miR-625 affects AKT2 expression in glioma, we analyzed miR-625-overexpressing U87 and U251 cells. These analyses showed that levels of AKT2 mRNA (Figure 3B) and protein (Figure 3D) were both increased by miR-625 expression in glioma cell lines. To verify that AKT2 mRNA is an authentic target of miR-625, we tested U87 and U251 cells stably expressing miR-625 or miR-NC in luciferase reporter assays. As shown in Figure 3C, luciferase activity was significantly inhibited by miR-625 transduction in U87 and U251 cells expressing a reporter driven by the WT AKT2 3’-UTR, but not the mutated AKT2 3’-UTR. To determine whether the miR-625-AKT2 relationship also exists in human brain tissues, we first analyzed data from TCGA and GSE16011 databases. Both datasets showed that AKT2 mRNA levels were significantly higher in GBM samples than in NBTs (Figure 3E, 3F). Next, we measured AKT2 protein levels in the cell lines and brain tissues. Consistent with the mRNA analyses, AKT2 levels were higher in glioma cell lines than in NHAs (Figure 3G) and also higher in glioma tissues (n = 26) than in NBTs (n = 5, P = 0.0037, Figure 3H). Spearman’s rank correlation analysis showed that miR-625 and AKT2 levels were inversely correlated in 26 glioma specimens (Spearman, r = -0.4756, P = 0.0004, Figure 3I). Taken together, these results suggest that AKT2 mRNA is a target of miR-625.
Figure 3.

AKT2 mRNA is a direct target of miR-625, and AKT2 protein expression is inversely correlated with that of miR-625 in glioma tissues. (A) Predicted miR-625 target sequence in the wild-type (WT) 3’-UTR of AKT2 mRNA and the mutated construct (mut). (B) Levels of miR-625 in U87 and U251 cells after ectopic expression of miR-625 or miR-NC. (C) Luciferase reporter assay of U87 and U251 cells co-transfected with miR-625 mimic and either pmiRNA-AKT2/3’-UTR-WT or pmiRNA-AKT2/3’-UTR-Mut. (D) Western blot analysis of AKT2 protein expression levels in U87 and U251 cells transfected with miR-NC or miR-625. GAPDH served as the loading control. (E, F) Analysis of AKT2 expression in glioblastoma multiforme (GBM) and normal brain tissue (NBT) from the TCGA (E) and GSE16011 (F) datasets. (G) Western blot analysis of AKT2 protein expression in normal human astrocytes (NHAs) and U87, LN229, U251, A172, and U118 glioma cell lines. (H) Western blot analysis of AKT2 protein expression in five NBTs and 26 glioma specimens. Expression levels were normalized to GAPDH levels. P = 0.0033. (I) Spearman’s correlation analysis of AKT2 protein and miR-625 expression levels in human glioma specimens (r = −0.6035, P < 0.001). **P < 0.01.
MiR-625 suppresses tumor growth and angiogenesis in vivo
Accumulating evidence has shown that miRNA mimics are promising anti-cancer treatments [23]. To evaluate the effects of miR-625 on tumor progression and angiogenesis in vivo, we employed an ectopic transplantation model of human glioma in nude mice. We generated lentiviral constructs to stably express miR-NC and miR-625 in U87 cells, and then injected the cells subcutaneously into the flanks of athymic mice. Tumor sizes were measured every 3 d for 30 d, at which point the mice were euthanized and tumors were removed for analysis. We found that the tumors derived from miR-NC U87 cells were much larger (Figure 4A, 4B) and weighed more (P < 0.01, Figure 4C) than the tumors formed from miR-625-overexpressing cells. Tumor tissues were analyzed for AKT2 expression by immunohistochemical staining. As shown in Figure 4D, the AKT2 expression was lower in miR-625-U87 tumors compared with control cells, which was consistent with the in vitro data. MiR-625-U87 tumors also showed enhanced expression of the apoptotic marker cleaved caspase 3 and lower expression of the proliferation marker Ki-67 compared with control tumors (Figure 4D). Similarly, immunofluorescence microscopy of tumor sections revealed that AKT2 expression was lower in the miR-625-U87 group, and nuclear AKT2 localization was less prominent that in the control tumors (Figure 4E). Consistent with these findings, western blot analysis showed that AKT2 protein expression was significantly lower in tumors derived from miR-625-U87 cells than miR-NC-U87 cells (Figure 4F). Collectively, these results suggest that miR-625 plays a tumor suppressor role in vivo by targeting AKT2 mRNA.
