

Abstract
Non-Small Cell Lung Cancer (NSCLC) is driven by a variety of deregulated kinases and the development of multi-target inhibitor for multiple signaling pathways or multiple steps is required. Here, we reported that ZWM026, an indolocarbazoles analogue, derived from mangrove in coastal marine wetland, exhibited selectivity and reversibility against T790M mutant over wild-type EGFR in naturally occurring NSCLC cells and constructed NIH-3T3 cells. It simultaneously inhibited activities of HER2, HER3, HER4 and RET but was different from current multi-target kinase inhibitors. There was no activity in protein kinase C (PKC) family which is generally recognized as molecule target of indolocarbazoles. ZWM026 had more potent activities against gefitinib sensitizing, non-sensitizing and rare EGFR mutant NSCLC cells and constructed NIH-3T3 cells. ZWM026 induced apoptosis and exerted a synergistic effect by combining with cisplatin in NCI-H1975 cells. In summary, we identified a novel reversible multi-target inhibitor which could serve as a promising lead compound of drug development for NSCLC.
Keywords: EGFR T790M, ZWM026, multi-target inhibitor, NSCLC
Introduction
It has been reported that lung cancer is one of the most common cancers and the non-small cell lung cancer (NSCLC) accounts for 80-85% of all lung cancers [1]. Although great efforts have been made to improve the survival of patients with NSCLC, especially single target drugs offered hope of changing the dim situation of the disease, secondary resistance is still occurred inevitably. Therefore, alternative treatment strategies and new agents are urgently needed to be developed and explored.
During the past few years, the mutations in the kinase domain of EGFR gene have been targeted to treat NSCLC in the clinical therapy. EGFR belongs to a member of ErbB family in which HER2, HER3 and HER4 are also included [2]. The first-generation EGFR-TKIs are found to be most effective in patients with activating EGFR mutations, including small deletions at exon 19 and L858R mutation at exon 21, however, majority of patients who initially respond to therapy ultimately developed disease progression and relapsed after about 12 months of treatment due to secondary resistance [3]. Genetic mutations and other signaling aberrations that drive resistance include HER2 amplification, K-ras mutation, and MET amplification, T790M mutation, etc [4]. The T790M mutation in the EGFR tyrosine kinase domain accounts for half of secondary resistance mechanisms to first-generation EGFR-TKIs [5]. For some patients, this mutation is also detected as a primary event before drug exposure [6].
ErbB family plays a key role in lung carcinogenesis, and more evidences have shown that the other members of the family Her3 and Her4 also contribute to a more aggressive tumor phenotype and attenuate their response to targeted therapy [7]. In tumor development, HER3 may function as an oncogenic unit together with other ROS1 rearrangements [8]. c-Met/HER3/PI3K signaling pathway was found to correlate with acquired resistance of NSCLC in recent years [9]. HER4 overexpressed in 91.4% of NSCLC and was associated with cell proliferation and lymph node metastasis [10]. In addition to ErbB family, NSCLC was characterized by more activating gene rearrangements. For example, RET rearrangements which are present in 1-2% of NSCLC related to aberrant activation of RET kinase, and is considered to be a new driver mutation of NSCLC [11].
As seen above, lung cancer is a heterogeneous disease and many activated kinases concern multiple pathways of signal transduction to drive oncogenic behavior of NSCLC. Single target inhibitor that blocks only one of these pathways may be powerlessness [12]. Most promising treatments are use of agents that target the one more pathways involved in cancer development. Vandetanib [13,14], sorafenib [15] and sunitinib [16] are multi-target receptor tyrosine kinase inhibitors against different molecules, and favorable for the treatment of NSCLC. In this paper, we described a novel reversible multi-target inhibitor ZWM026 (Figure 1A), a kind of indolocarbazoles derived from mangrove in marine wetland, with a feature of selectivity against T790M mutant over wild-type EGFR, and HER2, HER3, HER4 as well as RET, without activity to protein kinase C (PKC) family which is generally recognized as molecule target of indolocarbazoles [17]. ZWM026 can more potently and selectively inhibit growth of EGFR T790M mutant cells, including EGFR-activating mutant or rare EGFR mutant NSCLC cells. Our findings suggest ZWM026 is a promising lead compound of multi-target drug development for NSCLC.
Figure 1.
Structures of ZWM026 and PKC412.
Materials and methods
Cell culture and reagents
EGFR mutant NSCLC cell lines NCI-H1975 (L858R/T790M) and HCC827 (del E746_A750), and EGFR wild-type cell lines including NCI-H3122, NCI-H1299 and PC14 were cultured in RPMI-1640 (Life Technologies). EGFR wild-type cell lines A549 were grown in F12K (Life Technologies). A431 (EGFR-WT), 293T and NIH-3T3 cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM; Life Technologies). All these cell lines were obtained from American Type Culture Collection (ATCC) and maintained and supplemented with 10% FBS (GIBCO), 2 mM L-glutamine, and 1% penicillin-streptomycin and propagated as monolayer cultures at 37°C in a humidified 5% CO2 incubator.
ZWM026 and PKC412 were provided by School of Medicine and Pharmacy, Ocean University of China. The purity of these compounds was determined to be more than 99% by HPLC (Agilent 1260 HPLC system, Agilent Technologies, CA, USA), Gefitinib and Vandetanib were purchased from the J&K Scientific Ltd. Cisplatin was obtained from Sigma-Aldrich. All the compounds were dissolved in dimethyl sulfoxide (DMSO) with a final concentration of 10 mM and stored at 20°C for the in vitro studies.
Assays for activitities of various kinases
The activities of tyrosine kinases (EGFR-T790M/L858R, EGFR-WT, HER2, HER3, HER4, RET) were detected by enzyme-linked immunosorbent assays (ELISA) with purified recombinant proteins as follows [19]: In brief, 20 µg/mL poly (Glu, Tyr) was pre-coated in 96-well plate as a substrate and 10 µmol/L ATP diluted in kinase reaction buffer (50 mmol/L HEPES [pH 7.4], 50 mmol/L MgCl2, 0.5 mmol/L MnCl2, 0.2 mmol/L Na3VO4, and 1 mmol/L DTT) was added; 1 µL of various concentrations of ZWM026 diluted in 1% DMSO were then added. Subsequently, the kinase reaction was initiated by the addition of purified tyrosine kinase proteins. After incubation for 60 min at 37°C, the plate was washed three times with PBS containing 0.1% Tween 20 (T-PBS). Anti-phosphotyrosine (PY99) antibody (1:500) was then added. After 30-min incubation at 37°C, the plate was washed three times, and goat anti-rabbit IgG was added. The plate was analyzed using a multi-well spectrophotometer (Spectra-MAX 190, Molecular Devices) at 490 nm. The inhibition rate (%) was calculated using the following equation: [1-(A490/A490 control)] × 100%.
