

Abstract
In this study, we conducted a genome-wide scan of single nucleotide polymorphisms (SNPs) to identify coding variants that is associated with the risk of glioblastoma (GBM), the most common and most malignant subtype of glioma. We genotyped 1038 GBM cases and 1008 controls in a Chinese Han population using Illumina HumanExome Beadchip v1.0. A missense variant, rs8957 (E[GAG]233D[GAU], SLC2A4RG, 20q13.33), was found being associated with GBM risk, with an odd ratio (OR) of 1.43 (95% confidence interval (CI) = 1.25-1.64, P = 1.72E-07). The G>T transversion at rs8957 leading to changes of subcellular localization of SLC2A4RG, possibly due to the impairment of its nuclear export signal or protein folding. Moreover, the amino acid substitution compromised the function of SLC2A4RG as a cancer suppressor by promoting cell growth through de-inhibition of CDK1 in U87 and U251 cell lines. These results suggest SLC2A4RG plays an important role in the etiology of GBM and may be a potential therapeutic target.
Keywords: Glioblastoma (GBM), rs8957 (E[GAG]233D[GAU]), SLC2A4RG, coding variants, cyclin dependent kinase 1 (CDK1)
Introduction
Genome-wide association study (GWAS) have identified 15 common SNPs in 7 loci to be associated glioma risk till recently [1-6] and most of the SNPs were consistently confirmed by each another GWAS or by following replication studies [7,8]. These findings advanced our understandings of glioma etiology. However, common SNPs identified by GWAS altogether only accounted for a fraction of glioma heritability, leaving the majority unexplained [9-11]. Furthermore, all of these variants map to noncoding intergenic and intronic regions [12] leaving their functional nature a great challenging to assess. Coding variants, on the other hand, affecting either protein sequence, translation rate, or alternative splicing, would possibly influence protein function directly. As a result, they may cause a more severe outcome [13,14].
Glioma is a highly heterogeneous cancer. It can be categorized into different subtypes, which have their distinct molecular profiles [15-17]. GBM, accounted for ~50% of all gliomas, is the most common and most malignant CNS (central nervous system) tumor (WHO grade IV) with a median survival of 12-14 months [9]. The dismal prognosis is partly caused by functional alterations of crucial molecules, which are induced by certain high penetrant mutations including coding variants [9]. However, the role of the coding variant has not yet been systematically investigated. Therefore, in this study we conducted a genome-wide interrogation of the associations of variants, over 50% of which lie in coding regions, with GBM risk. We carried out an exome array-based genotyping using Illumina HumanExome chip v1.0 on 1038 cases and 1008 controls in a Chinese population.
Materials and methods
Study population
Cases were pathologically confirmed GBM patients, newly diagnosed and previously untreated (no tumor resection, chemotherapy, or radiotherapy) who were consecutively recruited from the Department of Neurosurgery at Huashan Hospital (Shanghai, China) from March 2005 to August 2011. Those who reported spinal gliomas, previous cancer history and metastasized cancer from other organs were excluded. In total, we recruited 1038 case subjects. Detailed recruitment criteria were defined previously [11,18,19].
The 1008 control subjects were recruited from four districts and counties in Shanghai between April and November in 2010. The detailed recruit criteria was described elsewhere [20,21]. Briefly, all controls had no known central nervous system-related diseases, self-reported history of any cancer or history of radiotherapy/chemotherapy at the time of recruitment. All participants were genetically unrelated ethnic Han Chinese, most of whom are from the southern part of China. Written informed consents were given by all participants. This study was approved by the Ethics Board of School of Life Sciences, Fudan University.
Genotyping and quality control
Genomic DNA were extracted from blood samples and were genotyped using the Illumina HumanExome Beadchip v1.0 (N = 247,870 variants). Detailed description about SNP contents and selection strategies for this array is available at http://genome.sph.umich.edu/wiki/Exome_Chip_Design. Illumina’s GenTrain version 2.0 clustering algorithm within GenomeStudio v2011.1 was used for genotyping calling.
For quality control (QC), we first excluded samples with < 97% genotype call rate (No = 29 for cases, No = 2 for controls), evidence of duplicates, contamination or genetic relationship determined by identity-by-decent (No = 43 for cases, No = 33 for controls), population outliers determined by identity-by-state based on common SNPs (minor allele frequency [MAF] >0.05) (No = 0). Next, we excluded monomorphic variants (No = 199,530), variants with a genotype call rate < 0.97 (No = 803), MAF < 0.01 (With a relatively small sample size, we might not have sufficient power to detect the associations of rare variants with GBM risk. No = 16,568), located in X and Y chromosomes (No = 76), deviation from Hardy-Weinberg equilibrium (HWE) (P < 1.0E-4, No = 74). See Supplementary Figure 1 for detailed QC step.
Cell culture and transfection
Hela cell line, and two Human GBM cell lines, U87 and U251 were obtained from the Japanese Cancer Research Resources Bank. All cell lines were cultured in DMEM with 10% fetal bovine serum (FBS) and penicillin (100 units/ml)/streptomycin (100 μg/ml) (Life Technologies), at 37°C in 5% CO2. Detailed transfection procedures were described previously [22]. Briefly, plasmids were introduced into appropriate cells using Lipofectamine 2000 (Invitrogen) according to manufacturer’s instructions.
Plasmid construction
The cDNA of SLC2A4RG (RefSeq NG_046992.1) containing rs8957[G] allele was amplified from U251 cell line by PCR. Site-specific mutation was performed on the resulting plasmid (designated p-G, wild type SLC2A4RG) to generate the p-T construct (mutated SLC2A4RG), which contains the rs8957[T] allele. Both plasmids were confirmed by DNA sequencing analyses. All plasmid construction primers are shown in Supplementary Table 3.