Figure 4.
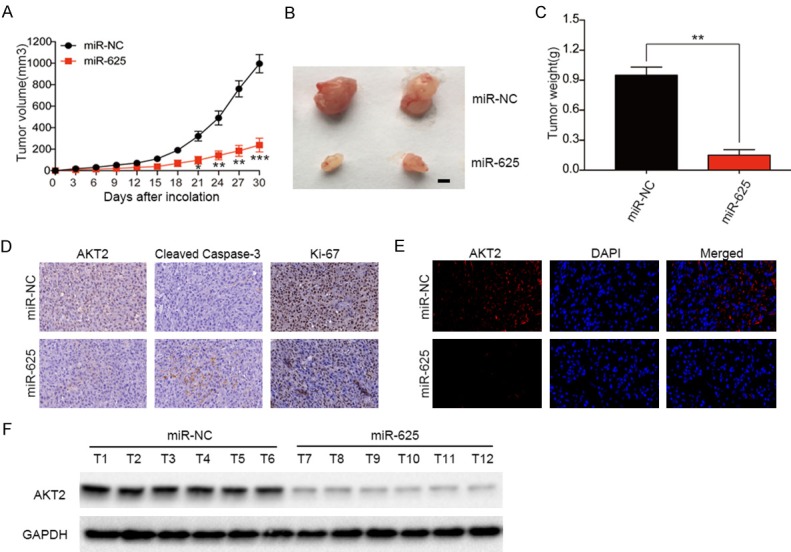
MiR-625 overexpression suppresses glioma growth in vivo. (A) Tumor growth curves after subcutaneous injection of nude mice with U87 cells stably expressing miR-NC or miR-625 (n = 4). Tumor volumes were measured every 3 d from days 3 to 30. (B-F) Analysis of miR-NC- and miR-625-expressing U87 tumors on day 30 after injection: representative tumor images (B); tumor weights (C); immunohistochemical staining of AKT2, Ki-67, and cleaved caspase 3 (D); immunofluorescent staining of AKT2 protein (E); and western blot analysis of AKT2, with GAPDH as the loading control (F). *P < 0.01, **P < 0.01, ***P < 0.001.
AKT2 overexpression reverses the inhibitory effects of miR-625 in glioma cell lines
Previous studies have shown that AKT2 plays an important role in modulating the progression of various human cancers. To determine whether AKT2 plays a similar role in glioma growth and development, U251 and U87 cells were infected with AKT2-expressing or control lentiviruses (Figure 5A). We found that AKT2 overexpression promoted the proliferation (Figure 5E) and colony-forming efficiency (Figure 5C) of both cell lines. Conversely, siRNA-mediated knockdown of AKT2 (Figure 5B) dramatically inhibited the colony-forming ability of U251 and U87 cells (Figure 5D). These results are consistent with the miR-625 overexpression data and suggest that miR-625 may regulate glioma behavior through modulation of AKT2. Therefore, we investigated whether forced AKT2 expression could reverse the effects of miR-625 overexpression. For this, we transfected stable miR-625- or miR-NC-expressing U87 and U251 cells with a plasmid encoding AKT2 lacking a 3’-UTR, which prevents miR-625-mediated AKT2 downregulation. As shown in Figure 5E-G, restoration of AKT2 expression partially abolished the miR-625-mediated suppression of cell proliferation, colony formation ability, and DNA synthesis. Similarly, AKT2 expression reduced the number of miR-625-transfected U87 and U251 cells in G0/G1 cell cycle arrest and increased the S+G2/M population (Figure 5H). Moreover, western blot analysis showed that while miR-625 suppressed Cyclin D1 protein expression, this effect was abolished by forced AKT2 expression (Figure 5I). CDK4 and Cyclin E1 protein levels were unaffected. Collectively, these data confirm that AKT2 is a functional target of miR-625 in glioma cells.
Figure 5.