The evaluations of compounds against the rest kinases were completed by The Kinexus target profiling service (www.kinexus.ca, Canada). The protein kinase assays were performed using the ADP-GloTM assay kit from Promega which measures the generation of ADP by the kinase. The protein kinase assays were performed at 30°C for 15 minutes in a final volume of 25 µl solution. The assay reaction receipe includes 5 µl of diluted active protein kinase, 5 µl of 125 µM stock solution of indicated substrate, 5 µl of kinase assay buffer, 5 µl of various concentrations of ZWM026, and 5 µl of 250 µM ATP stock solution. The assay was started by incubating the reaction mixture in a 96-well plate and was terminated by the addition of 25 µl of ADPGloTM Reagent. The 96-well plate was shaken and then incubated for 40 minutes at ambient temperature. Then 50 µl of Kinase Detection Reagent was added and then the 96-well reaction plate was read using the ADP-GloTM Luminescence Protocol on a GloMax plate reader (Promega; Catalog# E7031).
Modeling the binding mode of ZWM026
EGFR-T790M mutant protein kinase (PDB code 3IKA) was used for the modeling of potential binding modes of ZWM026 and the molecular modeling was conducted using Sybyl × 1.10. The active site of EGFR kinase was defined by using the bound ligand, and the covalent docking protocol was used to model potential binding modes. These hydrophobic bonds were ranked using the assigned colors and scores.
Cell viability assay
A modified tetrazolium salt assay was used to measure the inhibition of cancer cell growth. Cancer cells were plated into a 96-well microtiter plate and treated with ZWM026, gefitinib, PKC412, and vandetanib at indicated concentrations for 72 h. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT, Sigma) was added to each well of a 96-well plate. After 4 h incubation, the formazan product was dissolved and quantitated spectrophotometrically at a wavelength of 570 nm. All experiments were repeated at least three times. Inhibition rate of each sample was calculated from the A570 values as follows: % inhibition rate = (A570 nm control-treated cells/A570 nm control × 100%). The IC50 was defined as the concentration that gave a rise to 50% inhibition of cell viability.
Western blotting assay
Cells were incubated with ZWM026, gefitinib, PKC412, and vandetanib at indicated concentrations and time, then washed with PBS twice, lysed on ice for 30 min in loading buffer and then boiled about 10 min. Protein concentration was detected with BCA reagent. Equal amounts of protein were run on 6% to 10% SDS-PAGE gels, then transferred to nitrocellulose membranes (Pall), probed with the antibodies and detected by chemiluminescence with the enhanced chemiluminescence detection reagents (PIERCE). For level of HER4 phosphorylation, cells were serum-starvated for 10 hours followed by treatments of indicated compounds. Before the analysis by western blotting, cell lines were stimulated by EGF (250 ng/ml) for 15 minutes. Blots were quantified using Image J software and expressed by graphs. All statistical comparisons made to control and normalized to 1.
Antibodies to detect EGFR, phosphor-EGFR (pY1068), phosphor-ErbB3 (Tyr1222), HER4, phosphor-HER4 (Tyr984), Akt, phosphor-Akt (Ser473), Ret, phosphor-Ret (Tyr905), phosphor-ERK1/2 (pT185/pY187), ERK1/2, and GAPDH were obtained from Cell Signaling Technology. Anti-HER2 and anti-phosphor HER2 (Tyr1248) antibodies were obtained from Santa Cruz Biotechnology. ErbB3 monoclonal antibody was purchased from ImmunoWay. Recombinant Human EGF was obtained from Pepro Tech. Anti-rabbit and anti-mouse (1:5000) HRP-conjugated secondary antibodies were used as secondary antibodies. Antibodies to detect cleaved-PARP, LC3B and phosphor-PKCα/βII were obtained from Cell Signaling Technology.
Retroviral plasmids propagation and infections
The following retroviral vectors were used: pBabe EGFR-L858R/T790M (Plasmid #32073), EGFR-WT (Plasmid #11011), pBabe EGFR-Del1/T790M (Plasmid #32072), pBabe EGFR-Del2/T790M (Plasmid #32075), pBabe EGFR-Del3/T790M (Plasmid #32071), pBabe EGFR-Del4/T790M (Plasmid #32074), pBabe EGFR-Del5/T790M (Plasmid #32069), pBabe EGFR-L858R (Plasmid #11012), pBabe EGFR-Del1 (Plasmid #32062), pBabe EGFR-Del2 (Plasmid #32065) and pBabe puro-HER2 (Plasmid #40978). All of them were obtained from Addgene. EGFR D770-N771 insNPG, gag/pol and VSV-G were kind gifts from Dr. Heidi Greulich in Harvard Medical School [18]. We isolated single colonies using oblique thorn bacteria onto the plate. Then a liquid culture was used to grow enough bacteria for plasmid DNA extraction and purification. We extracted and purified the plasmid DNA using the E.Z.N.A.TM Endo-free Plasmid Mini Kit I (OMEGA, D6948-01) according to the manufacturer’s instructions. Retroviruses encoding the gene of interest were produced by transfection of the 293T cells with packaging plasmids encoding VSVG, gag-pol. Retroviral supernatants were collected after transfection of 48 h and 72 h, filtered with a 0.45 μm filter and applied to the target cells (NIH-3T3) in the presence of polybrene (Sigma-Aldrich) according to standard protocols [18]. The cell lines stably expressed the specific gene were obtained by selection in median containing 8 μg/mL puromycin (Sigma-Aldrich).
Reversibility assessments for ZWM026
NCI-H1975 cells were treated with PKC412 and ZWM026 for 16 hours at the concentration of 1 μM. Culture supernatant with the residual compounds was washed out with PBS twice and cells were further incubated with 10% FBS for the indicated times. The recovery of EGFR signaling was assessed by western blotting to detect the phosphorylation of EGFR [20].
Synergism analyses
H1975 cell were seeded in 96-well plates and treated with ZWM026 or cisplatin alone or with the combination of ZWM026 and cisplatin at the indicated doses. MTT assays were performed after 72 h of treatment. The synergism effectiveness of the inhibitors was analyzed by using CompuSyn software. CI < 1 indicates synergy, CI > 1 means antagonism, and CI = 1 means addictive between the two drugs.
Statistical analysis
Data were represented as mean ± SD. P < 0.05 was considered as being statistically significant according to t test.
Results
ZWM026 is a selectively multipotent inhibitor of kinase in vitro
To explore the kinase inhibition of ZWM026, we assessed this compound against a series of human kinases in a cell-free system using the Kinexus profiling platform and ELISA assay (Figure 1 and Table 1). The results showed that ZWM026 potently inhibited the kinase activity of EGFR-L858R/T790M with the IC50 at 183.7 nM, 13-fold stronger compared with wild-type EGFR (EGFR-WT) (IC50 is 2429 nM). Similarly, ZWM026 demonstrated a preferable selectivity when profiled against HER2, HER3, HER4, which are all the oncoproteins related to NSCLC, with IC50 at 190.7 nM, 160.3 nM, 180.1 nM respectively. ZWM026 also showed an apparent IC50 of 279.6 nM against RET kinase.
Table 1.
