

Abstract
Increased methylation levels at cytosines proximal to guanines (CpG) in the promoter regions of tumor suppressor genes have been reported to play an important role in the development and progression of bladder cancer. In this study, we conducted a genome-wide analysis using data from The Cancer Genome Atlas to better characterize CpG methylation and mRNA expression patterns in urothelial carcinomas and to identify new epigenetic biomarkers of survival. Across 408 tumors, we identified 223 genes that displayed significant relationships between CpG methylation and mRNA expression levels. Hypermethylation within 200 base pairs upstream of the transcription start site and hypomethylation within the 3’ untranslated region and body region were associated with gene silencing. These 223 genes were functionally enriched for their role in glutamate receptor signaling and among them was a novel, tumor-stage-independent epigenetic biomarker of overall mortality, GRIA1. GRIA1 hypermethylation and elevated mRNA expression levels were associated with significantly worse survival outcomes in patients with basal-like urothelial carcinomas. Furthermore, 70 genes associated with glutamate receptor signaling were differentially expressed between basal (n = 203 tumors) and luminal (n = 205 tumors) subtypes of bladder cancer, including genes involved in glutamate receptor-mediated activation of the calmodulin, PI3K/Akt, and EGFR signaling pathways. The majority of genes displayed increased expression levels in basal-like subtypes. This research highlights glutamate receptors as targets for investigation in the development and pharmacological treatment of urothelial cancer.
Keywords: Bladder cancer, CpG methylation, epigenetics, glutamate receptors, The Cancer Genome Atlas
Introduction
Urothelial bladder carcinoma is a highly prevalent cancer in the United States, and its incidence is on the rise [1]. As the public health burden from this cancer increases, better understanding of the biological mechanisms underlying its etiology is critical [2]. Mechanistic studies have identified several biological pathways that may be targeted in the development of these cancers, and genetic analyses have identified somatic mutations in multiple genes that are associated with bladder cancer tumors [2,3]. Research has also implicated that epigenetic mechanisms play a key role in bladder carcinogenesis, and chromatin-modifying genes are frequently mutated in bladder cancer [2,4].
Methylation of cytosines proximal to guanines (CpG) is an epigenetic mechanism that is known to be involved in carcinogenesis [5]. Large clusters of CpG sites, CpG islands, are often found in the promoter regions of genes [6]. In tumors, global hypomethylation has been implicated in a loss of cell cycle control and other cellular processes, thus leading to malignant growth [2]. In some instances, increases in CpG methylation in promoter regions of genes have been associated with gene silencing [5]. Numerous studies have identified genes that display promoter hypermethylation in bladder cancer (reviewed in [7]), although few studies report corresponding gene expression levels, and recent research has implicated that methylation in other regions of the gene may play an important role in epigenetic regulation [5,6,8].
In this study, we aimed to better characterize the epigenomic profiles of CpG methylation in urothelial bladder tumors across all intragene regions and to identify functional epigenetic biomarkers of bladder cancer. Therefore, methylation and mRNA expression data sets from The Cancer Genome Atlas (TCGA) were analyzed to identify genes that are both differentially expressed and methylated in bladder cancer and that display a significant relationship between CpG methylation and mRNA expression levels.
Materials and methods
Data acquisition
All available data files from Infinium Human-Methylation 450 BeadChip arrays (n = 440) were downloaded from the TCGA and were read into SAS V9.3 [9]. Data were merged on probe identifier, and, for quality control purposes, probes where approximately 1% of the data were missing were removed. Data were log-transformed and probes corresponding to single nucleotide polymorphism (SNPs) were removed [10]. The final data set consisted of n = 412 methylation arrays of genome-wide DNA methylation for tumor tissue and n = 21 methylation arrays of genome-wide DNA methylation for non-tumor tissue, each containing 332,950 genomic sites.
All available RNASeqV2 normalized count data files (n = 427), each containing data across 20,531 genes, were downloaded from the TCGA. These data were normalized using the RSEM method by the TCGA [11]. Data were then imported into SAS V9.3 and normalized count values were summarized by gene. There were 408 data files that corresponded to tumor samples and 19 data files that corresponded to non-tumor samples. Gene expression levels across all samples for each gene were log-transformed, as in prior publications from the TCGA [12].
All available clinical data files (n = 417), each containing 54 variables from bladder cancer tumors were downloaded from the TCGA. These included demographic factors, including subject sex (male vs. female), age at initial diagnosis (continuous variable), tumor pathologic subtype (papillary vs. non-papillary), smoking status (ever vs. never), and race (white vs. non-white), and clinical data, such as the American Joint Committee on Cancer (AJCC) tumor pathologic stage and days until death. Of the 408 subjects that had both CpG methylation and RNASeq data available for tumor samples, 381 also had clinical data files.
Identifying differentially expressed genes (DEGs) between urothelial tumor and non-tumor tissue
All possible subjects were identified that had RNASeq data available for matched tumor and non-tumor tissue (n = 19). Individual fold changes (FC) were calculated using the log-transformed values in the formula FC = RNASeq value (tumor)/RNASeq value (non-tumor) for each subject across all genes available for analysis and the median FC per gene was calculated across subjects. In addition, ANCOVA of the log-transformed RNASeq values across the 20,531 genes was conducted contrasting on tumor vs. non-tumor tissue, controlling for sex, age at initial diagnosis, pathologic subtype, smoking status, and race. FDR (false discovery rate) q-values were computed. DEGs were defined by the following criteria: (i) FDR q-value < 0.05 and (ii) the median absolute FC for the gene between tumor and non-tumor tissue across all matched subjects was ≥ |2.0|.
Identifying differentially methylated genes (DMGs) between urothelial tumor and non-tumor tissue
Subjects were identified that had DNA methylation data available for matched tumor and non-tumor tissue (n = 21). ANCOVA analysis of the DNA methylation beta-values across all CpG sites was conducted contrasting on tumor vs. non-tumor tissue controlling for sex, age at initial diagnosis, pathologic subtype, smoking status, and race. FDR q-values were computed. The Infinium HumanMethylation 450 BeadChip array annotates each probe to a gene and to one of six intragene sites- (i) from 200-1500 base pairs upstream of the gene transcription start site (TSS1500), (ii) within 200 base pairs upstream of the gene transcription start site (TSS200), (iii) in the 5’ untranslated region of the gene (5’ UTR), (iv) in the first exon of the gene (1st Exon), (v) in the body of the gene (Body), and (vi) in the 3’ untranslated region of the gene (3’ UTR) [13]. Individual beta-differences were calculated using the formula beta-difference = beta-value (tumor) - beta-value (non-tumor) for each subject across all CpG sites available for analysis and the median beta difference per gene was calculated across subjects across all CpG sites associated with a gene and within each of the six intragene sites. DMGs were defined by the following criteria: (i) at least one probe associated with the gene had a FDR q-value < 0.05 and (ii) the median beta difference across all CpG sites associated with a gene or at least one of the intragene sites between tumor and non-tumor tissue across all matched subjects was ≥ |0.10|, representing a 10% difference in methylation, as this methylation difference threshold resulted in approximately a 95% true positive rate using the beta-value method of detecting differences in CpG methylation values [14]. Permutation testing using R was used to test if the number of genes hypermethylated versus hypomethylated in the promoter regions (TSS1500 and TSS200) in tumor versus non-tumor tissue differed from a pure 0.5 probability of methylation directionality.