AKT2 overexpression reverses the suppressive effects of miR-625 on glioma cells in vitro. (A) Western blot analysis of AKT2 expression in U87 and U251 cells expressing control (Ctrl) or AKT2 overexpression vectors. GAPDH served as the loading control. (B) Western blot analysis of AKT2 expression in U87 and U251 cells after transfection with control (si-Ctrl) or AKT2-targeting siRNA (si-AKT2). GAPDH served as the loading control. (C) Representative images and quantification of colony-forming assays of control or AKT2-overexpressing U87 and U251 cells. (D) Representative images and quantification of colony-forming assays of U87 and U251 cells transfected with si-AKT2 or si-Ctrl. (E-H) CCK-8 viability assay (E), colony-forming assay (F), EDU proliferation assay (G), and cell cycle distribution assay (H) of U87 and U251 cells transfected with miR-NC, miR-625, or miR-625+AKT2. (I) Western blot analysis of AKT2 and downstream effector proteins Cyclin D1, CDK4, and Cyclin E1 in U87 and U251 cells transfected with miR-NC, miR-625, or miR-625+AKT2. GAPDH was used as the loading control. **P < 0.01.
MiR-625 overexpression increases the chemosensitivity of glioma cells to TMZ by targeting AKT2
TMZ is an alkylating drug currently used as the first-line chemotherapy for glioma [24]. However, a major obstacle to effective treatment is primary and secondary resistance to TMZ. To determine whether miR-625 affects the chemosensitivity of glioma cells, we stably expressed miR-625 or miR-NC in U87 or U251 cells and treated them with various concentrations of TMZ. Our results revealed that miR-625-overexpressing cells were significantly more sensitive to TMZ than the control cells (Figure 6A). Furthermore, the sensitivity of both cell lines to 100 μM TMZ was blocked by forced expression of AKT2 (Figure 6B). Similarly, while miR-625-overexpressing U87 and U251 cells showed a significant increase in apoptosis after treatment with TMZ compared with vehicle, forced AKT2 expression also partially abolished this effect (Figure 6C). Finally, we found that expression of MGMT, an enzyme that repairs damaged DNA, was substantially lower in the miR-625-overexpressing TMZ-treated cells compared with either the miR-625-overexpressing or TMZ-treated cells alone. However, MGMT expression was restored by co-transfection of cells with AKT2 (Figure 6D). Taken together, these results suggest that miR-625 enhances glioma cell sensitivity to TMZ treatment and that the combination of miR-625 overexpression and TMZ treatment induced apoptosis of glioma cells by targeting AKT2.
Figure 6.

Glioma cell chemosensitivity to TMZ is increased by miR-625, and AKT2 overexpression partially reverses this effect. A. CCK-8 assay of U87 and U251 cells transfected with miR-NC or miR-625 and cultured with the indicated concentrations of TMZ for 48 h. B. CCK-8 assay of U87 and U251 cells transfected with miR-NC, miR-625, or miR-625+AKT2 and cultured with 100 µM TMZ for the indicated times. C. Flow cytometric Annexin V-FITC/PI apoptosis assay of U87 and U251 cells transfected with miR-NC, miR-625, or miR-625+AKT2 and cultured with 100 µM TMZ for 48 h. D. Western blot analysis of O-6-methylguanine-DNA methyltransferase (MGMT) expression in U251 cells stably expressing miR-NC, miR-625, or miR-625+AKT2 and cultured with 100 μM TMZ. GAPDH was used as the loading control. *P < 0.05, **P < 0.01.
Discussion
The proliferative ability of GBM is a key factor that leads to poor prognosis, but the underlying mechanisms remain unclear. MiRNAs are a class of small regulatory molecules that function as post-transcriptional gene regulators in many pathophysiological processes [25]. Human gliomas are known to display aberrant miRNA expression profiles [26-28], and the biological functions of a number of these miRNAs have been studied extensively. For example, miR-146-5p has been shown to regulate MMP16 expression, and its overexpression suppresses the migration and invasion of glioma cells [29]. Dysregulated miRNA expression is a common feature of human cancers [30]. In general, miRNAs regulate the expression levels of target genes by binding to specific sites in mRNAs. Accumulating data suggests that miRNAs are involved in cancer development and may act as tumor suppressors or oncogenes by regulating many cellular processes. Here, we showed that miR-625 functions as an antiangiogenic regulator in human gliomas. However, the role of miR-625 in cancer development and progression is still unclear. In this study, we found that miR-625 was expressed at lower levels in glioma tissues and cell lines compared with NBTs and NHAs. Restoring miR-625 expression reduced glioma cell proliferation in vitro by blocking the G1/S transition. In a subcutaneous glioma mouse xenograft model, tumor growth was reduced by overexpression of miR-625, suggesting that manipulation of miR-625 levels may have therapeutic potential for glioma patients.