Kinase selectivity profile of ZWM026
| Kinases | IC50 (mean ± SD)/nM |
|---|---|
| EGFR-T790M/L858R | 183.7±30.7 |
| EGFR-WT | 2429.0±396.1 |
| HER2 | 190.6±66.9 |
| HER3 | 160.3±32.2 |
| HER4 | 180.1±78.6 |
| RET | 279.6±58.6 |
| KDR | > 10000.0 |
| PDGFR-β | > 10000.0 |
| Flt-1 | > 10000.0 |
| ABL | > 10000.0 |
| EPH-A2 | > 10000.0 |
| RON | > 10000.0 |
| PDK1 | > 1000.0 |
| PDHK2 | > 1000.0 |
| PKBα | > 1000.0 |
| PI3K (p110α/p85α) | > 1000.0 |
| PKAcα | > 1000.0 |
| GSK3β | > 1000.0 |
| PKCα | > 1000.0 |
| PKCβI | > 1000.0 |
| PKCβII | > 1000.0 |
| PKCδ | > 1000.0 |
| PKCε | > 1000.0 |
| PKCη | > 1000.0 |
| PKCγ | > 1000.0 |
| PKCι | > 1000.0 |
| PKCμ | > 1000.0 |
| PKCν | > 1000.0 |
| PKCθ | > 1000.0 |
| PKCζ | > 1000.0 |
Note: shown are inhibitory activities of ZWM026 against a variety of kinases using an in vitro ELASA assay or provided by the Kinexus target profiling service. Some values are expressed as IC50 (mean ± SD) from three or more independent experiments.
Indolocarbazoles have been generally identified as inhibitors of PKC. Hence, PKC inhibitory activities of ZWM026 were further evaluated. To our surprise, unlike PKC412, any member of PKC families was hardly observed to be inhibited by ZWM026 with the IC50 greater than 1000 nM.
Besides PKC families, as an indolocarbazoles compound, PKC412 exhibited an extensive kinase inhibition profile, such as PKB, PKA, FLT, etc. [21,22]. Thus, we further examined ZWM-026 at 1000 nM across more other kinases. ZWM026 showed minimal off-target kinase activity, as no significant activity on other kinases was observed even at 1000 nM, and we designated the activity of ZWM026 on other kinase as IC50 > 1000 nM. All these results suggest ZWM026 is a multipotent kinase inhibitor that can selectively target EGFR-L858R/T790M, HER2, HER3, HER4 and RET in vitro.
ZWM026 selectively inhibits EGFR-T790M sparing wild-type EGFR in cells
As previously described, ZWM026 potently inhibited the recombinant kinase of EGFR-L858R/T790M over wild-type EGFR in vitro. We further investigated the inhibitory effects of ZWM026 on cells haboring EGFR-WT and EGFR-T790M. Phosphorylation of EGFR and downstream signaling pathway were determined by western blotting analysis in NCI-H1975 and A549 cell lines. As shown in Figure 2A, ZWM026 more potently inhibited phosphor-EGFR and downstream signaling phosphor-Akt and phosphor-ERK in H1975 cell lines with T790M mutant EGFR compared with A549 cell lines with EGFR-WT (Figure 2A). In addition, we also assessed the ability of ZWM026 to block ligand (EGF)-induced EGFR phosphorylation. As shown in Figure 2B, ZWM026 displayed more potent ability to inhibit the ligand-induced EGFR phosphorylation as well as the downstream phosphor-Akt and phosphor-ERK in H1975 than A549 in cell lines.
Figure 2.

ZWM026 selectively inhibits EGFR-T790M and spares with EGFR-WT in cells. (A) ZWM026 inhibits EGFR phosphorylation and downstream signaling pathways in NCI-H1975 cells (left) and A549 cells (right). Cells were treated with increasing concentrations of ZWM026 for 30 minutes and lysed as described Methods. The resulting extracts were probed with the indicated antibodies. (B) ZWM026 inhibits EGF-induced EGFR phosphorylation and downstream signaling pathways in NCI-H1975 cells (left) and A549 cells (right). Cell lines were cultured in serum-starved medium for 10 hours followed by indicated compounds treatment. Cell lines were stimulated by EGF (250 ng/ml) for 15 minutes and then were analyzed by western blotting as above. (C, D) Efficacy comparisons of ZWM026, gefitinib, vandetanib and PKC412 against phosphor-EGFR in NCI-H1975 cells and A549 cells. (E, F) Efficacy comparisons of ZWM026, gefitinib, vandetanib and PKC412 against phosphor-EGFR in constructed NIH-3T3 stably expressed EGFR-L858R/T790M and EGFR-WT. (G, H) Efficacy comparisons of ZWM026, gefitinib, vandetanib and PKC412 against HCC827 cell lines harboring the sensitive mutant EGFR. Blots are quantified in Figure 2 (D), (F) and (H), respectively. (I) Molecule docking of ZWM026 binding to EGFR-T790M. EGFR x-ray structure was retrieved from the Protein Data Bank (PDB code 3IKA) and the molecular modeling was conducted using Sybyl × 1.10. Compound ZWM026 was docked into the active site of the T790M mutant EGFR. The rings of ZWM026 formed hydrophobic interaction with the LEU718, VAL726, ALA743, MET790 GLU791, MET793 and LEU844. (J) Reversibility assessments for ZWM026 and PKC412. Cells were treated with ZWM026 and PKC412 at 1 μM for 16 hours. Then removed the culture medium containing compounds, and EGFR expression was determined by Western Blotting after washing the culture at 0, 1, 3, 6, 24 hours.
To extend the comparison in phosphor-EGFR inhibition, we further assessed the abilities of other various EGFR kinase inhibitors on cells with EGFR-WT and EGFR-T790M. Gefitinib, the first-generation EGFR TKIs, has been limited the use due to the secondary resistance of EGFR T790M. Vandetanib is a novel multitarget tyrosine kinase inhibitor that inhibits EGFR, with additional inhibition of RET and VEGFR-2, which has shown promising clinical efficacy for advanced NSCLC. PKC412, an indolocarbazoles compound in development as a PKC inhibitor, is found to selectively inhibit EGFR-T790M over EGFR-WT in a non-covalent fashion. We firstly tested phosphorylation levels of EGFR in NCI-H1975 and A549 cells by these inhibitors. Significantly, ZWM026 was more potently inhibited the phosphorylation of EGFR in NCI-H1975 cells compared with gefitinib and vandetanib. While in A549 cells, ZWM026 was less potent at inhibiting phosphorylation of EGFR compared with vandetanib, but was similar to gefitinib. Vandetanib was more potently inhibited the phosphor-EGFR at lower concentration in A549 cells compared with NCI-H1975 cells. In line with reported results in the literature [20], PKC412 was also detected to selectively inhibit the phosphor-EGFR in NCI-H1975 cells over A549 cells (Figure 2C and 2D).