Cell proliferation assay and cell cycle analysis
Transfected cell lines were seeded in 96-well plates (10000 cells/well) in sextuple and cell proliferation analysis were evaluated by Cell Counting Kit-8 (CCK-8; Dojindo Laboratories) assay at different time points according to the manufacturer’s instructions. Cell cycle analysis was analyzed as described previously [23]. Briefly, glioma cells were harvested, washed with 1 × PBS and fixed with 70% ice-cold ethanol. Before being analyzed by flow cytometry, fixed cells were incubated with propidium iodide (PI, 25 μg/ml) containing 10 mg/mL RNase for 30 min in the dark.
All assays were performed in three independent experiments.
Quantitative real-time RT-PCR and western blot
Total RNA was isolated from U87 and U251 cell lines using Trizol reagent (Invitrogen) according to manufacturer’s instructions and converted to cDNA using ReverTra Ace (Toyobo). Quantitative RT-PCR was performed in triplicate using SYBR-Green dye (Toyobo) on ABI 7900HT Sequence Detection System (Applied Biosystems). Melting curve analysis was used to check the specificity of amplification. Actin was used as the internal reference gene. Relative genes expression level of each sample was calculated using the 2-ΔΔCT method. Total proteins were extracted by radio-immunoprecipitation assay (RIPA) buffer containing protease inhibitors cocktail (Sigma). Equal amounts of proteins were separated on 8% or 10% SDS-PAGE and were electrotransferred onto PVDF membranes (Millipore). After blocking with 5% nonfat milk for 1 hour at room temperature, membranes blotted with primary antibodies at 4°C overnight. Next, membranes incubated with horseradish peroxidase-labeled anti-rabbit IgG or anti-mouse IgM as the secondary antibody (Epitomics; 1:1000). Antibody against actin (1:3000; Vazyme) was used as a loading control. Antibodies against CDK1, CDK2, E2F1, Myc and Actin were purchased from Proteintech (1:1000).
Immunofluorescence assay
To investigate the subcellular localization of SLC2A4RG, Hela cells were transiently transfected with p-G or p-T expression plasmids and were fixed with 4% paraformaldehyde 36 hours after transfection. Then cells were analyzed for subcellular localization of EGFP fusion protein with a LSM700 confocal microscope (Carl Zeiss).
Statistical analysis
For each of the 30,819 SNPs, we performed unconditional logistic regression analysis as implemented in PLINK v. 1.07 assuming an additive genetic model with adjustment for sex, age and the first 4 principal components. Bonferroni correction was employed to control for multiple testing issues. Therefore, the significant level for this study was 1.62 × 10-6. To avoid performing a large number of underpowered tests due to relative small sample size, we excluded all variants with MAF < 0.01 from further analyses in QC step. The power to detect associations between variants and GBM risk under an additive model was estimated using the Quanto software (http://biostats.usc.edu/software). Assuming alpha level of 10-6 (2-sided), sample size of 1000 cases and 1000 controls, we can have an 88% power to detect an odds ratio (OR) of 4.0, for variants with a MAF of 0.01 and a 99% power to detect an OR of 2.4, for variants with a MAF of 0.05 (Supplementary Table 1).
Pairwise Multi-dimensional scaling (MDS) analysis implemented in PLINK was used to calculate the 1-10 principal components derived from common variants. All biochemical experiments were performed in triplicate with means and standard error subjected to Student’ t test for multivariate analyses.
Results
GWAS for GBM risk
After QC, a total of 30819 SNPs with a minor allele frequency (MAF) >0.01 in 878 cases and 973 controls remained for further analyses, with an average genotype call rate of 99.4% for cases and 99.8% for controls, respectively. Over 50% of the SNPs located in coding region (Supplementary Table 2). MDS analysis indicated that the cases and controls were genetically well matched for all samples (Supplementary Figure 2). The genomic inflation factor (λGC) was 1.04, indicating that there was a low possibility of false positive associations resulting from population stratification (Supplementary Figure 3).
Six variants exceeded the predefined Bonferroni-corrected significance threshold as shown in Table 1. Besides the well-established GWAS common SNP rs2853676, we identified one novel low-frequency coding variants (rs331537) and four common SNPs that were associated with GBM risk. Of the four common SNPs, three were intronic SNPs (rs2315008, rs4809330 and rs12325410) and one, rs8957, lied in the exonic region of SLC2A4RG.
Table 1.
Top SNPs of exome chip-wide analyses survived in Bonferroni correction
| SNP | Location (nearest Gene) | Context | Position | Alleles Minor major | MAF | P | OR (95% CI) | ||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Cases | Controls | Logistic regression | Bonferroni corrected | ||||||
| rs331537 | 11p15.4 (OR52K2) | Exon | 4471276 | A/G | 0.013 | 0.049 | 3.94E-08 | 1.22E-03 | 0.26 (0.16-0.41) |
| rs2315008 | 20q13.3 (ZGPAT) | Intron | 62343956 | C/A | 0.428 | 0.342 | 2.99E-07 | 9.22E-03 | 1.44 (1.26-1.65) |
| rs4809330 | 20q13.3 (ZGPAT) | Intron | 62349586 | G/A | 0.427 | 0.342 | 3.91E-07 | 0.012 | 1.44 (1.26-1.64) |
| rs12325410 | 16p13.2 (LOC101927026) | Intron | 9675246 | A/C | 0.518 | 0.434 | 4.64E-07 | 0.014 | 1.40 (1.23-1.60) |
| rs2853676a | 5p15.33 (TERT) | Intron | 1288547 | A/G | 0.228 | 0.163 | 5.16E-07 | 0.016 | 1.52 (1.29-1.79) |
| rs8957 | 22q13.33 | Exon | 62373707 | G/T | 0.436 | 0.352 | 1.72E-07 | 0.005 | 1.42 (1.25-1.64) |
reported by previous GWAS.
The logistic P value and OR, 95CI were adjusted for age, sex and first 4 principal components.
Among all the SNPs, rs331537 showed the strongest association (P = 3.94 × 10-8, OR = 0.26, 95% CI 0.16-0.41). Although we have >99% power to detect such an association signal, this is a protective low-frequent mutation with the minor allele A decreasing GBM risk by 74% compared with the major allele G (OR = 0.26, 95% CI [0.16-0.41]), which goes against the hypothesis that rare/low frequent mutation are tend to be deleterious. Therefore, rs331537 and other rare/low frequent mutation with GBM risk needs further validation in larger populations. We then focused on the genomic region encompassing rs8957 (E[GAG]233D[GAU]), 20q13.33 (RTEL1), for further deep and refined analysis.