To date, few miR-625 targets have been experimentally validated. In our study, luciferase reporter assays and western blot analysis demonstrated that AKT2 is a direct miR-625 target. AKT2 is a pro-survival protein activated by phosphatidylinositol 3-kinase (PI3K) [31]. The PI3K/AKT2/mTOR signaling pathway contributes to gallbladder carcinoma cell proliferation [32], and AKT2 can also promote tumor angiogenesis in pancreatic cancer [33], prostate cancer [34], and squamous cell carcinoma of the oral cavity [35]. Our results also support previous studies showing that inhibiting AKT2 can decrease proliferation in a wide variety of tumor cells, including pancreatic, ovarian, and breast cancer [33,34]. However, the specific molecular mechanisms underlying the observed AKT2 increases in glioma have been unclear. In this report, we showed for the first time that miR-625 specifically targets AKT2 in human glioma cells. AKT2 expression was upregulated in glioma tissues compared with NBTs, and the levels were inversely correlated with miR-625 expression in clinical glioma tumor tissues. AKT2 expression was significantly decreased in miR-625-overexpressing glioma cells, while forced AKT2 expression reversed the phenotypes associated with miR-625 overexpression. Thus, this study provides the first evidence that miR-625 suppresses glioma cell growth by inhibiting AKT2 translation.
We also observed that AKT2 knockdown significantly suppressed glioma cell proliferation, similar to the phenotype observed following miR-625 overexpression. More importantly, reintroducing AKT2 into miR-625 overexpressing cells blocked the effect of miR-625 on glioma cell proliferation. Additionally, luciferase reporter assays revealed that miR-625 could directly target the 3’-UTR of AKT2 mRNA. Collectively, our results demonstrate for the first time that AKT2 mediates the anti-proliferative functions of miR-625 in glioma. The upregulated AKT2 expression induced by decreased miR-625 levels appears to facilitate glioma growth and consequently drive disease progression.
Our results not only provide insight into the molecular mechanisms that regulate human glioma, but also offer an explanation for the challenges associated with treating glioma. Several studies have indicated that miRNAs regulate the growth of chemoresistant tumors [36,37]. A recent study showed that aberrant miR-211 expression significantly increased apoptosis and DNA fragmentation in glioma cells [38]. In this study, we found that miR-625 overexpression reduced the resistance of human glioma cells to TMZ and increased apoptosis by targeting AKT2. MiR-625 overexpression may therefore be an effective therapeutic strategy for overcoming TMZ chemoresistance in human gliomas.
In conclusion, our study provides new evidence of an important connection between miR-625 and tumorigenesis in gliomas. Our data clearly demonstrate the role of miR-625 in suppressing glioma progression and development by directly targeting the AKT2 3’-UTR. These findings suggest that loss of miR-625 leads to overexpression of the AKT2 oncogene, which in turn favors glioma proliferation. We demonstrated that miR-625 acts as a tumor suppressor through multiple mechanisms, including induction of cell cycle arrest. We also showed that miR-625 and AKT2 expression are inversely correlated in glioma tissues, and that miR-625 overexpression enhances glioma cell sensitivity to TMZ treatment. Although miRNA-based combination therapies are still in the initial stages of development, targeting this novel miR-625-AKT2 relationship may be a useful strategy for treating patients with malignant gliomas.
Acknowledgements
This work was supported by grants from the Research Special Fund for Public Welfare Industry of Health (201402008), National Natural Science Foundation of China (81472362, 81372709, 81302185), Jiangsu Province’s Natural Science Foundation (BK20131019, BK20151585), the Program for Advanced Talents within Six Industries of Jiangsu Province (2015-WSN-036, 2016-WSW-023), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Disclosure of conflict of interest
None.