Considering the influence of other oncogene within the NSCLC cells, we further constructed the NIH-3T3 cells stably expressing EGFR-T790M or EGFR-WT. NIH-3T3 cells do not contain a large number of endogenous ErbB family members. A similar observation was made in these constructed NIH-3T3 cell lines (Figure 2E and 2F). EGFR-L858R/T790M-3T3 cells also displayed significant sensitivity to ZWM026 at the start concentration of 0.625 μM, which was about 16 folds greater than that in EGFR-WT-3T3 cells at the concentration of 10 μM. Vandetanib more potently inhibited the phosphor-EGFR in EGFR-WT-3T3 cells compared with EGFR-L858R/T790M-3T3 cells. Similarly, PKC412 was detected to potently inhibit the phosphor-EGFR in EGFR-L858R/T790M-3T3 cells at the start concentration of 2.5 μM over EGFR-WT-3T3 cell lines (10 μM). Furthermore, in these NIH-3T3 cell lines, ZWM026 was more effective in inhibiting the growth of cells containing EGFR-T790M resistance mutations than EGFR-WT-3T3 cells. Gefitinib had much lower activity than ZWM026 whether in EGFR-L858R/T790M-3T3 cells or EGFR-WT-3T3 cells. Vandetanib was more sensitive to EGFR-WT-3T3 cells than EGFR-L858R/T790M-3T3 cells. Like ZWM026, PKC412 also showed more obvious anti-proliferation activity in EGFR-L858R/T790M-3T3 cells than EGFR-WT-3T3 cells based on the MTT assay (Table 2). All these results suggest that ZWM026 has greater selectivity than gefitinib and higher sensitivity than vandetanib in EGFR-T790M mutant cells over EGFR-WT cells, and has similar activity to PKC412. Moreover, ZWM026 also potently inhibited phosphor-EGFR in the HCC827 cell lines harboring the common EGFR-activating mutant EGFR whereas PKC412 did not exhibit more activity than ZWM026 (Figure 2G and 2H).
Table 2.
Efficacy comparisons of ZWM026, gefitinib, vandetanib and PKC412 against EGFR-WT and EGFR-L858R/T790M in cell proliferation
| EGFR-L858R/T790M IC50 (mean ± SD)/μM | EGFR-WT IC50 (mean ± SD)/μM | |
|---|---|---|
| ZWM026 | 1.08±0.12 | 5.92±0.55 |
| Gefitinib | 13.69±0.69 | 8.37±0.40 |
| Vandetanib | 9.45±0.91 | 2.93±0.93 |
| PKC412 | 2.54±0.55 | 9.33±0.85 |
Note: shown are the IC50 values of each drugs using MTT assay. All values are shown in μM and expressed as mean ± SD from three or more independent experiments.
To further prove whether ZWM026 could bind to the EGFR-T790M kinase, we performed the molecule docking experiment using the structure-based approach. Compound ZWM026 was docked into the active site of the T790M mutant EGFR. From the docking results (Figure 2I), we found that there was a small hydrophobic pocket between the indolocarbazole ring and the Met790 side chain, the hydrophobicity conferred by this mutation likely contributed to the potency of these compounds against the T790M mutant. The indolocarbazole rings of ZWM026 formed hydrophobic interaction with the Leu718, Val726, Ala743, Met790, Glu791, Met793 and Leu844 of the kinase.
In addition, we studied the reversible nature of ZWM026 as well as PKC412 on EGFR-T790M kinase inhibition activity in NCI-H1975 cells by examining the recovery of phosphor-EGFR after treatment in a drug “wash-out” assay. The results showed that, similar to PKC412, the expression of phosphor-EGFR was recovered as the extension of elution time (Figure 2J). These suggested that ZWM026 was also a reversible inhibitor of EGFR-T790M kinase.
Activities of ZWM026 against other kinases in vitro
By viewing of the kinase profiling data in vitro, we further examined the kinase inhibitory activities of ZWM026 on other oncoproteins including HER2, HER3, HER4 and RET in H1975 cells, and compared the inhibition activity of ZWM026 with those of gefitinib, vandetanib and PKC412, respectively. ZWM026 showed moderate a inhibition of HER2 and was slightly evident than gefitinib and vandetanib, but it displayed a significant sensitivity against the HER2 than PKC412 (Figure 3A and 3C), which was reappeared in the NIH-3T3 cell lines engineered to stably express wild-type HER2 (Figure 3I). ZWM026 more potently inhibited the phosphorylation of HER3 similar to vandetanib, while gefitinib was less potent at inhibiting phosphorylation of HER3. PKC412 hardly inhibited the phosphor-HER3 even though at high concentration of 20 μM (Figure 3B and 3D). Next, the inhibition activity of ZWM026 against HER4 was explored. ZWM026 could inhibit the phosphorylation of HER4 potently at the lower concentration of 0.3125 μM (Figure 3E and 3G) stimulated by EGF. Vandetanib was less potent against HER4 with the obvious start concentration at 2.5 μM, and gefitinib and PKC412 flabbily inhibited the phosphorylation of HER4.
Figure 3.

Activities of ZWM026 against other kinases in H1975 cells. (A-H) Efficacy comparisons of ZWM026, gefitinib, vandetanib and PKC412 against phosphorylation HER2 (A), HER3 (B), HER4 (E) and RET (F) in NCI-H1975 cell lines. Cells were treated with increasing concentrations of indicated compounds for 30 minutes and the cell extracts were probed with the indicated antibodies. Blots are quantified in Figure 3 (C), (D), (G) and (H), respectively. (I) Efficacy comparisons of ZWM026 and PKC412 on phosphorylation of HER2 in constructed NIH-3T3 stably expressed HER2-WT. The experiment methods are same as above.
In addition, we evaluated the inhibition activity of ZWM026 against RET oncoprotein (Figure 3F and 3H). ZWM026 showed a moderate inhibition of RET phosphorylation and was comparable with those of vandetanib and PKC412. Gefitinib was hardly inhibited the phosphorylation of RET even at high concentration. In a word, it was consistent with the previous kinase activity assay in cell-free system. These data suggest that ZWM026 is a multi-target inhibitor, and has different characteristics of kinase profile from vandetanib and PKC412.
ZWM026 potently and selectively inhibits growth of naturally occurring NSCLC cells and constructed NIH-3T3 cells
Next, we explored how the pharmacologic activity against multiple kinase signaling is translated into cell proliferation effects (Figure 4). ZWM026 potently inhibited proliferations of NCI-H1975 and HCC827 cells with IC50 at 0.26 and 0.41 μM respectively, which indicated that ZWM026 had equivalent activity against sensitizing and non-sensitizing mutant EGFR. For the EGFR-WT, the IC50 values in cell lines (PC14, NCI-H3122) were 6.44 and 9.02 μM respectively, and they were weaker than that of cells with mutant EGFR. However, ZWM026 displayed a higher level of activity on two cell lines expressing EGFR-WT in the presence of a KRAS or NRAS mutation (A549 and NCI-H1299) with IC50 of 1.764 and 2.123 μM, respectively. The underlying mechanism was not known. However, it is noticed that ZWM026 has advantage over early generation TKIs gefitinib and afatinib, which are induced drug resistance by activated Ras in cell harboring Ras mutant. ZWM026 was also associated with greater potency toward A431 cells harboring amplified EGFR and HER2 than gefitinib. This may be attributed to inhibitory activities of ZWM026 on the mutant EGFR and HER2 kinases. In line with the HER2 recombinant enzyme assays, ZWM026 also exhibited great a potency against NIH-3T3 cells constructed with cDNA of HER2 (Table 3).
Figure 4.