Deep and refined analysis at 22q13.33 (RTEL1)
There are 28 SNPs at 22q13.33 (RTEL1), detailed information and their association results with GBM risk are presented in Table 2. Two GWAS variants, rs6010620 and rs3809324 were in moderate LD (pairwise D’ = 0.96, r2 = 0.30), and their associations with GBM were confirmed in our dataset (P = 3.40E-07 for rs6010620, P = 2.02E-03 for rs3809324). Conditioned on rs6010620, the association of rs4809324 was not significant any more (Pconditional = 0.765). While conditioned on rs4809324, the association of rs6010620 remains significant (Pconditional = 7.18E-05), suggesting rs4809324 was actually a tag for GBM risk in Chinese population and rs6010620 was causal or causal variant was in strong LD with rs6010620. Hereinafter, we conducted conditional analysis on rs6010620 for variants encompassing RTEL1. The association of two novel variants, rs2315008 and rs4809330, exceeded the genome-wide significance threshold (P = 3.79E-08 and 4.77E-08, respectively). They are in strong-to-complete LD (pair-wise D’ = 0.99, r2 = 0.98). In conditional analysis, association significances reduced to 9.12E-03 and 0.011 for rs2315008 and rs4809330, respectively, while the association of rs6010620 was no more significant (Pconditional = 0.161 and 0.139). Rs6010620 was in moderate-to-high LD with rs2315008 and rs4809330 (D’ = 0.89, r2 = 0.55 for both pairs). These results indicated the GWAS variants identified in Caucasian population were tag signals in Chinese population. Rs8957 and rs6010620 were in moderate-to-high LD (pair-wise D’ = 0.83, r2 = 0.46). When conditioned on rs6010620, the association signal of rs8957 remained significant (Pconditional = 0.011).
Table 2.
Results of association analyses at 20q13.33 (RTEL1) before and after conditioning on rs6010620 or rs4809324
| SNP | Position | SNP Type | Minor/Major Allele | MAF | Unadjusted | Conditional analysis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Conditioned on rs6010620 | D’ | r2 | Association Pattern | |||||||||
|
| ||||||||||||
| Cases | Controls | P | OR (95% CI) | Conditional P | P for rs6010620 | |||||||
| rs3827022 | 62195220 | Missense | G/A | 0.420 | 0.412 | 0.647 | 1.03 (0.91-1.17) | 0.300 | 4.87E-07 | 0.36 | 0.08 | |
| rs114497943 | 62195426 | Missense | G/C | 0.0054 | 0.0005 | 4.33E-03 | 10.89 (1.39-85.15) | 0.013 | 1.86E-07 | NA | NA | ↓pair |
| rs115211910 | 62195617 | Missense | A/G | 0.0016 | 0.0010 | 0.586 | 1.64 (0.27-9.80) | 0.588 | 5.24E-07 | NA | NA | |
| rs3810487 | 62196033 | Missense | G/A | 0.465 | 0.475 | 0.531 | 0.96 (0.85-1.09) | 0.031 | 5.46E-08 | 0.38 | 0.07 | |
| rs117986816 | 62196096 | Missense | A/G | 0.011 | 0.012 | 0.958 | 0.98 (0.54-1.78) | 0.469 | 3.09E-07 | 0.74 | 0.02 | |
| rs310631 | 62196253 | Missense | A/C | 0.143 | 0.148 | 0.616 | 0.95 (0.80-1.14) | 0.892 | 1.24E-06 | 0.27 | 0.01 | |
| rs201951872 | 62196600 | Missense | C/A | 0.0011 | 0.0036 | 0.114 | 0.3 (0.06-1.46) | 0.200 | 1.29E-06 | 1.00 | 0.00 | |
| rs310632 | 62196807 | Missense | A/G | 0.0005 | 0.0005 | 0.951 | 1.09 (0.07-17.46) | 0.971 | 5.80E-07 | 0.00 | ||
| rs200473388 | 62200017 | Missense | A/G | 0.0044 | 0.0104 | 0.030 | 0.42 (0.18-0.94) | 0.012 | 4.40E-07 | 0.86 | 0.01 | ↑pair |
| rs6090457 | 62200576 | Missense | G/A | 0.442 | 0.448 | 0.690 | 0.97 (0.86-1.11) | 0.052 | 1.19E-07 | 0.38 | 0.08 | |
| rs2039369 | 62253821 | Intron | G/A | 0.417 | 0.416 | 0.980 | 1.00 (0.88-1.14) | 0.803 | 6.02E-07 | 0.07 | 0.00 | |
| rs3848668 | 62293272 | Missense | G/A | 0.0027 | 0.0005 | 0.081 | 5.47 (0.64-46.89) | 0.202 | 1.12E-06 | NA | NA | |
| rs199685200 | 62294237 | Missense | G/A | 0.0016 | 0.0005 | 0.276 | 3.28 (0.34-31.56) | 0.359 | 7.10E-07 | NA | NA | |
| rs6010620a | 62309839 | Intron | G/A | 0.340 | 0.265 | 3.40E-07 | 1.43 (1.25-1.64) | NA | NA | |||
| rs4809324b | 62318220 | Intron | G/A | 0.140 | 0.107 | 2.02E-03 | 1.35 (1.12-1.64) | 0.765 | 7.18E-05 | 0.96 | 0.30 | |