Identifying genes demonstrating a significant relationship between CpG methylation and mRNA expression in urothelial tumor tissue
In order to identify genes whose expression was associated with methylation levels in tumors, Spearman rank correlations were run on all genes that were both differentially methylated and expressed between tumor and non-tumor tissue. Individual median methylation values for all CpG sites associated with a gene and for the six intragene regions were calculated for all individuals who had both methylation and RNASeq tumor data available (n = 408). In separate analyses, these methylation values were log-transformed and then tested via Spearman rank analysis with the individuals’ log-transformed RNASeq values. A significant relationship between CpG methylation and mRNA expression was defined as a Spearman rank correlation p-value < 0.05. In order to integrate these three analyses, we determined that genes that met the following criteria could serve as potential epigenetic biomarkers of bladder carcinogenesis: (i) differentially expressed between matched tumor and non-tumor tissue, (ii) differentially methylated between matched tumor and non-tumor tissue, and (iii) displayed a significant association between CpG methylation and mRNA expression in tumor tissue.
Network analysis of potential epigenetic biomarkers of urothelial cancer
In order to examine the higher-level biological processes related to the genes identified as both differentially methylated and expressed between bladder cancer tumor and non-tumor tissue and with a significant relationship between CpG methylation and gene expression in tumor tissue, we analyzed these genes in Ingenuity Network Analysis (IPA) (Ingenuity Systems®, Redwood City, CA, USA). Canonical pathways were identified as enriched using the right-tailed Fisher’s Exact test, where significance was set at p-value < 0.001 [15].
Assessment of prognostic potential of epigenetic biomarkers in urothelial tumors
ANCOVA was used as an initial screen to identify specific genes among the 223 that were differentially methylated and expressed among survivors and non-survivors of bladder cancer. ANCOVA of the log-transformed RNASeq values across the 223 genes and of the log-transformed median CpG methylation beta-values of all differentially methylated regions (DMRs) associated with gene expression was performed, contrasting on survival status in the 408 bladder cancer tumors, controlling for sex, age at initial diagnosis, pathologic subtype, smoking status, and race. Genes that were differentially expressed and methylated between survivors and non-survivors at an alpha level of 0.10 were further analyzed using Kaplan-Meier analysis with log-rank statistics. For genes significant in Kaplan-Meier analysis, a Cox regression model was run, with AJCC Pathologic Tumor Stage incorporated as a co-predictor of survival. For both analyses, subjects were stratified into two groups of (i) mean - SD and (ii) mean + SD based on CpG methylation or gene expression level and significance was defined as p-value < 0.05.
Validation of a prognostic epigenetic biomarker in an independent cohort
In order to validate prognostic indicators of overall mortality in urothelial bladder tumors, expression levels originally reported in Choi et al. 2014 were obtained from the Gene Expression Omnibus (GSE48277) (n = 146) [16]. As the sample size of this cohort was approximately one-third the size of the TCGA cohort, individuals were stratified into two groups of mean ± [SD]/2, in order to have a sufficient number of individuals within each stratification group. Kaplan-Meier analysis with log-rank statistics was used to test for differences in survival. A Cox regression model was used to test if observed differences were independent of AJCC Pathologic Tumor Stage. For both analyses, significance was defined as a p-value < 0.05.
Analysis of the prognostic potential of an epigenetic biomarker in basal and luminal subtypes of urothelial tumors
In order to elucidate potential biological roles of the identified epigenetic prognostic biomarker, we tested if this gene had significantly different expression and methylation levels in basal- versus luminal subtypes of bladder cancer. Methodology of tumor subtyping is described in detail elsewhere [17]. ANCOVA of the log-transformed RNASeq values and of the log-transformed median CpG methylation beta-values of the significant prognostic intragene region was performed, contrasting on basal (n = 203) versus luminal (n = 205) subtype, controlling for sex, age at initial diagnosis, smoking status, and race. Tumor pathologic subtype and tumor stage were not controlled for in this analysis as it is likely correlated with basal and luminal subtype classification. The median FC and median beta difference of the log-transformed RNASeq values of the gene and of the log-transformed median CpG methylation beta-values of the significant prognostic intragene region were also calculated between basal and luminal subtypes of urothelial cancers. Then, the prognostic power of mRNA expression and CpG methylation levels of the identified gene was assessed separately in basal and luminal subtypes using Kaplan-Meier analysis with log-rank statistics independently among the basal and luminal subtypes. Cox regression models were used to test if observed differences were independent of AJCC Pathologic Tumor Stage. For all analyses, subjects were stratified into two groups of (i) mean - SD and (ii) mean + SD based on CpG methylation or gene expression level, and significance was defined as p-value < 0.05.
Assessment of DEGs between basal and luminal subtypes of urothelial cancer
To assess whether there is a difference in glutamate receptor signaling in basal-like versus luminal-like bladder cancers, we tested for differential expression between basal and luminal subtypes of bladder cancer in a total of n = 1,776 genes associated with glutamate receptor signaling. ANCOVA of log-transformed RNASeq values was performed, contrasting on basal versus luminal subtype in the 408 bladder cancer tumors, controlling for sex, age at initial diagnosis, smoking status, and race. Tumor pathologic subtype was not controlled for in this analysis as it is likely correlated with basal and luminal subtype classification. FDR q-values were generated to control for multiple tests. Significance for DEGs was defined as a FDR q-value < 0.05 and a median FC (basal/luminal) ≥ |2.00|.
Results
Gene expression and CpG methylation differences between urothelial tumor and non-tumor tissue
A total of 413 DEGs were identified between matched tumor and non-tumor tissue samples (n = 38) (Figure 1). The majority of genes (261/413 = 63.2%) displayed decreased expression in tumor tissue versus non-tumor tissue, while 152/413 (36.8%) displayed increased expression in tumor versus non-tumor tissue.
Figure 1.
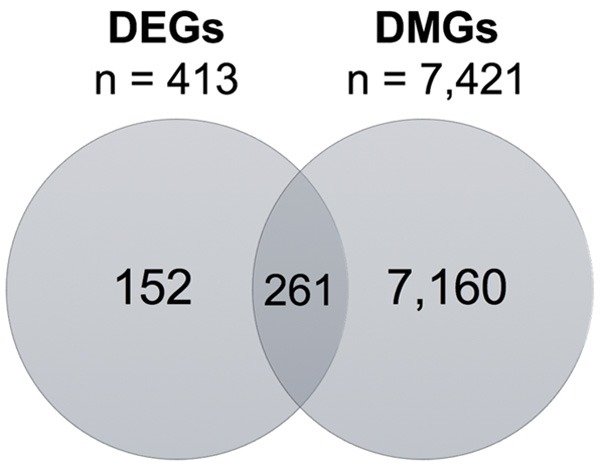
Venn Diagram of the overlapping DEGs and DMGs between matched tumor and non-tumor tissues (n = 38 and n = 42, respectively) in the TCGA.