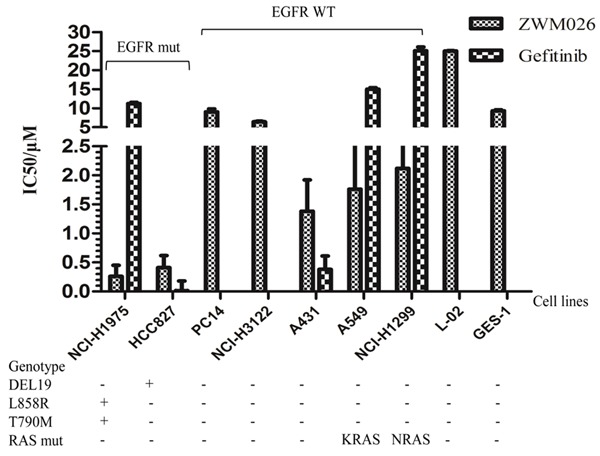
ZWM026 potently and selectively inhibits growths of several different NSCLC cells. ZWM026 inhibits growths in various tumor cell lines, including NSCLC cells with EGFR-mutant and EGFR-WT, as well as some normal cell lines. Cell viability was analyzed using MTT assay after 72 hours of treatment. The data are expressed as mean ± SD from three or more independent experiments.
Table 3.
Proliferation inhibition comparisons of ZWM026 and gefitinib against HER2-WT in constructed NIH-3T3 cells
| Cell lines | ZWM026 IC50/μM (mean ± SD) | Gefitinib IC50/μM (mean ± SD) |
|---|---|---|
| NIH-3T3 | 3.89±1.11 | 10.88±0.78 |
| HER2-WT-3T3 | 1.09±0.19 | 20.08±0.95 |
Note: shown are the IC50 values of each drugs using MTT assay. All values are shown in μM and expressed as mean ± SD from three or more independent experiments.
In addition, it was found that ZWM026 had low toxicity on two noncancerous cells L-02 and GES-1 (Figure 4); The IC50 values were 25.04 and 9.33 μM, respectively. Moreover, activities of ZWM026 against other EGFR mutants were investigated in cells (Table 4). Mutant cDNAs were transfected into NIH-3T3 cells, including L858R mutant, exon 19 deletion mutants, exon 19 deletion mutants/T790M and insNPG (exon 20) mutant. Compared with EGFR-WT (Table 2), ZWM026 has more efficacy to inhibit proliferation of these cells, and more potency than gefitinib. Especially, ZWM026 also displayed a higher sensitivity to cells with L858R/T790M than with L858R, and to cells with exon 19 deletion mutants/T790M than with exon 19 deletion mutants.
Table 4.
Proliferation inhibition comparisons of ZWM026 and gefitinib against other mutant EGFR in constructed NIH-3T3 cells
| EGFR mutation (constructed NIH-3T3) | ZWM026 IC50 (mean ± SD)/μM | Gefitinib IC50 (mean ± SD)/μM | |
|---|---|---|---|
| EGFR-activating mutation | EGFR-L858R | 2.1±0.87 | 12.86±1.23 |
| EGFR-del1 | 3.25±1.52 | 12.71±0.97 | |
| EGFR-del2 | 3.09±0.67 | 15.37±1.52 | |
| EGFR-resistant mutation | EGFR-del1/T790M | 2.51±1.62 | 8.05±1.87 |
| EGFR-del2/T790M | 1.76±0.92 | 38.22±1.96 | |
| EGFR-del3/T790M | 1.69±1.14 | 11.85±1.08 | |
| EGFR-del4/T790M | 1.41±0.16 | 35.9±1.42 | |
| EGFR-del5/T790M | 2.02±0.17 | 18.21±0.85 | |
| EGFR-insNPG (exon 20) | 1.42±1.33 | 12.61±0.59 |
Note: shown are the IC50 values of each drugs using MTT assay. All values are shown in μM and expressed as mean ± SD from three or more independent experiments.
ZWM026 induces apoptosis in NCI-H1975 cell lines and exerts synergistic effect by combining with cisplatin in NCI-H1975 cell lines
Lee [20] reported that the PKC inhibitor Gö6976 induced apoptosis in EGFR mutant NSCLC cells independently of PKC inhibition by Gö6976. We next explored the death way of ZWM026 in different treated cells. Treatment with ZWM026 for 48 h induced apoptosis in H1975 cells as demonstrated by an increase of cleaved PARP. However, this change was not observed in A549 cells (Figure 5A and 5B). Western blotting assays also showed that expressions of phosphor-EGFR and phosphor-PKCα/βII were inhibited by ZWM026 in H1975 cells and A549 cells. However, we speculated that inhibition of phosphor-PKCα/βII was not probably accompanied by the decrease of phosphor-EGFR, rather due to inhibitory effects on other members of eERB family by ZWM026. In order to demonstrate this hypothesis, ZWM026 was exposed to the constructed NIH3T3 cells with EGFR-WT and EGFR-L858R/T790M for short term (30 min). It was found that the expression of phosphor-PKCα/βII was not altered following by the level of phosphor-EGFR whether in cells with EGFR-WT or with EGFR-L858R/T790M. In contrast, PKC412, a PKC inhibitor, inhibited phosphor-PKCα/βII expression in the two cell lines all the time. These results suggested that ZWM026 had not potency to inhibit activity of PKC in cells, confirmed findings in recombinant kinase assay (Figure 5C). This indicated that ZWM026 induced apoptosis in H1975 but not in A549. In fact, ZWM026 was found to induce autophagy in A549 cells by detecting the autophagy marker protein LC3B, because the light chain LC3-II was increased with an increase of ZWM026.
Figure 5.