| rs141717966 | 62324183 | Missense | A/G | 0.0016 | 0.0035 | 0.259 | 0.47 (0.12-1.81) | 0.143 | 3.68E-07 | 1.00 | 0.01 | |
| rs3208008 | 62326110 | Missense | C/A | 0.406 | 0.334 | 3.75E-06 | 1.36 (1.20-1.55) | 0.232 | 0.026 | 0.90 | 0.59 | |
| rs115303435 | 62326159 | Missense | A/G | 0.047 | 0.028 | 2.03E-03 | 1.71 (1.21-2.42) | 0.059 | 1.25E-05 | 0.85 | 0.07 | |
| rs137950052 | 62329672 | Missense | A/G | 0.0005 | 0.0005 | 0.951 | 1.09 (0.07-17.46) | 0.832 | 4.57E-07 | NA | NA | |
| rs2315008 | 62343956 | Intron | C/A | 0.426 | 0.340 | 3.79E-08 | 1.44 (1.27-1.64) | 9.12E-03 | 0.161 | 0.89 | 0.55 | |
| rs4809330 | 62349586 | Intron | G/A | 0.425 | 0.340 | 4.77E-08 | 1.44 (1.26-1.64) | 0.011 | 0.139 | 0.89 | 0.55 | |
| rs199550529 | 62364653 | Missense | A/C | 0.0044 | 0.0119 | 0.011 | 0.37 (0.17-0.83) | 4.12E-03 | 8.17E-07 | 0.94 | 0.02 | ↑pair |
| rs200051356 | 62366916 | Missense | A/G | 0.0005 | 0.0005 | 0.950 | 1.09 (0.07-17.47) | 0.833 | 6.08E-07 | NA | NA | |
| rs116980580 | 62372798 | Synonymous | A/G | 0.0016 | 0.0040 | 0.172 | 0.41 (0.11-1.54) | 0.104 | 3.66E-07 | 1.00 | 0.01 | |
| rs8957 | 62373707 | Missense | A/C | 0.436 | 0.352 | 9.58E-08 | 1.43 (1.25-1.63) | 0.011 | 0.032 | 0.83 | 0.46 | ↓pair |
| rs932826 | 62380516 | Intron | A/C | 0.322 | 0.279 | 3.78E-03 | 1.23 (1.07-1.41) | 0.177 | 1.24E-05 | 0.31 | 0.10 | |
| rs2281929 | 62422080 | Missense | G/A | 0.407 | 0.446 | 0.013 | 0.85 (0.75-0.9) | 0.356 | 8.43E-06 | 0.54 | 0.09 | |
| rs2281534 | 62492922 | Missense | T/A | 0.131 | 0.134 | 0.784 | 0.97 (0.81-1.17) | 0.287 | 3.30E-07 | 0.26 | 0.02 | |
SNP reported by published GWAS;
SNP reported by published GWAS;
NA, MAF too small to calculate accurate D’ or r2; 3, down arrow indicates a SNP pair for which significance decreases after conditioning on the other SNP.
Three rare variants were also detected for association with GBM risk in RETL1 coding region, rs114497943, rs200473388 and rs199550529, each with a MAF less than 0.01. The small sample size of our study and the rareness of these variants limited statistical power for firm conclusions.
Therefore, based on the association strength, the significance after conditioning on rs6010620 and the potential functional plausibility, we hereinafter focused on rs8957 for further functional analysis.
G>T at SLC2A4RG-rs8957 E[GAG]233D[GAU] alters the distribution pattern of SLC2A4RG
SLC2A4RG is a transcriptional activator shuttling between nucleus and cytoplasm [24]. And the localization of nucleus-cytoplasm shuttling proteins play a key role in tumorigenesis [25,26]. To investigate the role of SLC2A4RG in GBM cells, we first examined whether the G>T transversion at rs8957 could change the subcellular localization of SLC2A4RG. To this end, we generated N terminal fusion of SLC2A4RG-rs8957-G and SLC2A4RG-rs8957-T to N2-EGFP (p-G and p-T). Immunofluorescence staining demonstrates that both fusion proteins localize in cytoplasm and in nuclei and the majority is nuclear localization (Figure 1A), which is consistent with published findings that SLC2A4RG acts as a transcriptional factor [24,27,28]. Furthermore, SCL2A4RG occasionally aggregated in foci within nuclei in cells transfected with p-G, whereas the number of nuclear foci was significantly increased in cells transfected with p-T (Figure 1B).
Figure 1.
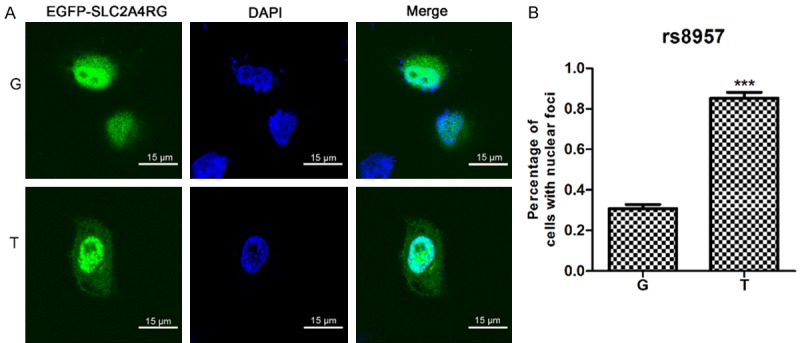
G>T transition at rs8957 E[GAG]233D[GAU] alters the subcellular distribution pattern of SLC2A4RG. (A)Subcellular localization of SLC2A4RG-rs8957-EGFP polymorphism expressed transiently in HeLa cells. Scale bars, 15 μm. (B) Percentage of cells with nuclear speckles after Hela cells were transiently transfected with expression plasmids of SLC2A4RG-rs8957-G-EGFP and SLC2A4RG-rs8957-T-EGFP. Values are means ± SD. All images were obtained at a magnification of × 630 (A). *, p < 0.05; **, p < 0.01, ***, p < 0.001.