A total of 7,421 genes were significantly differentially methylated between tumor and non-tumor tissue between matched tumor and non-tumor tissue samples (n = 42) (Figure 1). This high number of DMGs observed between tumor and non-tumor tissue in this analysis has also been described in other types of cancer [18,19]. Of these 7,421 genes, 2,666 (35.9%) had a median beta difference across all CpG probes ≥ |0.10|, representing a 10% increase or decrease in methylation in the entire gene.
In further analyses of these data by intragene region, the TSS1500 and TSS200 regions, which are predicted to contain gene promoters, comprised 3,081 (41.5%) and 2,054 (27.7%) DMGs, respectively. Furthermore, 631 (20.5%) genes in the TSS1500 region and 696 (33.9%) genes in the TSS200 regions were hypermethylated. Permutation testing revealed that this was significantly fewer hypermethylated genes than would be expected to result from chance alone in both the TSS1500 and TSS200 regions (p-values < 0.0001), a surprising result as promoter CpG hypermethylation is often discussed in the context of bladder carcinogenesis [7,20,21]. Some known tumor suppressor genes were hypermethylated in their promoter regions in tumor versus non-tumor tissue, including DBC1, PAX6, RUNX3, and WT1, although these methylation changes were not associated with decreases in gene expression. Others, such as BRCA1, PTEN, TP53, and RB1 were not present among the DMGs. There were 261 genes that overlapped between the lists of DMGs and DEGs (Figure 1).
CpG methylation is associated with mRNA expression in urothelial tumor tissue
An association measure was calculated between CpG methylation and mRNA expression for the 261 DMGs and DEGs to assess whether CpG methylation in tumor tissue had a functional effect on mRNA expression. Of the 261 overlapping DMGs and DEGs, 223 displayed a significant relationship between DNA methylation and gene expression. The majority of these genes (n = 161, 72%) were decreased in expression in tumor tissue versus non-tumor tissue (Figure 2A). Interestingly, only 69 genes (31%) displayed significant promoter-associated hypermethylation. In addition, 160 (72%) DMRs that displayed the strongest correlation with gene expression for each gene in tumor tissue demonstrated a loss of methylation in tumor versus non-tumor tissue. An inverse relationship between mRNA expression levels and CpG methylation levels was not observed among these samples (Figure 2B).
Figure 2.
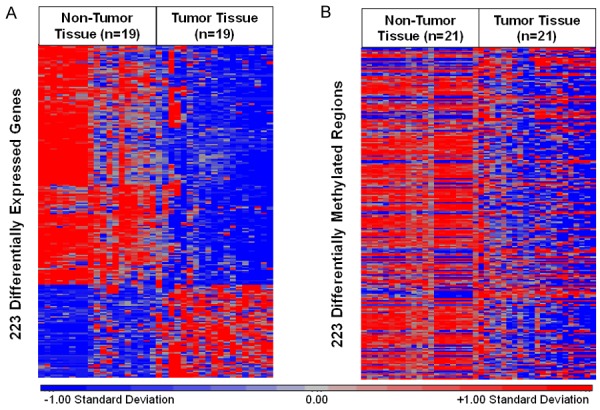
A. A total of 223 DEGs in n = 19 matched controls and cases. Red indicates relatively higher expression. Blue indicates relatively lower expression. B. A total of 223 DMRs with strongest correlation to gene expression in n = 21 matched non-tumor and tumor tissues. Red indicates relatively higher levels of methylation. Blue indicates relatively lower levels of methylation.
Further analysis of these patterns by intragene locality revealed several interesting findings. First, a consistent trend in gene suppression via promoter hypermethylation was observed in the TSS200 region, but not in the TSS1500 region. These results suggest that proximal promoter hypermethylation (e.g. TSS200) may have a greater role in cancer-associated gene silencing than hypermethylation at more distal nucleotides (e.g. TSS1500). Second, the majority of hypomethylated DMRs in the TSS1500, 5’ UTR, gene body, and 3’ UTR regions displayed gene activation (Figure 3). These results support that intragene location of methylation is a critical determinant of gene expression.
Figure 3.
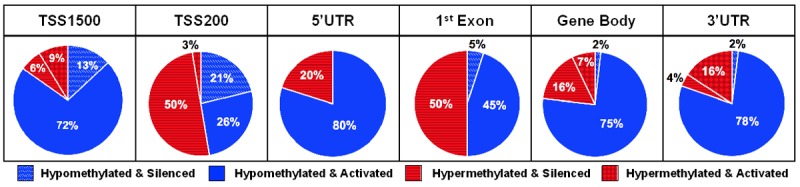
Intragene regional distribution of hypo- and hypermethylation of DMRs and DEGs and directionality of CpG methylation and mRNA expression correlation among TCGA urothelial tumors (n = 408).
Genes epigenetically dysregulated in urothelial tumors are associated with glutamate receptor signaling
In order to examine the function of these 223 genes, they were analyzed for enriched canonical pathways (Table 1). The most significantly enriched canonical pathway was glutamate receptor signaling. The seven genes identified in this pathway included CALML5, GRIA1, GRIK3, GRIN2A, GRM4, GRM7, and SLC1A6. Three genes, namely CALML5, GRM4, and SLC1A6, displayed increased expression levels in tumor tissue. GRIA1, GRIK3, GRIN2A, and GRM7 displayed decreased expression levels in tumor tissue.
Table 1.
Canonical pathways enriched among N = 223 DMGs and DEGs
| Canonical Pathways | P-Value | Associated Genes |
|---|---|---|
| Glutamate Receptor Signaling | 1.29e-6 | CALML5, GRIA1, GRIK3, GRIN2A, GRM4, GRM7, SLC1A6 |
| Transcriptional Regulatory Network in Embryonic Stem Cells | 2.29e-6 | CDX2, FOXD3, ISL1, LHX5, OTX1, SIX3 |
| cAMP-Mediated Signaling | 1.12e-5 | ADRB3, CALML5, CHRM2, CNGA3, GPR17, GRM4, GRM7, HTR1B, PDE1C, SLC1A6, TULP2, VIPR2 |
| G-Protein Coupled Receptor Signaling | 3.24e-4 | ADRA1D, ADRB3, CHRM2, GPR17, GRM4, GRM7, HTR1B, PDE1C, TULP2, VIPR2 |
Significance was defined as a right-tailed Fisher’s Exact test p-value < 0.001.