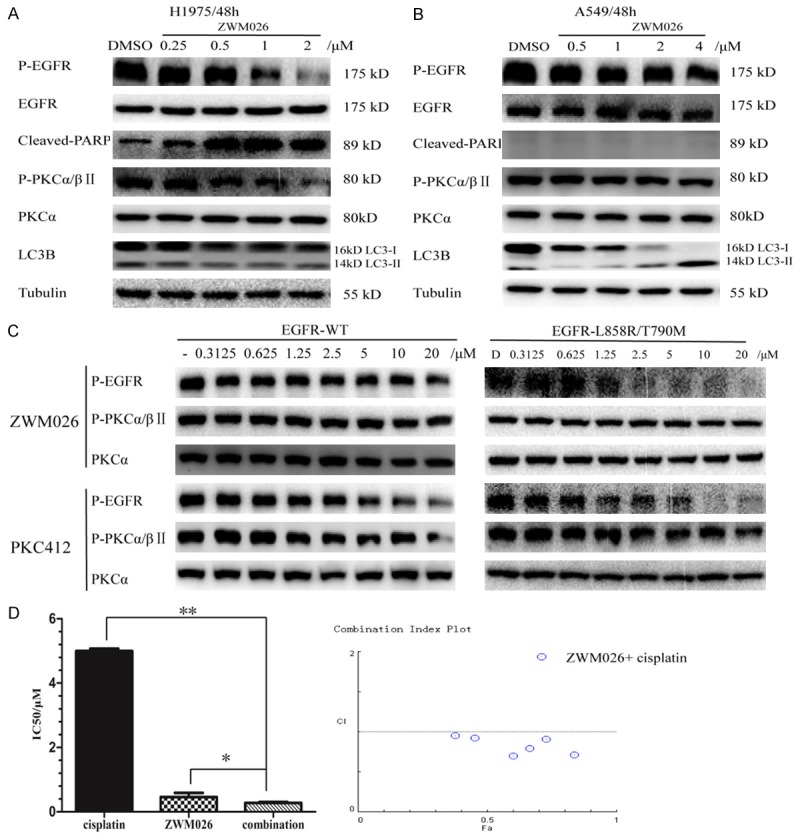
ZWM026 induces different death way of cells and exerts synergistic effect combined with cisplatin. (A, B) Activities of ZWM026 against cleaved-PARP, LC3B and phosphor-PKCα/βII in H1975 and A549 cell lines. Cells were treated with ZWM026 and gefitinib at indicated concentration for 48 hours. The resulting extracts were probed with the indicated antibodies. (C) Efficacy comparisons of ZWM026 and PKC412 against phosphor-EGFR and phosphor-PKCα/βII in constructed NIH-3T3 stably expressed EGFR-L858R/T790M and EGFR-WT. Cells were treated with the compounds for 30 minutes and lysed as described methods. (D) ZWM026 in combination with cisplatin induces the synergistic inhibition of proliferation. We examined the effects of ZWM026 and cisplatin, either alone or in combination, on growth inhibition of H1975 cells by the MTT assay (The CI value was calculated according to the CompuSyn software). The CI > 1 is antagonistic, CI = 1 is additive and CI < 1 is synergistic. *p < 0.05, combination vs. ZWM026, **p < 0.01, combination vs. cisplatin.
A prospective issue to improve the efficacy against NSCLC is the integration of EGFR-TKI with cytotoxic chemotherapy [23]. As we all know, cisplatin is the first-line treatment drug of advanced NSCLC patients. We further examined the effects of ZWM026 and cisplatin, either alone or in combination, on growth inhibition of H1975 with the MTT assay. As shown in Figure 5D, IC50 values of growth inhibition on H1975 cells by ZWM026 and cisplatin were 0.42 and 4.86 μM, respectively, while co-treatment with ZWM026 and cisplatin greatly enhanced the cells proliferation inhibition compared to the treatment with ZWM026 or cisplatin alone (IC50 value was 0.38 μM). CI value were calculated to be 0.95 less than 1. This suggests that ZWM026 is synergistic with cisplatin and has potency to improve the treatment effect of cisplatin against NSCLC.
Discussion
It is conceived that NSCLC cells are driven by deregulated kinases involving in multiple signaling pathways, such as ALK, BRAF, EGFR, HER2, KRAS, MEK1, MET, NRAS, PIK3CA, RET, ROS1 and so on [24]. Therefore, development of multi-target agents is pursued to inhibit multiple signaling pathways or multiple steps in the same pathway. Presently, it has been observed that several small molecules multiple tyrosine kinase inhibitors have been studied in clinic trail, and have demonstrated favorable clinical activity with dose limiting toxicity. Sorafenib inhibits the kinase activities of C-RAF and B-RAF, VEGFR, PDGFR-β and KIT [15]. Sunitinib is a multi-target inhibitor of PDGFR, KIT, RET, and VEGFR [16]. Vandetanib inhibits the kinase activities of VEGFR, EGFR and RET [13]. In this paper, we reported a novel small molecule multiple tyrosine kinase inhibitor ZWM026 which inhibits proliferation of NSCLC cells by targeting mutant EGFR, RET, as well as other ErbB family members such as HER2, HER3 and HER4.
Most of NSCLC patients characterized by EGFR activated mutations show a high response rate and a survival benefit when they were treated with the early first-generation EGFR TKIs, gefitinib or erlotinib. However, the T790M secondary mutation has been proved to be the most frequent mechanism for acquired resistance. The second-generation of EGFR TKI that covalently modifies EGFR, such as afatinib, irreversibly binds to EGFR and HER2 and potently suppresses the kinase activity of wild-type and activated EGFR mutants [25,26]. It can lead to consequent dose-limiting toxicities and limited therapeutic window related to nonselective inhibition of WT EGFR. Therefore, there remains a significant need for the development of third-generation EGFR TKIs to target T790M and sensitize mutations more selectively than wild-type EGFR. AZD9291 [4,27,28], a novel oral, irreversible, third-generation TKI with the potently and selectively inhibition of T790M over WT, was recently approved by the US Food and Drug Administration for patients with EGFR-T790M positive NSCLC. Here, we showed that ZWM026 reversibly and selectively inhibited the EGFR-T790M mutation sparing WT EGFR. Consistent with the results of recombinant kinase experiment in cell-free system, using either NSCLC cell lines with endogenous T790M mutation or NIH-3T3 cells engineered to express EGFR-T790M, the inhibitions of ZWM026 against cell proliferation and phosphorylation of EGFR in cells with T790M mutation were found to be more potent compared with cells with wild-type EGFR. Our studies also revealed that ZWM026 was effective against the other activating mutation such as L858R and exon 19 deletions (Figure 2G and Table 4), and more sensitive to double mutant comprising T790M. Additionally, it has been reported that currently approved EGFR TKIs are ineffective against the EGFR exon 20 insertion [29,30]. But, to our surprise, ZWM026 also displayed to be effective against rare gefitinib-resistant EGFR-insNPG mutants (Table 4).
Overexpression of the ErbB family of receptor tyrosine kinases has been found in a variety of tumors including lung, breast and prostate [31]. Identified inhibitors to target multiple ErbB family members may provide a therapeutic benefit to cancer patients. Indeed, a number of pan-ErbB inhibitors that target two, three or four members of ErbB family have been developed for treatment of epithelial cancers [32]. Our data showed that ZWM026 was active against mutant EGFR and other three members of the ErbB family, which suggested that ZWM026 can be thought as a special panErbB tyrosine kinase inhibitor.
RET fusions have been found in lung adenocarcinoma, where KIF5B-RET is the most prevalent [33]. Several related clinical trials for NSCLC with KIF5B-RET rearrangements using existing RET inhibitors, such as lenvatinib, vandetanib, sunitinib, and AUY922, have been swiftly initiated and displayed favorable response [34]. In our study, ZWM026 inhibited the kinase activity of RET in vitro as well as in cells detected by western blotting assay, which may be beneficial in treating patients with KIF5B-RET fusions.
Conventional wisdom suggests that PKC functions as a tumor promoter and subsequent result is the surge of inhibitors against PKC [35]. Surprisingly, recent study indicates that PKC plays a tumor suppressive role and its activity requires restoring than inhibiting [36]. PKC412, a well-characterized classical PKC inhibitor with structurally related indolocarbazoles compound, could potently inhibit EGFR T790M reversibly, while sparing wild-type EGFR [21]. In the current work, ZWM026 was revealed to be a reversible EGFR-T790M kinase inhibitor similar to PKC412, but hardly inhibited any isotype of PKC family. These results suggest that ZWM026 will exhibit more efficacy and low toxicity without PKC inhibition than PKC412.