G>T at SLC2A4RG-rs8957 E[GAG]233D[GAU] increases GBM cell proliferation rate
According to gene expression data of The Cancer Genome Atlas (TCGA), SLC2A4RG was upregulated in GBM specimens. Hence, we postulate that SLC2A4RG may influence GBM cell growth. Moreover, we further evaluated the changes of cell proliferation upon amino acid substitution of G>T at rs8957. We found that overexpression of p-G and p-T in both U87 and U251 cell lines markedly inhibited cell growth compared with the empty vector controls (Figure 2A). This result indicates that SLC2A4RG may function as a tumor suppressor gene in GBM etiology. In addition, we then studied the impact of G>T at rs8957 on GBM cell proliferation using Cell Counting Kit 8 assay within a 4-day period monitoring. The results showed that cells transfected with p-T significantly promoted the U87 and U251 cells proliferation compared with cells transfected with p-G at day 3 and day 4 after plating, which suggested that the transversion at rs8957 compromised the tumor suppressor role of SLC2A4RG in GBM (Figure 2A).
Figure 2.
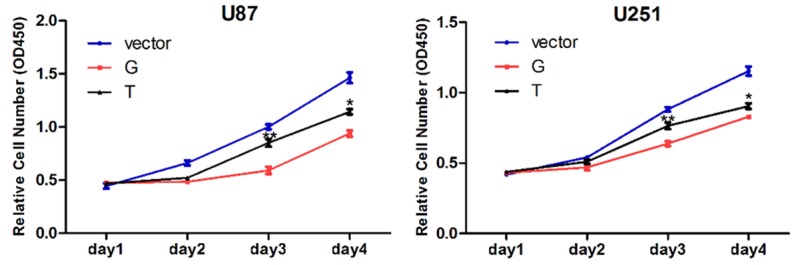
G>T at SLC2A4RG-rs8957 E[GAG]233D[GAU] increases GBM cell proliferation rate. The cell growth curves of U87 and U251 cells with empty vector, SLC2A4RG-rs8957-G-EGFP (p-G) and SLC2A4RG-rs8957-T-EGFP (p-T) overexpression were determined by CCK-8 assays. Values are means ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
SLC2A4RG regulates cell cycle progression in GBM cell lines
To gain further insight of SLC2A4RG-regulated glioma cell proliferation, we next examined whether proliferation changes were associated with specific cell cycle distribution using flow cytometric analyses. When compared with cells transfected with wild type p-G, both U87 and U251 cells lines showed dramatic increase in the percentage of S phase and decrease in G2/M phase after overexpression of p-T (Figure 3A). To further explore this finding, we measured the expression levels of several important cell cycle related genes and proteins by qRT-PCR and western blot in U87 cells. While the expression levels of CDK2, E2F1 and Myc had no significant changes, CDK1, which plays vital roles in both regulating the progression of the cell cycle and oncogenesis [29,30], is dramatically down-regulated in cells with SLC2A4RG overexpression (Figure 3B and 3C). However, the overexpression of the mutated SLC2A4RG partly failed to suppress the expression of CDK1, which may explain their higher cell proliferation rates (Figure 3B and 3C).
Figure 3.
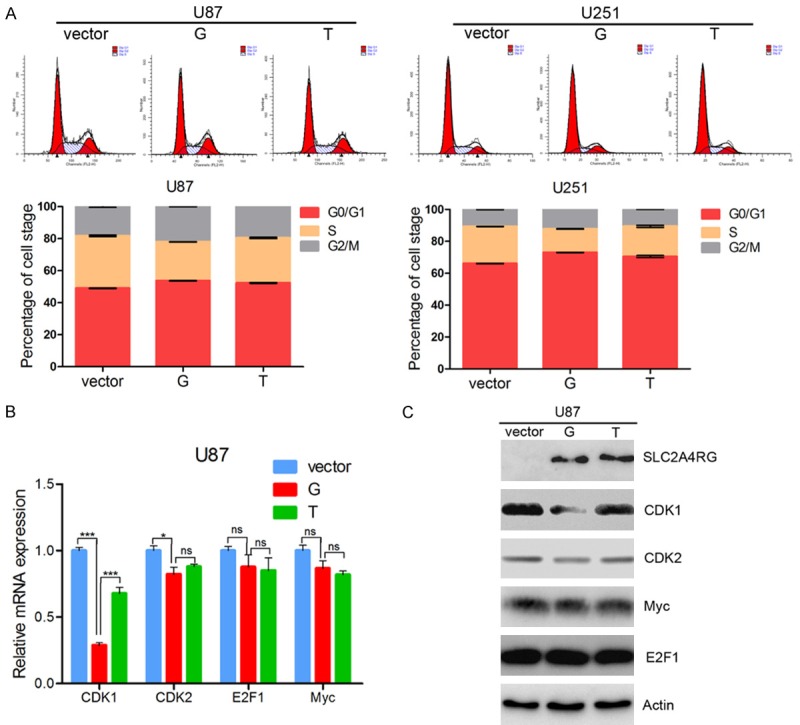
SLC2A4RG down-regulates CDK1 in GBM cell lines. (A) The cell cycle distribution of U87 and U251 glioma cells after empty vector, p-G and p-T overexpression. The level of cell cycle-related genes (CDK1, CDK2, E2F1 and Myc) were determined by q-PCR (B) and western blot (C) analyses. Actin was used as an endogenous control. Values are means ± SD. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Discussion
To investigate the contributions of coding variants to GBM susceptibility, we evaluated 30819 variants across the genome for their associations with GBM risk. A novel common missense SNP, rs8957, encoding E(GAG)233D(GAU) of SLC2A4RG at 20q13.33 was identified at genome-wide significance level. Subsequent functional study demonstrate the G>T transversion at rs8957 changed the subcellular localization of SLC2A4RG. Moreover, the amino acid substitution compromised the tumor suppressor role of SLC2A4RG in GBM, which resulted in a higher cell proliferation rate probably due to de-inhibition of CDK1 expression.