GRIA1 is prognostic indicator of overall survival independent of tumor stage
To further explore the functional significance of the 223 DMGs and DEGs, all genes were analyzed for prognostic significance of overall patient survival and potential targets were validated in an independent cohort of urothelial tumors (n = 146) [16]. Analysis of all 223 genes revealed one gene whose CpG methylation and mRNA expression levels displayed significant relationships to overall survival independent of tumor stage. Specifically, it was found that increased mRNA expression levels of GRIA1 and increased CpG methylation in the TSS1500 region were significantly associated with overall mortality in bladder cancer tumors (Figure 4A and 4B). These findings remained significant when tumor stage was included as a co-predictor of mortality (Wald chi-squared p-values = 1.34e-3; 1.05e-2, respectively). Tumor grade was not assessed as a co-predictor as all tumors were high-grade urothelial bladder tumors. These findings are supported by the significant positive correlations observed among tumor tissues between GRIA1 TSS1500 CpG methylation and mRNA expression values among tumors. However, GRIA1 was found to be both hypomethylated in the TSS1500 region and decreased in expression in tumor versus non-tumor tissue (TSS1500 Median Beta Difference = -0.24; RNASeq FC = -2.51). GRIA1 TSS1500 methylation levels were significantly correlated with GRIA1 expression levels, suggesting that hypermethylation of the TSS1500 region may activate GRIA1 mRNA expression (Figure 5). To note, a fraction of the samples displayed low-level expression of GRIA1. These findings underscore the complex relationship between CpG methylation and mRNA expression patterns in tumor tissue. Both GRIA1 TSS1500 methylation levels and GRIA1 mRNA levels are novel biological endpoints associated with mortality in bladder cancer patients.
Figure 4.

Differences in TCGA patient survival associated with GRIA1 mRNA expression and TSS1500 CpG methylation levels. In all plots, blue represents low levels of expression or methylation and red represents high levels of expression or methylation. A. Kaplan-Meier plot of overall survival in subjects with low versus high GRIA1 mRNA expression. B. Kaplan-Meier plot of overall survival in subjects with low versus high GRIA1 TSS1500 methylation levels. C. Kaplan-Meier plot of overall survival in subjects with basal-like bladder cancer with low versus high GRIA1 mRNA expression. D. Kaplan-Meier plot of overall survival in subjects with basal-like bladder cancer with low versus high GRIA1 TSS1500 methylation levels. E. Kaplan-Meier plot of overall survival in subjects with luminal-like bladder cancer with low versus high GRIA1 mRNA expression. F. Kaplan-Meier plot of overall survival in subjects with luminal-like bladder cancer with low versus high GRIA1 TSS1500 methylation levels.
Figure 5.
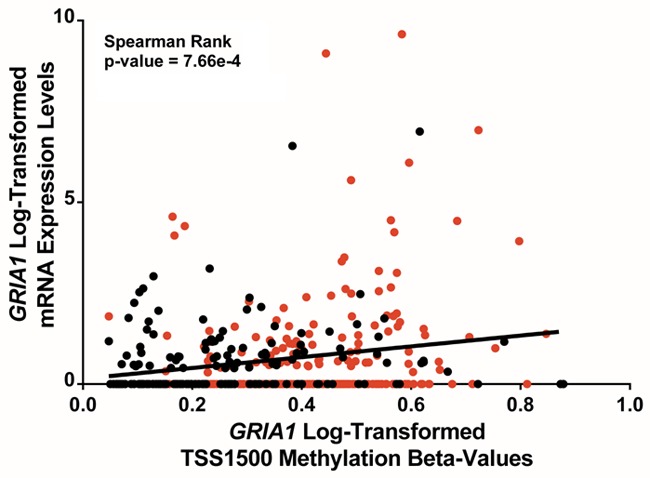
GRIA1 mRNA expression versus GRIA1 TSS1500 methylation. Log-transformed GRIA1 RNASeq mRNA expression values are plotted against log-transformed GRIA1 CpG methylation beta-values. Red points indicate values from basal-like subtypes and black points indicate values from luminal-like subtypes.
Validation of GRIA1 mRNA levels as a prognostic indicator of overall survival independent of tumor stage
The prognostic significance of increased GRIA1 mRNA expression levels in predicting overall survival of patients with bladder cancer tumors was confirmed using data from an independent cohort of 146 tumors published in a previous study by Choi et al. 2014 (Figure 6) [16]. In a Cox regression analysis, the association was also confirmed to be independent of tumor stage (p-value = 2.98e-2).
Figure 6.
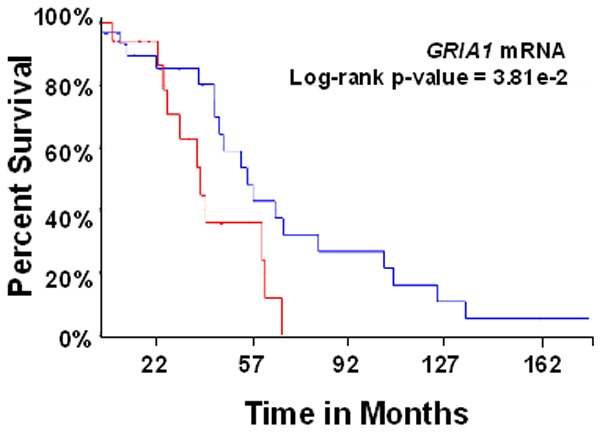
Kaplan-Meier plot of overall survival in subjects with low (blue) versus red (high) GRIA1 mRNA expression in the MD Anderson Cancer Center discovery and validation cohorts.
GRIA1 prognostic power is specific to basal-like urothelial cancers
As two distinct molecular subtypes of urothelial bladder cancers have been classified, we sought to examine whether GRIA1 was a subtype-specific prognostic biomarker [16]. GRIA1 mRNA expression levels were found to be higher in basal versus luminal subtype bladder cancers (FDR q-value = 2.66e-3, FC = 1.80). CpG methylation levels in the TSS1500 region of GRIA1 were also significantly greater in basal-like subtypes (FDR q-value = 2.61e-14, median beta difference (basal-luminal) = 0.17). Stratified Kaplan-Meier analyses between basal and luminal subtypes of urothelial tumors revealed that the prognostic power of GRIA1 is significant in basal subtypes of bladder cancer, but not in luminal subtypes. Specifically, increased mRNA expression levels of GRIA1 and increased CpG methylation in the TSS1500 region remained significantly associated with overall mortality in basal-like bladder cancer tumors (Figure 4C and 4D), while neither expression levels or TSS1500 methylation levels were significant among luminal-like bladder cancers (Figure 4E and 4F). In addition, higher transcript levels of GRIA1 and increased CpG methylation in the TSS1500 region remained significantly associated with overall mortality in basal-like bladder independent of tumor stage (Wald chi-squared p-values = 6.30e-5; 1.86e-2, respectively). These results potentially suggest a unique role for dysregulation of glutamate receptor signaling in basal-like bladder cancers.