References
- 1.Kim YW, Liu TJ, Koul D, Tiao N, Feroze AH, Wang J, Powis G, Yung WK. Identification of novel synergistic targets for rational drug combinations with PI3 kinase inhibitors using siRNA synthetic lethality screening against GBM. Neuro Oncol. 2011;13:367–75. doi: 10.1093/neuonc/nor012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vredenburgh JJ, Desjardins A, Reardon DA, Friedman HS. Experience with irinotecan for the treatment of malignant glioma. Neuro Oncol. 2009;11:80–91. doi: 10.1215/15228517-2008-075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linz U. Commentary on effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial (Lancet Oncol. 2009;10:459-466) Cancer. 2010;116:1844–1846. doi: 10.1002/cncr.24950. [DOI] [PubMed] [Google Scholar]
- 4.Prasad G, Sottero T, Yang X, Mueller S, James CD, Weiss WA, Polley MY, Ozawa T, Berger MS, Aftab DT, Prados MD, Haas-Kogan DA. Inhibition of PI3K/mTOR pathways in glioblastoma and implications for combination therapy with temozolomide. Neuro Oncol. 2011;13:384–392. doi: 10.1093/neuonc/noq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li RY, Chen LC, Zhang HY, Du WZ, Feng Y, Wang HB, Wen JQ, Liu X, Li XF, Sun Y, Yang DB, Jiang T, Li YL, Jiang CL. MiR-139 inhibits Mcl-1 expression and potentiates TMZ-induced apoptosis in glioma. CNS Neurosci Ther. 2013;19:477–483. doi: 10.1111/cns.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 7.Kloosterman WP, Plasterk RH. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–450. doi: 10.1016/j.devcel.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Hwang HW, Mendell JT. MicroRNAs in cell proliferation, cell death, and tumorigenesis. Br J Cancer. 2006;94:776–780. doi: 10.1038/sj.bjc.6603023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang L, Li Q, Wang Q, Jiang Z, Zhang L. Silencing of miRNA-218 promotes migration and invasion of breast cancer via Slit2-Robo1 pathway. Biomed Pharmacother. 2012;66:535–540. doi: 10.1016/j.biopha.2012.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Esquela-Kerscher A, Slack FJ. Oncomirs - microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 11.Sana J, Hajduch M, Michalek J, Vyzula R, Slaby O. MicroRNAs and glioblastoma: roles in core signalling pathways and potential clinical implications. J Cell Mol Med. 2011;15:1636–1644. doi: 10.1111/j.1582-4934.2011.01317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsuchiya N, Nakagama H. MicroRNA, SND1, and alterations in translational regulation in colon carcinogenesis. Mutat Res. 2010;693:94–100. doi: 10.1016/j.mrfmmm.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 13.Toker A, Yoeli-Lerner M. Akt signaling and cancer: surviving but not moving on. Cancer Res. 2006;66:3963–3966. doi: 10.1158/0008-5472.CAN-06-0743. [DOI] [PubMed] [Google Scholar]
- 14.Cheung M, Testa JR. Diverse mechanisms of AKT pathway activation in human malignancy. Curr Cancer Drug Targets. 2013;13:234–244. doi: 10.2174/1568009611313030002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Villegas-Comonfort S, Castillo-Sanchez R, Serna-Marquez N, Cortes-Reynosa P, Salazar EP. Arachidonic acid promotes migration and invasion through a PI3K/Akt-dependent pathway in MDA-MB-231 breast cancer cells. Prostaglandins Leukot Essent Fatty Acids. 2014;90:169–177. doi: 10.1016/j.plefa.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Guo X, Yang M, Yu L, Li Z, Lin N. Identification of AKT kinases as unfavorable prognostic factors for hepatocellular carcinoma by a combination of expression profile, interaction network analysis and clinical validation. Mol Biosyst. 2014;10:215–222. doi: 10.1039/c3mb70400a. [DOI] [PubMed] [Google Scholar]
- 17.Kong X, Chang X, Cheng H, Ma R, Ye X, Cui H. Human epididymis protein 4 inhibits proliferation of human ovarian cancer cells via the mitogen-activated protein kinase and phosphoinositide 3-kinase/AKT pathways. Int J Gynecol Cancer. 2014;24:427–436. doi: 10.1097/IGC.0000000000000078. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Shi Z, Chen Q, Li C, Wang L, Qian X, Jiang C, Liu X, Wang X, Li H, Kang C, Jiang T, Liu LZ, You Y, Liu N, Jiang BH. MiR-124 governs glioma growth and angiogenesis and enhances chemosensitivity by targeting R-Ras and N-Ras. Neuro Oncol. 