SLC2A4RG encodes a nuclear transcription factor involved in the activation of the solute carrier family 2 member 4 gene, whereas its involvement in tumorigenesis was rarely studied and understood. CDK1 is a cyclin-dependent kinase, which plays a vital role in the transition phases of the cell cycle [31]. In the present study, we found that overexpression of SLC2A4RG could downregulate CDK1 expression and inhibit GBM cell growth. Besides, recent studies reveal that the up-regulation of CDK1 is associated with the development of various cancer including GBM [32-34], while it keeps a low detectable level in non-cancerous tissues. Therefore, we postulate that SLC2A4RG functions as a tumor suppressor in GBM etiology and as is illustrated here in this study, dysfunction of SLC2A4RG caused by missense mutation could increase the risk of GBM. Moreover, we found the missense mutation at rs8957 caused subcellular distribution alteration of SLC2A4RG, resulting in excess accumulation of unphysiological SLC2A4RG in nuclear. We implicated that it was because the E(GAG)233D(GAU) damaged the nuclei exporting signal and folding of SLC2A4RG, which led to nuclear accumulation of unphysiological SLC2A4RG. And it was possibly the nuclear accumulation of unphysiological SLC2A4RG that abated the suppression of CDK1 expression.
In this study, we used the exome chip launched by Illumina as the research tool. In fact, this chip was the earliest version and was not designed specifically for Chinese population. Therefore, coding variants represented in Chinese population may not be adequately captured. Moreover, common coding variants only constitute a fraction of this chip, most of which are low-frequent and rare coding variants. Taken together, it may be not an ideal tool to investigate the associations between common coding variants and GBM risk, highlighting the needs for an exhaustive interrogation of coding variants in larger samples through whole exome sequencing. However, taken the cost into consideration, the exome chip is still a valuable interim.
Glioma is highly heterogeneous, encompassing a wide spectrum of subtypes (astrocytoma, oligodendroglioma, mixed oliogoastrocytoma, ependymoma and GBM) [35]. Studies have suggested different subgroups of glioma may represent distinct pathological entities [15-17,36]. In this study, we focused on GBM, the most common and most malignant subtype of glioma and recruited 1038 GBM cases and 1008 controls in a Chinese Han population for the initial genome-wide association stage. GBM accounted for approximately 40% of all malignant brains tumors, which rank 10th in malignant tumor incidences (5.03 per 100,000 individuals) according to Cancer registration center in China, 2011 [37]. Taken the relative rareness into consideration, it is a great effort to recruit such a large sample. Moreover, it is the largest in Chinese population.
To the best of our knowledge, this is the first genome-wide interrogation of the coding variant for GBM risk. We have identified a novel functional missense variant, rs8957, E(GAG)233D(GAU), located in the exon region of SLC2A4RG as genetic risk factor affecting GBM tumorigenesis. Functional study indicates this missense mutation caused the subcellular localization alterations of SLC2A4RG and diminished its tumor suppressor role via abolishing the downregulation of CDK1.
Acknowledgements
We would like to thank all participates recruited in this study. This study was supported by the National Natural Science Funds of China (81372706, 81372235 and 81071739).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Sanson M, Hosking FJ, Shete S, Zelenika D, Dobbins SE, Ma Y, Enciso-Mora V, Idbaih A, Delattre JY, Hoang-Xuan K, Marie Y, Boisselier B, Carpentier C, Wang XW, Di Stefano AL, Labussière M, Gousias K, Schramm J, Boland A, Lechner D, Gut I, Armstrong G, Liu Y, Yu R, Lau C, Di Bernardo MC, Robertson LB, Muir K, Hepworth S, Swerdlow A, Schoemaker MJ, Wichmann HE, Müller M, Schreiber S, Franke A, Moebus S, Eisele L, Försti A, Hemminki K, Lathrop M, Bondy M, Houlston RS, Simon M. Chromosome 7p11.2 (EGFR) variation influences glioma risk. Hum Mol Genet. 2011;20:2897–2904. doi: 10.1093/hmg/ddr192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang TH, Kon M, Hung JH, DeLisi C. Combinations of newly confirmed glioma-associated loci link regions on chromosomes 1 and 9 to increased disease risk. BMC Med Genomics. 2011;4:4–63. doi: 10.1186/1755-8794-4-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kinnersley B, Labussiere M, Holroyd A, Di Stefano AL, Broderick P, Vijayakrishnan J, Mokhtari K, Delattre JY, Gousias K, Schramm J, Schoemaker MJ, Fleming SJ, Herms S, Heilmann S, Schreiber S, Wichmann HE, Noethen MM, Swerdlow A, Lathrop M, Simon M, Bondy M, Sanson M, Houlston RS. Genome-wide association study identifies multiple susceptibility loci for glioma. Nat Commun. 