We also compared inhibitory effects of ZWM026 on kinase EGFR/T790M, HER2, HER3, HER4 and RET with gefitinib, vandetanib, and PKC412. ZWM026 showed more potency to inhibit T790M over WT EGFR than gefitinib, vandetanib, and similar to PKC412. For HER2, ZWM026 exhibited much efficiency than PKC412, and didn’t make itself stand out of gefitinib and vandetanib. Also, ZWM026 was revealed to be more capable to inhibit activity of HER3 and HER4 than gefitinib or PKC412, and exhibited efficacy in inhibiting phosphor-RET, while gefitinib hardly blocked the activity of RET.
In summary, our findings characterize ZWM026 as a novel multi-target inhibitor with selective inhibition EGFR T790M over EGFR-WT, It is different from current multi-target kinase inhibitors because it is simultaneously against HER2, HER3, HER4 and RET. Based on the findings that ZWM026 induced apoptosis and exerted synergistic effect in combination with cisplatin in NCI-H1975 cell lines, we believe that ZWM026 is a promising lead compound of drug development for NSCLC. Although currently imperative challenge is to enhance the solubility and quantity of ZWM026 to further perform the anti-tumor study in vivo, our study offers a possibility to explore more multi-target inhibitors with different characteristics from indolocarbazoles.
Acknowledgements
We appreciate Dr. Heidi Greulich’s generous help for providing the plasmids. This work was supported by the Nation Natural Science Foundation of China [No. 81373323], NSFC-Shandong Joint Fund [U1406402] and the Scientific and Technological Innovation Project Financially Supported by Qingdao National Laboratory for Marine Science and Technology [No. 2015ASKJ02]. We thanks Dr. Ming-yi Sun from Georgia Uni. for the excellent and professional revision of our manuscript.
Disclosure of conflict of interest
None.
Abbreviations
- NSCLC
non-small cell lung cancer
- EGFR
epidermal growth factor receptor
- HER2
human epidermal growth factor receptor 2
- HER3
human epidermal growth factor receptor 3
- HER4
human epidermal growth factor receptor 4
- RET
rearranged during transfection
- PKC
protein kinase C
- EGFR-TKIs
EGFR tyrosine kinase inhibitors
- ELISA
enzyme linked immunosorbent assay
References
- 1.Yang J, Lin J, Liu T, Chen T, Pan S, Huang W, Li S. Analysis of lncRNA expression profiles in non-small cell lung cancers (NSCLC) and their clinical subtypes. Lung Cancer. 2014;85:110–115. doi: 10.1016/j.lungcan.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 2.Doebele RC, Oton AB, Peled N, Camidge DR, Bunn PA Jr. New strategies to overcome limitations of reversible EGFR tyrosine kinase inhibitor therapy in non-small cell lung cancer. Lung Cancer. 2010;69:1–12. doi: 10.1016/j.lungcan.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, Kris MG, Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. doi: 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitorsin lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ohashi K, Maruvka YE, Michor F, Pao W. Epidermal growth factor receptor tyrosine kinase inhibitor-resistant disease. J. Clin. Oncol. 2013;31:1070–1080. doi: 10.1200/JCO.2012.43.3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, Suehisa H, Ouchida M, Aoe K, Aoe M, Kiura K, Shimizu N, Date H. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer Res. 2006;66:7854–7858. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- 7.Bellezza G, Del Sordo R, Colella R, Ludovini V, Ragusa M, Bianconi F, Ferri I, Borri F, Chiari R, Puma F, Crinò L, Sidoni A. Co-expression of receptors of the HER family correlates with clinical outcome in non-small cell lung cancer (NSCLC) Virchows Archiv. 2013;463:663–671. doi: 10.1007/s00428-013-1445-x. [DOI] [PubMed] [Google Scholar]
- 8.Engelman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive nonsmall cell lung cancer cell lines. Proc Natl Acad Sci U S A. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelman JA, Zejnullahu K, Gale CM, Lifshits E, Gonzales AJ, Shimamura T, Zhao F, Vincent PW, Naumov GN, Bradner JE, Althaus IW, Gandhi L, Shapiro GI, Nelson JM, Heymach JV, Meyerson M, Wong KK, Jänne PA. PF00299804, an irreversible pan-ERBB inhibitor, is effective in lung cancer models with EGFR and ERBB2 mutations that are resistant to gefitinib. Cancer Res. 2007;67:11924–11932. doi: 10.1158/0008-5472.CAN-07-1885. [DOI] [PubMed] [Google Scholar]
- 10.Deng Z, Yu W, Hu G, Zheng R, Zhang D, Tan Y, Xu Y, Jiang W. A study on the expression of erbB4/HER4 in non-small cell lung cancer. Zhongguo Fei Ai Za Zhi. 2002;5:177–179. doi: 10.3779/j.issn.1009-3419.2002.03.06. [DOI] [PubMed] [Google Scholar]
- 11.Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, Nammo T, Sakamoto H, Tsuta K, Furuta K, Shimada Y, Iwakawa R, Ogiwara H, Oike T, Enari M, Schetter AJ, Okayama H, Haugen A, Skaug V, Chiku S, Yamanaka I, Arai Y, Watanabe S, Sekine I, Ogawa S, Harris CC, Tsuda H, Yoshida T, Yokota J, Shibata T. KIF5B-RET fusions in lung adenocarcinoma. Nature Medicine. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.De Jonge MJ, Verweij J. Multiple targeted tyrosine kinase inhibition in the clinic: all for one or one for all? Eur J Cancer. 2006;42:1351–1356. doi: 10.1016/j.ejca.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Ichihara E, Ohashi K, Takigawa N, Osawa M, Ogino A, Tanimoto M, Kiura K. Effects of vandetanib on lung adenocarcinoma cells harboring epidermal growth factor receptor T790M mutation in vivo. Cancer Res. 2009;69:5091–5098. doi: 10.1158/0008-5472.CAN-08-4204. [DOI] [PubMed] [Google Scholar]
- 14.Wu W, Onn A, Isobe T, Itasaka S, Langley RR, Shitani T, Shibuya K, Komaki R, Ryan AJ, Fidler IJ, Herbst RS, O’Reilly MS. Targeted therapy of orthotopic human lung cancer by combined vascular endothelial growth factor and epidermal growth factor receptor signaling blockade. Mol Cancer Ther. 2007;6:471–483. doi: 10.1158/1535-7163.MCT-06-0416. [DOI] [PubMed] [Google Scholar]
- 15.Je Y, Schutz FA, Choueiri TK. Risk of bleeding with vascular endothelial growth factor receptor tyrosine-kinase inhibitors sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. Lancet Oncol. 2009;10:967–974. doi: 10.1016/S1470-2045(09)70222-0. [DOI] [PubMed] [Google Scholar]