2014;16:1341–1353. doi: 10.1093/neuonc/nou084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang H, Wu W, Wang HW, Wang S, Chen Y, Zhang X, Yang J, Zhao S, Ding HF, Lu D. Analysis of specialized DNA polymerases expression in human gliomas: association with prognostic significance. Neuro Oncol. 2010;12:679–686. doi: 10.1093/neuonc/nop074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–761. doi: 10.1128/mcb.18.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 23.Bader AG, Brown D, Winkler M. The promise of microRNA replacement therapy. Cancer Res. 2010;70:7027–7030. doi: 10.1158/0008-5472.CAN-10-2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plowman J, Waud WR, Koutsoukos AD, Rubinstein LV, Moore TD, Grever MR. Preclinical antitumor activity of temozolomide in mice: efficacy against human brain tumor xenografts and synergism with 1,3-bis(2-chloroethyl)-1-nitrosourea. Cancer Res. 1994;54:3793–3799. [PubMed] [Google Scholar]
- 25.Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle. 2005;4:1179–1184. doi: 10.4161/cc.4.9.2032. [DOI] [PubMed] [Google Scholar]
- 26.Yan W, Zhang W, Sun L, Liu Y, You G, Wang Y, Kang C, You Y, Jiang T. Identification of MMP-9 specific microRNA expression profile as potential targets of anti-invasion therapy in glioblastoma multiforme. Brain Res. 2011;1411:108–115. doi: 10.1016/j.brainres.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 27.Lavon I, Zrihan D, Granit A, Einstein O, Fainstein N, Cohen MA, Cohen MA, Zelikovitch B, Shoshan Y, Spektor S, Reubinoff BE, Felig Y, Gerlitz O, Ben-Hur T, Smith Y, Siegal T. Gliomas display a microRNA expression profile reminiscent of neural precursor cells. Neuro Oncol. 2010;12:422–433. doi: 10.1093/neuonc/nop061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang L, Mao P, Song L, Wu J, Huang J, Lin C, Yuan J, Qu L, Cheng SY, Li J. miR-182 as a prognostic marker for glioma progression and patient survival. Am J Pathol. 2010;177:29–38. doi: 10.2353/ajpath.2010.090812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin F, Wang X, Jie Z, Hong X, Li X, Wang M, Yu Y. Inhibitory effects of miR-146b-5p on cell migration and invasion of pancreatic cancer by targeting MMP16. J Huazhong Univ Sci Technolog Med Sci. 2011;31:509–514. doi: 10.1007/s11596-011-0481-5. [DOI] [PubMed] [Google Scholar]
- 30.Croce CM, Calin GA. miRNAs, cancer, and stem cell division. Cell. 2005;122:6–7. doi: 10.1016/j.cell.2005.06.036. [DOI] [PubMed] [Google Scholar]
- 31.Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- 32.Zhang P, Guo Z, Wu Y, Hu R, Du J, He X, Jiao X, Zhu X. Histone deacetylase inhibitors inhibit the proliferation of gallbladder carcinoma cells by suppressing AKT/mTOR signaling. PLoS One. 2015;10:e0136193. doi: 10.1371/journal.pone.0136193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, Altomare DA, Wan M, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Benedetti Panici P, Mancuso S, Neri G, Testa JR. Molecular alterations of the AKT2 oncogene in ovarian and breast carcinomas. Int J Cancer. 1995;64:280–285. doi: 10.1002/ijc.2910640412. [DOI] [PubMed] [Google Scholar]
- 34.Liao Y, Grobholz R, Abel U, Trojan L, Michel MS, Angel P, Mayer D. Increase of AKT/PKB expression correlates with gleason pattern in human prostate cancer. Int J Cancer. 2003;107:676–680. doi: 10.1002/ijc.11471. [DOI] [PubMed] [Google Scholar]
- 35.Nakayama H, Ikebe T, Beppu M, Shirasuna K. High expression levels of nuclear factor kappaB, IkappaB kinase alpha and Akt kinase in squamous cell carcinoma of the oral cavity. Cancer. 2001;92:3037–3044. doi: 10.1002/1097-0142(20011215)92:12<3037::aid-cncr10171>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 36.Boren T, Xiong Y, Hakam A, Wenham R, Apte S, Chan G, Kamath SG, Chen DT, Dressman H, Lancaster JM. MicroRNAs and their target messenger RNAs associated with ovarian cancer response to chemotherapy. Gynecol Oncol. 2009;113:249–255. doi: 10.1016/j.ygyno.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Xia L, Zhang D, Du R, Pan Y, Zhao L, Sun S, Hong L, Liu J, Fan D. miR-15b and miR-16 modulate multidrug resistance by targeting BCL2 in human gastric cancer cells. Int J Cancer. 2008;123:372–379. doi: 10.1002/ijc.23501. [DOI] [PubMed] [Google Scholar]
- 38.Asuthkar S, Velpula KK, Chetty C, Gorantla B, Rao JS. Epigenetic regulation of miRNA-211 by MMP-9 governs glioma cell apoptosis, chemosensitivity and radiosensitivity. Oncotarget. 2012;3:1439–1454. doi: 10.18632/oncotarget.683. [DOI] [PMC free article] [PubMed] [Google Scholar]