2015;6:8559. doi: 10.1038/ncomms9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajaraman P, Melin BS, Wang Z, McKean-Cowdin R, Michaud DS, Wang SS, Bondy M, Houlston R, Jenkins RB, Wrensch M, Yeager M, Ahlbom A, Albanes D, Andersson U, Freeman LE, Buring JE, Butler MA, Braganza M, Carreon T, Feychting M, Fleming SJ, Gapstur SM, Gaziano JM, Giles GG, Hallmans G, Henriksson R, Hoffman-Bolton J, Inskip PD, Johansen C, Kitahara CM, Lathrop M, Liu C, Le Marchand L, Linet MS, Lonn S, Peters U, Purdue MP, Rothman N, Ruder AM, Sanson M, Sesso HD, Severi G, Shu XO, Simon M, Stampfer M, Stevens VL, Visvanathan K, White E, Wolk A, Zeleniuch-Jacquotte A, Zheng W, Decker P, Enciso-Mora V, Fridley B, Gao YT, Kosel M, Lachance DH, Lau C, Rice T, Swerdlow A, Wiemels JL, Wiencke JK, Shete S, Xiang YB, Xiao Y, Hoover RN, Fraumeni JF Jr, Chatterjee N, Hartge P, Chanock SJ. Genome-wide association study of glioma and meta-analysis. Hum Genet. 2012;131:1877–1888. doi: 10.1007/s00439-012-1212-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wrensch M, Jenkins RB, Chang JS, Yeh RF, Xiao Y, Decker PA, Ballman KV, Berger M, Buckner JC, Chang S, Giannini C, Halder C, Kollmeyer TM, Kosel ML, LaChance DH, McCoy L, O’Neill BP, Patoka J, Pico AR, Prados M, Quesenberry C, Rice T, Rynearson AL, Smirnov I, Tihan T, Wiemels J, Yang P, Wiencke JK. Variants in the CDKN2B and RTEL1 regions are associated with high-grade glioma susceptibility. Nat Genet. 2009;41:905–908. doi: 10.1038/ng.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shete S, Hosking FJ, Robertson LB, Dobbins SE, Sanson M, Malmer B, Simon M, Marie Y, Boisselier B, Delattre JY, Hoang-Xuan K, El Hallani S, Idbaih A, Zelenika D, Andersson U, Henriksson R, Bergenheim AT, Feychting M, Lonn S, Ahlbom A, Schramm J, Linnebank M, Hemminki K, Kumar R, Hepworth SJ, Price A, Armstrong G, Liu Y, Gu X, Yu R, Lau C, Schoemaker M, Muir K, Swerdlow A, Lathrop M, Bondy M, Houlston RS. Genome-wide association study identifies five susceptibility loci for glioma. Nat Genet. 2009;41:899–904. doi: 10.1038/ng.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh KM, Anderson E, Hansen HM, Decker PA, Kosel ML, Kollmeyer T, Rice T, Zheng S, Xiao Y, Chang JS, McCoy LS, Bracci PM, Wiemels JL, Pico AR, Smirnov I, Lachance DH, Sicotte H, Eckel-Passow JE, Wiencke JK, Jenkins RB, Wrensch MR. Analysis of 60 reported glioma risk SNPs replicates published GWAS findings but fails to replicate associations from published candidate-gene studies. Genet Epidemiol. 2013;37:222–228. doi: 10.1002/gepi.21707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Chen Y, Zhao Y, Fan W, Zhou K, Liu Y, Zhou L, Mao Y, Wei Q, Xu J, Lu D. Association of sequence variants on chromosomes 20, 11, and 5 (20q13.33, 11q23.3, and 5p15.33) with glioma susceptibility in a Chinese population. Am J Epidemiol. 2011;173:915–922. doi: 10.1093/aje/kwq457. [DOI] [PubMed] [Google Scholar]
- 9.Wirsching HG, Weller M. The role of molecular diagnostics in the management of patients with gliomas. Curr Treat Options Oncol. 2016;17:17–51. doi: 10.1007/s11864-016-0430-4. [DOI] [PubMed] [Google Scholar]
- 10.Ostrom QT, Bauchet L, Davis FG, Deltour I, Fisher JL, Langer CE, Pekmezci M, Schwartzbaum JA, Turner MC, Walsh KM, Wrensch MR, Barnholtz-Sloan JS. The epidemiology of glioma in adults: a “state of the science” review. Neuro Oncol. 2014;16:896–913. doi: 10.1093/neuonc/nou087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Zhou K, Zhang H, Shugart YY, Chen L, Xu Z, Zhong Y, Liu H, Jin L, Wei Q, Huang F, Lu D, Zhou L. Polymorphisms of LIG4 and XRCC4 involved in the NHEJ pathway interact to modify risk of glioma. Hum Mutat. 2008;29:381–389. doi: 10.1002/humu.20645. [DOI] [PubMed] [Google Scholar]
- 12.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:1001–1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang QY. Genetic study of complex diseases in the oost-GWAS Era. J Genet Genomics. 2015;42:87–98. doi: 10.1016/j.jgg.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X, Bailey SD, Lupien M. Laying a solid foundation for Manhattan-‘setting the functional basis for the post-GWAS era’. Trends Genet. 2014;30:140–149. doi: 10.1016/j.tig.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mistry M, Zhukova N, Merico D, Rakopoulos P, Krishnatry R, Shago M, Stavropoulos J, Pole J, Ray P, Remke M, Buczkowicz P, Ramaswamy V, Shlien A, Rutka J, Dirks P, Taylor M, Malkin D, Bouffet E, Hawkins C, Tabori U. Braf mutation and Cdkn2a deletion define a clinically distinct subgroup of childhood secondary highgrade glioma. Neuro Oncology. 2015;17:18–18. doi: 10.1200/JCO.2014.58.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckel-Passow JE, Kollmeyer TM, Sarkar G, Chada A, Decker PA, Kosel ML, Caron AA, Sicotte H, Nandakumar K, Prodduturi N, O’Neill BP, Lachance DH, Jenkins RB. The association of glioma germline risk SNPs with mutationbased molecular subgroups. Cancer Research. 2014;74 [Google Scholar]
- 17.Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K Cancer Genome Atlas Research Network. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Zhang H, Zhou K, Chen L, Xu Z, Zhong Y, Liu H, Li R, Shugart YY, Wei Q, Jin L, Huang F, Lu D, Zhou L. Tagging SNPs in non-homologous end-joining pathway genes and risk of glioma. Carcinogenesis. 2007;28:1906–1913. doi: 10.1093/carcin/bgm073. [DOI] [PubMed] [Google Scholar]
- 19.Zhou K, Liu Y, Zhang H, Liu H, Fan W, Zhong Y, Xu Z, Jin L, Wei Q, Huang F, Lu D, Zhou L. XRCC3 haplotypes and risk of gliomas in a Chinese population: a hospital-based case-control study. Int J Cancer. 2009;124:2948–2953. doi: 10.1002/ijc.24307. [DOI] [PubMed] [Google Scholar]