Genes involved in glutamate receptor signaling are differentially expressed between basal and luminal subtypes of urothelial cancer
We identified 70 DEGs associated with glutamate receptor signaling between basal and luminal molecular subtypes of bladder cancer (Table 2). Of these, 42 (60%) displayed increased levels and 28 (40%) genes displayed decreased levels of mRNA in basal-like subtypes. Among these 70 genes was one gene encoding for another glutamate ionotropic receptor, GRIA2, and two genes encoding for metabotropic glutamate receptors, namely GRM3 and GRM5. GRIA2 and GRM5 displayed increased levels of mRNA expression in basal-like subtypes (FDR q-values = 2.17e-3; 2.36e-11, FC = 35.40; 2.11, respectively), while GRM3 displayed decreased levels of mRNA expression in basal-like subtypes (FDR q-value = 8.00e-19, FC = -2.50). Additionally, the DEGs that displayed increased expression levels in basal-like subtypes included two genes encoding downstream effector proteins of glutamate receptors. Specifically, these were CAMK2A and PIK3C2G (FDR q-values = 9.86e-20; 4.46E-6, FC = 2.49; 66.02, respectively) [22]. Finally, two regulators of metabotropic glutamate receptor signaling were also among the 70 DEGs. GDNF displayed increased expression levels in basal-like subtypes (FDR q-value = 4.17e-7, FC = 36.14), and F2 displayed decreased expression levels in basal-like subtypes (FDR q-value = 4.45e-5, FC = -60.69) [23,24]. These results provide further evidence of differential activity of glutamate receptors in basal and luminal subtypes of urothelial cancer.
Table 2.
Genes associated with glutamate receptor signaling differentially expressed in basal-like and luminal-like subtypes of bladder cancer
| Gene | ANCOVA FDR q-value | Median FC (Basal/Luminal) |
|---|---|---|
| ASTL | 2.65E-10 | -2.07 |
| CACNA2D1 | 3.18E-11 | 2.06 |
| CALB2 | 3.75E-17 | 2.09 |
| CAMK2A | 9.86E-20 | 2.49 |
| CASP14 | 1.31E-08 | 2.43 |
| CASP5 | 1.18E-21 | 3.64 |
| CBLN4 | 6.88E-04 | 49.23 |
| CEACAM3 | 2.14E-14 | 2.21 |
| CHGA | 3.17E-04 | 2.17 |
| CR2 | 1.45E-04 | 2.16 |
| CRH | 1.58E-21 | -358.55 |
| CSF3 | 8.47E-12 | 2.05 |
| CYP1A1 | 1.77E-11 | -2.45 |
| DAB1 | 2.65E-21 | -6.50 |
| DLGAP2 | 7.75E-04 | 44.44 |
| DRD1 | 2.88E-09 | -2.04 |
| ERBB4 | 1.41E-15 | -2.86 |
| F2 | 4.45E-05 | -60.69 |
| FMN1 | 5.06E-22 | 2.17 |
| FOLR3 | 6.13E-13 | 76.61 |
| GABBR2 | 1.13E-18 | -2.46 |
| GABRP | 2.33E-13 | 2.15 |
| GAL | 4.87E-10 | 3.51 |
| GAP43 | 4.82E-11 | 2.07 |
| GDNF | 4.17E-07 | 36.14 |
| GJB1 | 3.40E-10 | -3.64 |
| GNRH2 | 3.69E-06 | -33.66 |
| GRIA2 | 2.17E-03 | 35.40 |
| GRM3 | 8.00E-19 | -2.50 |
| GRM5 | 2.36E-11 | 2.11 |
| GRP | 1.89E-07 | 3.67 |
| HBE1 | 6.12E-06 | 29.59 |
| HNF1B | 3.63E-27 | -3.18 |
| HRH3 | 1.07E-17 | -125.54 |
| HTR2A | 6.52E-07 | 3.13 |
| HTR3A | 1.35E-06 | 66.51 |
| IFNG | 3.37E-14 | 3.06 |
| IL13 | 1.23E-05 | 39.43 |
| IL5RA | 9.57E-03 | 39.70 |
| KCNA2 | 6.49E-05 | 30.02 |
| KCNJ6 | 2.39E-05 | 49.11 |
| KLK11 | 1.28E-06 | 2.03 |
| L1CAM | 3.48E-20 | 2.01 |
| LHX1 | 1.71E-12 | 106.28 |
| LPA | 1.10E-06 | -3.06 |
| MUC2 | 1.10E-13 | -3.12 |
| NR1H4 | 3.94E-12 | -5.06 |
| NR2E1 | 2.44E-06 | -2.03 |
| NRXN1 | 3.98E-04 | 2.25 |
| PENK | 9.12E-06 | 71.70 |
| PIK3C2G | 4.46E-06 | 66.02 |
| PRKCG | 5.03E-07 | 44.46 |
| PTPRN | 3.47E-17 | 2.37 |
| PVALB | 3.43E-14 | -2.77 |
| RETN | 4.66E-09 | 2.73 |
| RNASE3 | 2.02E-05 | 2.21 |
| SGK2 | 8.93E-29 | -2.43 |
| SHH | 1.47E-14 | -5.66 |
| SLC26A5 | 9.52E-12 | -88.02 |
| SLC5A1 | 2.43E-07 | 2.31 |
| SLC6A4 | 8.57E-21 | -2.50 |
| SLCO1B3 | 8.89E-16 | 188.44 |
| SST | 2.83E-03 | -28.11 |
| TAC1 | 4.19E-07 | -52.37 |
| TFF2 | 4.53E-12 | -4.13 |
| TH | 7.99E-15 | -2.31 |
| TPO | 7.51E-04 | 42.51 |
| TTR | 1.73E-15 | -187.68 |
| TUBA4B | 6.44E-04 | 36.59 |
| UGT1A1 | 1.56E-23 | -2.25 |
Discussion
Bladder cancer is highly prevalent throughout the world and the incidence is increasing [1]. Furthermore, bladder carcinomas are the most expensive cancer to treat over the course of a patient’s lifetime [25]. While CpG methylation has been associated with bladder carcinogenesis, the role of CpG methylation in the development and progression of bladder tumors is largely unknown [2,5,7]. We identified a set of 223 DMGs and DEGs where CpG methylation levels were associated with gene expression levels. The identified genes are enriched for their role in glutamate receptor signaling. One of these genes, GRIA1, is a significant biomarker of overall mortality in patients with basal-like urothelial bladder tumors that is independent of tumor stage.
Our data corroborate several other studies in reporting that the intragene region of methylation is tied to functional changes in gene expression [5,6,8]. Specifically, we found that hypermethylation within the TSS200 region was most often associated with gene silencing, a finding that has been reported in both normal and cancerous tissues [5,26,27]. In addition, several genes previously reported to display promoter hypermethylation in bladder cancer, including DBC1, PAX6, RUNX3, and WT1 also displayed promoter hypermethylation in the present study, although none were found to have altered expression [7]. This apparent incongruity may be explained by unmeasured effects of CpG methylation at distal-acting enhancer and silencer regions and demonstrates that our knowledge of the role of CpG methylation in tumor tissues remains incomplete [28-31]. Finally, our observation of a large number of hypomethylated genes in the gene body and 3’ UTR regions is consistent with data from methylation profiling of colorectal cancers [32].
The data also demonstrate the surprising finding that many genes previously identified to be hypermethylated in their promoter regions in bladder cancer tumors are not hypermethylated in the TCGA samples. Our result that BRCA1 showed promoter hypomethylation in tumor vs. non-tumor samples contrasts with previous publications that the BRCA1 promoter is hypermethylated in bladder cancer tumors [20,21]. In addition, other tumor suppressor genes that are commonly reported to display promoter hypermethylation in bladder cancer, including PTEN, TP53, and RB1, displayed minimal changes in methylation (< 1%) between tumor and non-tumor tissue in their promoter regions in the present analysis [20,21]. A possible explanation for this discrepancy is that not all studies compare methylation status between tumor and non-tumor tissue to determine the change in methylation levels at the promoter regions. Similarly, not all studies used matched non-tumor control tissues, thus possibly failing to control for the interindividual differences in methylation [33,34]. Our analysis indicates that promoter hypermethylation may only constitute part of an epigenetic-mediated bladder carcinogenesis signature and further contributes to the emerging picture of the complex relationship between CpG methylation and gene expression.