- 16.Haas NB, Manola J, Uzzo RG, Flaherty KT, Wood CG, Kane C, Jewett M, Dutcher JP, Atkins MB, Pins M, Wilding G, Cella D, Wagner L, Matin S, Kuzel TM, Sexton WJ, Wong YN, Choueiri TK, Pili R, Puzanov I, Kohli M, Stadler W, Carducci M, Coomes R, DiPaola RS. Adjuvant sunitinib or sorafenib for high-risk, non-metastatic renal-cell carcinoma (ECOG-ACRIN E2805): a double-blind, placebo-controlled, randomised, phase 3 trial. Lancet. 2016;387:2008–2016. doi: 10.1016/S0140-6736(16)00559-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prudhomme M. Biological targets of antitumor indolocarbazoles bearing a sugar moiety. Curr Med Chem Anticancer Agents. 2004;4:509–521. doi: 10.2174/1568011043352650. [DOI] [PubMed] [Google Scholar]
- 18.Greulich H, Chen TH, Feng W, Jänne PA, Alvarez JV, Zappaterra M, Bulmer SE, Frank DA, Hahn WC, Sellers WR, Meyerson M. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313. doi: 10.1371/journal.pmed.0020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xing W, Ai J, Jin S, Shi Z, Peng X, Wang L, Ji Y, Lu D, Liu Y, Geng M, Hu Y. Enhancing the cellular anti-proliferation activity of pyridazinones as c-met inhibitors using docking analysis. Eur J Med Chem. 2015;95:302–312. doi: 10.1016/j.ejmech.2015.03.041. [DOI] [PubMed] [Google Scholar]
- 20.Lee HJ, Schaefer G, Heffron TP, Shao L, Ye X, Sideris S, Malek S, Chan E, Merchant M, La H, Ubhayakar S, Yauch RL, Pirazzoli V, Politi K, Settleman J. Non-covalent wild-type-sparing inhibitors of EGFR T790M. Cancer Discov. 2013;3:168–181. doi: 10.1158/2159-8290.CD-12-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabbro D, Ruetz S, Bodis S, Pruschy M, Csermak K, Man A, Campochiaro P, Wood J, O’Reilly T, Meyer T. PKC412-a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug Des. 2000;15:17–28. [PubMed] [Google Scholar]
- 22.Weisberg E, Boulton C, Kelly LM, Manley P, Fabbro D, Meyer T, Gilliland DG, Griffin JD. Inhibition of mutant FLT3 receptors in leukemia cells by the small molecule tyrosine kinase inhibitor PKC412. Cancer Cell. 2002;1:433–443. doi: 10.1016/s1535-6108(02)00069-7. [DOI] [PubMed] [Google Scholar]
- 23.Cheng H, An SJ, Zhang XC, Dong S, Zhang YF, Chen ZH, Chen HJ, Guo AL, Lin QX, Wu YL. In vitro sequence-dependent synergism between paclitaxel and gefitinib in human lung cancer cell lines. Cancer Chemother Pharmacol. 2011;67:637–646. doi: 10.1007/s00280-010-1347-4. [DOI] [PubMed] [Google Scholar]
- 24.Kohno T, Nakaoku T, Tsuta K, Tsuchihara K, Matsumoto S, Yoh K, Goto K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl Lung Cancer Res. 2015;4:156–164. doi: 10.3978/j.issn.2218-6751.2014.11.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janjigian YY, Smit EF, Groen HJ, Horn L, Gettinger S, Camidge DR, Riely GJ, Wang B, Fu Y, Chand VK, Miller VA, Pao W. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4:1036–1045. doi: 10.1158/2159-8290.CD-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kosaka T, Tanizaki J, Paranal RM, Endoh H, Lydon C, Capelletti M, Repellin CE, Choi J, Ogino A, Calles A, Ercan D, Redig AJ, Bahcall M, Oxnard GR, Eck MJ, Janne PA. Response heterogeneity of EGFR and HER2 exon 20 insertions to covalent EGFR and HER2 inhibitors. Cancer Res. 2017;77:2712–2721. doi: 10.1158/0008-5472.CAN-16-3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eberlein CA, Stetson D, Markovets AA, Al-Kadhimi KJ, Lai Z, Fisher PR, Meador CB, Spitzler P, Ichihara E, Ross SJ, Ahdesmaki MJ, Ahmed A, Ratcliffe LE, O’Brien EL, Barnes CH, Brown H, Smith PD, Dry JR, Beran G, Thress KS, Dougherty B, Pao W, Cross DA. Acquired resistance to the mutant-selective EGFR Inhibitor AZD9291 is associated with increased dependence on RAS signaling in preclinical models. Cancer Res. 2015;75:2489–2500. doi: 10.1158/0008-5472.CAN-14-3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, Xu C, Rhee K, Chen T, Zhang H, Palakurthi S, Jang J, Lelais G, DiDonato M, Bursulaya B, Michellys PY, Epple R, Marsilje TH, McNeill M, Lu W, Harris J, Bender S, Wong KK, Jänne PA, Eck MJ. Overcoming EGFR (T790M) and EGFR (C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–132. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arcila ME, Nafa K, Chaft JE, Rekhtman N, Lau C, Reva BA, Zakowski MF, Kris MG, Ladanyi M. EGFR exon 20 insertion mutations in lung adenocarcinoma as: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013;12:220–229. doi: 10.1158/1535-7163.MCT-12-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jia Y, Juarez J, Li J, Manuia M, Niederst MJ, Tompkins C, Timple N, Vaillancourt MT, Pferdekamper AC, Lockerman EL, Li C, Anderson J, Costa C, Liao D, Murphy E, DiDonato M, Bursulaya B, Lelais G, Barretina J, McNeill M, Epple R, Marsilje TH, Pathan N, Engelman JA, Michellys PY, McNamara P, Harris J, Bender S, Kasibhatla S. EGF816 exerts anticancer effects in non-small cell lung cancer by irreversibly and selectively targeting primary and acquired activating mutations in the EGF receptor. Cancer Res. 2016;76:1591–1602. doi: 10.1158/0008-5472.CAN-15-2581. [DOI] [PubMed] [Google Scholar]
- 31.Shih AJ, Telesco SE, Radhakrishnan R. Analysis of somatic mutations in cancer: molecular mechanisms of activation in the ErbB family of receptor tyrosine kinases. Cancers (Basel) 2011;3:1195–1231. doi: 10.3390/cancers3011195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, Kosaka T, Holmes AJ, Rogers AM, Cappuzzo F, Mok T, Lee C, Johnson BE, Cantley LC, Jänne PA. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 33.Huang Q, Schneeberger VE, Luetteke N, Jin C, Afzal R, Budzevich MM, Makanji RJ, Martinez GV, Shen T, Zhao L, Fung KM, Haura EB, Coppola D, Wu J. Preclinical modeling of KIF5BRET fusion lung adenocarcinoma. Mol Cancer Ther. 2016;15:2521–2529. doi: 10.1158/1535-7163.MCT-16-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song M, Kim SH, Yoon SK. Cabozantinib for the treatment of non-small cell lung cancer with KIF5B-RET fusion. An example of swift repositioning. Arch Pharm Res. 2015;38:2120–2123. doi: 10.1007/s12272-015-0660-1. [DOI] [PubMed] [Google Scholar]
- 35.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 36.Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, Hunter T, Brognard J, Newton AC. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]