- 20.Xu J, Mo Z, Ye D, Wang M, Liu F, Jin G, Xu C, Wang X, Shao Q, Chen Z, Tao Z, Qi J, Zhou F, Wang Z, Fu Y, He D, Wei Q, Guo J, Wu D, Gao X, Yuan J, Wang G, Xu Y, Wang G, Yao H, Dong P, Jiao Y, Shen M, Yang J, Ou-Yang J, Jiang H, Zhu Y, Ren S, Zhang Z, Yin C, Gao X, Dai B, Hu Z, Yang Y, Wu Q, Chen H, Peng P, Zheng Y, Zheng X, Xiang Y, Long J, Gong J, Na R, Lin X, Yu H, Wang Z, Tao S, Feng J, Sun J, Liu W, Hsing A, Rao J, Ding Q, Wiklund F, Gronberg H, Shu XO, Zheng W, Shen H, Jin L, Shi R, Lu D, Zhang X, Sun J, Zheng SL, Sun Y. Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nature Genetics. 2012;44:1231–1235. doi: 10.1038/ng.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang P, Ye D, Guo J, Liu F, Jiang H, Gong J, Gu C, Shao Q, Sun J, Zheng SL, Yu H, Lin X, Xia G, Fang Z, Zhu Y, Ding Q, Xu J. Genetic score of multiple risk-associated single nucleotide polymorphisms is a marker for genetic susceptibility to bladder cancer. Genes Chromosomes Cancer. 2014;53:98–105. doi: 10.1002/gcc.22121. [DOI] [PubMed] [Google Scholar]
- 22.Zhang S, Chen H, Zhao X, Cao J, Tong J, Lu J, Wu W, Shen H, Wei Q, Lu D. REV3L 3’UTR 460 T>C polymorphism in microRNA target sites contributes to lung cancer susceptibility. Oncogene. 2013;32:242–250. doi: 10.1038/onc.2012.32. [DOI] [PubMed] [Google Scholar]
- 23.Li X, Wan X, Chen H, Yang S, Liu Y, Mo W, Meng D, Du W, Huang Y, Wu H, Wang J, Li T, Li Y. Identification of miR-133b and RB1CC1 as independent predictors for biochemical recurrence and potential therapeutic targets for prostate cancer. Clinical Cancer Research. 2014;20:2312–2325. doi: 10.1158/1078-0432.CCR-13-1588. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka K, Shouguchi-Miyata J, Miyamoto N, Ikeda JE. Novel nuclear shuttle proteins, HDBP1 and HDBP2, bind to neuronal cell-specific cis-regulatory element in the promoter for the human Huntington’s disease gene. J Biol Chem. 2004;279:7275–7286. doi: 10.1074/jbc.M310726200. [DOI] [PubMed] [Google Scholar]
- 25.Wang SC, Hung MC. Cytoplasmic/nuclear shuttling and tumor progression. In: ElDeiry WS, editors. Tumor progression and therapeutic resistance. Ann N Y Acad Sci. 2005;1059:11–5. doi: 10.1196/annals.1339.002. [DOI] [PubMed] [Google Scholar]
- 26.Chu CT, Plowey ED, Wang Y, Patel V, Jordan-Sciutto KL. Location, location, location: altered transcription factor trafficking in neurodegeneration. J Neuropathol Exp Neurol. 2007;66:873–883. doi: 10.1097/nen.0b013e318156a3d7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oshel KM, Knight JB, Cao KT, Thai MV, Olson AL. Identification of a 30-base pair regulatory element and novel DNA binding protein that regulates the human GLUT4 promoter in transgenic mice. J Biol Chem. 2000;275:23666–23673. doi: 10.1074/jbc.M001452200. [DOI] [PubMed] [Google Scholar]
- 28.Knight JB, Eyster CA, Griesel BA, Olson AL. Regulation of the human GLUT4 gene promoter: interaction between a transcriptional activator and myocyte enhancer factor 2A. Proc Natl Acad Sci U S A. 2003;100:14725–14730. doi: 10.1073/pnas.2432756100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paruthiyil S, Cvoro A, Tagliaferri M, Cohen I, Shtivelman E, Leitman DC. Estrogen receptor beta causes a G2 cell cycle arrest by inhibiting CDK1 activity through the regulation of cyclin B1, GADD45A, and BTG2. Breast Cancer Res Treat. 2011;129:777–784. doi: 10.1007/s10549-010-1273-5. [DOI] [PubMed] [Google Scholar]
- 30.Fan XL, Chen JJ. Role of Cdk1 in DNA damage-induced G1 checkpoint abrogation by the human papillomavirus E7 oncogene. Cell Cycle. 2014;13:3249–3259. doi: 10.4161/15384101.2014.953879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmitt E, Beauchemin M, Bertrand R. Nuclear colocalization and interaction between bcl-xL and cdk1(cdc2) duringG(2)/M cell-cycle checkpoint. Oncogene. 2007;26:5851–5865. doi: 10.1038/sj.onc.1210396. [DOI] [PubMed] [Google Scholar]
- 32.Liu WT, Chen C, Lu IC, Kuo SC, Lee KH, Chen TL, Song TS, Lu YL, Gean PW, Hour MJ. MJ-66 induces malignant glioma cells G2/M phase arrest and mitotic catastrophe through regulation of cyclin B1/Cdk1 complexE. Neuropharmacology. 2014;86:219–227. doi: 10.1016/j.neuropharm.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu WT, Chen C, Lu IC, Kuo SC, Lee KH, Chen TL, Song TS, Lu YL, Gean PW, Hour MJ. MJ-66 induces malignant glioma cells G2/M phase arrest and mitotic catastrophe through regulation of cyclin B1/Cdk1 complex. Neuropharmacology. 2014;86:219–227. doi: 10.1016/j.neuropharm.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen H, Huang Q, Zhai DZ, Dong J, Wang AD, Lan Q. [CDK1 expression and effects of CDK1 silencing on the malignant phenotype of glioma cells] . Zhonghua Zhong Liu Za Zhi. 2007;29:484–488. [PubMed] [Google Scholar]
- 35.Bigner DD. Biology of gliomas-potential clinical implications of glioma cellular heterogeneity. Neurosurgery. 1981;9:320–326. [PubMed] [Google Scholar]
- 36.You G, Sha ZY, Yan W, Zhang W, Wang YZ, Li SW, Sang L, Wang Z, Li GL, Li SW, Song YJ, Kang CS, Jiang T. Seizure characteristics and outcomes in 508 Chinese adult patients undergoing primary resection of low-grade gliomas: a clinicopathological study. Neuro Oncology. 2012;14:230–241. doi: 10.1093/neuonc/nor205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen W, Zheng R, Zeng H, Zhang S, He J. Annual report on status of cancer in China, 2011. Chin J Cancer Res. 2015;27:2–12. doi: 10.3978/j.issn.1000-9604.2015.01.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.