In the present study, we found that the DEGs and DMGs were enriched for their role in the glutamate receptor signaling pathway. Numerous genes related to glutamate receptor signaling were found to be differentially expressed between basal and luminal subtypes of bladder cancer. In addition, GRIA1, which encodes for glutamate ionotropic receptor AMPA type subunit 1, was found to significantly predict prognosis among basal-like urothelial bladder cancers. Glutamate is a neurotransmitter that also functions as a growth factor to stimulate proliferation in both normal and cancerous cells, and glutamate signaling has been found to be dysregulated in numerous cancers via changes in expression of glutamate receptors [22,35-38]. Genes involved in glutamate receptor signaling have been reported to be aberrantly methylated in other malignant neoplasms [22,35-37]. This is the first study to provide evidence of dysregulation of CpG methylation and gene expression of glutamate receptors in bladder cancer. Interestingly, glutamate receptors are reported to contribute to carcinogenesis through activation of the calmodulin, PI3K/Akt, and EGFR signaling pathways [39,40]. These results highlight altered CpG methylation and/or mRNA expression of several genes involved in glutamate receptor signaling and these three downstream pathways, including CAMK2A, PIK3C2G, GDNF, and F2 [22-24]. Activation of these pathways is known to contribute to cell growth and proliferation through various mechanisms, including activation of p63, a hallmark feature of basal-like bladder cancers [16,41].
Several factors should be considered when interpreting the data from this study. First, intratumor sample location was unknown, and thus cellular heterogeneity within the tumor samples may be a confounding variable in our analysis. Second, there was not an available dataset from an independent cohort in which we could validate our finding that elevated levels of GRIA1 TSS1500 methylation predict worse overall survival outcomes in urothelial bladder patients. Finally, the literature does not currently have an explanation for the apparent paradox of methylation and expression directionality from non-tumor to tumor states to tumor progression and more severe clinical outcomes (e.g. GRIA1 mRNA expression was decreased in tumor tissue, but patients with increased mRNA expression had a worse survival outcome). This inconsistency has been observed in previous studies of associations between hypermethylation of another ionotropic glutamate receptor and mutations of critical enzymes and prognostic outcomes in non-small cell lung cancer and glioblastomas, respectively [42,43].
In summary, we conclude that epigenetic profiling of urothelial bladder carcinomas increases the understanding of the development and progression of this highly prevalent neoplasm. Although bladder cancer is primarily a disease of somatic mutations, our study supports the growing body of evidence that implicates epigenetic mechanisms in urothelial bladder carcinogenesis [2,3]. Importantly, epigenetic modifications may be reversible, and thus represent potential targets to halt tumor progression by restoring normal tissue function through epigenetic-directed pharmaceuticals [44]. As demonstrated in the present study, identification of these targets is an important first step in the development of novel cancer therapies.
Acknowledgements
This research was supported by the National Institutes of Health (http://www.nih.gov) (R01 ES019315 and P42ES005948) and the National Institute for Occupational Safety and Health (T42/OH-008673). The funders of this study had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Disclosure of conflict of interest
None.
References
- 1.Ploeg M, Aben KK, Kiemeney LA. The present and future burden of urinary bladder cancer in the world. World J Urol. 2009;27:289–293. doi: 10.1007/s00345-009-0383-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knowles MA, Hurst CD. Molecular biology of bladder cancer: new insights into pathogenesis and clinical diversity. Nat Rev Cancer. 2015;15:25–41. doi: 10.1038/nrc3817. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein JN, Akbani R, Broom BM, Wang W, Verhaak RGW, McConkey D, Lerner S, Morgan M, Creighton CJ, Smith C, Kwiatkowski DJ, Cherniack AD, Kim J, Sekhar Pedamallu C, Noble MS, Al-Ahmadie HA, Reuter VE, Rosenberg JE, Bajorin DF, Bochner BH, Solit DB, Koppie T, Robinson B, Gordenin DA, Fargo D, Klimczak LJ, Roberts SA, Au J, Laird PW, Hinoue T, Schultz N, Ramirez R, Hansel D, Hoadley KA, Kim WY, Damrauer JS, Baylin SB, Mungall AJ, Gordon Robertson A, Chu A, Kwiatkowski DJ, Sougnez C, Cibulskis K, Lichtenstein L, Sivachenko A, Stewart C, Lawrence MS, Getz G, Lander E, Gabriel SB, Creighton CJ, Donehower L, Cherniack AD, Kim J, Carter SL, Saksena G, Schumacher SE, Sougnez C, Freeman SS, Jung J, Sekhar Pedamallu C, Bhatt AS, Pugh T, Getz G, Beroukhim R, Gabriel SB, Meyerson M, Mungall AJ, Gordon Robertson A, Chu A, Ally A, Balasundaram M, Butterfield YSN, Dhalla N, Hirst C, Holt RA, Jones SJM, Lee D, Li HI, Marra MA, Mayo M, Moore RA, Schein JE, Sipahimalani P, Tam A, Thiessen N, Wong T, Wye N, Bowlby R, Chuah E, Guin R, Jones SJM, Marra MA, Hinoue T, Shen H, Bootwalla MS, Triche T Jr, Lai PH, Van Den Berg DJ, Weisenberger DJ, Laird PW, Hansel D, Hoadley KA, Balu S, Bodenheimer T, Damrauer Alan P, Hoyle JS, Jefferys SR, Meng S, Mose LE, Simons JV, Soloway MG, Wu J, Kim WY, Parker JS, Neil Hayes D, Roach J, Buda E, Jones CD, Mieczkowski PA, Tan D, Veluvolu U, Waring S, Todd Auman J, Perou CM, Wilkerson MD, Santoso N, Parfenov M, Ren X, Pantazi A, Hadjipanayis A, Seidman J, Kucherlapati R, Lee S, Yang L, Park PJ, Baylin SB, Wei Xu A, Protopopov A, Zhang J, Bristow C, Mahadeshwar HS, Seth S, Song X, Tang J, Zeng D, Chin L, Guo C, Weinstein JN, Akbani R, Broom BM, McConkey D, Casasent TD, Liu W, Ju Z, Motter T, Peng B, Ryan M, Wang W, Verhaak RGW, Su X, Yang JY, Lorenzi PL, Yao H, Zhang N, Zhang J, Mills GB, Kim J, Noble MS, Cho J, DiCara D, Frazer S, Gehlenborg N, Heiman DI, Lin P, Liu Y, Stojanov P, Voet D, Zhang H, Zou L, Chin L, Getz G, Bernard B, Kreisberg D, Reynolds S, Rovira H, Shmulevich I, Ramirez R, Schultz N, Gao J, Jacobsen A, Arman Aksoy B, Antipin Y, Ciriello G, Dresdner G, Gross B, Lee W, Reva B, Shen R, Sinha R, Onur Sumer S, Weinhold N, Ladanyi M, Sander C, Benz C, Carlin D, Haussler D, Ng S, Paull EO, Stuart J, Zhu J, Liu Y, Zhang W, Taylor BS, Lichtenberg TM, Zmuda E, Barr T, Black AD, George M, Hanf B, Helsel C, McAllister C, Ramirez NC, Tabler TR, Weaver S, Wise L, Bowen J, Gastier-Foster JM, Weinstein JN, Lerner S, Jian W, Tello S, Ittman M, Castro P, McClenden WD, Morgan M, Gibbs R, Liu Y, Saller C, Tarvin K, DiPiero JM, Owens J, Bollag R, Li Q, Weinberger P, Czerwinski C, Huelsenbeck-Dill L, Iacocca M, Petrelli N, Rabeno B, Swanson P, Shelton T, Curley E, Gardner J, Mallery D, Penny R, Van Bang N, Thi Hanh PT, Kohl B, Van Le X, Phu BD, Thorp R, Tien NV, Vinh LQ, Sandusky G, Burks E, Christ K, Gee J, Holway A, Moinzadeh A, Sorcini A, Sullivan T, Al-Ahmadie HA, Bajorin DF, Bochner BH, Garcia-Grossman IR, Regazzi AM, Solit DB, Rosenberg JE, Reuter VE, Koppie T, Boice L, Kimryn Rathmell W, Thorne L, Bastacky S, Davies B, Dhir R, Gingrich J, Hrebinko R, Maranchie J, Nelson J, Parwani A, Bshara W, Gaudioso C, Morrison C, Alexopoulou V, Bartlett J, Engel J, Kodeeswaran S, Antic T, O’Donnell PH, Smith ND, Steinberg GD, Egea S, Gomez-Fernandez C, Herbert L, Jorda M, Soloway M, Beaver A, Carter S, Kapur P, Lewis C, Lotan Y, Robinson B, Hansel D, Guo C, Bondaruk J, Czerniak B, Akbani R, Broom BM, Liu Y, Zhang W, Weinstein JN, Lerner S, Morgan M, Kim J, Cherniack AD, Freeman SS, Sekhar Pedamallu C, Noble MS, Kwiatkowski DJ, Al-Ahmadie HA, Bajorin DF, Bochner BH, Solit DB, Rosenberg JE, Reuter VE, Koppie T, Robinson B, Skinner E, Ramirez R, Schultz N, Hansel D, Kim WY, Guo C, Bondaruk J, Aldape K, Czerniak B, Jensen MA, Kahn AB, Pihl TD, Pot DA, Srinivasan D, Wan Y, Ferguson ML, Claude Zenklusen J, Davidsen T, Demchok JA, Mills Shaw KR, Sheth M, Tarnuzzer R, Wang Z, Yang L, Hutter C, Ozenberger BA, Sofia HJ, Eley G. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature. 2014;507:315–322. doi: 10.1038/nature12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, Wu R, Chen C, Li X, Zhou L, He M, Li Z, Sun X, Jia W, Chen J, Yang S, Zhou F, Zhao X, Wan S, Ye R, Liang C, Liu Z, Huang P, Liu C, Jiang H, Wang Y, Zheng H, Sun L, Liu X, Jiang Z, Feng D, Chen J, Wu S, Zou J, Zhang Z, Yang R, Zhao J, Xu C, Yin W, Guan Z, Ye J, Zhang H, Li J, Kristiansen K, Nickerson ML, Theodorescu D, Li Y, Zhang X, Li S, Wang J, Yang H, Wang J, Cai Z. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–92. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 6.Hernando-Herraez I, Garcia-Perez R, Sharp AJ, Marques-Bonet T. DNA methylation: insights into human evolution. PLoS Genet. 2015;11:e1005661. doi: 10.1371/journal.pgen.1005661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sánchez-Carbayo M. Hypermethylation in bladder cancer: biological pathways and translational applications. Tumor Biol. 2012;33:347–361. doi: 10.1007/s13277-011-0310-2. [DOI] [PubMed] [Google Scholar]
- 8.Rojas D, Rager JE, Smeester L, Bailey KA, Drobná Z, Rubio-Andrade M, Stýblo M, García-Vargas G, Fry RC. Prenatal arsenic exposure and the epigenome: identifying sites of 5-methylcytosine alterations that predict functional changes in gene expression in newborn cord blood and subsequent birth outcomes. Toxicol Sci. 2015;143:97–106. doi: 10.1093/toxsci/kfu210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.TCGA. The cancer genome atlas: charting a new course for cancer prevention, diagnosis and treatment. 2014 [Google Scholar]
- 10.Singhal SK, Usmani N, Michiels S, Metzger-Filho O, Saini KS, Kovalchuk O, Parliament M. Towards understanding the breast cancer epigenome: a comparison of genome-wide DNA methylation and gene expression data. Oncotarget. 2016;7:3002–3017. doi: 10.18632/oncotarget.6503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics. 2009;26:493–500. doi: 10.1093/bioinformatics/btp692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Necela BM, Crozier JA, Andorfer CA, Lewis-Tuffin L, Kachergus JM, Geiger XJ, Kalari KR, Serie DJ, Sun Z, Aspita AM, O’Shannessy DJ, Maltzman JD, McCullough AE, Pockaj BA, Cunliffe HE, Ballman KV, Thompson EA, Perez EA. Folate receptor-α (FOLR1) expression and function in triple negative tumors. PLoS One. 2015;10:e0122209. doi: 10.1371/journal.pone.0122209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Infinium ® HumanMethylation450 BeadChip. 2012 [Google Scholar]
- 14.Du P, Zhang X, Huang CC, Jafari N, Kibbe WA, Hou L, Lin SM. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics. 2010;11:587. doi: 10.1186/1471-2105-11-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rager JE, Yosim A, Fry RC. Prenatal exposure to arsenic and cadmium impacts infectious disease-related genes within the glucocorticoid receptor signal transduction pathway. Int J Mol Sci. 2014;15:22374–22391. doi: 10.3390/ijms151222374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi W, Porten S, Kim S, Willis D, Plimack ER, Hoffman-Censits J, Roth B, Cheng T, Tran M, Lee I, Melquist J, Bondaruk J, Majewski T, Zhang S, Pretzsch S, Baggerly K, Siefker-Radtke A, Czerniak B, Dinney CP, McConkey DJ. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25:152–165. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aine M, Eriksson P, Liedberg F, Sjödahl G, Höglund M. Biological determinants of bladder cancer gene expression subtypes. Sci Rep. 2015;5:10957. doi: 10.1038/srep10957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fleischer T, Frigessi A, Johnson KC, Edvardsen H, Touleimat N, Klajic J, Riis M, Haakensen VD, Wärnberg F, Naume B, Helland Å, Børresen-Dale AL, Tost J, Christensen BC, Kristensen VN, Sultan N, Baig SM, Sheikh MA, Jamil A. Genome-wide DNA methylation profiles in progression to in situ and invasive carcinoma of the breast with impact on gene transcription and prognosis. Genome Biol. 2014;15:435. doi: 10.1186/s13059-014-0435-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen J, Wang S, Zhang YJ, Wu HC, Kibriya MG, Jasmine F, Ahsan H, Wu DP, Siegel AB, Remotti H, Santella RM. Exploring genome-wide DNA methylation profiles altered in hepatocellular carcinoma using Infinium HumanMethylation 450 BeadChips. Epigenetics. 2013;8:34–43. doi: 10.4161/epi.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabello MJ, Grau L, Franco N, Orenes E, Alvarez M, Blanca A, Heredero O, Palacios A, Urrutia M, Fernández JM, López-Beltrán A, Sánchez-Carbayo M. Multiplexed methylation profiles of tumor suppressor genes in bladder cancer. J Mol Diagn. 2011;13:29–40. doi: 10.1016/j.jmoldx.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agundez M, Grau L, Palou J, Algaba F, Villavicencio H, Sanchez-Carbayo M. Evaluation of the methylation status of tumour suppressor genes for predicting bacillus Calmette-Guerin response in patients with T1G3 highrisk bladder tumours. Eur Urol. 2011;60:131–140. doi: 10.1016/j.eururo.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 22.Willard SS, Koochekpour S. Glutamate, glutamate receptors, and downstream signaling pathways. Int J Biol Sci. 2013;9:948–959. doi: 10.7150/ijbs.6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller S, Sehati N, Romano C, Cotman CW. Exposure of astrocytes to thrombin reduces levels of the metabotropic glutamate receptor mGluR5. J Neurochem. 1996;67:1435–1447. doi: 10.1046/j.1471-4159.1996.67041435.x. [DOI] [PubMed] [Google Scholar]
- 24.Okada Y, Eibl G, Duffy JP, Reber HA, Hines OJ. Glial cell-derived neurotrophic factor upregulates the expression and activation of matrix metalloproteinase-9 in human pancreatic cancer. Surgery. 2003;134:293–299. doi: 10.1067/msy.2003.239. [DOI] [PubMed] [Google Scholar]
- 25.Sievert KD, Amend B, Nagele U, Schilling D, Bedke J, Horstmann M, Hennenlotter J, Kruck S, Stenzl A. Economic aspects of bladder cancer: What are the benefits and costs? World J Urol. 2009;27:295–300. doi: 10.1007/s00345-009-0395-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–590. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wagner JR, Busche S, Ge B, Kwan T, Pastinen T, Blanchette M. The relationship between DNA methylation, genetic and expression inter-individual variation in untransformed human fibroblasts. Genome Biol. 2014;15:R37. doi: 10.1186/gb-2014-15-2-r37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griffiths A, Miller J, Suzuki D, Lewontin R, Gelbart W. An introduction to genetic analysis. 7th edition. New York: W. H. Freeman; 2000. Transcription: an overview of gene regluation in eukaryotes. [Google Scholar]
- 29.Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. doi: 10.1126/science.1063852. [DOI] [PubMed] [Google Scholar]
- 30.Aran D, Sabato S, Hellman A. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol. 2013;14:R21. doi: 10.1186/gb-2013-14-3-r21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Estécio MR, Issa JP. Dissecting DNA hypermethylation in cancer. FEBS Lett. 2011;585:2078–2086. doi: 10.1016/j.febslet.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Naumov VA, Generozov EV, Zaharjevskaya NB, Matushkina DS, Larin AK, Chernyshov SV, Alekseev MV, Shelygin YA, Govorun VM. Genome-scale analysis of DNA methylation in colorectal cancer using Infinium Human-Methylation450 BeadChips. Epigenetics. 2013;8:921–934. doi: 10.4161/epi.25577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fisel P, Schaeffeler E, Schwab M. DNA methylation of ADME genes ADME genes and interindividual variability of drug response. Clin Pharmacol Ther. 2016;99:512–27. doi: 10.1002/cpt.343. [DOI] [PubMed] [Google Scholar]
- 34.Sandovici I, Kassovska-Bratinova S, Loredo-Osti JC, Leppert M, Suarez A, Stewart R, Bautista FD, Schiraldi M, Sapienza C. Interindividual variability and parent of origin DNA methylation differences at specific human Alu elements. Hum Mol Genet. 2005;14:2135–2143. doi: 10.1093/hmg/ddi218. [DOI] [PubMed] [Google Scholar]
- 35.Stepulak A, Rola R, Polberg K, Ikonomidou C. Glutamate and its receptors in cancer. J Neural Transm. 2014;121:933–944. doi: 10.1007/s00702-014-1182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luksch H, Uckermann O, Stepulak A, Hendruschk S, Marzahn J, Bastian S, Staufner C, Temme A, Ikonomidou C. Silencing of selected glutamate receptor subunits modulates cancer growth. Anticancer Res. 2011;31:3181–92. [PubMed] [Google Scholar]
- 37.Chang HJ, Yoo BC, Lim SB, Jeong SY, Kim WH, Park JG. Metabotropic glutamate receptor 4 expression in colorectal carcinoma and its prognostic significance. Clin Cancer Res. 2005;11:3288–3295. doi: 10.1158/1078-0432.CCR-04-1912. [DOI] [PubMed] [Google Scholar]
- 38.Wu CS, Lu YJ, Li HP, Hsueh C, Lu CY, Leu YW, Liu HP, Lin KH, Hui-Ming Huang T, Chang YS. Glutamate receptor, ionotropic, kainate 2 silencing by DNA hypermethylation possesses tumor suppressor function in gastric cancer. Int J Cancer. 2010;126:2542–2552. doi: 10.1002/ijc.24958. [DOI] [PubMed] [Google Scholar]
- 39.de Groot J, Sontheimer H. Glutamate and the biology of gliomas. Glia. 2011;59:1181–1189. doi: 10.1002/glia.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brocke KS, Staufner C, Luksch H, Geiger KD, Stepulak A, Marzahn J, Schackert G, Temme A, Ikonomidou C. Glutamate receptors in pediatric tumors of the central nervous system. Cancer Biol Ther. 2010;9:455–68. doi: 10.4161/cbt.9.6.10898. [DOI] [PubMed] [Google Scholar]
- 41.Yoh K, Prywes R. Pathway regulation of p63, a director of epithelial cell fate. Front Endocrinol (Lausanne) 2015;6:1–9. doi: 10.3389/fendo.2015.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tamura H, Suzuki M, Moriya Y, Hoshino H, Okamoto T, Yoshida S, Yoshino I. Aberrant methylation of N-methyl-D-aspartate receptor type 2B (NMDAR2B) in non-small cell carcinoma. BMC Cancer. 2011;11:220. doi: 10.1186/1471-2407-11-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Labussiere M, Sanson M, Idbaih A, Delattre JY. IDH1 gene mutations: a new paradigm in glioma prognosis and therapy? Oncologist. 2010;15:196–199. doi: 10.1634/theoncologist.2009-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2009;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]